

Background: Eukaryotic translation initiation factor 5A (eIF5A) regulates pancreatic cancer pathogenesis.
Results: eIF5A was shown to control the expression of a set of key signaling molecules including RhoA and ROCK2, and to promote the invasive potential of pancreatic cancer cells.
Conclusion: Hypusine/eIF5A/RhoA/ROCK cascade promotes pancreatic cancer cell metastasis.
Significance: eIF5A may be a novel druggable target to treat metastatic pancreatic cancer.
Keywords: pancreatic cancer, Rho (Rho GTPase), translation control, translation initiation factor, tumor cell biology, tumor metastasis
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest cancers with an overall survival rate of less than 5%. The poor patient outcome in PDAC is largely due to the high prevalence of systemic metastasis at the time of diagnosis and lack of effective therapeutics that target disseminated cells. The fact that the underlying mechanisms driving PDAC cell migration and dissemination are poorly understood have hindered drug development and compounded the lack of clinical success in this disease. Recent evidence indicates that mutational activation of K-Ras up-regulates eIF5A, a component of the cellular translational machinery that is critical for PDAC progression. However, the role of eIF5A in PDAC cell migration and metastasis has not been investigated. We report here that pharmacological inhibition or genetic knockdown of eIF5A reduces PDAC cell migration, invasion, and metastasis in vitro and in vivo. Proteomic profiling and bioinformatic analyses revealed that eIF5A controls an integrated network of cytoskeleton-regulatory proteins involved in cell migration. Functional interrogation of this network uncovered a critical RhoA/ROCK signaling node that operates downstream of eIF5A in invasive PDAC cells. Importantly, eIF5A mediates PDAC cell migration and invasion by modulating RhoA/ROCK protein expression levels. Together our findings implicate eIF5A as a cytoskeletal rheostat controlling RhoA/ROCK protein expression during PDAC cell migration and metastasis. Our findings also implicate the eIF5A/RhoA/ROCK module as a potential new therapeutic target to treat metastatic PDAC cells.
Introduction
Despite recent advances in cancer research, pancreatic cancer remains one of the deadliest diseases to date, representing the fourth leading cause of cancer death in the United States. Pancreatic ductal adenocarcinoma (PDAC)3 is the most common type of pancreatic cancer that accounts for up to 90% of all pancreatic cancer cases, with the overall 5-year survival rate of only 5% (1). This is largely due to the rapid dissemination of tumors, which is commonly observed at the time of diagnosis. Indeed, a major cause of the poor prognosis of PDAC is its frequent metastasis to the liver, lungs, and other organs, through extensive local invasion as well as lymphatic and hematogenous metastases (2, 3).
Recent studies indicate that metastasis occurs at the earliest stage of PDAC pathogenesis (4). In mouse models, hematogenous dissemination of PDAC was shown to even precede primary PDAC tumor formation (4). Oncogenic K-Ras is believed to be the major driver in this process as its withdrawal resulted in regression of metastasized PDAC cells in mouse models (5–7). These findings indicate that even advanced stage metastatic tumors remain largely dependent on oncogenic K-Ras expression (7). Although the mechanisms whereby activated K-Ras drives PDAC malignancy are complex and are not fully understood, they likely involve reprogramming of cell metabolism and protein synthesis pathways (8, 9). In fact, mutationally activated K-Ras has been shown to reprogram mRNA translation to enable efficient production of pro-oncogenic proteins and enhance tumor growth in glioblastoma (10). On the other hand, mTOR (mammalian target of rapamycin), a regulator of translation commonly implicated in a broad spectrum of human malignancies, may not be critical for oncogenic translation in PDAC, as mTOR inhibition failed to show efficacy in clinical trials (11, 12). These findings suggest that alternative mechanisms operate to modulate oncogenic protein synthesis in PDAC.
We recently identified the eukaryotic translation initiation factor 5A (eIF5A) as a critical translational regulator and a novel druggable target involved in K-Ras-mediated human PDAC pathogenesis (13). eIF5A is highly conserved across a broad spectrum of species, and is indispensable for normal mammalian development (14–16). Notably, eIF5A is the only protein post-translationally activated by the synthesis of a unique amino acid, hypusine (N-(4-amino-2-hydroxybutyl)lysine) (14, 15). Hypusine formation is catalyzed by two enzymes, deoxyhypusine synthase (DHPS) and deoxyhypusine hydroxylase (DOHH). To achieve this unique posttranslational modification, the butylamine portion of the polyamine spermidine is transferred to a specific lysine residue of eIF5A by DHPS. Subsequently, carbon 2 of the transferred 4-aminobutyl moiety is hydroxylated by DOHH. Importantly, each step in the enzymatic reaction is amenable to specific pharmacological inhibition. N(1)-guanyl-1,7,-diamineoheptane (GC7) is an inhibitor of DHPS, and ciclopirox olamine (CPX) is a bidentate iron chelator that inhibits DOHH (14, 15).
Important recent work has shown that eIF5A and its bacterial homolog, EF-P (translation elongation factor P), both enhance the synthesis of polyproline stretch-containing proteins (17–19). As polyproline stretches facilitate protein-protein interactions relevant to a range of biological events including signal transduction and cytoskeletal remodeling (20, 21), this raises the possibility that eIF5A has the capability to control cellular processes by modulating the expression of subsets of proteins with polyproline motifs. Emerging work supports this idea as eIF5A does not regulate general protein synthesis, but appears to have evolved to fine-tune the production of subsets of proteins in a contextual manner (22, 23).
The above findings have important implications for the role of eIF5A in PDAC, as eIF5A protein amplification may be responsible for fine-tuning the expression of a set of proteins to drive PDAC cell migration and metastasis (13). In fact, increased demands for migration-associated proteins may explain why eIF5A expression is increased in several cancers with a high propensity to metastasize including PDAC, liver, and colorectal cancer (13, 24, 25). The fact that eIF5A is selectively up-regulated in tumors, modulates translation of mRNAs encoding polyproline-containing proteins, and is the only protein known to be hypusine-modified, makes it an attractive therapeutic target to treat metastatic cancer. Such new therapeutic targets and strategies are sorely needed to target the highly metastatic nature of human PDAC. Therefore, we undertook studies to determine the role of eIF5A in mediating PDAC cell migration and metastasis. We show here that pharmacological inhibition of eIF5A hypusination or genetic knockdown of eIF5A inhibits PDAC cell migration, invasion, and metastasis in vitro and in vivo. Proteomic profiling and informatics analyses of eIF5A-depleted human PDAC cell lines revealed networks of cytoskeleton regulatory proteins involved in cell migration and uncovered a functional RhoA/ROCK signaling node that operates downstream of eIF5A in invasive cancer cells. These results indicate that eIF5A activation serves as a rheostat that controls the protein expression of RhoA and ROCK in PDAC cells. Our findings also implicate the hypusine/eIF5A/RhoA/ROCK module as a possible new therapeutic target in metastatic PDAC cells.
Experimental Procedures
Reagents and Antibodies
Dulbecco's modified Eagle's medium (DMEM) was from Gibco (Invitrogen). Porcine trypsin (modified sequencing grade) was from Promega. Acetonitrile and HPLC grade distilled water were from Merck. Ammonium bicarbonate was purchased from Sigma. Antibodies for eIF5A (ab32443) and ZO-1 (33-9100) were acquired from Abcam and Invitrogen, respectively. RasGAP (610040) and Rac1 (610650) antibodies were from BD Biosciences, and RhoA (number 2117), tubulin (number 3873), MYPT (myosin phosphatase targeting protein; number 2634), phospho-MYPT (Thr-853; number 4563), and phospho-ribosomal S6 (number 4858) antibodies were from Cell Signaling Technology. ROCK2 (GTX108247) and TRIM29 (GTX115749) antibodies were purchased from GeneTex. Hypusinated eIF5A antibody (NIH353) and Xrn1 antibody were kindly provided by Dr. Myung-Hee Park (National Institutes of Health) and Dr. Jens Lykke-Andersen (University of California, San Diego), respectively. N(1)-guanyl-1,7-diaminoheptane (GC7) and rapamycin were purchased from EMD Millipore, and Y-27632 was purchased from Selleck Chemicals.
Cell Culture, eIF5A Knockdown, and Pharmacological Inhibition of Hypusination
Human PDAC cells (PANC1, 779E) and mouse PDAC cells (PDA4964 cells: derived from Pdx1-Cre;LSL-KRasG12D/+;p53R172H/+ pancreas (26), and kindly provided by Dr. Andrew Lowy (UCSD Moores Cancer Center)) were used in this study. 779E is a patient-derived human PDAC cell line obtained from moderate-to-poorly differentiated PDAC tissue by Dr. Andrew Lowy (13). The cells were maintained in DMEM supplemented with 10% FBS (fetal bovine serum), penicillin/streptomycin, gentamycin/glutamine, and sodium pyruvate, and cultured in a humidified incubator at 37 °C with 5% CO2. Cells stably expressing shRNAs or overexpressing eIF5A were generated and maintained as described previously (13). For knockdown of 779E with an independent eIF5A shRNA construct, lentiviral particles from Sigma were used (TRCN0000062548; 106 transducing units/ml). For knockdown of mouse eIF5A in murine PDA4964 cells, lentiviral particles containing shRNA against mouse eIF5A were used (Sigma; TRCN0000308824; 106 transducing units/ml). For pharmacological inhibition of eIF5A hypusination, PANC1 cells were grown to 50% confluence in 6-well plates and treated with 25 μm GC7 for 24 h before being processed for Western blotting or transwell assays.
