

Abstract
Endothelial cells release prostacyclin (PGI2) and nitric oxide (NO) to inhibit platelet functions. PGI2 and NO effects are mediated by cyclic nucleotides, cAMP- and cGMP-dependent protein kinases (PKA, PKG), and largely unknown PKA and PKG substrate proteins. The small G-protein Rac1 plays a key role in platelets and was suggested to be a target of cyclic nucleotide signaling. We confirm that PKA and PKG activation reduces Rac1-GTP levels. Screening for potential mediators of this effect resulted in the identification of the Rac1-specific GTPase-activating protein ARHGAP17 and the guanine nucleotide exchange factor ARHGEF6 as new PKA and PKG substrates in platelets. We mapped the PKA/PKG phosphorylation sites to serine 702 on ARHGAP17 using Phos-tag gels and to serine 684 on ARHGEF6. We show that ARHGAP17 binds to the actin-regulating CIP4 protein in platelets and that Ser-702 phosphorylation interferes with this interaction. Reduced CIP4 binding results in enhanced inhibition of cell migration by ARHGAP17. Furthermore, we show that ARHGEF6 is constitutively linked to GIT1, a GAP of Arf family small G proteins, and that ARHGEF6 phosphorylation enables binding of the 14-3-3 adaptor protein to the ARHGEF6/GIT1 complex. PKA and PKG induced rearrangement of ARHGAP17- and ARHGEF6-associated protein complexes might contribute to Rac1 regulation and platelet inhibition.
Keywords: cyclic AMP (cAMP), cyclic GMP (cGMP), phosphorylation, platelet, Rac (Rac GTPase)
Introduction
As key components of the hemostatic system platelets rapidly seal the sites of vascular injury to prevent blood loss by forming a localized thrombus. To avoid vessel occlusion endothelial cells restrict thrombus formation by releasing prostacyclin (PGI2)2 and nitric oxide (NO), two powerful endogenous inhibitors of platelet activation (1, 2). In diseased vessels, where endothelial function is compromised, platelet activation can lead to arterial thrombosis and subsequently to life-threatening myocardial infarction or ischemic stroke, which collectively account for more than 25% of all deaths worldwide (3). The essential role of endothelium in preventing thrombosis is apparent in patients undergoing percutaneous coronary intervention and stent placement where a defective endothelial layer results in increased rates of late stent thrombosis (4). Endothelial PGI2 and NO keep platelets in a non-adherent state by elevating the levels of intracellular cAMP and cGMP which leads to the activation of cAMP- and cGMP-dependent protein kinases (PKA, PKG), respectively (5). In turn, PKA and PKG phosphorylate serine and threonine residues in a diverse set of substrate proteins to regulate their function. In general, a newly added phosphate group can either alter protein conformation or its interaction potential which can functionally translate to a change in protein activity, subcellular localization or stability (6). Although the list of potential PKA substrates in platelets is growing (7), the fundamental question of how phosphorylated substrates mechanistically inhibit specific aspects of platelet activation remains unanswered. Detailed information is only available for a handful of substrates (5).
Cyclic nucleotide pathways have been suggested to inhibit Rac1 activation in platelets (8, 9). Rac1 belongs to the Rho family of small G-proteins and operates as a molecular switch by cycling between an inactive, GDP-bound, and an active, GTP-bound form. Collagen, fibrinogen, von Willebrand factor (VWF), thrombin, ADP and thromboxane A2 are all able to activate Rac1 which integrates these distinct platelet activation pathways and drives downstream signaling events (for review see Ref. 10). Rac1 fulfils critical roles in platelet function: mouse platelets lacking Rac1 are unable to form lamellipodia on multiple surfaces (11) and display major defects in platelet activation downstream of GPVI and GPIb (12, 13). Rac1 is essential for in vivo thrombus formation as evidenced in three different thrombosis models (11, 13, 14). Little is known about the mechanisms of Rac1 regulation in platelets. In general, small G-proteins of the Rho family are controlled by GTPase-activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs). Rho family GAPs accelerate GTP hydrolysis by Rac1, thus interrupting the interaction with their effectors and terminating signaling. Rho family GEFs facilitate the dissociation of GDP and rebinding of GTP, thereby activating Rac1.
In this study we provide evidence that PKA and PKG phosphorylate the Rho family GAP ARHGAP17 (also called Nadrin or Rich1) and the Rho family GEF ARHGEF6 (α-PIX, Cool-2) resulting in a reorganization of signaling complexes involving CIP4 and 14-3-3 proteins.
Experimental Procedures
Antibodies
The following antibodies were used in this study: mouse anti-Rac1 clone 23A8 (05–389, Merck Millipore), rabbit anti-ARHGAP17 (ab74454, Abcam), mouse anti-CIP4 clone 21/CIP4 (612556, BD Biosciences), mouse anti-GIT1 clone 13/GIT1 (611396, BD Biosciences), rabbit anti-Cool2/αPIX antibody (C23D2, Cell Signaling Technology), mouse anti-14-3-3 gamma clone 3F8 (ab14118, Abcam), mouse anti-c-Myc clone 9E10 (sc-40, Santa Cruz Biotechnology), mouse anti-HA clone 16B12 (mms-101r, BioLegend), mouse anti-FLAG clone M2 (F3165, Sigma-Aldrich). Horseradish peroxidase-coupled donkey anti-mouse IgG (715–035-150, Jackson ImmunoResearch Europe), donkey anti-rabbit IgG (711–035-152, Jackson ImmunoResearch Europe) antibodies were used as secondary antibodies for Western blot analysis visualized by enhanced chemiluminescence method. For Western blot analysis visualized by the Odyssey imaging system, IRDye 680LT goat anti-mouse IgG (926–68020, LI-COR) and IRDye 800CW goat anti-rabbit IgG (926-32211, LI-COR) secondary antibodies were used.
DNA Constructs
Human HA-Rac1 construct was obtained from the Missouri S&T cDNA Resource Center. Human ARHGAP17 (IRATp970D04105D, Source BioScience) was expressed without a tag in pcDNA4/TO (Invitrogen). FLAG-ARHGAP17 and ARHGAP17-EGFP were obtained through sub-cloning into pCMV-3Tag-1A (Agilent Technologies) and EGFP-N1 (Takara Clontech) respectively using EcoRI/BamHI restriction sites. Rabbit NHERF1 was kindly provided by Mark Donowitz (15). GST-NHERF1 was generated using pGEX-4T3 (GE Healthcare) and BamHI/EcoRI restriction sites. The human CIP4 (isoform 2)-GST was from DNASU Plasmid Repository (clone HsCD00078923 in pANTT7_cGST). Human Myc-ARHGEF6 was kindly provided by Richard Cerione (16). GST-14-3-3 γ was described previously (17). Site-directed mutagenesis was performed by PCR amplification using mutagenic primer pairs, Pfu DNA polymerase (Fermentas), followed by digestion with DpnI restriction enzyme (Fermentas) and transformation into One Shot TOP10 bacteria (Thermo Fisher Scientific). Constructs were verified by DNA sequencing.
Cell Preparation and Lysis
HEK293T cells were cultured using in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C and 5% CO2. Cells were transiently transfected using PolyJet in vitro DNA transfection reagent (SignaGen Laboratories) according to manufacturer's protocol. 24 h after transfection, HEK293T cells were washed twice with ice-cold Dulbecco's phosphate-buffered saline, or with Tris-buffered saline in case of sample preparation for Phos-tag gels.
