

Background: Prostaglandin E2 (PGE2) is an inflammatory mediator produced in cancer.
Results: B16 melanoma cells injected into mice metastasized to bone and soft tissues by activating PGE2-EP4 signaling in stromal cells, inducing osteoclast activation, angiogenesis, and cancer cell proliferation.
Conclusion: Stromal cell PGE2 is a key mediator of melanoma tumorigenesis and metastasis.
Significance: EP4 receptor blockade is a new potential therapy for tumor metastasis.
Keywords: angiogenesis, bone, metastasis, prostaglandin, tumor microenvironment, B16 melanoma, Bone resorption, EP4 receptor, host-cancer interaction, prostaglandin E
Abstract
The stromal cells associated with tumors such as melanoma are significant determinants of tumor growth and metastasis. Using membrane-bound prostaglandin E synthase 1 (mPges1−/−) mice, we show that prostaglandin E2 (PGE2) production by host tissues is critical for B16 melanoma growth, angiogenesis, and metastasis to both bone and soft tissues. Concomitant studies in vitro showed that PGE2 production by fibroblasts is regulated by direct interaction with B16 cells. Autocrine activity of PGE2 further regulates the production of angiogenic factors by fibroblasts, which are key to the vascularization of both primary and metastatic tumor growth. Similarly, cell-cell interactions between B16 cells and host osteoblasts modulate mPGES-1 activity and PGE2 production by the osteoblasts. PGE2, in turn, acts to stimulate receptor activator of NF-κB ligand expression, leading to osteoclast differentiation and bone erosion. Using eicosanoid receptor antagonists, we show that PGE2 acts on osteoblasts and fibroblasts in the tumor microenvironment through the EP4 receptor. Metastatic tumor growth and vascularization in soft tissues was abrogated by an EP4 receptor antagonist. EP4-null Ptger4−/− mice do not support B16 melanoma growth. In vitro, an EP4 receptor antagonist modulated PGE2 effects on fibroblast production of angiogenic factors. Our data show that B16 melanoma cells directly influence host stromal cells to generate PGE2 signals governing neoangiogenesis and metastatic growth in bone via osteoclast erosive activity as well as angiogenesis in soft tissue tumors.
Introduction
Prostaglandin E2 (PGE2)2 is an inflammatory mediator produced by many cells and is known to have effects on aspects of tumorigenesis, including cell proliferation, survival, and invasiveness (1). PGE production is regulated by three metabolic steps: the release of arachidonic acid from membrane phospholipids by phospholipase A2 (PLA2), the conversion of arachidonic acid to PGH2 by cyclooxygenases (COX), and the synthesis of PGE2 by PGE synthases (PGES). There are two COX enzymes, COX-1 and COX-2, and three PGES, membrane-bound PGE synthase (mPGES)1, mPGES-2, and cytosolic PGES, in the metabolic pathway of PGE synthesis, and increased expression of COX-2 and mPGES-1 have been implicated in various inflammatory diseases (2, 3). Previous studies have also shown a correlation between tumor growth and PG synthesis. Blocking PG synthesis by nonsteroidal anti-inflammatory drugs has been reported to reduce the risk of breast cancer and colon carcinogenesis (4, 5). Cancer cells from breast, skin, kidney, gastric, lung, and colon cancers express COX-2 highly, and COX-induced PGE2 production enhances their tumorigenesis, suggesting that PGE2 acts directly on these cells to enhance cell proliferation, invasion, and tumor metastasis (6–8). On the other hand, tumor cell PGE2 may act on non-tumor cells, such as stromal cells, vascular endothelial cells, and natural killer cells, to regulate angiogenesis and natural killer cell function, which are critical for tumorigenesis and metastasis (9, 10). We have shown that the expression of COX-2 was elevated in the surrounding stromal cells and osteoblasts in the region of bone metastasis of cancer (11) and that PGE2 is mainly produced by osteoblasts in bone tissue and may act as an inducer of bone resorption associated with the metastasis of cancer to bone (12). However the precise importance of PGE2 derived from other stromal cells has not been determined.
We previously generated mPGES-1-deficient (mPges1−/−) mice to examine the role of mPGES-1 in PGE production and its biological significance and found that PGE2 production by macrophages treated with LPS was impaired in mPges1−/− mice (13). In addition, we have reported that the bone resorption associated with inflammation was attenuated in mPges1−/− mice because of the lack of PGE production by osteoblasts (14). The outstanding question, therefore, is the role of mPGES-1/PGE in the host stromal cells in driving tumorigenesis and bone metastasis.
The metastasis of cancer to bone is accompanied by severe osteolysis with enhanced osteoclastic bone resorption. Previous studies have identified the receptor activator of NF-κB ligand (RANKL) as a pivotal factor in osteoclast differentiation and bone resorption (15–18). Osteoblasts express RANKL in response to bone-resorbing factors and interact with osteoclast precursors expressing RANK, inducing their differentiation into osteoclasts (17–19). An anti-RANKL antibody clearly suppressed bone metastasis and the occurrence of skeleton-related events in both animals and humans (20, 21). We have reported that direct contact between human breast cancer cells and osteoblasts induced RANKL expression in osteoblasts to stimulate osteoclastogenesis (22). We therefore wished to assess the mechanism of melanoma-induced osteolysis during bone metastasis through the RANK-RANKL axis.
The effects of PGE2 are mediated through a family of G-protein-coupled receptor subtypes identified as EP1, EP2, EP3, and EP4 in individual target cells (23). Using EP4-null mice (Ptger4−/−) and specific agonists for the respective EPs, we have demonstrated previously that PGE2 produced by osteoblasts in bone binds to the EP4 receptor of osteoblasts to initiate RANKL-dependent osteoclast formation (24, 25). EP-related signaling appears to play a key role in both cancer and stromal cells of the microenvironment and needs to be better understood to gain an insight into the mechanisms of PGE2-dependent tumor growth and metastasis.
In this study, we used mPges1−/− mice, Ptger4−/− mice, and specific antagonists of EPs to examine the role of PGE2 derived from normal host cells on the growth and metastasis of malignant melanoma. PGE2 production and EP4 receptor signaling in host stromal lineage cells in the tumor microenvironment may play a key role in tumor growth, bone metastasis with severe osteolysis, and also metastasis to various soft tissues.