LC-MS/MS Sample Preparation and Tryptic Digestion
779E cells stably expressing control or eIF5A shRNA were cultured in 10-cm diameter dishes until they reached 70 to 80% confluence and were washed with ice-cold phosphate-buffered saline (PBS). The cells were lysed in a RIPA buffer followed by sonication and soluble fractions were isolated by centrifugation at 14,000 rpm for 15 min. Each of the 80 μg of cell lysates in SDS-PAGE loading buffer were boiled for 5 min, and loaded on a NuPAGE 4–12% BisTris gel (Invitrogen). After separation, the gel was stained with Coomassie Blue G-250 (Sigma) for 3 h. Each gel lane was divided into 16 fractions and each gel fraction was destained by 40% methanol. To remove the cysteine disulfide bond, the samples were treated with 10 mm dithiothreitol (DTT) for 60 min at 56 °C, then 100 mm iodoacetamide was added and further incubated for 60 min in the dark. Each of the reduced and alkylated fractions was digested with 500 ng of trypsin (Promega) at 37 °C overnight. After digestion, the peptide samples were dried and stored at −20 °C.
LC-MS/MS Analysis
All MS/MS experiments for peptide identification were performed by Agilent 1200 series nano-flow liquid chromatography system and a LTQ linear ion-trap mass spectrometry system (Thermo Scientific) equipped with a nano-electrospray ionization source. The peptides were trapped by the pre-column (inner diameter, 300 μm; length, 1 mm; particle size; 5 μm) for pre-concentration and desalting by solvent A (100% distilled water, 0.1% formic acid, 0.05% acetic acid). The trapped peptides were then eluted from pre-column using a mobile phase gradient (solvent B, 100% acetonitrile, 0.1% formic acid, 0.05% acetic acid) directly onto a reversed phase (RP) analytical column (15 cm × 75 μm inner diameter) packed with C18 (particle size 3 μm). The gradient began at 5% solvent B then ramped to 60% solvent B for 80 min, and finally, ramped 80% solvent B for 20 min at a flow rate of 250 nl/min. The eluent was then introduced into the LTQ mass spectrometer. The LTQ was operated by 2.25 kV spray voltage, 200 °C ion transfer capillary, and 35% collision energy. The analysis method consisted of a full MS scan with a range of 400–1600 m/z and data-dependent MS/MS (MS2) on the nine most intense ions from the full MS scan. The previous fragmented ions were excluded for 60 s. The LC-MS/MS analysis was performed in triplicate for each biological sample.
Data Processing
The RAW files were converted to mzXML files by BioWorks 3.3.1 and searched using Swiss-Prot HUMAN database (release 2013) with the MASCOT search engine (version 2.2.04, Matrix Science). The parameters for identification of MS/MS data were 2.0 Da for MS and 0.8 Da for MS/MS, allowing up to one missed cleavage. Carbamidomethylation of cysteine was considered as a fixed modification and oxidation of methionine as a variable modification. The MASCOT scores of individual ions were in the confidence range of 95% probability (significance threshold p < 0.05). Initial peptide filtering was performed using a 1% false discovery rate, which was calculated by a decoy method (27). Proteins with more than 2-fold differences in abundance in both biological replicates, as determined by spectral counts, were considered significantly affected by eIF5A knockdown and used for subsequent bioinformatic analyses as described below. To enhance the reliability of our profiling, we set the minimum average spectral count cutoff to two per MS analysis. A complete list of proteins significantly affected by eIF5A knockdown by MS analysis is provided in supplemental Table S1.
Bioinformatic Analysis
Proteins whose expression levels were significantly altered were subjected to bioinformatics gene ontology (GO) and network analysis. Selected proteins were classified based on protein interaction and biological function using DAVID (Database for Annotation, Visualization and Integrated Discovery: david.abcc.ncifcrf.gov). DAVID calculates a modified Fisher Exact test score using the Expression Analysis Systematic Explorer (EASE) program (EASE score) to measure gene enrichment indicating over-representation within GO terms or pathways defined by PANTHER (28). Parameters used for significance for DAVID annotations on pathways were counted threshold = 2 (minimum number of genes for the corresponding GO term) and EASE threshold = 0.1 (maximum EASE score/p value). Network analyses of eIF5A-regulated proteins were performed by STRING program (Search Tool for the Retrieval of Interacting Genes/Proteins), which performs network analysis based on protein-protein interactions.
Western Blotting and Quantitative PCR (qPCR)
Equal amounts of lysates from control and eIF5A knockdown cells dissolved in LDS sample buffer were separated and transferred onto a nitrocellulose membrane (Whatman). After blocking with 5% bovine serum albumin (BSA) in PBS for 1 h, the membranes were probed with appropriate antibodies and visualized using the enhanced chemiluminescence (Thermo Scientific). qPCR was performed as described previously (13).
Cell Migration and Invasion Assays
Cell migration assay was performed using Corning transwell inserts (Sigma) with a 8.0-μm pore-sized membrane as described previously (29). Cell invasion assay was performed using transwell inserts with 8.0-μm pore size, precoated with basal membrane extract (Trevigen). 1 × 105 cells in 200 μl of serum-free medium were added to the upper chamber and the lower chamber was filled with 400 μl of DMEM containing 10% FBS as a chemoattractant. Cells were allowed to migrate for 16 h and subsequently, fixed by 100% methanol and stained by 0.1% crystal violet (Sigma). After staining, a cotton swab was used to remove non-migrated cells in the upper chamber. The number of migrated cells was counted manually in 5 high power fields (×40). To rule out the possibility that the recorded change in cell numbers on the lower surface of the membrane is not simply due to overall changes in cell numbers, we quantified the total number of cells that attached to and survived on the transwell membranes at the end of the migration period.
Cell Spreading Assay
Cell spreading assays were performed by monitoring cell attachment to type I collagen in real time using xCELLigence system (Acea Bioscience). Briefly, a gold-plated 16-well microtiter plate (Acea Bioscience) was coated with 10 μg/ml of type I collagen (Gibco) for 1 h at room temperature. Subsequently, cells suspended in cell adhesion/spreading assay medium (DMEM + 0.5% BSA) were plated at 1 × 105 cells/well, and their attachment to type I collagen was monitored using xCELLigence system, a real-time, automated electro-sensing platform (30). The impedance of each well surface, which is proportional to the cell spreading area and is displayed as arbitrary units (Cell Index), was recorded at 3-min intervals and plotted against time to generate cell adhesion/spreading curves.
Chicken Embryo Metastasis Assay
Chick CAM (chorioallantoic membrane) metastasis assay was performed as described previously (31). Briefly, a 1-cm2 square window was created in the shell of fertilized chicken eggs to expose the underlying vasculature. PANC1 cells labeled with CellTracer CFSE kit (Invitrogen) were suspended in sterile PBS and 3 × 105 tumor cells (100 μl/embryo) were injected into the main CAM vein. After incubating the embryos for 48 h, the chicken livers were removed and imaged using Nikon C1-si confocal microscope (×10 objective). For quantification, the number of metastasized cells per ×10 field was counted from 10 different fields of view. A total of 7 chicken embryos were used for the injection of each cell line.
Syngeneic Mouse Tail Vein Metastasis Assays
Mouse PDA4964 cells containing control or eIF5A shRNA were suspended in PBS and injected through the tail vein (5 × 105 cells/mouse) into immunocompatible 4–6-week-old B6/129 mice (Jackson Laboratory), and allowed to metastasize for 2 weeks. Subsequently, the mice were sacrificed and their lungs were isolated, and processed for hematoxylin and eosin (H&E) staining to assess metastatic burden in the lungs. Metastatic burden was calculated as the percentage of tumor area compared with the total lung area using ImageJ software (NIH). Total of 7 mice were used for the injection of each cell line.
Statistical Analysis
All data represent mean ± S.D. p values were determined by GraphPad Prism, using Student's t test and, where indicated, a χ square test.