Blood was obtained from healthy volunteers who gave their informed consent according to the Declaration of Helsinki. Ethical approval (LS-08-13-Smolenski) was granted by The Human Research Ethics Committee of University College Dublin. Venous blood was collected into 20% CCD-EGTA buffer (100 mm tri-sodium citrate, 7 mm citric acid, 140 mm glucose, 15 mm EGTA). The tube was gently mixed and immediately centrifuged at 180 × g, room temperature for 15 min without brake. Platelet-rich plasma (PRP) was collected then platelets were pelleted at 600 × g, room temperature for 10 min. After resuspension in pre-warmed resuspension buffer (145 mm NaCl, 5 mm KCl, 1 mm MgCl2, 10 mm HEPES, 10 mm glucose, pH 7.4) platelets were incubated at 37 °C for at least 25 min and up to 1 h before stimulation. Platelets were incubated at 37 °C with thrombin (Roche) at 0.1 units/ml for 30 s or 1 min, collagen (Bio/Data Corporation) at 95 μg/ml for 1 min, forskolin (Sigma) at 10 μm for 10 min, prostaglandin E1 (PGE1, Sigma) at 0.5 μm for 1 min, sodium nitroprusside (SNP, Sigma) at 10 μm for 10 min, cAMP analog 5,6-dichloro-1-b-d-ribofuranosylbenzimidazole-3′, 5′-cyclic monophosphorothioate, Sp-isomer (Sp-5,6-DCI-cBIMPS, Biolog) at 300 μm for 30 min or cGMP analog 8-(4-chlorophenylthio) guanosine-3′,5′-cyclic monophosphate (8-pCPT-cGMP, Biolog) at 300 μm for 30 min.
HEK293T cells and platelets were lysed in buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm MgCl, 1% Triton X-100 supplemented with Complete Mini protease inhibitor mixture and PhosSTOP phosphatase inhibitor mixture tablets (Roche). For Rac1-GTP assays a lysis buffer containing 1% (v/v) Nonidet P-40, 10% (v/v) glycerol, 50 mm Tris-HCl, pH 7.4, 200 mm NaCl, 2.5 mm MgCl2 supplemented with protease and phosphatase inhibitors was used. To remove insoluble material, lysates were centrifuged at 13000 × g at 4 °C for 10 min.
Protein Purification, Pull-Down Experiment, and Immunoprecipitation
For pull-down experiments recombinant GST-PAKI-CRIB, GST-NHERF1, GST-14-3-3, and CIP4-GST fusion proteins were expressed in BL21 (DE3) Escherichia coli and purified using glutathione-Sepharose 4B suspension (GE Healthcare). 5 μl of beads saturated with GST proteins were added to cell lysates and incubated overnight, or, in case of GST-PAKI-CRIB, for 50 min at 4 °C.
For immunoprecipitations tagged cDNA constructs were expressed in HEK293T cells, cells were lysed and tagged proteins were immunoprecipitated by adding 5 μl of ANTI-FLAG M2 affinity gel for 3 h or overnight, 6 μl of GFP-Trap_A (Chromotek) for 1 h, or 10 μl of anti-Myc antibody overnight followed by 5 μl of protein A/G plus agarose for 1 h at 4 °C. Endogenous GIT1 was immunoprecipitated using 40 μl of polyclonal GIT1 antibody. Precipitates were washed, boiled in loading buffer, and analyzed by SDS-PAGE and Western blotting.
Phos-tag SDS-PAGE
Phos-tag gels were prepared as described in the manufacturer's protocol. Briefly, 25 μm Phos-tag acrylamide (Wako Chemicals) and 50 μm ZnCl2 were added to the separating gel solution (350 mm Bis-Tris-HCl, pH 6.8, 7% polyacrylamide) prior to polymerization. Samples were prepared without EDTA in lysis buffer and subjected to a Zn2+-Phos-tag SDS-PAGE protocol under neutral pH conditions as described (18). Electrophoresis was performed at constant 30 mA/gel (running buffer: 100 mm Tris, 100 mm MOPS, 0.1% SDS and 5 mm sodium bisulfite). To remove Zn2+ ions before Western blotting, gels were incubated in transfer buffer containing 1 mm EDTA for 2 times 10 min, followed by a wash step in transfer buffer without EDTA for 20 min. Western blotting was performed using wet-tank transfer and PVDF membrane.
Radioactive Phosphate Incorporation Assay
Immunoprecipitated FLAG-ARHGAP17 was incubated with the catalytic subunit of PKA in the presence of 50 mm Tris/HCl pH 7.5, 10 mm MgCl2 and 50 μm cold ATP mixed with 1 μCi [γ-32P]ATP at 30 °C for 4 min. The reaction was stopped by addition of 3× SDS-sample buffer, subjected to SDS-PAGE and blotted onto membrane. Radiolabeled proteins were detected by autoradiography whereas total protein levels were determined by Western blotting with anti-FLAG.
Mass Spectrometry
For the identification of ARHGAP17 binding proteins a novel quantitative interaction proteomic protocol was applied which combines immunoprecipitation, in-solution digestion and label-free quantification mass spectrometry as described (19).
For the identification of proteins in Coomassie Blue-stained gels in gel digestion was performed as described (20). The trypsin digested samples were analyzed using a Thermo Scientific Q Exactive mass spectrometer connected to a Dionex Ultimate 3000 (RSLCnano) chromatography system. The raw data were de novo sequenced and searched against the Human subset of the Uniprot/Swissprot database using the search engine PEAKS Studio 7 (Bioinformatics Solutions Inc.).
Migration Assay
HeLa cells were seeded into both wells of culture-inserts (80209, ibidi) placed on 35 mm microscopy dishes (80136, ibidi), and transfected using Polyjet reagent (SignaGen Laboratories). On the following day the culture-inserts were removed, and cells were allowed to migrate into the cell-free open area between the wells. Images were taken at defined time-points using a Olympus X41 microscope with tablet camera and UPlanFLN 4×/0.13Php objective. The open cell-free area was determined using NIH Image software, and data were evaluated using Prism6 software (GraphPad).
Results
Previous studies have indicated that cyclic nucleotide pathways inhibit Rac1 activation in platelets. Prostacyclin was shown to block thromboxane A2 (8), and thrombin (9)-mediated Rac1 activation, and cyclic nucleotide analogs were shown to inhibit thromboxane A2-induced Rac1 activation (8). To confirm and expand these data we measured Rac1-GTP levels by pull-down assay in thrombin and collagen-treated platelets. PGE1 (agonist of the prostacyclin receptor) and SNP (NO donor), both inhibited thrombin induced Rac1 activation (Fig. 1A). These effects were mimicked by membrane-permeable cyclic nucleotide analogs (Fig. 1B) pointing toward a role for PKA and PKG as mediators of PGE1 and SNP effects. We show for the first time that collagen-induced Rac1 activation is also blocked by PGE1 and SNP (Fig. 1C).
FIGURE 1.

Cyclic nucleotide signaling inhibits thrombin and collagen induced Rac1 activation. A, effects of PGE1 and SNP on thrombin-induced Rac1 activation. Washed human platelets were pre-incubated without or with PGE1 (0.5 μm, 1 min) or SNP (10 μm, 10 min) followed by thrombin (0.1 unit/ml, 1 min). Platelets were lysed, and pull-down assays were performed using GST-PAKI-PBD followed by Western blotting. Quantitative data on Rac1-GTP (upper panel) and total Rac1 (lower panel) levels were obtained using an Odyssey Classic scanner. Below the panels the ratios of active versus total Rac1 normalized to the untreated controls are shown for three independent experiments (n1–3). B, effects of cAMP and cGMP analogues on thrombin-induced Rac1 activation. Platelets were incubated with Sp-5,6-DCl-cBIMPS (cAMP analogue, 300 μm, 30 min) or 8-pCPT-cGMP (cGMP analogue, 300 μm, 30 min) followed by thrombin (0.1 units/ml, 30 s). Rac1-GTP and total Rac1 levels were determined as described in A, and Rac1-GTP/total Rac1 ratios are shown below the panels. C, effects of PGE1 and SNP on collagen-induced Rac1 activation. Platelets were incubated with PGE1 (0.5 μm, 1 min) or with SNP (10 μm, 10 min) followed by collagen (95 μg/ml, 1 min). Rac1 levels were analyzed as described in A.