Experimental Procedures
Animals, Cells, and Reagents
The mPges1−/−, Ptger4−/−, and littermate wild-type mice were established by gene targeting as described previously (13, 26). The mouse malignant melanoma cell line B16 was obtained from the RIKEN Cell Bank (Ibaraki, Japan), and we isolated a clone that consistently underwent a high frequency (∼100%) of bone metastasis (12). B16 cells were cultured in DMEM with 10% FBS at 37 °C under 5% CO2 in air.
Metastasis Model of B16 Cells in Mice
B16 cells (2 × 105 cells) were suspended in 0.1 ml of PBS and injected into the tail vein of 6-week-old male mice. To quantitate the metastases, the numbers of black tumor nodules in the lungs, liver, and kidneys were counted. The EP1 antagonist (catalog no. 8713), EP3 antagonist (catalog no. AE5-599), and EP4 antagonist (AE3-208) were prepared by Ono Pharmaceutical Co., Ltd. (12). The EP2 antagonist (catalog no. TG4-155) was obtained from Cayman Chemical Co. Ltd. (Ann Arbor, MI). The respective EP antagonists (10 mg/kg of body weight/day) were administered by oral gavage to mice. As a control group, mice were administered distilled water. All procedures were performed in accordance with the institutional guidelines for animal research at the Tokyo University of Agriculture and Technology.
Implantation of B16 Tumors in Mice
B16 cells (1.2 × 105 cells) were suspended in 0.06 ml of PBS and injected into the dorsal subcutaneous tissue of 6-week-old male mice under anesthesia with pentobarbital. The size of the tumor was determined by direct measurement of the tumor dimensions using calipers. To quantitate the tumor volume, the following equation was used: = [length/2 × width/2 × height/2] × 4/3.
Fluorescence Imaging of New Blood Vessels
The imaging reagent AngioSense750 (PerkinElmer Life Sciences) is a near infrared-labeled fluorescent macromolecule that remains localized in the vasculature for extended periods of time and enables imaging of blood vessels and angiogenesis. The mice were injected with AngioSense750 on day 15. After 24 h, the fluorescence was detected using the In-Vivo Imaging System FX (Kodak).
Measurement of Bone Mineral Density
The bone mineral density (BMD) of the femora was measured by dual x-ray absorptiometry (model DCS-600R, Aloka). The bone mineral content of the femora was closely correlated with the ash weight. The BMD was calculated as the bone mineral content of the measured area.
Micro CT Analysis
CT scanning of the femora was performed using a microfocus x-ray CT system (inspeXio SMX-90T, Shimadzu). Three-dimensional microstructural image data were reconstructed, and structural indices (bone volume/tissue volume, trabecular thickness, and trabecular separation) were calculated using TRI/3D-BON software (Ratoc System Engineering Co., Ltd).
Preparation of Bone Marrow Supernatant
To obtain the bone marrow supernatant from mice, bone marrow cells and cancellous bone fragments were collected with 0.5 ml of PBS from the tibiae, as reported previously (27). After centrifugation to remove the cells and bone fragments, the supernatant was collected for the measurement of PGE2.
RT-PCR Analysis
Total RNA was extracted from B16 tumors and cultured cells, and cDNA was synthesized from total RNA and amplified by PCR (9). The PCR primers for the mouse mPGES-1, mPGES-2, cytosolic PGES, RANKL, VEGF-A, bFGF, and GAPDH genes were used as reported previously (22). The PCR product was run on a 1.5% agarose gel and stained with ethidium bromide. The band intensity was measured by densitometric analysis using ImageJ.
Culture of Primary Mouse Osteoblastic Cells and B16 Cells
Primary osteoblastic cells were isolated from 2-day-old mouse calvariae as described previously (24). In co-culture experiments, B16 cells were fixed with 4% paraformaldehyde and washed three times with PBS. Osteoblasts were cultured for 6 h on the layer of fixed B16 cells.
Osteoclast Formation in Co-cultures of Mouse Bone Marrow Cells and Osteoblasts
Bone marrow cells (3 × 106 cells) were isolated from 6-week-old mouse tibiae and co-cultured with primary osteoblastic cells (1 × 104 cells) in 1 ml of α minimum Eagle's medium containing 10% FBS on 24-well plates. To examine the effects of B16 cells on osteoclast formation, fixed B16 cells were added to the co-culture system. After culturing for 7 days, the cells adhering to the well surface were stained for tartrate-resistant acid phosphatase, and tartrate-resistant acid phosphatase-positive multinucleated cells containing three or more nuclei per cell were counted as osteoclasts.
Culture of Primary Mouse Dermal Fibroblasts
Primary mouse dermal fibroblasts were isolated from 6- to 8-week-old mouse dorsal skin. Skin tissue was collected from each mouse and incubated in DMEM at 37 °C for 1 h, chopped into small pieces, and cultured in DMEM supplemented with 10% FBS at 37 °C under 5% CO2 in air. After 14 days, the outgrown dermal fibroblasts were collected by trypsin treatment.
Measurement of the PGE2, VEGF-A, and bFGF Content
The concentration of PGE2 in the bone marrow supernatant or conditioned medium was determined using an enzyme immunoassay (GE Healthcare). The concentration of VEGF-A in the conditioned medium was determined by ELISA (R&D Systems). The concentration of bFGF in the conditioned medium was also determined using an ELISA (R&D Systems).
Statistical Analysis
The data are expressed as the mean ± S.E. The significance of differences was analyzed using Student's t test.
Results
B16 Melanoma Tumorigenesis and Angiogenesis Are Attenuated in mPges1−/− Mice
To ascertain the contribution of PGE2 produced by cells in the microenvironment to the growth of primary melanomas and to tumor-associated angiogenesis, B16 cells were implanted on the backs of wild-type and mPges1−/− mice. A palpable tumor could clearly be detected in the wild-type mice from days 6–16, whereas tumor growth in mPges1−/− mice was reduced significantly (Figs. 1, A and B). The tumor growth was accompanied by the substantial formation of new blood vessels around the dorsal subcutaneous tumor in wild-type mice, but tumor-associated angiogenesis was attenuated in mPges1−/− mice (Fig. 1C). The reduced angiogenesis in mPges1−/− mice was confirmed by fluorescence imaging of new blood vessels around the subcutaneous tumor on day 16 after B16 injection (Fig. 1D). We detected an overall survival benefit in mPges1−/− mice compared with wild-type mice (data not shown).
FIGURE 1.