Results
eIF5A Knockdown Suppresses PDAC Cell Migration and Invasion in Vitro
Advanced PDAC cells show an exceptionally high propensity to metastasize to the liver and other organs (1–3). As eIF5A expression is significantly increased in high grade PDAC tissues (13), we sought to determine whether eIF5A plays a role in PDAC cell migration and metastasis, which has not been investigated to date. To this end, we performed transwell cell migration and invasion assays using PANC1 cells in which eIF5A protein had been stably knocked down by ∼80% (Fig. 1A). We found that both cell migration and invasion were significantly reduced upon eIF5A knockdown compared with control cells expressing a scramble shRNA (Fig. 1A). Importantly, the reduced cell migration and invasion of eIF5A-depleted cells was not simply due to a deficiency in cell loading or cell adhesion to the transwell membrane, or changes in overall cell numbers, as similar numbers of knockdown and control cells were detected on the transwell membrane at the end of the migration period (Fig. 1C).
FIGURE 1.

eIF5A promotes PDAC cell migration and invasion. A, PANC1 cells containing control (shCtrl) or eIF5A shRNA (sh5A) were subjected to transwell migration assays (left) or invasion assays (right) as described under “Experimental Procedures” (n = 4). Western blot shows efficient knockdown of eIF5A in cells expressing eIF5A shRNA. B, PANC1 cells treated without (Control) or with (GC7) 25 μm GC7 for 24 h were subjected to migration assays (left) or invasion assays (right) as described under “Experimental Procedures” (n = 4). Western blot validates efficient inhibition of eIF5A hypusination upon GC7 treatment, as demonstrated by a significantly decreased amount of hypusinated eIF5A (Hyp-5A). C, control experiments for migration assays in A and B. 1 × 105 PANC1 cells containing control or eIF5A shRNA (left panel) or PANC1 cells without or with GC7 treatment (right panel) were seeded on transwell inserts and cultured for 16 h. Subsequently, the number of cells attached to transwell membranes was quantified as described under “Experimental Procedures” (n = 4). Statistical analysis revealed that eIF5A knockdown or GC7 treatment did not significantly affect the number of cells growing on transwell membranes. D, cell spreading assays. PANC1 (left) or 779E (right panel) cells were seeded on collagen (type I) and allowed to adhere and spread on the substratum. The kinetics of cell spreading were determined using the xCELLigence system and presented as Cell Index (n = 4). E, PANC1 cells expressing control (shCtrl) or eIF5A shRNA (sh5A) were fluorescently labeled and injected to the CAM vein of chicken embryos and allowed to invade into the liver for 48 h. Representative images of cells in the liver from embryos injected with cells containing control shRNA (shCtrl) or eIF5A shRNA (sh5A) are shown on the left. Bar represents 200 μm. The bar graph shows the quantification of cells invaded to the liver (n = 7). F, mouse PDA4964 cells were injected into the tail vein of immunocompatible B6/129 mice and allowed to metastasize to the lung. Representative H&E staining of lung tissue sections from mice injected with cells containing control shRNA (shCtrl) or eIF5A shRNA (sh5A) are shown on the left (n = 7). Bar represents 200 μm. The bar graph shows the quantification of metastatic burden in the lung. * represents p values of <0.05 as determined by Student's t test.
Pharmacological Inhibition of eIF5A Hypusination Inhibits Cell Migration and Invasion in Vitro
We subsequently sought to determine the effect of pharmacological inhibition of eIF5A hypusination on PDAC cell migration and invasion. GC7 is a spermidine analogue and a competitive inhibitor that specifically blocks eIF5A hypusination by inhibiting DHPS (14, 15). We found that GC7 treatment effectively blocked eIF5A hypusination (Fig. 1B), and significantly reduced migration and invasion of PANC1 cells (Fig. 1B), phenocopying eIF5A knockdown (Fig. 1A). Importantly, the reduced cell migration and invasion of GC7-treated cells was not simply due to a deficiency in cell loading or cell adhesion to the transwell membrane, or changes in overall cell numbers, as similar numbers of control and GC7-treated cells were detected on the transwell membrane at the end of the migration period (Fig. 1C).
eIF5A Regulates PDAC Cell Spreading on Type I Collagen
In many cases, the ability of cancer cells to migrate and invade tissues requires cell spreading on collagen matrices (32, 33). In fact, PDAC is characterized by a dense collagen-rich desmoplastic reaction, which contributes to the highly invasive and metastatic nature of the disease (1, 34). Therefore, we determined the ability of eIF5A to regulate the kinetics of PDAC cell spreading on type I collagen using the xCELLigence electrical impedance system. This system is beneficial over end point assays in that it allows for full kinetic monitoring of cell spreading over the surface of electrode arrays, which is directly proportional to changes in the electrical impedance. As shown in Fig. 1D, eIF5A knockdown significantly delayed PANC1 cell spreading compared with control cells. Similarly, knockdown of eIF5A in 779E cells strongly inhibited cell spreading. Together these findings indicate that hypusinated eIF5A controls cell spreading, migration, and invasion of PDAC cells in vitro.
eIF5A Is Necessary for PDAC Cell Metastasis in Vivo
The ability of eIF5A to mediate cell spreading, migration, and invasion suggests that it may contribute to PDAC metastasis in vivo. To investigate this possibility, we used an established chicken embryo metastasis assay (31) to monitor cell dissemination to the liver, the most common site of human PDAC metastasis (1–3). This model primarily addresses the late stages of tumor metastasis, such as tumor cell dissemination through the circulation, vessel extravasation, organ colonization, and microtumor formation (31). PANC1 cells expressing control or eIF5A shRNA were labeled with a fluorescent dye and injected into the large chorioallantoic vein in the CAM. Cells were allowed to disseminate through the circulation, extravasate, and colonize the liver for 48 h before being quantified by confocal microscopy. As shown in Fig. 1E, eIF5A knockdown significantly reduced PANC1 cell metastasis to the liver compared with control cells expressing eIF5A.
The KCP (Pdx1-Cre;LSL-KRasG12D/+;p53R172H/+) mouse model of PDAC faithfully recapitulates human disease progression from early stage PanIN (pancreatic intraepithelial neoplasia) development to late state systemic metastasis (5). PDA4964 is a cell line isolated from the tumors of these mice that form new tumors in syngeneic recipients, which recapitulates the original tumor progression and metastatic potential (26). Therefore, we used these cells to determine the role of eIF5A in mediating metastasis in immune-competent mice, by conducting a well established tail vein injection assay of cancer metastasis. Similar to the chicken embryo model (Fig. 1E), this assay primarily addresses the late steps in the metastatic cascade (35). PDA4964 cells transduced with control or eIF5A-specific shRNA were injected into the tail vein of syngeneic B6/129 mice. Cells were allowed to colonize the lungs for 2 weeks and the metastatic burden quantified by hematoxylin and eosin (H&E) staining. As shown in Fig. 1F, eIF5A knockdown significantly inhibited PDA4964 cell metastasis to the lungs compared with control cells. Altogether, these results indicate that eIF5A promotes PDAC cell metastasis in both the chicken CAM and syngeneic mouse models. Collectively, these findings indicate that eIF5A plays a critical role in mediating PDAC cell migration, invasion, and metastasis in vivo.
Identification and Network Analyses of eIF5A-regulated Proteins in PDAC Cells
Current research suggests that eIF5A does not just regulate global mRNA translation, but rather fine-tunes the production of subsets of proteins, including polyproline-tract containing proteins (17–19, 22, 23). However, specific downstream effectors of eIF5A responsible for the observed migratory/invasive behavior of PDAC cells remain unclear. To systematically identify the downstream effectors of eIF5A in PDAC cells, we profiled protein expression changes in 779E cells with or without eIF5A knockdown using label-free comparative LC-MS/MS. Fig. 2A shows a schematic of the experimental design. It is notable that two biological replicates were analyzed by mass spectrometry in triplicate to ensure reliable protein identification. Also, only proteins with more than 2-fold differences in spectral counts and identified in both biological replicates, were considered significantly affected by eIF5A knockdown and used for subsequent bioinformatics analysis as described below. Using this approach, a total of 3108 proteins were identified. Of these 147 proteins were significantly down-regulated by eIF5A depletion, whereas 112 proteins were up-regulated in both biological replicates (Fig. 2B). A complete list of identified proteins after filtration is shown in supplemental Table S1.
FIGURE 2.
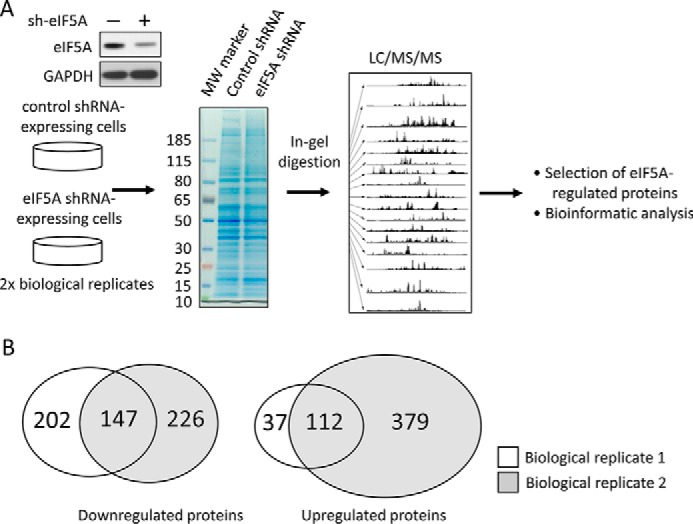
Overview of comparative proteomic profiling between control and eIF5A knockdown PDAC cells. A, schematics of experimental design. Cell lysates from 779E cells stably expressing control or eIF5A shRNA were resolved by SDS-PAGE and subjected to LC-MS/MS analysis after tryptic digestion. Western blot validates efficient depletion of eIF5A in the eIF5A shRNA-expressing cells. Identified proteins were filtered and subjected to bioinformatics analysis. B, Venn diagram showing the overlap of proteins significantly down- or up-regulated by eIF5A knockdown from two biological replicates.