ARHGAP17 Is a New Substrate of PKA and PKG in Platelets
Since Rac1 does not contain any consensus site for PKA or PKG phosphorylation (-R-R/K-X-S/T-, according to (21)) in its amino acid sequence, we hypothesized that phosphorylation of a regulatory GAP or GEF protein could be involved in cyclic nucleotide effects on Rac1, as described for the small G-protein Rap1 in platelets previously (22–25). Screening of quantitative platelet proteome data (26) revealed that at least 22 Rho family GAPs and 24 GEFs are expressed in human platelets. We scanned the GAPs for consensus sites for PKA/PKG phosphorylation, which had also been detected by mass spectrometry. The initial list of candidate GAPs included ARHGAP4, ARHGAP15, ARHGAP17, ARHGAP21, ARHGAP35, BCR, HMHA1, and MYO9B. We excluded GAPs that had been shown not to regulate Rac1, as well as BCR, because of its complex structure (containing GAP, GEF, and kinase domains), resulting in a final list of ARHGAP4, ARHGAP15, ARHGAP17, and HMHA1. We expressed these candidate proteins in HEK293T cells and performed radioactive phosphate incorporation assays using purified catalytic subunit of PKA in vitro (Fig. 2A and data not shown). All proteins except HMHA1 could be phosphorylated in vitro. To test phosphorylation by endogenous PKA in intact cells we expressed candidate proteins in HEK293T cells and treated the cells with forskolin, an activator of adenylyl cyclase. Phosphorylation was detected by phosphate-affinity electrophoresis (Phos-tag SDS-PAGE) and Western blotting similarly as described (17, 27). The Phos-tag compound permits efficient separation of phosphorylated and non-phosphorylated proteins on SDS-PAGE resulting in a band shift. Using an improved protocol with Zn2+-Phos-tag complex and neutral pH conditions (18) a clear shift was observed in the mobility of ARHGAP17 in response to forskolin treatment (Fig. 2B) compared with the non-treated sample. No difference was detected in the phosphorylation pattern of ARHGAP4, ARHGAP15, or HMHA1 (data not shown). We concluded that transfected ARHGAP17 can be phosphorylated by endogenous PKA in intact cells.
FIGURE 2.
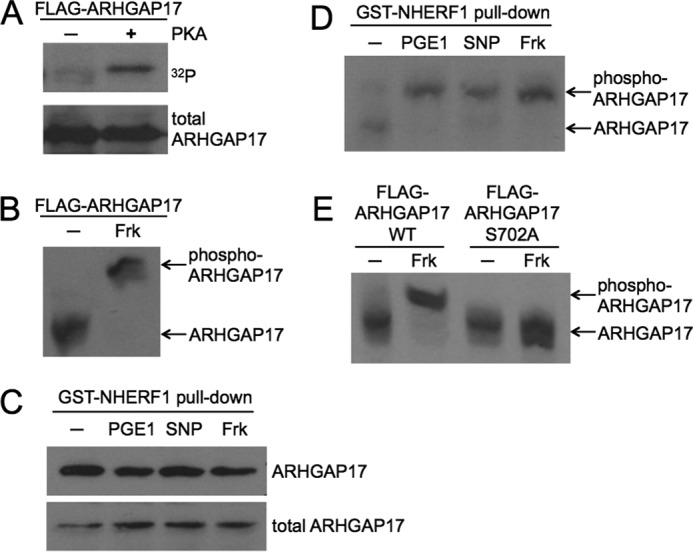
The GTPase-activating protein ARHGAP17 is a target of cyclic nucleotide signaling. A, ARHGAP17 is phosphorylated by PKA in vitro. FLAG-ARHGAP17 was expressed in HEK293T cells, immunoprecipitated using anti-FLAG beads, and incubated with purified catalytic subunit of PKA in the presence of [γ-32P]ATP for 4 min. The reaction was stopped by addition of SDS-sample buffer, subjected to SDS-PAGE and blotting. Radiolabeled ARHGAP17 was detected by autoradiography (upper panel) and total ARHGAP17 levels were determined by Western blotting with anti-FLAG antibody (lower panel). Data are representative of three independent experiments. B, ARHGAP17 is phosphorylated by PKA in intact cells. HEK293T cells expressing FLAG-ARHGAP17 were treated without or with forskolin (Frk, 10 μm, 10 min) to activate endogenous PKA and lysed. Samples were subjected to Zn2+-Phos-tag SDS-PAGE as described under “Experimental Procedures” followed by Western blotting using an anti-FLAG antibody. The shifted, phosphorylated form of ARHGAP17 is indicated. Data are representative of three independent experiments. C, NHERF1 precipitates endogenous ARHGAP17 from platelet lysates. Washed human platelets were incubated with PGE1 (0.5 μm, 1 min), SNP (10 μm, 10 min), or forskolin (10 μm, 15 min). Platelets were lysed and purified GST-NHERF1 was used to pull down endogenous ARHGAP17. Samples were analyzed by Western blotting using anti-ARHGAP17 antibody to determine precipitated (upper panel) and total ARHGAP17 levels (lower panel). Data are representative of two independent experiments. D, endogenous ARHGAP17 is phosphorylated by endogenous PKA and PKG in platelets. Washed human platelets were incubated with PGE1 (0.5 μm, 1 min) or forskolin (10 μm, 15 min) to activate PKA, or with the NO-donor SNP (10 μm, 10 min) to activate PKG, lysed, and GST-NHERF1 pull-down assays were performed to precipitate ARHGAP17 as described in C. Samples were subjected to Zn2+-Phos-tag SDS-PAGE and Western blotting as described in B and endogenous ARHGAP17 was detected by anti-ARHGAP17 antibody. The shifted, phosphorylated form of ARHGAP17 is indicated. Data are representative of three independent experiments. E, Ser-702 is the PKA phosphorylation site on ARHGAP17. HEK293T cells expressing wild type FLAG-ARHGAP17 or mutant FLAG-ARHGAP17-S702A were incubated without or with forskolin (10 μm, 10 min) and lysed. Samples were subjected to Zn2+-Phos-tag SDS-PAGE followed by Western blotting using an anti-FLAG antibody. The shifted, phosphorylated form of ARHGAP17 is indicated. Data are representative of two independent experiments.
To analyze endogenous ARHGAP17 protein in platelets we first confirmed the expression of ARHGAP17 in platelets by Western blotting. Various commercial ARHGAP17 antibodies showed a band of about 120 kDa (Fig. 2C, lower panel) confirming previous protein (26, 28) and mRNA data (29). The detected band most likely corresponds to isoform 1 of UniProt number Q68EM7 (calculated mass of 95.4 kDa). We did not find any evidence for additional isoforms of ARHGAP17 in total lysates or in NHERF1 pull-downs (see below). Initial attempts at using Phos-tag gels to study endogenous ARHGAP17 phosphorylation failed presumably because of low sensitivity of the available antibodies. However, literature data suggested that ARHGAP17 could bind to the first PDZ domain of the adaptor protein NHERF1 (also called EBP50) via its C-terminal PDZ-binding motif (Fig. 3D, (30)). We decided to use this interaction with NHERF1 to enrich ARHGAP17 from platelet lysates prior to Phos-tag gel blotting. Purified GST-NHERF1 fusion protein was indeed able to pull down endogenous ARHGAP17 from human platelets efficiently, and treatment of platelets with PGE1, forskolin, or SNP did not interfere with the interaction of ARHGAP17 with NHERF1 (Fig. 2C, upper panel). On Phos-tag gels a clear shift of the ARHGAP17 band could be detected upon treatment with PGE1, forskolin, and SNP confirming the phosphorylation of endogenous ARHGAP17 by endogenous PKA and PKG in platelets (Fig. 2D). ARHGAP17 contains only one site, serine 702, that matches the consensus sequence for PKA substrates exactly (21). To test if Ser-702 is a phosphorylation site, we generated a point mutant of ARHGAP17 with Ser-702 mutated to alanine. FLAG-ARHGAP17 WT and S702A mutant transfected cells were treated with forskolin and analyzed by Phos-tag SDS-PAGE. Mutation of Ser-702 completely abolished the shift induced by cAMP signaling indicating that Ser-702 is the main PKA phosphorylation site on ARHGAP17 (Fig. 2E).