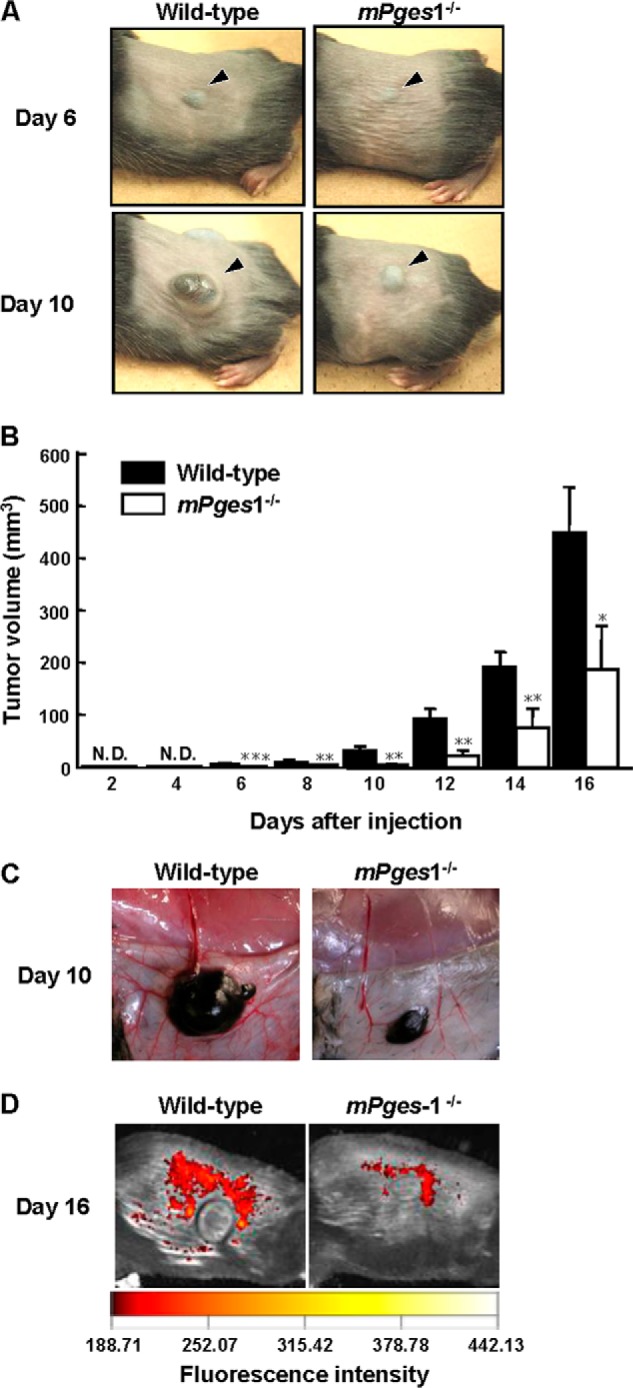
Tumor growth was attenuated in mPges1−/− mice injected with B16 cells. A, B16 cells were injected into the dorsal subcutaneous tissue of wild-type and mPges1−/− mice. Representative pictures of the tumors on days 6 and 10 after injection are shown. The arrowheads indicate the subcutaneous solid tumor. B, the tumor volume was determined using calipers. *, p < 0.05; **, p < 0.01; ***, p < 0.001. N.D., not detected. Data are mean ± S.E. of wild-type (n = 9) or mPges1−/− (n = 6) mice. C, representative pictures of new blood vessels around dorsal subcutaneous tumors in mPges1−/− or wild-type mice on day 10 after injection. D, fluorescence imaging of new blood vessels around dorsal subcutaneous tumors in mPges1−/− or wild-type mice on day 16 after injection.
Reduced Metastasis of B16 Melanoma in mPges1−/− Mice
To evaluate the role of mPGES-1 in melanoma metastasis to bone and subsequent osteolysis, B16 cells were injected into the tail vein of wild-type and mPges1−/− mice, and the femora were collected from the mice to measure the BMD and analyze bone morphometrics by micro CT. Because B16 cells actively produce melanin, the metastatic region could be detected as a black area in the femora on day 18 after the injection of B16 cells into wild-type mice. In mPge1−/− mice, the metastasis of B16 cells could be detected, but the area of metastasis was reduced markedly compared with that in the wild-type mice (Fig. 2A). The BMD measured by dual x-ray absorptiometry was decreased significantly in the presence of B16 metastases in the femora of wild-type mice, but the femoral BMD did not decrease in mPges1−/− mice injected with tumor cells (Fig. 2B). The levels of PGE2 in bone marrow supernatants collected from wild-type mice were elevated by the metastasis of B16 cells, but the PGE2 level in the bone marrow supernatants collected from mPges1−/− mice was significantly lower than that in wild-type mice and was not elevated by the injection of B16 cells (Fig. 2C).
FIGURE 2.

Metastases and osteolysis of B16 melanoma were attenuated in mPges1−/− mice. A, wild-type and mPges1−/− mice were injected with B16 cells or PBS into the tail vein. Representative pictures of melanoma metastases in the femora are shown after 18 days. B, BMD was measured in the total area of the femur. Data show mean ± S.E. of 13–27 mice. *, p < 0.01. C, the concentration of PGE2 in bone marrow supernatants was determined by enzyme immunoassay. Data are mean ± S.E. of 7–10 mice. *, p < 0.01; **, p < 0.001. D, micro CT analyses of the distal femur of mPges1−/− or wild-type mice, with or without B16 injection, were conducted, and three-dimensional images of trabecular bones were constructed. E, bone morphometric analyses of the distal femur were performed by micro CT to calculate the bone volume/tissue volume (BV/TV), trabecular thickness (Tb.Th), and trabecular separation (Tb.Sp). Data are mean ± S.E. of six mice.*, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, no significance. F, wild-type and mPges1−/− mice were injected with B16 cells, and the lungs, kidneys, and liver were collected on day 18 after injection. Representative pictures of tumors in each organ are shown in the top panels. The bottom panels show the number of cancer colonies. Data are mean ± S.E. of wild-type (n = 17) or mPges1−/− (n = 32) mice. *, p < 0.05; **, p < 0.001.