To independently validate the protein expression changes detected by LC-MS/MS, a sample set of 6 proteins from 779E knockdown and control cells were examined for changes in expression levels by Western blotting. We confirmed reduced expression of RhoA, ROCK2, TRIM29 (also known as ATDC), Xrn1 and ZO-1 (TJP-1) as well as the increased expression of RasGAP (Fig. 3A). These results, along with the fact that eIF5A itself was identified as one of the significantly down-regulated proteins (supplemental Table S1), underscore the reliability of the label-free spectral count methodology used in this study.
FIGURE 3.

Validation and bioinformatic analyses of proteins down- or up-regulated by eIF5A knockdown. A, validation of proteomic profiling by Western blots. Cell lysates from 779E cells expressing control- or eIF5A-shRNA were probed for the indicated proteins. GAPDH served as a loading control, and mean relative signal intensities for the probed proteins relative to GAPDH are shown below the blots (n = 3). B, functional classification of proteins down- (left) or up-regulated (right) by eIF5A knockdown based on gene ontology (GO) analysis. Dotted lines represent significance threshold. C, functional classification of proteins down- (left) or up-regulated (right) by eIF5A knockdown based on biological pathway analysis. Left panel shows statistical significance, whereas the right panel shows enrichment. Dotted lines represent significance threshold.
We next annotated the biological relevance of the eIF5A-regulated proteome using gene ontology (GO) analysis and the website-based database, DAVID (Fig. 3B). GO categories significantly enriched in the down-regulated proteins include protein metabolism and modification (GO:00060), protein targeting (GO:00138), nuclear transport (GO:00133), intracellular protein traffic (GO:00125), protein targeting and localization (GO:00137), carbohydrate metabolism (GO:00001), cell structure and motility (GO:00285), and protein modification (GO:00063). For the up-regulated proteins, significantly enriched GO categories were: protein metabolism and modification (GO:00060), pre-mRNA processing (GO:00047), mRNA splicing (GO:00048), protein biosynthesis (GO:00061), and stress response (GO:00178).
To identify functional modules of proteins regulated by eIF5A, cluster analyses of the down- or up-regulated proteins were performed using DAVID (Fig. 3C). Cytoskeletal regulation by RhoGTPase, 5-hydroxytryptamine degradation, and axon guidance mediated by semaphorins were identified as key modules present in the eIF5A down-regulated protein group. On the other hand, ubiquitin-proteasome pathway, Parkinson disease, and cell cycle pathways were identified as key modules in the group of up-regulated proteins. These data from GO analyses and cluster analysis indicate that eIF5A may be involved in a broader spectrum of biological processes than previously thought, possibly by controlling translation elongation of gene transcripts involved in diverse cellular functions.
Because eIF5A and its bacterial homologue, EF-P, have been reported to facilitate the synthesis of proteins with polyproline-stretches consisting of more than three consecutive proline residues (PPP) (17–19), we determined the proportion of polyproline stretch-containing proteins in our eIF5A-regulated proteome. Based on a previously published analysis on polyproline sequences in the human proteome (21), 4564 proteins of 18666 proteins (24%) analyzed possess one or more polyproline stretches. We found that one or more polyproline tracts are present in 54 of 147 proteins (37%) down-regulated upon eIF5A knockdown, whereas 24 of 112 (21%) up-regulated proteins also possess polyproline stretches (supplemental Table S2). The χ square test revealed that proteins with the polyproline stretch are enriched in the down-regulated group (p < 0.01), whereas their prevalence was not significantly altered in the up-regulated group (p > 0.45). These results indicate that the presence of polyproline stretches represents an important signature of eIF5A-regulated proteins, but is not the sole determinant of eIF5A dependence in protein expression. This is in line with a recent report showing that protein motifs other than polyproline can also confer EF-P dependence in protein synthesis in bacteria (36).
To understand how eIF5A affects known regulators of PDAC pathogenesis, we identified eIF5A-regulated proteins with reported involvement in PDAC using the PubMed literature search program (Table 1). We found that a total of 32 proteins with relevance to PDAC were down-regulated by eIF5A knockdown, which suggests that eIF5A drives PDAC malignancy through multiple downstream effectors. Bioinformatic network analysis using the STRING program identified the presence of two functional modules among eIF5A-regulated proteome with relevance to PDAC (Fig. 4A); epigenetic and transcriptional regulation (Module 1), and cell motility and the cytoskeleton (Module 2). These results suggest that eIF5A-driven PDAC pathogenesis may primarily involve these two biological processes.
TABLE 1.
List of eIF5A-regulated proteins implicated in PDAC
Proteins implicated in PDAC malignancy in PubMed were manually picked from the list of proteins down-regulated by eIF5A knockdown.
| Official symbol | Official full name |
|---|---|
| ALDH1A2 | Aldehyde dehydrogenase 1 family, member A2 |
| ARHGAP5 | Rho GTPase-activating protein 5 |
| ATG7 | Autophagy related 7 |
| BIRC6 | Baculoviral IAP repeat-containing protein 6 |
| CASP8 | Caspase-8 |
| CHD3 | Chromodomain helicase DNA binding protein 3 |
| CMPK1 | Cytidine monophosphate (UMP-CMP) kinase 1, cytosolic |
| CTBP2 | C-terminal-binding protein 2 |
| ECH1 | Enoyl-CoA hydratase 1, peroxisomal |
| EPHX1 | Epoxide hydrolase 1 |
| EPPK1 | Epiplakin |
| GALNT3 | Polypeptide N-acetylgalactosaminyltransferase 3 |
| GCLC | Glutamate-cysteine ligase catalytic subunit |
| GRB7 | Growth factor receptor-bound protein 7 |
| HDAC1 | Histone deacetylase 1 |
| HUWE1 | HECT, UBA, and WWE domain containing 1 |
| IDH2 | Isocitrate dehydrogenase [NADP], mitochondrial |
| MUC5AC | Mucin-5AC, oligomeric mucus/gel-forming |
| PRKDC | DNA-dependent protein kinase catalytic subunit |
| PSMB4 | Proteasome subunit beta type-4 |
| RHOA | Ras homolog gene family A |
| RHOC | Ras homolog gene family C |
| ROCK1 | Rho-associated protein kinase 1 |
| ROCK2 | Rho-associated protein kinase 2 |
| SDC1 | Syndecan-1 |
| SIN3A | SIN3 transcription regulator family member A |
| SMG1 | SMG1 phosphatidylinositol 3-kinase-related kinase |
| TJP1 | Tight junction protein 1 |
| TP53BP1 | Tumor protein p53-binding protein 1 |
| TRIM29 | Tripartite motif-containing protein 29 |
| TRIP10 | Thyroid hormone receptor interactor 10 |
| TRRAP | Transformation/transcription domain-associated protein |
FIGURE 4.

Network analysis of proteins presented in Tables 1 (A) and 2 (B). Functional network of proteins listed in Tables 1 or 2 were generated using STRING database. Line thickness represents stronger associations. Dashed circles represent functional modules of proteins; epigenetics and transcription (Module 1), cell motility, and the cytoskeleton (Module 2).
Table 2 shows the list of eIF5A-regulated proteins associated with the cytoskeleton and cell motility, as identified by GO analysis and PubMed search. Importantly, of these proteins, the small GTPase RhoA and its downstream effector ROCK (Rho-associated kinase) were identified as a central node in the eIF5A-controlled cytoskeletal network (Fig. 4B). RhoA activates ROCK proteins, which in turn phosphorylates LIM kinase, myosin light chain, and myosin light chain phosphatase to promote stress fiber formation, contractile forces, and motility (37).
TABLE 2.