FIGURE 3.

Phosphorylation of ARHGAP17 by PKA on Ser-702 leads to the dissociation of an ARHGAP17-CIP4 complex. A, ARHGAP17 is a GTPase-activating protein of Rac1. HEK293T cells were transfected with ARHGAP17 (with no tag) and 24 h later endogenous levels of Rac1-GTP were analyzed by pull-down assay followed by SDS-PAGE and Western blotting. Rac1 antibody was used to determine Rac1-GTP (upper panel) and total Rac1 (middle panel) levels and ARHGAP17 antibody was used to determine total ARHGAP17 levels (lower panel). Data are representative of five independent experiments. B, Ser-702 phosphorylation of ARHGAP17 results in the dissociation of CIP4, Toca-1, and PACSIN2 from ARHGAP17 in transfected cells. HEK293T cells expressing EGFP, ARHGAP17-EGFP or ARHGAP17-S702A-EGFP were incubated without or with forskolin (10 μm, 10 min). ARHGAP17 proteins were immunoprecipitated with anti-GFP antibody and co-precipitated proteins were identified by mass spectrometry. Label free quantification (LFQ) intensities of three co-immunoprecipitated proteins CIP4, Toca-1, and PACSIN2 are shown. This experiment was performed once. C, ARHGAP17-S702E mutation mimics phosphorylation-dependent CIP4 dissociation in transfected cells. HEK293T cells expressing EGFP, ARHGAP17-EGFP, ARHGAP17-S702A-EGFP, or ARHGAP17-S702E-EGFP were incubated without or with forskolin (10 μm, 10 min) as indicated. ARHGAP17 proteins were immunoprecipitated with anti-GFP antibody and co-precipitated endogenous CIP4 was analyzed by Western blotting using anti-CIP4 antibody. Anti-ARHGAP17 antibody was used to determine the levels of transfected ARHGAP17. Data are representative of three independent experiments. D, domain composition of ARHGAP17 and CIP4 with the possible interaction site indicated. ARHGAP17 consists of an N-terminal Bin/Amphiphysin/Rvs (N-BAR) domain, a RhoGAP domain, a long proline-rich region with 4 predicted SH3-binding regions and a PDZ-binding motif. Ser-702 is located in the proline-rich region. CIP4 consists of an Fes/CIP4 homology-Bin/Amphiphysin/Rvs (F-BAR) domain and a C-terminal Src homology 3 (SH3) domain. E, endogenous ARHGAP17 dissociates from CIP4 in response to PKA activation in platelets. Washed human platelets were incubated without or with PGE1 (0.5 μm, 1 min). Platelets were lysed and purified GST or CIP4-GST was used to pull-down endogenous ARHGAP17. Samples were analyzed by Western blotting using anti-ARHGAP17 antibody to determine precipitated (upper panel) and total ARHGAP17 levels (lower panel). Data are representative of four independent experiments.
Consequences of ARHGAP17 Phosphorylation
ARHGAP17 has been described to stimulate the GTPase activity of Rac1 (31). To confirm the specificity of ARHGAP17 toward Rac1 we measured endogenous Rac1-GTP levels in cells transfected with ARHGAP17. A clear reduction of Rac1-GTP was observed in ARHGAP17 expressing cells compared with controls (Fig. 3A). To test if ARHGAP17 phosphorylation had a direct effect on GAP activity we expressed wild type, a phosphorylation-deficient mutant (S702A), and a phospho-mimetic mutant (S702E). Expression of ARHGAP17 mutants led to a similar reduction of Rac1-GTP levels compared with wildtype (data not shown). ARHGAP17 has been proposed to be tyrosine phosphorylated after thrombin treatment of mouse platelets (32). To investigate a possible impact of Ser-702 phosphorylation on tyrosine phosphorylation we analyzed ARHGAP17 after GST-NHERF1 pull-down using phospho-tyrosine antibody for blotting. We could confirm tyrosine phosphorylation of ARHGAP17 after treatment of platelets with thrombin, but pre-incubation with activators of cyclic nucleotide pathways had no effect on the levels of tyrosine-phosphorylated ARHGAP17 (data not shown). Ser-702 is located in the middle of a proline-rich region, C-terminal of the catalytic GAP domain of ARHGAP17 (Fig. 3D). This unique 296 residue long sequence contains 82 proline residues and is predicted to be intrinsically disordered (IUPred (33)). Intrinsically disordered regions are often found to mediate protein-protein interactions (34). To identify ARHGAP17 binding partners that might be regulated by Ser-702 phosphorylation we treated cells expressing EGFP-tagged wild-type ARHGAP17 and the S702A mutant with forskolin to stimulate PKA. ARHGAP17 and associated proteins were precipitated using anti-GFP antibody and the precipitates were analyzed by quantitative mass spectrometry as described (19). 172 potential ARHGAP17 interacting proteins were identified, of which 40 changed significantly after forskolin treatment. However, the change of only 3 interaction partners was lost in the S702A mutant, indicating that the binding of these proteins was regulated by Ser-702 phosphorylation (Fig. 3B). These proteins were CIP4 (Cdc42-interacting protein 4 or TRIP10), FNBP1L (Formin-binding protein 1-like or Toca-1) and PACSIN2 (protein kinase C and casein kinase substrate in neurons protein 2 or Syndapin-2). All three exhibit a similar domain organization containing an Fes/CIP4 homology-Bin/Amphiphysin/Rvs (F-BAR) domain and a C-terminal Src homology 3 (SH3) domain. CIP4 has been described to bind to ARHGAP17 via its SH3 domain before and the second polyproline stretch of the C-terminal proline-rich region of ARHGAP17 was identified as SH3 binding site for CIP4 (31). This SH3 binding site (amino acids 708–718 of ARHGAP17) is in close proximity to the Ser-702 phosphorylation site suggesting that Ser-702 phosphorylation might interfere with CIP4 binding. To confirm this finding we tested the interaction of ARHGAP17 and CIP4 by co-immunoprecipitation and Western blotting. Indeed wild-type ARHGAP17 was shown to bind to CIP4, which could be inhibited by activation of PKA (Fig. 3C). Mutation of Ser-702 to the phosphomimetic amino acid glutamic acid led to detachment of CIP4, whereas mutation to non-phosphorylatable alanine abolished this effect. Finally, we confirmed the regulation of CIP4 binding in platelets. Using a GST fusion protein of CIP4 we were able to pull-down endogenous ARHGAP17, and treatment of platelets with PGE1 resulted in an inhibition of the interaction of ARHGAP17 with CIP4 (Fig. 3E). To determine the functional role of the ARHGAP17/CIP4 interaction we performed migration assays in cells expressing either the S702A mutant of ARHGAP17 that binds CIP4 more strongly or the phosphomimetic S702E mutant that binds CIP4 less effectively. Expression of ARHGAP17 inhibited cell migration as expected. However, the S702E mutant inhibited migration more potently compared with the S702A mutant (Fig. 4). These data suggest that phosphorylation of Ser-702 of ARHGAP17 and dissociation of CIP4 might stimulate ARHGAP17 function leading to altered Rac1 dynamics.
FIGURE 4.