Using micro CT scans, we analyzed the three-dimensional microstructure of the trabecular bone in the femur. The trabecular bone in the femoral distal metaphysis was reduced by the metastasis of B16 cells in wild-type mice, but the microstructure of the trabecular bone was similar in mPges1−/− mice regardless of whether they had been injected with the B16 cells (Fig. 2D). Using the micro CT images, we measured the structural indices of trabecular bone, bone volume/tissue volume, trabecular thickness, and trabecular separation. In wild-type mice, the bone volume/tissue volume and trabecular thickness were reduced markedly, and the trabecular separation was elevated by the metastasis of B16 melanoma, but these changes did not occur in mPges1−/− mice (Fig. 2E). These data indicate that the bone metastasis of B16 cells induced osteolytic bone resorption via mPGES-1-dependent PGE2 production by non-tumor cells in the tumor microenvironment.
In this model, B16 cells injected into the tail veins metastasized simultaneously to bone and to various tissues, including the lung, kidney, and liver. To detect the incidence of B16 metastasis into soft tissues, we collected the lungs, kidneys, and liver from each mouse and measured the number of black colonies present. In wild-type mice, numerous metastatic colonies were detected in the tissues after B16 injection, but their number was reduced markedly in mPges1−/− mice (Fig. 2F). This indicates that PGE2 production mediated by mPGES-1 in the tumor microenvironment regulates melanoma metastasis to soft tissues.
Reduced Metastatic Tumor-associated Angiogenesis in mPges1−/− Mice
Angiogenesis plays a critical role in the metastasis of tumor cells, but the role of PGE2 in metastasis-associated angiogenesis is unknown. Using a fluorescence imaging technique with AngioSense750, new blood microvessel formation was analyzed in bone and other soft tissues. In wild-type mice, numerous metastatic foci were detected in the femora and tibiae, as shown by the presence of black spots, and red fluorescence, as an indicator of angiogenesis, was detected in bone with B16 metastases (Fig. 3A). On the other hand, neither bone metastasis nor angiogenesis was detectable in the mPges1−/− mice using any method, including visual inspection, x-rays, fluorescence imaging, or their combination (Fig. 3A).
FIGURE 3.

Reduced angiogenesis in B16 metastases in bone and soft tissues in mPges1−/− mice. A, wild-type and mPges1−/− mice were injected with B16 melanoma as in Fig. 2A, and new blood vessels were measured by fluorescence imaging with AngioSense750 in the femur and tibia of mice. Bone metastases detected in visible panels were co-localized with angiogenesis detected by AngioSense750 (Merge). B, fluorescence imaging of new blood vessels was performed in the lungs, liver, and kidneys as in A. Angiogenesis was clearly detected in all organs with B16 metastasis in wild-type mice (Merge), but it was reduced in mPges1−/− mice.
As shown in Fig. 2F, the metastasis of B16 melanoma to soft tissues, including the lungs, liver, and kidneys, was detected in wild-type mice, but the number of metastatic foci was reduced in all tissues of mPges1−/− mice. Angiogenesis associated with B16 metastasis was detected in the affected tissues of wild-type mice, but the fluorescent signals were reduced markedly in the tissues of mPges1−/− mice (Fig. 3B). We concluded that metastasis-related systemic neovascularization was dependent on PGE2 where the synthesis was mediated by mPGES-1 activity in each tissue, including bone.
Interaction between B16 Tumor Cells and Host Fibroblasts Modulates mPGES-1-mediated PGE2 Production and VEGF Expression
The contribution of secreted factors such as IL-1 from cancer cells to the growth and metastasis of cancer is well documented (28). On the other hand, we have shown previously that direct interactions between breast cancer cells and osteoblasts stimulated the production of PGE2 by the latter (12). Because this study indicated that host cell-derived PGE2 promotes melanoma growth, angiogenesis, and metastasis, we assessed the potential of B16 melanoma cells to modulate PGE2 production by the cells in their microenvironment, including fibroblasts and osteoblasts.
Dermal fibroblasts collected from mPges1−/− or wild-type mice were added to a layer of fixed B16 cells or untreated culture wells, and conditioned media were collected after 24 h to measure the concentration of PGE2. Fibroblasts from wild-type mice constitutively produced PGE2, but PGE production was enhanced greatly when the cells were cultured on a fixed B16 cell layer (Fig. 4A). By contrast, only a small amount of PGE2 was produced by dermal fibroblasts from mPges1−/− mice in the cultures, regardless of the presence of the fixed B16 cells (Fig. 4A). These results suggest that mPGES-1-dependent production of PGE2 by dermal fibroblasts plays a key role in melanoma growth and neovascularization.
FIGURE 4.

Reduced PGE production and expression of VEGF and FGF in dermal fibroblasts from mPges1−/− mice. A, dermal fibroblasts from mPges1−/− or wild-type mice were cultured on fixed B16 cells or control wells, and the concentration of PGE2 in the conditioned medium was determined after 24 h by enzyme immunoassay. Data are mean ± S.E. of 3–4 wells. *, p < 0.01; **, p < 0.001. B, dermal fibroblasts from mPges1−/− or wild-type mice were cultured with or without PGE2 (10 μm). After 48 h, total RNA was extracted, and the expression of VEGF-A and bFGF mRNA was determined by RT-PCR. The graphs show the relative intensity of VEGF-A188, VEGF-A164, VEGF-A120, and bFGF expression compared with the wild-type control. C, dermal fibroblasts from mPges1−/− or wild-type mice were cultured with or without PGE2 (10 μm). The concentrations of VEGF-A and bFGF in the conditioned media were determined after 48 h by ELISA. Data are mean ± S.E. of three wells. *, p < 0.05; **, p < 0.01; ***, p < 0.001. N.D., not detected.
Previous studies have shown that both VEGF and bFGF are essential for the formation of new blood vessels associated with tumors (29). We therefore examined the role of PGE2 in the production of VEGF and bFGF by dermal fibroblasts. The expression of VEGF-A and bFGF mRNA was enhanced by adding PGE2 to the cultures of dermal fibroblasts from wild-type mice, but the expression levels were attenuated in fibroblasts from mPges1−/− mice (Fig. 4B). The production of VEGF-A and bFGF was elevated significantly by adding PGE2 to dermal fibroblasts collected from wild-type mice. In fibroblasts collected from mPges1−/− mice, VEGF-A and bFGF levels were enhanced by adding PGE2 (Fig. 4C). However, the levels were still lower than those in cells from wild-type mice (Fig. 4C), probably because of the lack of autoamplification of the PGE-induced PGE production in mPges1−/− mice.