List of eIF5A-regulated proteins involved in cytoskeletal organization and cell motility by Panther Pathway analysis
| Official symbol | Official full name |
|---|---|
| AKAP13 | A kinase (PRKA) anchor protein 13 |
| ARHGAP5 | Rho GTPase-activating protein 5 |
| CASP8 | Caspase-8 |
| CORO1A | Coronin, actin-binding protein, 1A |
| DPYSL2 | Dihydropyrimidinase-like 2 |
| DYNC1H1 | Dynein, cytoplasmic 1, heavy chain 1 |
| EPPK1 | Epiplakin 1 |
| EVPL | Envoplakin |
| FKBP15 | FK506-binding protein 15 |
| MYO18A | Myosin XVIIIA |
| PPP1CA | Protein phosphatase 1, catalytic subunit, alpha isoform |
| RAP1GDS1 | Rap1 GTPase-GDP dissociation stimulator 1 |
| RHOA | Ras homolog gene family, member A |
| RHOC | Ras homolog gene family, member C |
| ROCK1 | Rho-associated, coiled-coil containing protein kinase 1 |
| ROCK2 | Rho-associated, coiled-coil containing protein kinase 2 |
| SDC1 | Syndecan 1 |
| SPTBN2 | Spectrin, β, non-erythrocytic 2 |
| TJP1 | Tight junction protein 1 (zona occludens 1) |
| TRIP10 | Thyroid hormone receptor interactor 10 |
| UBR4 | E3 ubiquitin-protein ligase UBR4 |
eIF5A Regulates RhoA and ROCK2 Protein Expression in PDAC Cells
Although Rho/ROCK signaling has been linked to cell motility and metastasis in various cancers including PDAC, little is known about how Rho/ROCK protein levels are regulated in tumor cells, especially in regard to translation (37–41). Therefore, we decided to focus on RhoA and ROCK2, established components of the Rho/ROCK pathway whose protein levels were reduced upon eIF5A knockdown (Fig. 3A). The reduction in RhoA and ROCK2 protein expression is most likely due to changes in post-transcriptional regulation, as neither RhoA nor ROCK2 mRNA expression was significantly altered by eIF5A knockdown (RhoA and ROCK2 mRNA levels were 91 ± 4 and 69 ± 14% of those in control cells, respectively, in eIF5A knockdown cells (p > 0.06)). These findings suggest that eIF5A regulates RhoA and ROCK2 expression post-transcriptionally, possibly at the level of translation.
To validate this hypothesis, we first confirmed the ability of eIF5A to control RhoA and ROCK2 protein expression using independently designed shRNAs. We determined whether RhoA and ROCK2 protein levels are reduced upon eIF5A knockdown in 779E cells expressing an independent eIF5A shRNA (Fig. 5A), and also in PDA4964 cells expressing a murine-specific shRNA against eIF5A (Fig. 5A). In both cases RhoA and ROCK2 expression levels were significantly reduced in eIF5A-depleted cells compared with control cells (Fig. 5A). Therefore, the reduction in RhoA/ROCK2 expression was not due to off-target effects of eIF5A-targeting shRNA (Fig. 3A). Also, eIF5A knockdown did not alter protein expression of the Rho family member protein, Rac1, indicating that not all Rho family proteins are regulated by eIF5A (Fig. 5B). In addition, inhibition of mTORC1 (mammalian target of rapamycin complex 1) activity by rapamycin, as indicated by the loss of ribosomal S6 protein phosphorylation (42), did not affect RhoA or ROCK2 protein expression (Fig. 5C). Taken together these findings indicate that eIF5A is necessary for the proper expression of RhoA and ROCK2 proteins in PDAC cells.
FIGURE 5.
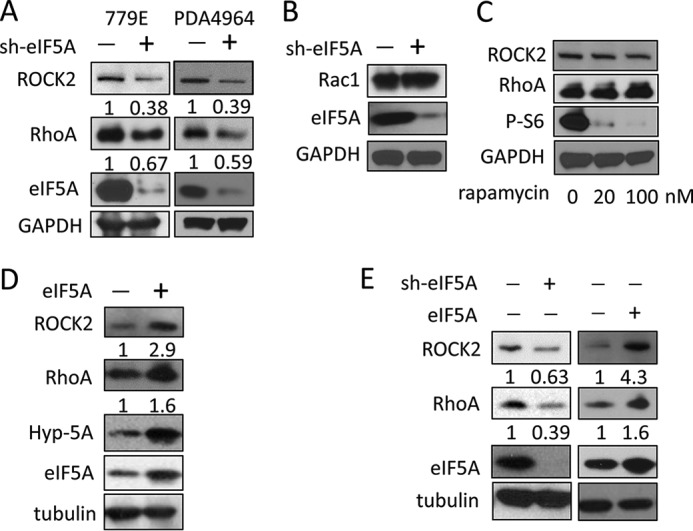
Hypusinated eIF5A positively regulates the expression of RhoA and ROCK2 proteins in PDAC cells. A, Western blot analysis of eIF5A, RhoA, and ROCK2 in 779E cells expressing control or an independent eIF5A shRNA construct (left) and in mouse PDA4964 cells expressing control or murine eIF5A-specific shRNA (right). GAPDH served as a loading control. Mean relative signal intensities for RhoA and ROCK2 relative to GAPDH are shown below the blots (n = 3). B, Western blot analysis of Rac1 in 779E cells containing control or eIF5A shRNA. GAPDH served as a loading control. C, Western blot analysis of RhoA and ROCK2 in PANC1 cells treated with 0, 20, and 100 nm rapamycin for 72 h. GAPDH served as a loading control, whereas phospho-S6 was used to verify inhibition of mTORC1 activity. D, Western blot analysis of RhoA and ROCK2 expression in 779E cells stably overexpressing eIF5A (left panel). Increased expression of eIF5A and hypusinated eIF5A (Hyp-5A) was also validated. Tubulin served as a loading control. Mean relative signal intensities for RhoA and ROCK2 relative to tubulin are shown below the blots (n = 3). E, Western blot analysis of RhoA and ROCK2 expression in PANC1 cells expressing control or eIF5A shRNA (left), or PANC1 cells containing empty vector or eIF5A-encoding vector (right). Tubulin served as a loading control. Mean relative signal intensities for RhoA and ROCK2 relative to tubulin are shown below the blots (n = 3).
eIF5A protein expression is amplified in PanINs and PDAC tissues compared with uninvolved tissues and normal pancreatic ducts (13). Therefore, we wanted to determine whether eIF5A overexpression in 779E cells was sufficient to drive increased RhoA and ROCK2 protein expression. Exogenous expression of eIF5A increased RhoA and ROCK2 expression in 779E cells (Fig. 5D). Similar findings were obtained in PANC1 cells overexpressing or depleted of eIF5A (Fig. 5E). These findings indicate that eIF5A is sufficient and necessary for proper expression of RhoA and ROCK2 proteins in PDAC cells.
eIF5A/RhoA/ROCK2 Signaling Regulate Actin-myosin-mediated PDAC Cell Spreading and Migration
MYPT (myosin phosphatase targeting subunit) is an established substrate and critical downstream effector of ROCK (36). The RhoA-ROCK-MYPT pathway controls the phosphorylation of myosin light chain, which in turn facilitates actin-myosin force generation needed for cell spreading and migration (41). eIF5A knockdown reduced MYPT phosphorylation, whereas eIF5A overexpression promoted phosphorylation of MYPT (Fig. 6A). Finally, eIF5A knockdown or inhibition of eIF5A hypusination by GC7 reduced cell migration and spreading (Figs. 1, A, B, and D, and 6C), which was comparable with treatment with Y-27632, a well established ROCK kinase inhibitor (Fig. 6, B and D). Collectively, these data indicate that eIF5A regulates RhoA/ROCK2 expression to control cytoskeletal organization, cell migration, and metastatic potential of PDAC cells.
FIGURE 6.

eIF5A hypusination posttransciptionally controls Rho/ROCK signaling. A, Western blot analysis of phospho- or total MYPT levels in PANC1 cells expressing control or eIF5A shRNA (left) or 779E cells containing empty vector or eIF5A-encoding vector (right). eIF5A blots validate eIF5A knockdown or overexpression. GAPDH served as a loading control. B, effect of Rho/ROCK pathway inhibition on migration and invasion of PANC1 cells. PANC1 cells were subjected to transwell migration assay (left) or invasion assay (right) treated with DMSO or 10 μm Y-27362. Bar graphs show the number of migrated or invaded cells per field of view (n = 4). C, cell spreading assays for GC7-treated cells. PANC1 cells with or without GC7 pre-treatment (25 μm, 24 h) were seeded on collagen (type I) and their adhesion/spreading kinetics were monitored using the xCELLigence system, and presented as Cell Index (n = 4). * represents p < 0.05 as determined by Student's t test. D, cell spreading assays for Y27632-treated cells. PANC1 cells with or without Y-27632 treatment (10 μm) were seeded on collagen (type I) and their spreading kinetics were monitored using the xCELLigence system (n = 4). E, qPCR analysis of RhoA and ROCK2 mRNA levels in 779E cells stably depleted of eIF5A (n = 3). ** denotes p > 0.05 as determined by Student's t test. F, Western blot analysis of RhoA and ROCK2 expression in 779E or PANC1 cells treated without or with 25 μm GC7 for 24 h, where GAPDH served as a loading control (left panel). qPCR analysis of RhoA and ROCK2 mRNA levels in GC7-treated 779E or PANC1 cells normalized to control cells are shown on the right (n = 3).
eIF5A Regulates RhoA and ROCK2 Levels Post-transcriptionally
As described earlier, we determined whether eIF5A knockdown affects mRNA expression of RhoA and ROCK2 by qPCR in 779E cells. We found that the mRNA levels of neither RhoA nor ROCK2 were significantly affected by eIF5A knockdown, suggesting that eIF5A controls RhoA and ROCK2 expression at the post-transcriptional level (Fig. 6E). Furthermore, blockade of eIF5A hypusination by GC7 reduced RhoA and ROCK2 protein levels in both 779E and PANC1 cells without significantly reducing their mRNA levels (Fig. 6F). These findings indicate that RhoA and ROCK2 protein expression is regulated post-transcriptionally by eIF5A in a hypusination-dependent manner.