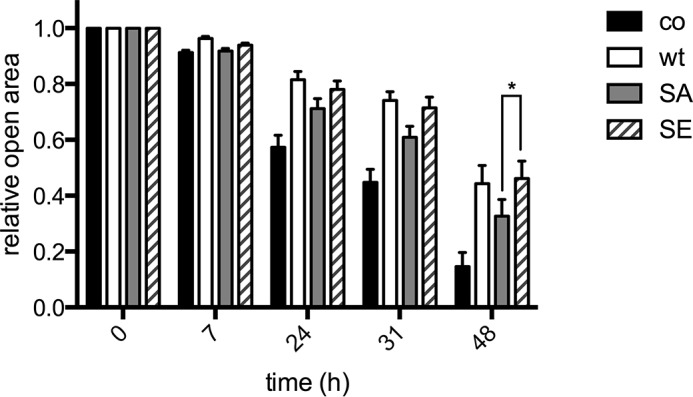
Dissociation of the ARHGAP17-CIP4 complex inhibits cell migration. HeLa cells were seeded onto culture inserts for wound healing assays. After transfection of ARHGAP17-EGFP (wt), ARHGAP17-S702A-EGFP (SA), or ARHGAP17-S702E-EGFP (SE) the inserts were removed resulting in a cell-free gap between cells. The size of this open area was measured by microscopy, and data were normalized relative to the size of the starting area. Shown are means +S.E. of five independent experiments. The asterisk indicates statistical significance between SA and SE mutants of ARHGAP17 (p < 0.05, ANOVA with Bonferroni correction).
ARHGEF6 Is a Substrate of PKA and PKG in Platelets
To identify GEFs that might contribute to the regulation of Rac1 by cyclic nucleotides we screened the 24 Rho family GEFs shown by proteome studies to be expressed in platelets for potential PKA/PKG phosphorylation sites. Only ARHGEF6, ARHGEF7, DOCK7, and DOCK8 emerged as potential candidates. A recently published mass spectrometry screening for PKA substrates provided further evidence that ARHGEF6 might be phosphorylated in platelets (7). To investigate the potential phosphorylation of ARHGEF6 by cyclic nucleotide pathways we expressed ARHGEF6 in HEK293T cells. Standard Western blot analysis revealed a shift of ARHGEF6 after PKA activation by forskolin consistent with ARHGEF6 phosphorylation (Fig. 5A, first two lanes). Analysis of the ARHGEF6 sequence predicted Ser-640, Thr-650, and Ser-684 as PKA phosphorylation sites and a mass spectrometry study provided evidence for phosphorylation of Ser-640 and Ser-684 in platelets (7). Therefore we mutated each of these sites to alanine and analyzed the migration pattern of mutant ARHGEF6 after forskolin treatment in transfected cells. Mutation of Ser-684 resulted in a loss of the PKA induced band shift confirming that Ser-684 is a phosphorylation site (Fig. 5A). To investigate ARHGEF6 phosphorylation in platelets we verified the expression of ARHGEF6 by Western blotting. A clear band of the expected molecular weight of about 85 kDa could be detected in platelet lysates (Fig. 5B, first lane). Treatment of platelets with activators of cyclic nucleotide pathways lead to a similar shift in the migration of ARHGEF6 as observed in transfected cells (Fig. 5B). These data confirm Ser-684 of ARHGEF6 as PKA/PKG phosphorylation site in platelets.
FIGURE 5.

Phosphorylation of the guanine nucleotide exchange factor ARHGEF6 by PKA or PKG on Ser-684 leads to the assembly of a GIT1-ARHGEF6–14-3-3 complex. A, ARHGEF6 is phosphorylated by PKA in intact cells. HEK293T cells expressing Myc-ARHGEF6, Myc-ARHGEF6-S640A, Myc-ARHGEF6-T650A, or Myc-ARHGEF6-S684A were incubated without or with forskolin (10 μm, 10 min). Cells were lysed, and lysates were analyzed by standard Western blotting using anti-Myc antibody. The shifted, phosphorylated form of ARHGEF6 is indicated (p-ARHGEF6). Data are representative of two independent experiments. B, ARHGEF6 is phosphorylated by PKA and PKG in intact platelets. Washed human platelets were incubated without or with PGE1 (0.5 μm, 1 min), SNP (10 μm, 10 min) or forskolin (10 μm, 15 min). Platelets were lysed and samples were analyzed by standard Western blotting using anti-ARHGEF6 antibody. Data are representative of three independent experiments. C, endogenous ARHGEF6 co-immunoprecipitates with GIT1 in platelets. Washed human platelets were incubated without or with PGE1 (0.5 μm, 1 min), lysed, and immunoprecipitated with anti-GIT1 antibody. Co-precipitated endogenous ARHGEF6 was analyzed by Western blotting using anti-ARHGEF6 antibody (middle panel). Anti-GIT1 antibody was used to determine precipitated (upper panel) and total GIT1 levels (lower panel). Data are representative of two independent experiments. D, NHERF1 precipitates endogenous GIT1 from platelet lysates. Washed human platelets were incubated with PGE1 (0.5 μm, 1 min), lysed, and incubated with purified GST-NHERF1 to pull down endogenous GIT1. Samples were analyzed by Western blotting using anti-GIT1 antibody to determine precipitated (upper panel) and total GIT1 levels (lower panel). Data are representative of three independent experiments. E, upstream activators of cyclic nucleotide production induce ARHGEF6 and GIT1 interaction with 14-3-3 in platelets. Washed human platelets were incubated with forskolin (10 μm, 15 min), PGE1 (0.5 μm, 1 min) or with SNP (10 μm, 10 min). Platelets were lysed, and purified GST-14-3-3γ was used to pull down endogenous ARHGEF6 and GIT1. Samples were analyzed by Western blotting using anti-ARHGEF6 and anti-GIT1 antibodies to determine precipitated ARHGEF6 (upper panel) and GIT1 (middle panel) levels, respectively. Anti-14-3-3γ antibody was used as loading control (lower panel). Data are representative of three independent experiments. F, domain composition of ARHGEF6 and GIT1 with the possible 14-3-3 interaction site. ARHGEF6 consists of an N-terminal Calponin-homology (CH) domain, an Src homology 3 (SH3) domain, a RhoGEF domain, and a Pleckstrin homology (PH) domain. Ser-640 and Ser-684 are indicated along with the GIT1 and potential 14-3-3 interaction sites. GIT1 consists of an N-terminal Arf-GAP domain and three ankyrin-repeats (labeled A).
Consequences of ARHGEF6 Phosphorylation
To investigate a potential role of PKA/PKG-mediated ARHGEF6 phosphorylation for ARHGEF6 function we measured Rac1-GTP levels in cells expressing wild-type and phosphorylation site mutants of ARHGEF6 (both Ser-640 and Ser-684 changed to alanine). However, we did not detect any significant effect of ARHGEF6 on global Rac1-GTP levels compared with controls (data not shown), which has been observed before and which might be due to the complex regulation of ARHGEF6 activity including feedback inhibition of ARHGEF6 by Rac1 (35).
Next, we studied if ARHGEF6 phosphorylation would affect interactions with known binding partners. A prominent interaction partner of ARHGEF6 is the adaptor protein GIT1 (G protein-coupled receptor kinase-interactor 1, also called ARF GTPase-activating protein GIT1, or Cool-associated and tyrosine-phosphorylated protein 1, CAT1). GIT1 has been shown to be expressed in platelets before (36, 37). Co-immunoprecipitation experiments confirmed that GIT1 binds to ARHGEF6 in platelets but this interaction was not affected by PKA activation (Fig. 5C). Interestingly, Coomassie staining of GST-NHERF1 pull-downs in platelets revealed a band of about 105 kDa which we identified as GIT1 by mass spectrometry. We confirmed the binding of GIT1 to NHERF1 by Western blotting, and we could show that PKA activation did not affect this interaction (Fig. 5D). Another potential interaction partner for ARHGEF6 is the 14-3-3 adaptor protein. The closely related ARHGEF7 protein has been shown to bind 14-3-3 in a PKA-dependent manner (38) and a screen for 14-3-3 binding partners had identified both, ARHGEF6 and ARHGEF7, as potential 14-3-3-interacting proteins (39). We investigated if ARHGEF6 might bind to 14-3-3 in platelets and performed pull-down assays using GST-14-3-3. In resting platelets no significant binding of ARHGEF6 to 14-3-3 was seen. However, in platelets treated with activators of cyclic nucleotide pathways a strong interaction between ARHGEF6 and 14-3-3 was readily detectable (Fig. 5E). Taken together we conclude that ARHGEF6 and GIT1 form a stable complex in platelets. PKA and PKG activation induce the phosphorylation of ARHGEF6 on Ser-684, and possibly also on Ser-640, resulting in the recruitment of 14-3-3 to the ARHGEF6/GIT1 complex (Fig. 5F).