Osteoclast Formation by B16 Melanoma-Osteoblast Contacts Requires PGE2 Production to Enhance RANKL Expression
To examine how PGE2 produced by non-tumor cells in the tumor microenvironment is associated with the bone metastasis of B16 melanoma, we assessed the role of B16 cells on osteoclast formation by co-culturing bone marrow cells and osteoblasts from wild-type and mPges1−/− mice in the presence of fixed B16 cells. The results showed that fixed B16 cells markedly induced osteoclast formation in the case of cells from wild-type mice without any stimuli, but no osteoclasts were observed in the cells from mPges1−/− mice (Figs. 5, A and B). RT-PCR analysis of the expression of RANKL mRNA on day 4 of co-culture showed that RANKL expression was enhanced by the presence of fixed B16 cells in the co-culture of cells derived from wild-type mice but not in co-cultures with cells from mPges1−/− mice (Fig. 5C). The level of PGE2 in the conditioned medium of the co-cultures with wild-type cells was elevated with fixed B16 cells, but PGE2 could not be detected in the co-cultures of cells from mPges1−/− mice, even after incubation with fixed B16 cells (Fig. 5D).
FIGURE 5.

mPGES-1 is essential for osteoclast formation and PGE2 production induced by co-culture of B16 melanoma with mouse bone marrow cells and osteoblasts. Mouse bone marrow cells and osteoblasts from mPges1−/− or wild-type mice were co-cultured with or without fixed B16 cells for 7 days. A, representative fields of the staining for tartrate-resistant acid phosphatase, a specific marker of osteoclasts. B, the number of tartrate-resistant acid phosphatase-positive osteoclasts, multinucleated cells containing three or more nuclei. Data are mean ± S.E. of four wells. *, p < 0.001. C, expression of RANKL mRNA determined by RT-PCR. Total RNA collected from the adherent cells on day 4 in the co-cultures was used for RT-PCR analysis. The graph shows the relative intensity of RANKL expression compared with the wild type. D, the concentration of PGE2 in conditioned medium determined by enzyme immunoassay. Data are mean ± S.E. of four wells. *, p < 0.001. E, osteoblasts from mPges1−/− or wild-type mice were added to a layer of fixed B16 cells or culture plates. The total RNA was extracted after 6 h and used to measure the expression of mPGES-1, mPGES-2, and cytosolic PGES by RT-PCR. The graphs show the relative intensity of each gene expression compared with the wild-type control.
To clarify the importance of cell-cell interactions between the osteoblasts and B16 cells, osteoblasts from wild-type mice were cultured on a layer of fixed B16 cells for 6 h. The expression of mPGES-1 mRNA was induced markedly in osteoblasts by contact with fixed B16 cells collected from wild-type mice but not in those from mPges1−/− mice (Fig. 5E). The expression of mPGES-2 and cytosolic PGES mRNAs in osteoblasts collected from either wild-type or mPges1−/− mice was not influenced by contact with fixed B16 cells (Fig. 5E). These results suggest that mPGES-1-mediated PGE2 production by osteoblasts is essential for the osteoclastogenesis induced by cell-to-cell interactions between cancer cells and osteoblasts.
EP4 Mediates the Actions of PGE2 in the Growth and Metastasis of B16 Melanoma
PGE2 is known to act on individual target cells through a family of receptor subtypes identified as EP1, EP2, EP3, and EP4. Using specific antagonists for respective EPs, we evaluated which EP mediated the PGE2 signaling in the B16 metastases to hard-tissue bone and other soft tissues. Wild-type mice were injected with B16 cells, and then an EP1 antagonist, EP2 antagonist, EP3 antagonist, or EP4 antagonist was administered for 17 days by oral gavage. The EP4 antagonist markedly suppressed bone metastasis compared with the control, but other EP (EP1, EP2, and EP3) antagonists had minimal effects (Fig. 6A). The number of metastatic colonies in the lungs was suppressed markedly by the EP4 antagonist, and both the EP2 and EP3 antagonist significantly suppressed lung metastasis (Fig. 6B).
FIGURE 6.

An EP4 antagonist inhibits the growth and metastasis of B16 tumors. A, wild-type mice were injected with B16 cells, and then EP antagonists were administered orally for 17 days. Representative pictures of the tumor growth in the femora are shown. B, lungs were collected 18 days after injection, and the tumor colonies were counted. Data are mean ± S.E. (vehicle, n = 8–10; EP1 antagonist, n = 9; EP2 antagonist, n = 8; EP3 antagonist, n = 9; EP4 antagonist, n = 10). *, p < 0.01; **, p < 0.001. C, wild-type mice were injected with B16 cells into the dorsal subcutaneous tissue and treated with either vehicle or an EP4 antagonist by subcutaneous injection around the tumor. Representative pictures of the tumors on day 16 after injection are shown in the top panel. The arrowheads indicate the subcutaneous solid tumor. The tumor volume was determined by measurement of the tumor dimensions using calipers on days 11–18. *, p < 0.01; **, p < 0.001. D, dermal fibroblasts collected from wild-type mice were cultured in the presence or absence of PGE2 (10 μm) and an antagonist of EP1, EP2, EP3, or EP4 (10 μm each). After 48 h, the concentration of VEGF-A in the conditioned medium was determined by ELISA. Data are mean ± S.E. of three wells. Significant differences from control (*, p < 0.01) and PGE2-treated (#, p < 0.05; ##, p < 0.01, ###, p < 0.001) cells are indicated.
To evaluate the role of EP4 in PGE-dependent tumor growth, wild-type mice were subcutaneously injected with B16 cells, and some mice were treated with the EP4 antagonist by subcutaneous injection into the tissue surrounding the tumor. Tumor growth was clearly found in control mice on days 11–18, but mice treated with the EP4 antagonist showed little change in the volume of the tumor, even on day 18 (Fig. 6C). In cultures of dermal fibroblasts, production of VEGF-A was elevated by PGE2 and suppressed completely by adding the EP4 antagonist. The EP1 or EP3 antagonist also partially suppressed the production of VEGF-A (Fig. 6D).
B16 cells express EP1 but not other EPs (EP2, EP3α, EP3β, EP3γ, and EP4) in RT-PCR (data not shown). To examine the effects of the respective EP antagonists on the proliferation of B16 cells, we added each antagonist to cultures of B16 cells and found that none of the antagonists affected the colony formation of B16 cells in vitro (data not shown). When PGE2 was added to the culture of B16 cells, the proliferation of B16 was not influenced by PGE2 in vitro (data not shown). The cell motility of B16 cells measured by the migration assay was not influenced by adding PGE2 in vitro (data not shown). These results indicate that both the metastasis and growth of B16 melanoma were regulated by the PGE2 produced by the host-derived cells in the tumor microenvironment, and subsequent signaling is mediated by the EP4 receptor in vivo.