Discussion
In this study, we demonstrate a pivotal role for eIF5A in regulating an integrated network of cytoskeleton-associated proteins involved in PDAC cell migration and metastasis of which RhoA/ROCK2 signaling played a central role. Hypusinated eIF5A was observed to modulate RhoA/ROCK2 protein levels to control actin-myosin-mediated cell spreading and migration. Genetic or pharmacological intervention of eIF5A activity inhibited RhoA/ROCK2 expression and inhibited PDAC migration, invasion, and metastasis in vivo. Although it cannot be excluded that the reduction of metastatic burden observed in our metastasis assays is due to reduced cell growth caused by eIF5A depletion (13), we believe that the suppression of liver metastasis induced by eIF5A knockdown in the chick CAM model is too significant to be explained simply by slower cell growth (Fig. 1E). Also, because the assay was conducted for only 48 h in which eIF5A knockdown leads to only a modest 20% decrease in PANC1 cell proliferation (13), we favor the notion that eIF5A depletion abrogated the ability of PANC1 cells to metastasize to the liver.
Our previous work demonstrated that mutational activation of K-Ras induces aberrant protein expression of hypusinated eIF5A (13). In fact, eIF5A protein expression is low in normal pancreatic ducts and significantly up-regulated in diseased tissues where it correlates with disease progression (13). This is in contrast to DHPS and DOHH, which are abundantly expressed in both normal ducts and diseased PDAC tissues. These findings suggest that unlike eIF5A, DHPS and DOHH expression is not regulated by mutationally activated K-Ras. Also, they are not likely limiting factors in eIF5A hypusination considering their abundant expression in normal pancreatic ductal tissues.
Based on our previous results (13) and results here, we present a working model for how eIF5A contributes to K-Ras-mediated PDAC malignancy (Fig. 7). Our previous work demonstrated that oncogenic K-Ras activation induces up-regulation of eIF5A protein and its hypusination (13). Here we show that up-regulation of eIF5A protein is sufficient and necessary to promote Rho/ROCK protein expression and downstream signaling (Figs. 3A, 5, and 6A). Together these findings are consistent with a model in which K-Ras activation drives the up-regulation of activated eIF5A, which in turn, promotes Rho/ROCK expression, leading to cytoskeletal reorganization as well as other pro-tumorigenic and pro-survival programs necessary for cancer metastasis. In support of this model, K-Ras has been shown to stimulate RhoA/ROCK signaling to promote tumor cell survival and metastasis (43–46). However, K-Ras-induced eIF5A expression also likely contributes to tumor metastasis by modulating the expression levels of other key signaling/cytoskeletal proteins including PEAK1 (pseudopodium-enriched atypical kinase 1), which is critically involved in PDAC progression (13).
FIGURE 7.
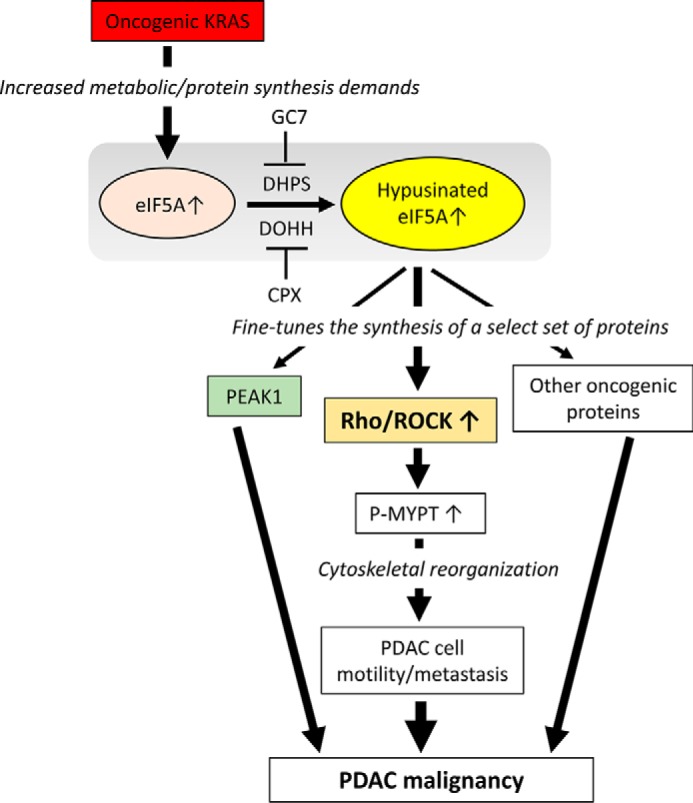
Model depicting the proposed role of eIF5A in PDAC malignancy. Oncogenic K-Ras activation induces eIF5A protein expression and its hypusination (13). Activated eIF5A promotes the expression of key cytoskeletal signaling molecules including Rho, ROCK, and PEAK1 (pseudopodium enriched atypical kinase 1) as well as other pro-oncogenic proteins to promote PDAC malignancy. In particular, eIF5A stimulates Rho/ROCK signaling to promote phosphorylation of MYPT (myosin phosphatase targeting protein) and subsequent cytoskeletal reorganization to enhance PDAC cell motility and metastasis. eIF5A hypusination can be targeted by small molecules including GC7 and CPX, an inhibitor of DHPS and DOHH, respectively.
The majority of work on Rho and ROCK has focused on the mechanisms that control their enzymatic activities (37). In fact, little attention has been given to understanding how Rho/ROCK protein levels are modulated in normal and malignant cells (47, 48). This is surprising as Rho/ROCK protein levels are significantly increased in multiple types of cancers, and their overexpression correlates with poor prognosis (49–52). It has also been reported that RhoA overexpression facilitates its own translocation from the cytosol to the plasma membrane leading to increased GTPase activity and invasive potential of tumor cells (53), indicating that the expression levels of RhoA is relevant for its activity. Altogether these findings implicate eIF5A-driven RhoA/ROCK amplification as an important mechanism contributing to metastatic tumor progression.
Our finding that eIF5A regulates the protein expression levels of Rho and ROCK, as well as other cytoskeleton-associated proteins (Table 2 and Figs. 3A and 5), has important implications for how the actin-myosin cytoskeleton becomes aberrantly activated in metastatic PDAC cells. During cell migration the physical process of cell body translocation requires extensive membrane protrusion, blebbing, and cytoskeletal remodeling (54). These processes are metabolically demanding, requiring significant amounts of energy and newly synthesized proteins (55). These components are assembled into and regulate the highly dynamic actin-myosin cytoskeleton, the central motor driving the protrusive and contractile forces necessary for cell body translocation (56). Our findings suggest that eIF5A may play a primary role in this process by specifically regulating translation of cytoskeleton-regulatory proteins, including RhoA and ROCK. Indeed, as mRNA levels of RhoA and ROCK2 were not significantly altered by eIF5A inhibition (Fig. 6, E and F), and pharmacological inhibition of the translational activity of eIF5A by GC7 reduces RhoA and ROCK2 protein expression (Fig. 6F). Thus, we conclude that eIF5A likely controls RhoA, and ROCK2 protein expression (as well as other cytoskeleton-associated proteins) at the translational level.
Although it is not precisely known how eIF5A mediates translation, recent work has shed new insight into this process. eIF5A and its bacterial homologue, EF-P, have been reported to play a prominent role in facilitating the synthesis of polyproline stretch-containing proteins (17–19). Notably, ROCK2 contains two polyproline stretches at amino acids 4–6 and amino acids 1287–1289, which conform to the eIF5A recognition motif (17–19). Other cytoskeleton proteins decreased in response to eIF5A knockdown with polyproline domains include AKAP13, TJP1, TRIP10 (Table 2), and PEAK1 (pseudopodium-enriched atypical kinase 1). PEAK1 is non-receptor tyrosine kinase that we have recently shown plays an important role in PDAC pathogenesis downstream of eIF5A (13). These findings are consistent with the concept that the presence of polyproline tracts confers eIF5A dependence for protein synthesis. However, eIF5A does not exclusively regulate the expression of proteins with polyproline stretches. In bacteria, EF-P was shown to control the expression of proteins with motifs other than polyproline tracts (36). Furthermore, a recent report by Memin et al. (57) showed that polyproline stretches were not enriched in DOHH-regulated proteins in cervical cancer cells. In fact, RhoA does not contain a polyproline tract, but is regulated by eIF5A (Figs. 3A, 5, and 6). Our proteomic profiling data also support this notion, as statistical analysis revealed that although the presence of the polyproline stretch represents a major signature of eIF5A-regulated proteins, it is not the only determinant for eIF5A dependence in protein expression. Additional work is warranted to identify the specific proteins modulated by eIF5A and to understand how this process is contextually regulated in normal and diseased cells including PDAC. Nevertheless, it is becoming more apparent that eIF5A fine-tunes the production of a select set of proteins and is not a general regulator of global protein synthesis (22, 23).