Discussion
Our findings provide novel insights into the regulation of GAP and GEF proteins of Rac1, which is a central G-protein in many platelet activation pathways. PKA and PKG inhibit Rac1 and phosphorylate the Rac1-specific ARHGAP17 and ARHGEF6 proteins in platelets. We mapped the phosphorylation sites to Ser-702 of ARHGAP17 and Ser-684 of ARHGEF6, and we show that ARHGAP17 phosphorylation leads to the detachment of the interacting protein CIP4, whereas ARHGEF6 phosphorylation induces 14-3-3 binding.
ARHGAP17 is one of 70 Rho family GAPs expressed by the human genome (40). These GAPs contain a conserved GAP domain but are very heterogenous in their remaining protein sequences. ARHGAP17 contains an N-BAR domain which might be involved in membrane targeting and in the regulation of GAP function (28, 41). The C-terminal part of ARHGAP17 contains proline-rich elements that have been shown to mediate protein-protein interactions. We show for the first time that ARHGAP17 binds to CIP4 in platelets. CIP4 is an effector of Rac1 and Cdc42 and coordinates lamellipodia and filopodia formation (31, 42) and recently CIP4 has been described to play a role in proplatelet formation (43). Our data indicate that Ser-702 phosphorylation interferes with CIP4 binding, which might be explained by the proximity of Ser-702 to the CIP4 binding site on ARHGAP17 (41). ARHGAP17 (Nadrin) has recently been studied in mouse platelets suggesting the presence of two major isoforms with different specificities toward Rac1, Cdc42, and RhoA (28). In human platelets we observed the expression of only one ARHGAP17 isoform, which showed activity toward Rac1 in transfected cells. The PKA/PKG phosphorylation site Ser-702 is conserved in mouse ARHGAP17 including the neighboring CIP4 binding region, but overall the C-terminal region of ARHGAP17 is only 65% identical between mouse and human proteins (EMBOSS program, (44)). In human platelets ARHGAP17 phosphorylation and detachment of CIP4 correlated with a decrease in Rac1-GTP levels (Figs. 1A, 2D, and 3E). In transfected cells detachment of CIP4 was associated with enhanced inhibition of cell migration by ARHGAP17. This indicates that CIP4 might modulate ARHGAP17 function and change the spatio-temporal pattern of active Rac1 (45, 46). Both, ARHGAP17 and CIP4, contain BAR domains that could link Rac1 regulation to plasma membrane events (47).
ARHGEF6 is another target of cyclic nucleotide pathways involved in controlling Rac1. We show that PKA and PKG phosphorylate ARHGEF6 on Ser-684 in platelets. ARHGEF6 is known to form a stable complex with the adaptor protein GIT1 (48, 49), and we were able to confirm the presence of the ARHGEF6/GIT1 complex in human platelets. Furthermore, we show that the ARHGEF6/GIT1-complex interacts with 14-3-3 upon stimulation of cyclic nucleotide signaling. This observation matches findings in other cells where PKA was observed to enhance the interaction of 14-3-3 with the related ARHGEF7 protein (38). 14-3-3 binding was suggested to reduce the GEF activity of ARHGEF7 toward Rac1, which would match our findings in platelets where PKA/PKG-induced binding of 14-3-3 to ARHGEF6/GIT1 correlates with decreased Rac1-GTP levels (38). GIT1 also contains a GAP domain that is specific for Arf6 (50). Thus GIT1 could contribute to the suppression of Arf6-GTP levels occurring during platelet activation (51–53). Interestingly, cyclic nucleotide pathways were shown to reverse the suppression of Arf6-GTP (52), which could potentially involve a modulation of the ARHGEF6/GIT1 complex by 14-3-3. The role of ARHGEF6/GIT1 could be even broader through further interaction partners. For example platelets have been shown to express the GIT1 binding proteins Hic-5, a paxillin-related adaptor protein (54), and GRK2, an interactor of G-protein-coupled receptors (55). We have identified NHERF1 as another potential binding partner of GIT1 as well as of ARHGAP17. NHERF1 might link ARHGEF6/GIT1 and ARHGAP17 to the P2Y12 receptor in platelets (56). However, NHERF1 is also an established binding partner of proteins of the ezrin/radixin/moesin family in platelets (57) and NHERF proteins are known to anchor PKA and PKG complexes to membrane proteins in other cells (15, 58). Thus NHERF1 has the potential to coordinate some of the protein interactions described including interactions with membrane receptors.
We conclude that cyclic nucleotide-dependent kinases inhibit Rac1, which probably involves the phosphorylation of multiple substrates including ARHGAP17 and ARHGEF6. Phosphorylation of both, a GAP and a GEF of the same small G-protein simultaneously, points toward a key role for Rac1 in cyclic nucleotide-mediated platelet regulation. The immediate consequence of ARHGAP17 and ARHGEF6 phosphorylation is a rearrangement of associated CIP4 and 14-3-3 proteins, respectively (Fig. 6). Similar findings have been made before regarding the inhibition of the small G-protein Rap1B by cyclic nucleotides in platelets. PKA- and PKG phosphorylate Rap1GAP2, leading to detachment of 14-3-3 (23, 24). At the same time the Rap1 specific CalDAG-GEFI is phosphorylated, although the direct molecular consequence is not known (22, 25). Another example for G-protein regulation by cyclic nucleotide pathways in platelets is RGS18, a GAP of the heterotrimeric G-proteins Gq and Gi. PKA and PKG phosphorylate RGS18 which results in the activation of protein phosphatase 1, the detachment of 14-3-3, and an inhibition of Gq signaling (17, 27). It is also interesting to note that PKA and PKG phosphorylation can lead to detachment of 14-3-3 proteins from targets like Rap1GAP2 and RGS18, whereas phosphorylation of ARHGEF6/GIT1 induces binding of 14-3-3. These data indicate that activation of cyclic nucleotide pathways might trigger a transfer of 14-3-3 proteins from Rap1GAP2 and RGS18 to ARHGEF6/GIT1. Moreover, we show for the first time, that the ability of platelet cyclic nucleotide signaling to control protein-protein interactions is not limited to 14-3-3-based interactions but has the potential to disrupt SH3 domain-based (and potentially other) interactions. Taken together our findings provide first insights into the coordination of multiple cyclic nucleotide induced phosphorylation events that collectively achieve the pronounced platelet inhibition characteristic of PGI2 and NO.
FIGURE 6.

Model of Rac1 inhibition during PKA and PKG activation. Upon platelet activation Rac1-GTP levels rise rapidly and Rac1-GTP is able to interact with its effector molecules to promote downstream signaling. Endothelial NO and PGI2, the two most potent endogenous platelet inhibitors described to date, are able to inhibit agonist induced Rac1-GTP formation. ARHGAP17 is a Rho GTPase-activating protein of Rac1 and is bound to the SH3 domain of CIP4 via its SH3 binding region in resting platelets. Endothelial PGI2 stimulates the activation of PKA and leads to the phosphorylation of Ser-702 in ARHGAP17, which results in the dissociation of the ARHGAP17-CIP4 complex. ARHGEF6 is a Rho guanine nucleotide exchange factor for Rac1 and constitutively bound to GIT1. NO and PGI2 activate PKG and PKA, respectively and both kinases phosphorylate ARHGEF6 on Ser-684 and possibly on Ser-640. Phosphorylation of ARHGEF6 results in the assembly of a GIT1-ARHGEF6–14-3-3 complex. These changes might contribute to PGI2- and NO-mediated Rac1 inhibition.