To confirm the role of the EP4 expressed in host-derived cells on the PGE2-mediated effects on the growth of B16 tumors, we used EP4-null Ptger4−/− mice. When Ptger4−/− and wild-type mice were injected with B16 cells into their dorsal subcutaneous tissue, tumor growth was attenuated significantly in Ptger4−/− mice on days 6–15 (Fig. 7A). An in vitro study showed that the production of VEGF-A was elevated by PGE2 in cultures of dermal fibroblasts isolated from wild-type mice, but fibroblasts from Ptger4−/− mice produced only a small amount of VEGF-A, and this level was not enhanced by PGE2 (Fig. 7B). Therefore, we conclude that PGE2 acts on the EP4 in stromal cells to regulate tumor growth.
FIGURE 7.

Tumor growth and VEGF production are attenuated in Ptger4−/− mice. A, Ptger4−/− or wild-type mice were injected with B16 cells into the dorsal subcutaneous tissue. Representative pictures of the subcutaneous tumors on day 9 after injection are shown in the top panel. The arrowheads indicate a subcutaneous solid tumor. The tumor volume was determined, and the data are shown in the bottom panel as mean ± S.E. of the wild-type (n = 9) or Ptger4−/− (n = 8) mice. *, p < 0.05. N.D., not detected. B, dermal fibroblasts from Ptger4−/− or wild-type mice were cultured in the presence or absence of PGE2 (10 μm). After 48 h, the concentration of VEGF-A in the conditioned media was determined by ELISA. Data are mean ± S.E. of three wells. *, p < 0.001. C, schematic of the PGE2/EP4-depedendent mechanism involved in angiogenesis, tumorigenesis, and osteoclastogenesis. In soft tissues, stromal fibroblasts interact with cancer cells and produce PGE2 via mPGES-1, and then PGE2 induces VEGF-A and bFGF production as a result of autocrine/paracrine signaling via EP4. In hard-tissue bone, the interaction with cancer cells elicits mPGES-1-dependent PGE2 production by osteoblasts, and then PGE2 induces the expression of RANKL on the surface of osteoblasts to induce osteoclast formation, which is critical for the osteolysis associated with the bone metastasis of cancer. AA, arachidonic acid.
Discussion
In this study, we have shown that direct interaction with tumor cells induced COX-2- and mPGES-1-dependent PGE2 production by stromal fibroblasts and osteoblasts in the tumor microenvironment and that PGE2 acts via the EP4 receptor to induce tumorigenesis, angiogenesis, and bone resorption. In soft tissues with cancer metastasis, PGE2-induced production of VEGF and bFGF by the stromal fibroblasts in the tumor microenvironment may be essential for angiogenesis to support tumor growth (Fig. 7C). In hard-tissue bone, PGE2-induced RANKL expression by osteoblasts is critical for osteoclast formation, which leads to the bone loss associated with bone metastasis (Fig. 7C).
Li et al. (28) examined the role of PGE2 produced by mesenchymal stromal cells in tumor progression and found that IL-1 produced by cancer cells acts on stromal cells to stimulate COX-2 expression and PGE production. PGE2 also acts on stromal cells in an autocrine fashion to trigger the production of cytokines such as IL-6 and IL-8 (28). Therefore, cancer-derived cytokines may be involved in PGE production by host cells driving tumor progression (28, 30). In this study, we have shown that cell-cell interactions between fixed melanoma cells and live osteoblasts elicit PGE2 production by osteoblasts and stimulate RANKL-dependent osteoclast formation. PGE2 could not be detected in the conditioned medium of B16 cultures, and the conditioned medium of B16 cultures could not induce RANKL-dependent osteoclast formation (data not shown). Therefore, cell surface molecule(s) in B16 may induce the expression of COX-2 and mPGES-1 in osteoblasts to produce PGE2 by cell-cell interaction, not by soluble factors. Brandner and Haass (31) showed the role of connexins (Cx) in direct cell-cell communication between melanoma cells and host cells in the tumor microenvironment. Melanoma cells expressing higher levels of Cx43 showed increased coupling to vascular endothelial cells and a high risk of metastasis. On the other hand, epidermal host cells expressed Cx26, and the expression of Cx26 in the adjacent non-cancer tissues may be useful to identify patients with a high risk of metastasis. Cx43 is expressed in osteoblasts as gap junction channels (32), and B16 cells express Cx26 (31). Further studies are needed to define a cell surface molecule involved in cell-cell interaction between B16 melanoma and osteoblasts.
It is also possible that the extracellular matrix produced by cancer cells is involved in cancer-related events. Osteoclast formation was induced markedly by fixed B16 cells. However, when bone marrow cells and osteoblasts were co-cultured on the extracellular matrix without B16 cells, osteoclast formation was not detected (data not shown), suggesting that a cell surface molecule in B16 cells, rather than B16-derived extracellular matrix, is responsible for osteoclast formation via cell-to-cell contact. Further studies are needed to examine the role of extracellular matrix derived from cancer cells and stromal cells in the growth and metastasis of cancer.
Previous studies have suggested that there is a correlation between cancer growth and PGs. There have been two conflicting reports of tumorigenesis in mPges1−/− Apc-mutant Min mice. Elander et al. (33) have shown that the genetic deletion of mPGES-1 accelerates intestinal tumorigenesis in Apc-mutant mice. However, Nakanishi et al. (34) have shown the suppression of intestinal tumorigenesis by genetic deletion of mPGES-1 in Apc-mutant mice. On the other hand, some prostate cancer cells express mPGES-1 highly, and knockdown of the mPges1 gene reduced the growth and colony formation of prostate cancer cells (35). These data suggest that some cancer cells produce PGE2 via their own mPGES-1 and that released PGE2 regulates the growth of cancer cells.