Our findings suggest that targeting Rho/ROCK protein expression by inhibiting eIF5A hypusination may be a feasible strategy to inhibit their cellular activities. Indeed, simultaneously down-regulating the expression levels of RhoA and its major downstream effector ROCK, as well as other cytoskeleton-associated proteins (Table 2, Fig. 4B), could serve as a potent means to target metastatic PDAC cells. In this regard, the DOHH inhibitor CPX, and polyamine biosynthesis inhibitor α-difluoromethylornithine are both already approved by the United States Food and Drug Administration as an anti-fungal agent and anti-hirsutism drug, respectively. Thus, CPX and/or α-difluoromethylornithine could be rapidly repurposed for PDAC treatment, alone or in combination with existing therapeutics like gemcitabine.
In summary, our findings indicate that eIF5A modulates the expression level of RhoA/ROCK2 proteins in PDAC cells to enhance their metastatic potential. The de novo development or repurposing of small molecule inhibitors that target the eIF5A hypusination pathway could be a promising strategy to combat this deadly disease.
Author Contributions
K. F. and S. C. designed and executed experiments and wrote the manuscript. M. W., J. S., and T. W. executed experiments. R. K. designed experiments and wrote the manuscript. All authors analyzed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank all the members of the Klemke laboratory for discussion, Dr. Jens Lykke-Andersen for kindly providing anti-Xrn1 antibody and Dr. Andrew Lowy for kindly providing 779E and PDA4964 cell line. We also thank Drs. Hartmut Hanauske-Abel (Rutgers University) and Myung-Hee Park (National Institute of Dental and Craniofacial Research) for kindly providing the NIH353 antibody and helpful comments.
This work was supported, in whole or in part, by National Institutes of Health Grants CA180374 (to K. F.), CA097022 and CA182495 (to R. K.). No author has an actual or perceived conflict of interest with the contents of this article.

This article contains supplemental Tables S1 and S2.
- PDAC
- pancreatic ductal adenocarcinoma
- PanIN
- pancreatic intraepithelial neoplasia
- ROCK
- Rho-associated kinase
- DHPS
- deoxyhypusine synthase
- DOHH
- deoxyhypusine hydroxylase
- GC7
- N(1)-guanyl-1,7-diaminoheptane
- MYPT
- myosin phosphatase targeting protein
- PEAK1
- pseudopodium-enriched atypical kinase 1
- mTOR
- mammalian target of rapamycin
- CPX
- ciclopirox olamine
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- EASE
- expression analysis systematic explorer
- qPCR
- quantitative PCR
- CAM
- chorioallantoic membrane.
References
- 1.Kern S. E., Shi C., and Hruban R. H. (2011) The complexity of pancreatic ductal cancers and multidimensional strategies for therapeutic targeting. J. Pathol. 223, 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruns C. J., Harbison M. T., Kuniyasu H., Eue I., and Fidler I. J. (1999) In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia 1, 50–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulke M. H. (2002) Metastatic pancreatic cancer. Curr. Treat. Options Oncol. 3, 449–457 [DOI] [PubMed] [Google Scholar]
- 4.Rhim A. D., Mirek E. T., Aiello N. M., Maitra A., Bailey J. M., McAllister F., Reichert M., Beatty G. L., Rustgi A. K., Vonderheide R. H., Leach S. D., and Stanger B. Z. (2012) EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hingorani S. R., Petricoin E. F., Maitra A., Rajapakse V., King C., Jacobetz M. A., Ross S., Conrads T. P., Veenstra T. D., Hitt B. A., Kawaguchi Y., Johann D., Liotta L. A., Crawford H. C., Putt M. E., Jacks T., Wright C. V., Hruban R. H., Lowy A. M., and Tuveson D. A. (2003) Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 [DOI] [PubMed] [Google Scholar]
- 6.Aguirre A. J., Bardeesy N., Sinha M., Lopez L., Tuveson D. A., Horner J., Redston M. S., and DePinho R. A. (2003) Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 17, 3112–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins M. A., Brisset J. C., Zhang Y., Bednar F., Pierre J., Heist K. A., Galbán C. J., Galbán S., and di Magliano M. P. (2012) Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS ONE 7, e49707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ying H., Kimmelman A. C., Lyssiotis C. A., Hua S., Chu G. C., Fletcher-Sananikone E., Locasale J. W., Son J., Zhang H., Coloff J. L., Yan H., Wang W., Chen S., Viale A., Zheng H., Paik J. H., Lim C., Guimaraes A. R., Martin E. S., Chang J., Hezel A. F., Perry S. R., Hu J., Gan B., Xiao Y., Asara J. M., Weissleder R., Wang Y. A., Chin L., Cantley L. C., and DePinho R. A. (2012) Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Son J., Lyssiotis C. A., Ying H., Wang X., Hua S., Ligorio M., Perera R. M., Ferrone C. R., Mullarky E., Shyh-Chang N., Kang Y., Fleming J. B., Bardeesy N., Asara J. M., Haigis M. C., DePinho R. A., Cantley L. C., and Kimmelman A. C. (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajasekhar V. K., Viale A., Socci N. D., Wiedmann M., Hu X., and Holland E. C. (2003) Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol. Cell 12, 889–901 [DOI] [PubMed] [Google Scholar]
- 11.Wolpin B. M., Hezel A. F., Abrams T., Blaszkowsky L. S., Meyerhardt J. A., Chan J. A., Enzinger P. C., Allen B., Clark J. W., Ryan D. P., and Fuchs C. S. (2009) Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J. Clin. Oncol. 27, 193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Javle M. M., Shroff R. T., Xiong H., Varadhachary G. A., Fogelman D., Reddy S. A., Davis D., Zhang Y., Wolff R. A., and Abbruzzese J. L. (2010) Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: results of two phase II studies. BMC Cancer 10, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujimura K., Wright T., Strnadel J., Kaushal S., Metildi C., Lowy A. M., Bouvet M., Kelber J. A., and Klemke R. L. (2014) A hypusine-eIF5A-PEAK1 switch regulates the pathogenesis of pancreatic cancer. Cancer Res. 74, 6671–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park M. H. (2006) The post-translational synthesis of a polyamine-derived amino acid, hypusine, in the eukaryotic translation initiation factor 5A (eIF5A). J. Biochem. 139, 161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maier B., Tersey S. A., and Mirmira R. G. (2010) Hypusine: a new target for therapeutic intervention in diabetic inflammation. Discov. Med. 10, 18–23 [PubMed] [Google Scholar]
- 16.Nishimura K., Lee S. B., Park J. H., and Park M. H. (2012) Essential role of eIF5A-1 and deoxyhypusine synthase in mouse embryonic development. Amino Acids 42, 703–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ude S., Lassak J., Starosta A. L., Kraxenberger T., Wilson D. N., and Jung K. (2013) Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science 339, 82–85 [DOI] [PubMed] [Google Scholar]
- 18.Doerfel L. K., Wohlgemuth I., Kothe C., Peske F., Urlaub H., and Rodnina M. V. (2013) EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 339, 85–88 [DOI] [PubMed] [Google Scholar]
- 19.Gutierrez E., Shin B. S., Woolstenhulme C. J., Kim J. R., Saini P., Buskirk A. R., and Dever T. E. (2013) eIF5A promotes translation of polyproline motifs. Mol. Cell 51, 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mandal A., Mandal S., and Park M. H. (2014) Genome-wide analyses and functional classification of proline repeat-rich proteins: potential role of eIF5A in eukaryotic evolution. PLoS ONE 9, e111800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan A. A., and Rubenstein E. (2013) Proline: the distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS ONE 8, e53785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li C. H., Ohn T., Ivanov P., Tisdale S., and Anderson P. (2010) eIF5A promotes translation elongation, polysome disassembly and stress granule assembly. PLoS ONE 5, e9942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathews M. B., and Hershey J. W. (2015) The translation factor eIF5A and human cancer. Biochim. Biophys. Acta 1849, 836–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee N. P., Tsang F. H., Shek F. H., Mao M., Dai H., Zhang C., Dong S., Guan X. Y., Poon R. T., and Luk J. M. (2010) Prognostic significance and therapeutic potential of eukaryotic translation initiation factor 5A (eIF5A) in hepatocellular carcinoma. Int. J. Cancer 127, 968–976 [DOI] [PubMed] [Google Scholar]