Author Contributions
Z. N., K. W., A. vK., S. G., and A. S. performed experiments and analyzed data. Z. N. and A. S. designed the research and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dimitri Scholz from the Imaging Core Facility of UCD Conway Institute for valuable help and suggestions. We thank Mark Donowitz for kindly providing the NHERF1 plasmid and Richard Cerione for the ARHGEF6 plasmid.
This work was supported by funding from the Science Foundation Ireland and the Irish Research Council (to A. S.). The authors declare that have no conflict of interest with the contents of this article.
- PGI2
- prostacyclin
- NO
- nitric oxide
- GAP
- GTPase-activating protein
- GEF
- guanine-nucleotide exchange factor
- PKA
- cAMP-dependent protein kinase
- PKG
- cGMP-dependent protein kinase
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1.Moncada S., Gryglewski R., Bunting S., and Vane J. R. (1976) An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature 263, 663–665 [DOI] [PubMed] [Google Scholar]
- 2.Radomski M. W., Palmer R. M., and Moncada S. (1987) Comparative pharmacology of endothelium-derived relaxing factor, nitric oxide and prostacyclin in platelets. Br. J. Pharmacol. 92, 181–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson S. P. (2011) Arterial thrombosis–insidious, unpredictable and deadly. Nat. Med. 17, 1423–1436 [DOI] [PubMed] [Google Scholar]
- 4.Otsuka F., Finn A. V., Yazdani S. K., Nakano M., Kolodgie F. D., and Virmani R. (2012) The importance of the endothelium in atherothrombosis and coronary stenting. Nat. Rev. Cardiol. 9, 439–453 [DOI] [PubMed] [Google Scholar]
- 5.Smolenski A. (2012) Novel roles of cAMP/cGMP-dependent signaling in platelets. J. Thromb. Haemost. 10, 167–176 [DOI] [PubMed] [Google Scholar]
- 6.Hunter T. (2012) Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2513–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beck F., Geiger J., Gambaryan S., Veit J., Vaudel M., Nollau P., Kohlbacher O., Martens L., Walter U., Sickmann A., and Zahedi R. P. (2014) Time-resolved characterization of cAMP/PKA-dependent signaling reveals that platelet inhibition is a concerted process involving multiple signaling pathways. Blood 123, e1-e10 [DOI] [PubMed] [Google Scholar]
- 8.Gratacap M. P., Payrastre B., Nieswandt B., and Offermanns S. (2001) Differential regulation of Rho and Rac through heterotrimeric G-proteins and cyclic nucleotides. J. Biol. Chem. 276, 47906–47913 [DOI] [PubMed] [Google Scholar]
- 9.Soulet C., Gendreau S., Missy K., Benard V., Plantavid M., and Payrastre B. (2001) Characterisation of Rac activation in thrombin- and collagen-stimulated human blood platelets. FEBS Lett. 507, 253–258 [DOI] [PubMed] [Google Scholar]
- 10.Aslan J. E., and McCarty O. J. (2013) Rho GTPases in platelet function. J. Thromb. Haemost. 11, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCarty O. J., Larson M. K., Auger J. M., Kalia N., Atkinson B. T., Pearce A. C., Ruf S., Henderson R. B., Tybulewicz V. L., Machesky L. M., and Watson S. P. (2005) Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J. Biol. Chem. 280, 39474–39484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delaney M. K., Liu J., Zheng Y., Berndt M. C., and Du X. (2012) The role of Rac1 in glycoprotein Ib-IX-mediated signal transduction and integrin activation. Arterioscl. Thromb. Vasc. Biol. 32, 2761–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pleines I., Elvers M., Strehl A., Pozgajova M., Varga-Szabo D., May F., Chrostek-Grashoff A., Brakebusch C., and Nieswandt B. (2009) Rac1 is essential for phospholipase C-γ2 activation in platelets. Pflugers Arch. 457, 1173–1185 [DOI] [PubMed] [Google Scholar]
- 14.Delaney M. K., Liu J., Kim K., Shen B., Stojanovic-Terpo A., Zheng Y., Cho J., and Du X. (2014) Agonist-induced platelet procoagulant activity requires shear and a Rac1-dependent signaling mechanism. Blood 124, 1957–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yun C. H., Oh S., Zizak M., Steplock D., Tsao S., Tse C. M., Weinman E. J., and Donowitz M. (1997) cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc. Natl. Acad. Sci. U.S.A. 94, 3010–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bagrodia S., Bailey D., Lenard Z., Hart M., Guan J. L., Premont R. T., Taylor S. J., and Cerione R. A. (1999) A tyrosine-phosphorylated protein that binds to an important regulatory region on the cool family of p21-activated kinase-binding proteins. J. Biol. Chem. 274, 22393–22400 [DOI] [PubMed] [Google Scholar]
- 17.Gegenbauer K., Elia G., Blanco-Fernandez A., and Smolenski A. (2012) Regulator of G-protein signaling 18 integrates activating and inhibitory signaling in platelets. Blood 119, 3799–3807 [DOI] [PubMed] [Google Scholar]
- 18.Kinoshita E., and Kinoshita-Kikuta E. (2011) Improved Phos-tag SDS-PAGE under neutral pH conditions for advanced protein phosphorylation profiling. Proteomics 11, 319–323 [DOI] [PubMed] [Google Scholar]
- 19.Turriziani B., Garcia-Munoz A., Pilkington R., Raso C., Kolch W., and von Kriegsheim A. (2014) On-beads digestion in conjunction with data-dependent mass spectrometry: a shortcut to quantitative and dynamic interaction proteomics. Biology 3, 320–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shevchenko A., Wilm M., Vorm O., and Mann M. (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 21.Kennelly P. J., and Krebs E. G. (1991) Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem. 266, 15555–15558 [PubMed] [Google Scholar]
- 22.Guidetti G. F., Manganaro D., Consonni A., Canobbio I., Balduini C., and Torti M. (2013) Phosphorylation of the guanine-nucleotide-exchange factor CalDAG-GEFI by protein kinase A regulates Ca+-dependent activation of platelet Rap1b GTPase. Biochem. J. 453, 115–123 [DOI] [PubMed] [Google Scholar]
- 23.Hoffmeister M., Riha P., Neumüller O., Danielewski O., Schultess J., and Smolenski A. P. (2008) Cyclic nucleotide-dependent protein kinases inhibit binding of 14-3-3 to the GTPase-activating protein Rap1GAP2 in platelets. J. Biol. Chem. 283, 2297–2306 [DOI] [PubMed] [Google Scholar]
- 24.Schultess J., Danielewski O., and Smolenski A. P. (2005) Rap1GAP2 is a new GTPase-activating protein of Rap1 expressed in human platelets. Blood 105, 3185–3192 [DOI] [PubMed] [Google Scholar]
- 25.Subramanian H., Zahedi R. P., Sickmann A., Walter U., and Gambaryan S. (2013) Phosphorylation of CalDAG-GEFI by protein kinase A prevents Rap1b activation. J. Thromb. Haemost. 11, 1574–1582 [DOI] [PubMed] [Google Scholar]
- 26.Burkhart J. M., Vaudel M., Gambaryan S., Radau S., Walter U., Martens L., Geiger J., Sickmann A., and Zahedi R. P. (2012) The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 120, e73–e82 [DOI] [PubMed] [Google Scholar]
- 27.Gegenbauer K., Nagy Z., and Smolenski A. (2013) Cyclic nucleotide dependent dephosphorylation of regulator of G-protein signaling 18 in human platelets. PloS one 8, e80251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beck S., Fotinos A., Lang F., Gawaz M., and Elvers M. (2013) Isoform-specific roles of the GTPase activating protein Nadrin in cytoskeletal reorganization of platelets. Cell. Signal. 25, 236–246 [DOI] [PubMed] [Google Scholar]