Using specific antagonists for different EP, previous studies have shown that PGE2 acts on cancer cells expressing EP1 and EP4. In cultures of oral squamous cell carcinoma, PGE2 promoted cell migration and ICAM-1 expression via EP1 (36). Several reports have shown that EP4 signaling regulates the migration of lung cancer cells, tumor-associated angiogenesis in prostate cancer, and the metastatic potential in breast and lung carcinomas (6, 36–39). The B16 cells used in this study do not express EP4, and treatment with the EP4 antagonist did not influence the proliferation of B16 cells in vitro (data not shown). Adding PGE2 did not influence the proliferation or migration of B16 cells in vitro (data not shown). However, administration of the EP4 antagonist to mice clearly suppressed tumor growth in vivo (Fig. 6). Furthermore, the growth of B16 tumors was attenuated in Ptger4−/− mice (Fig. 7), indicating that the PGE2 produced by stromal lineage cells may act on the EP4 expressed in these cells to promote cancer metastasis and tumor growth.
Regarding bone metastasis, the regulation of bone resorption by cancer cells is critical because bone metastasis is accompanied by osteolysis with enhanced osteoclastogenesis and skeletal phenomena with severe bone pain, which is a major clinical issue in patients with bone metastases. In this study, we found that cancer-induced osteoclastogenesis could not be induced in co-cultures of bone marrow cells and osteoblasts collected from mPges1−/− mice because of the lack of PGE2 production by osteoblasts and that the osteolysis associated with bone metastasis was attenuated in mPges1−/− mice. Therefore, cancer cells may induce PGE2 production by normal osteoblasts, and the released PGE2 subsequently acts on the osteoblasts via EP4 to elicit the expression of RANKL, which induces osteoclastic bone resorption in bone tissue with cancer metastasis.
Bone metastasis frequently occurs in breast and prostate cancers. In the clinical setting, anti-estrogen and anti-androgen therapies are crucial for breast and prostate cancers, but some cancer cells are resistant to hormone therapy, which leads to the recurrence and metastasis of the cancer. Bisphosphonate and a RANKL antibody could suppress the osteolysis associated with bone metastasis of cancer in clinical trials, but these agents mainly act on bone tissue (40). Blocking PGE2/EP4 signaling may be more useful for the prevention of both bone and soft tissue metastasis in several types of cancer, including hormone-resistant breast cancer, because an EP4 antagonist could inhibit not only osteolysis associated with bone metastasis but also tumor growth occurring via microenvironment-dependent mechanisms.
Angiogenesis with new blood vessel formation is essential for tumor growth to supply nutrition and oxygen for cancer cells, and VEGF is a pivotal factor required to induce tumor-associated angiogenesis (29). PGE2 has been reported to induce the expression of VEGF in stromal fibroblasts, and EP2 and/or EP4 signaling is considered to be involved in the induction of VEGF (41, 42). Using a gastric cancer mouse model, Guo et al. (42) have reported that the PGE2 and Wnt pathways play key roles in gastric tumorigenesis through the induction of VEGF production by stromal fibroblasts. In addition to VEGF, bFGF has been reported to induce angiogenesis associated with tumor growth. Previous studies have shown that bFGF is expressed in various types of human cancer, including prostate cancer, breast cancer, and lung cancer (43, 44), and that increased expression of bFGF can be detected in stromal cells within prostate tumors (45). Polnaszek et al. (46) have found that bFGF−/− mice exhibited increased survival, decreased metastasis, and inhibited progression of prostate tumors. In cultured endothelial cells, PGE2 stimulated proliferation by enhancing the signal for bFGF/FGFR-1, and PGE2-induced angiogenesis was attenuated in endothelial cells collected from bFGF−/− mice (47). Recent studies have shown that PGE2 regulates endothelial cell proliferation, migration, and tubulogenesis via EP4 (48). Further studies are needed to clarify the possible action of PGE2 in endothelial cells with regard to regulating tumor angiogenesis.
In conclusion, this study clearly showed that the growth and metastasis of malignant melanoma were attenuated in mPges1−/− mice and Ptger4−/− mice and that the administration of an EP4 antagonist inhibited the tumor growth and metastasis in various tissues, including bone, lung, kidney, and liver. Our data suggest that autocrine/paracrine PGE2/EP4 signaling has a critical role in the growth and metastasis of cancer and support the development of clinical trials to determine whether an EP4 antagonist can be useful as therapeutic agent in the management of primary and metastatic cancer.
Author Contributions
M. I., M. T., T. M., S. N., and C. Miyaura conceived and designed the study. M. I., M. T., T. M., and C. Miyaura developed the methodology. M. I., M. T., S. Y., K. W., T. T., C. Matsumoto, M. H., Y. S., S. N., S. U., S. A., and C. Miyaura acquired the data (provided animals, provided facilities, etc.). M. I., T. M., Y. S., S. N., S. U., G. M., H. N., and C. Miyaura analyzed and interpreted the data. M. I., Y. M., Y. S., S. N., S. U., G. M., H. N., and C. Miyaura wrote and reviewed the manuscript.
Acknowledgments
We thank Ms. Wakana Kaizuka for assistance.
This work was supported by Grants-in-Aid for Scientific Research 25460062 (to C. M.) and 23590069 (to M. I.), National Institute of Health Grant AR40994 (to H. N.), Arthritis Research UK (to H. N.), Cancer Research UK (to G. M.), and the Global Innovation Research Organization in Tokyo University of Agriculture and Technology (to M. I. and H. N.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PGE2
- prostaglandin E2
- Ep
- PGE2 receptor
- COX
- cyclooxygenase(s)
- PGES
- prostaglandin E synthase(s)
- mPGES
- membrane-bound prostaglandin E synthase(s)
- PG
- prostaglandin
- RANKL
- receptor activator of NF-κB ligand
- BMD
- bone mineral density
- CT
- computed tomography
- Cx
- connexin(s).