- 25.Tunca B., Tezcan G., Cecener G., Egeli U., Zorluoglu A., Yilmazlar T., Ak S., Yerci O., Ozturk E., Umut G., and Evrensel T. (2013) Overexpression of CK20, MAP3K8 and EIF5A correlates with poor prognosis in early-onset colorectal cancer patients. J. Cancer Res. Clin. Oncol. 139, 691–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reichert M., Takano S., von Burstin J., Kim S. B., Lee J. S., Ihida-Stansbury K., Hahn C., Heeg S., Schneider G., Rhim A. D., Stanger B. Z., and Rustgi A. K. (2013) The Prrx1 homeodomain transcription factor plays a central role in pancreatic regeneration and carcinogenesis. Genes Dev. 27, 288–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elias J. E., Haas W., Faherty B. K., and Gygi S. P. (2005) Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nature Methods 2, 667–675 [DOI] [PubMed] [Google Scholar]
- 28.Mi H., Lazareva-Ulitsky B., Loo R., Kejariwal A., Vandergriff J., Rabkin S., Guo N., Muruganujan A., Doremieux O., Campbell M. J., Kitano H., and Thomas P. D. (2005) The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 33, D284-D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y., Kelber J. A., Tran Cao H. S., Cantin G. T., Lin R., Wang W., Kaushal S., Bristow J. M., Edgington T. S., Hoffman R. M., Bouvet M., Yates J. R. 3rd, and Klemke R. L. (2010) Pseudopodium-enriched atypical kinase 1 regulates the cytoskeleton and cancer progression. Proc. Natl. Acad. Sci. U.S.A. 107, 10920–10925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irelan J. T., Wu M. J., Morgan J., Ke N., Xi B., Wang X., Xu X., and Abassi Y. A. (2011) Rapid and quantitative assessment of cell quality, identity, and functionality for cell-based assays using real-time cellular analysis. J. Biomol. Screen. 16, 313–322 [DOI] [PubMed] [Google Scholar]
- 31.Stoletov K., Strnadel J., Zardouzian E., Momiyama M., Park F. D., Kelber J. A., Pizzo D. P., Hoffman R., VandenBerg S. R., and Klemke R. L. (2013) Role of connexins in metastatic breast cancer and melanoma brain colonization. J. Cell Sci. 126, 904–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zetter B. R. (1993) Adhesion molecules in tumor metastasis. Semin. Cancer Biol. 4, 219–229 [PubMed] [Google Scholar]
- 33.Shintani Y., Hollingsworth M. A., Wheelock M. J., and Johnson K. R. (2006) Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH2-terminal kinase 1 and up-regulating N-cadherin expression. Cancer Res. 66, 11745–11753 [DOI] [PubMed] [Google Scholar]
- 34.Dunér S., Lopatko Lindman J., Ansari D., Gundewar C., and Andersson R. (2010) Pancreatic cancer: the role of pancreatic stellate cells in tumor progression. Pancreatology 10, 673–681 [DOI] [PubMed] [Google Scholar]
- 35.Saxena M., and Christofori G. (2013) Rebuilding cancer metastasis in the mouse. Mol. Oncol. 7, 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hersch S. J., Wang M., Zou S. B., Moon K. M., Foster L. J., Ibba M., and Navarre W. W. (2013) Divergent protein motifs direct elongation factor P-mediated translational regulation in Salmonella enterica and Escherichia coli. MBio 4, e00180–00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Narumiya S., Tanji M., and Ishizaki T. (2009) Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 28, 65–76 [DOI] [PubMed] [Google Scholar]
- 38.Li Z., Chang Z., Chiao L. J., Kang Y., Xia Q., Zhu C., Fleming J. B., Evans D. B., and Chiao P. J. (2009) TrkBT1 induces liver metastasis of pancreatic cancer cells by sequestering Rho GDP dissociation inhibitor and promoting RhoA activation. Cancer Res. 69, 7851–7859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel R. A., Forinash K. D., Pireddu R., Sun Y., Sun N., Martin M. P., Schönbrunn E., Lawrence N. J., and Sebti S. M. (2012) RKI-1447 is a potent inhibitor of the Rho-associated ROCK kinases with anti-invasive and antitumor activities in breast cancer. Cancer Res. 72, 5025–5034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vigil D., Kim T. Y., Plachco A., Garton A. J., Castaldo L., Pachter J. A., Dong H., Chen X., Tokar B., Campbell S. L., and Der C. J. (2012) ROCK1 and ROCK2 are required for non-small cell lung cancer anchorage-independent growth and invasion. Cancer Res. 72, 5338–5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riento K., and Ridley A. J. (2003) Rocks: multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 4, 446–456 [DOI] [PubMed] [Google Scholar]
- 42.Wang L., Lawrence J. C. Jr., Sturgill T. W., and Harris T. E. (2009) Mammalian target of rapamycin complex 1 (mTORC1) activity is associated with phosphorylation of raptor by mTOR. J. Biol. Chem. 284, 14693–14697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Castro Carpeño J., and Belda-Iniesta C. (2013) KRAS mutant NSCLC, a new opportunity for the synthetic lethality therapeutic approach. Transl. Lung Cancer Res. 2, 142–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basu Roy U. K., Henkhaus R. S., Loupakis F., Cremolini C., Gerner E. W., and Ignatenko N. A. (2013) Caveolin-1 is a novel regulator of K-RAS-dependent migration in colon carcinogenesis. Int. J. Cancer 133, 43–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xia M., and Land H. (2007) Tumor suppressor p53 restricts Ras stimulation of RhoA and cancer cell motility. (2007) Tumor suppressor p53 restricts Ras stimulation of RhoA and cancer cell motility. Nat. Struct. Mol. Biol. 14, 215–223 [DOI] [PubMed] [Google Scholar]
- 46.Konstantinidou G., Ramadori G., Torti F., Kangasniemi K., Ramirez R. E., Cai Y., Behrens C., Dellinger M. T., Brekken R. A., Wistuba I. I., Heguy A., Teruya-Feldstein J., and Scaglioni P. P. (2013) RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov. 3, 444–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xing L., Yao X., Williams K. R., and Bassell G. J. (2012) Negative regulation of RhoA translation and signaling by hnRNP-Q1 affects cellular morphogenesis. Mol. Biol. Cell 23, 1500–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H. R., Zhang Y., Ozdamar B., Ogunjimi A. A., Alexandrova E., Thomsen G. H., and Wrana J. L. (2003) Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 302, 1775–1779 [DOI] [PubMed] [Google Scholar]
- 49.Faried A., Faried L. S., Usman N., Kato H., and Kuwano H. (2007) Clinical and prognostic significance of RhoA and RhoC gene expression in esophageal squamous cell carcinoma. Ann. Surg. Oncol. 14, 3593–3601 [DOI] [PubMed] [Google Scholar]
- 50.Liu X., Chen D., and Liu G. (2014) Overexpression of RhoA promotes the proliferation and migration of cervical cancer cells. Biosci. Biotechnol. Biochem. 78, 1895–1901 [DOI] [PubMed] [Google Scholar]
- 51.Kamai T., Tsujii T., Arai K., Takagi K., Asami H., Ito Y., and Oshima H. (2003) Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin. Cancer Res. 9, 2632–2641 [PubMed] [Google Scholar]
- 52.Nakamori S., Tamura S., Arai I., Kameyama M., Furukawa F., Ishikawa O., Imaoka S., Yoshioka K., Mukai M., Shinkai K., and Akedo H. (1996) Increased expression of RhoAp21 protein is involved in progression and metastasis of colorectal carcinoma. Rec. Adv. Gastroenterol. Carcinogenesis 1, 901–904 [Google Scholar]
- 53.Yoshioka K., Nakamori S., and Itoh K. (1999) Overexpression of small GTP-binding protein RhoA promotes invasion of tumor cells. Cancer Res. 59, 2004–2010 [PubMed] [Google Scholar]
- 54.Bravo-Cordero J. J., Hodgson L., and Condeelis J. (2012) Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol. 24, 277–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen E. I., Hewel J., Krueger J. S., Tiraby C., Weber M. R., Kralli A., Becker K., Yates J. R. 3rd, and Felding-Habermann B. (2007) Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 67, 1472–1486 [DOI] [PubMed] [Google Scholar]
- 56.Le Clainche C., and Carlier M. F. (2008) Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol. Rev. 88, 489–513 [DOI] [PubMed] [Google Scholar]
- 57.Mémin E., Hoque M., Jain M. R., Heller D. S., Li H., Cracchiolo B., Hanauske-Abel H. M., Pe'ery T., and Mathews M. B. (2014) Blocking eIF5A modification in cervical cancer cells alters the expression of cancer-related genes and suppresses cell proliferation. Cancer Res. 74, 552–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.