- 29.Rowley J. W., Oler A. J., Tolley N. D., Hunter B. N., Low E. N., Nix D. A., Yost C. C., Zimmerman G. A., and Weyrich A. S. (2011) Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 118, e101–e111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reczek D., and Bretscher A. (2001) Identification of EPI64, a TBC/rabGAP domain-containing microvillar protein that binds to the first PDZ domain of EBP50 and E3KARP. J. Cell Biol. 153, 191–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richnau N., and Aspenström P. (2001) Rich, a rho GTPase-activating protein domain-containing protein involved in signaling by Cdc42 and Rac1. J. Biol. Chem. 276, 35060–35070 [DOI] [PubMed] [Google Scholar]
- 32.Beck S., Fotinos A., Gawaz M., and Elvers M. (2014) Nadrin GAP activity is isoform- and target-specific regulated by tyrosine phosphorylation. Cell. Signal. 26, 1975–1984 [DOI] [PubMed] [Google Scholar]
- 33.Dosztányi Z., Csizmok V., Tompa P., and Simon I. (2005) IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21, 3433–3434 [DOI] [PubMed] [Google Scholar]
- 34.Dunker A. K., Cortese M. S., Romero P., Iakoucheva L. M., and Uversky V. N. (2005) Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 272, 5129–5148 [DOI] [PubMed] [Google Scholar]
- 35.Baird D., Feng Q., and Cerione R. A. (2005) The Cool-2/α-Pix protein mediates a Cdc42-Rac signaling cascade. Curr. Biol. 15, 1–10 [DOI] [PubMed] [Google Scholar]
- 36.Aslan J. E., Baker S. M., Loren C. P., Haley K. M., Itakura A., Pang J., Greenberg D. L., David L. L., Manser E., Chernoff J., and McCarty O. J. (2013) The PAK system links Rho GTPase signaling to thrombin-mediated platelet activation. Am. J. Physiol. Cell Physiol. 305, C519–C528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato H., Suzuki-Inoue K., Inoue O., and Ozaki Y. (2008) Regulation of adaptor protein GIT1 in platelets, leading to the interaction between GIT1 and integrin α(IIb)β3. Biochem. Biophys. Res. Commun. 368, 157–161 [DOI] [PubMed] [Google Scholar]
- 38.Chahdi A., and Sorokin A. (2008) Protein kinase A-dependent phosphorylation modulates β1Pix guanine nucleotide exchange factor activity through 14-3-3β binding. Mol. Cell. Biol. 28, 1679–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Angrand P. O., Segura I., Völkel P., Ghidelli S., Terry R., Brajenovic M., Vintersten K., Klein R., Superti-Furga G., Drewes G., Kuster B., Bouwmeester T., and Acker-Palmer A. (2006) Transgenic mouse proteomics identifies new 14-3-3-associated proteins involved in cytoskeletal rearrangements and cell signaling. Mol. Cell. Proteomics 5, 2211–2227 [DOI] [PubMed] [Google Scholar]
- 40.Csépányi-Kömi R., Sáfár D., Grósz V., Tarján Z. L., and Ligeti E. (2013) In silico tissue-distribution of human Rho family GTPase activating proteins. Small GTPases 4, 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richnau N., Fransson A., Farsad K., and Aspenström P. (2004) RICH-1 has a BIN/Amphiphysin/Rvsp domain responsible for binding to membrane lipids and tubulation of liposomes. Biochem. Biophys. Res. Commun. 320, 1034–1042 [DOI] [PubMed] [Google Scholar]
- 42.Saengsawang W., Taylor K. L., Lumbard D. C., Mitok K., Price A., Pietila L., Gomez T. M., and Dent E. W. (2013) CIP4 coordinates with phospholipids and actin-associated proteins to localize to the protruding edge and produce actin ribs and veils. J. Cell Sci. 126, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Y., Aardema J., Kale S., Whichard Z. L., Awomolo A., Blanchard E., Chang B., Myers D. R., Ju L., Tran R., Reece D., Christensen H., Boukour S., Debili N., Strom T. S., Rawlings D., Vázquez F. X., Voth G. A., Zhu C., Kahr W. H., Lam W. A., and Corey S. J. (2013) Loss of the F-BAR protein CIP4 reduces platelet production by impairing membrane-cytoskeleton remodeling. Blood 122, 1695–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rice P., Longden I., and Bleasby A. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 [DOI] [PubMed] [Google Scholar]
- 45.Kurokawa K., Nakamura T., Aoki K., and Matsuda M. (2005) Mechanism and role of localized activation of Rho-family GTPases in growth factor-stimulated fibroblasts and neuronal cells. Biochem. Soc. Trans. 33, 631–634 [DOI] [PubMed] [Google Scholar]
- 46.Lawson C. D., and Burridge K. (2014) The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 5, e27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McMahon H. T., and Boucrot E. (2015) Membrane curvature at a glance. J. Cell Sci. 128, 1065–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Premont R. T., Perry S. J., Schmalzigaug R., Roseman J. T., Xing Y., and Claing A. (2004) The GIT/PIX complex: an oligomeric assembly of GIT family ARF GTPase-activating proteins and PIX family Rac1/Cdc42 guanine nucleotide exchange factors. Cell. Signal. 16, 1001–1011 [DOI] [PubMed] [Google Scholar]
- 49.Schlenker O., and Rittinger K. (2009) Structures of dimeric GIT1 and trimeric β-PIX and implications for GIT-PIX complex assembly. J. Mol. Biol. 386, 280–289 [DOI] [PubMed] [Google Scholar]
- 50.Vitale N., Patton W. A., Moss J., Vaughan M., Lefkowitz R. J., and Premont R. T. (2000) GIT proteins, A novel family of phosphatidylinositol 3,4, 5-trisphosphate-stimulated GTPase-activating proteins for ARF6. J. Biol. Chem. 275, 13901–13906 [DOI] [PubMed] [Google Scholar]
- 51.Choi W., Karim Z. A., and Whiteheart S. W. (2006) Arf6 plays an early role in platelet activation by collagen and convulxin. Blood 107, 3145–3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karim Z. A., Choi W., and Whiteheart S. W. (2008) Primary platelet signaling cascades and integrin-mediated signaling control ADP-ribosylation factor (Arf) 6-GTP levels during platelet activation and aggregation. J. Biol. Chem. 283, 11995–12003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van den Bosch M. T., Poole A. W., and Hers I. (2014) Cytohesin-2 phosphorylation by protein kinase C relieves the constitutive suppression of platelet dense granule secretion by ADP-ribosylation factor 6. J. Thromb. Haemost. 12, 726–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nishiya N., Shirai T., Suzuki W., and Nose K. (2002) Hic-5 interacts with GIT1 with a different binding mode from paxillin. J. Biochem. 132, 279–289 [DOI] [PubMed] [Google Scholar]
- 55.Premont R. T., Claing A., Vitale N., Freeman J. L., Pitcher J. A., Patton W. A., Moss J., Vaughan M., and Lefkowitz R. J. (1998) β2-Adrenergic receptor regulation by GIT1, a G protein-coupled receptor kinase-associated ADP ribosylation factor GTPase-activating protein. Proc. Natl. Acad. Sci. U.S.A. 95, 14082–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nisar S. P., Cunningham M., Saxena K., Pope R. J., Kelly E., and Mundell S. J. (2012) Arrestin scaffolds NHERF1 to the P2Y12 receptor to regulate receptor internalization. J. Biol. Chem. 287, 24505–24515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakamura F., Amieva M. R., and Furthmayr H. (1995) Phosphorylation of threonine 558 in the carboxyl-terminal actin-binding domain of moesin by thrombin activation of human platelets. J. Biol. Chem. 270, 31377–31385 [DOI] [PubMed] [Google Scholar]
- 58.Cha B., Kim J. H., Hut H., Hogema B. M., Nadarja J., Zizak M., Cavet M., Lee-Kwon W., Lohmann S. M., Smolenski A., Tse C. M., Yun C., de Jonge H. R., and Donowitz M. (2005) cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J. Biol. Chem. 280, 16642–16650 [DOI] [PubMed] [Google Scholar]