References
- 1.Wang D., and Dubois R. N. (2010) Eicosanoids and cancer. Nat. Rev. Cancer 10, 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jakobsson P.J., Thorén S., Morgenstern R., and Samuelsson B. (1999) Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. U.S.A. 96, 7220–7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murakami M., Naraba H., Tanioka T., Semmyo N., Nakatani Y., Kojima F., Ikeda T., Fueki M., Ueno A., Oh S., and Kudo I. (2000) Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 275, 32783–32792 [DOI] [PubMed] [Google Scholar]
- 4.Ristimäki A., Sivula A., Lundin J., Lundin M., Salminen T., Haglund C., Joensuu H., and Isola J. (2002) Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 62, 632–635 [PubMed] [Google Scholar]
- 5.Elder D. J., and Paraskeva C. (1998) COX-2 inhibitors for colorectal cancer. Nat. Med. 4, 392–393 [DOI] [PubMed] [Google Scholar]
- 6.Ma X., Kundu N., Rifat S., Walser T., and Fulton A. M. (2006) Prostaglandin E receptor EP4 antagonism inhibits breast cancer metastasis. Cancer Res. 66, 2923–2927 [DOI] [PubMed] [Google Scholar]
- 7.Rundhaug J. E., Simper M. S., Surh I., and Fischer S. M. (2011) The role of the EP receptors for prostaglandin E2 in skin and skin cancer. Cancer Metastasis Rev. 30, 465–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J., Zhang Y., Frilot N., Kim J. I., Kim W. J., and Daaka Y. (2011) Prostaglandin E2 regulates renal cell carcinoma invasion through the EP4 receptor-Rap GTPase signal transduction pathway. J. Biol. Chem. 286, 33954–33962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amano H., Hayashi I., Endo H., Kitasato H., Yamashina S., Maruyama T., Kobayashi M., Satoh K., Narita M., Sugimoto Y., Murata T., Yoshimura H., Narumiya S., and Majima M. (2003) Host prostaglandin E2-EP3 signaling regulates tumor-associated angiogenesis and tumor growth. J. Exp. Med. 197, 221–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holt D. M., Ma X., Kundu N., and Collin P. D., and Fulton A. M. (2012) Modulation of host natural killer cell functions in breast cancer via prostaglandin E2 receptors EP2 and EP4. J. Immunother. 35, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohshiba T., Miyaura C., and Ito A. (2003) Role of prostaglandin E produced by osteoblasts in osteolysis due to bone metastasis. Biochem. Biophys. Res. Commun. 300, 957–964 [DOI] [PubMed] [Google Scholar]
- 12.Takita M., Inada M., Maruyama T., and Miyaura C. (2007) Prostaglandin E receptor EP4 antagonist suppresses osteolysis due to bone metastasis of mouse malignant melanoma cells. FEBS Lett. 581, 565–571 [DOI] [PubMed] [Google Scholar]
- 13.Uematsu S., Matsumoto M., Takeda K., and Akira S. (2002) Lipopolysaccharide-dependent prostaglandin E2 production is regulated by the glutathione-dependent prostaglandin E2 synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J. Immunol. 168, 5811–5816 [DOI] [PubMed] [Google Scholar]
- 14.Inada M., Matsumoto C., Uematsu S., Akira S., and Miyaura C. (2006) Membrane-bound prostaglandin E synthase-1-mediated prostaglandin E2 production by osteoblast plays a critical role in lipopolysaccharide-induced bone loss associated with inflammation. J. Immunol. 177, 1879–1885 [DOI] [PubMed] [Google Scholar]
- 15.Anderson D.M., Maraskovsky E., Billingsley W.L., Dougall W.C., Tometsko M.E., Roux E.R., Teepe M.C., DuBose R.F., Cosman D., and Galibert L. (1997) A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390, 175–179 [DOI] [PubMed] [Google Scholar]
- 16.Wong B. R., Rho J., Arron J., Robinson E., Orlinick J., Chao M., Kalachikov S., Cayani E., Bartlett F. S. 3rd, Frankel W. N., Lee S. Y., and Choi Y. (1997) TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J. Biol. Chem. 272, 25190–25194 [DOI] [PubMed] [Google Scholar]
- 17.Lacey D. L., Timms E., Tan H. L., Kelley M. J., Dunstan C. R., Burgess T., Elliott R., Colombero A., Elliott G., Scully S., Hsu H., Sullivan J., Hawkins N., Davy E., Capparelli C., Eli A., Qian Y. X., Kaufman S., Sarosi I., Shalhoub V., Senaldi G., Guo J., Delaney J., and Boyle W. J. (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93, 165–176 [DOI] [PubMed] [Google Scholar]
- 18.Yasuda H., Shima N., Nakagawa N., Yamaguchi K., Kinosaki M., Mochizuki S., Tomoyasu A., Yano K., Goto M., Murakami A., Tsuda E., Morinaga T., Higashio K., Udagawa N., Takahashi N., and Suda T. (1998) Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. U.S.A. 95, 3597–3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morony S., Capparelli C., Sarosi I., Lacey D. L., Dunstan C. R., and Kostenuik P. J. (2001) Osteoprotegerin inhibits osteolysis and decreases skeletal tumor burden in syngeneic and nude mouse models of experimental bone metastasis. Cancer Res. 61, 4432–4436 [PubMed] [Google Scholar]
- 20.Zhang J., Dai J., Yao Z., Lu Y., Dougall W., and Keller E. T. (2003) Soluble receptor activator of nuclear factor κB Fc diminishes prostate cancer progression in bone. Cancer Res. 63, 7883–7890 [PubMed] [Google Scholar]
- 21.Lipton A., and Goessl C. (2011) Clinical development of anti-RANKL therapies for treatment and prevention of bone metastasis. Bone 48, 96–99 [DOI] [PubMed] [Google Scholar]
- 22.Ohshiba T., Miyaura C., Inada M., and Ito A. (2003) Role of RANKL-induced osteoclast formation and MMP-dependent matrix degradation in bone destruction by breast cancer metastasis. Br. J. Cancer 88, 1318–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugimoto Y., and Narumiya S. (2007) Prostaglandin E receptors. J. Biol. Chem. 282, 11613–11617 [DOI] [PubMed] [Google Scholar]
- 24.Miyaura C., Inada M., Suzawa T., Sugimoto Y., Ushikubi F., Ichikawa A., Narumiya S., and Suda T. (2000) Impaired bone resorption to prostaglandin E2 in prostaglandin E receptor EP4- knockout mice. J. Biol. Chem. 275, 19819–19823 [DOI] [PubMed] [Google Scholar]
- 25.Suzawa T., Miyaura C., Inada M., Maruyama T., Sugimoto Y., Ushikubi F., Ichikawa A., Narumiya S., and Suda T. (2000) The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology 141, 1554–1559 [DOI] [PubMed] [Google Scholar]
- 26.Segi E., Sugimoto Y., Yamasaki A., Aze Y., Oida H., Nishimura T., Murata T., Matsuoka T., Ushikubi F., Hirose M., Tanaka T., Yoshida N., Narumiya S., and Ichikawa A. (1998) Patent ductus arteriosus and neonatal death in prostaglandin receptor EP4-deficient mice. Biochem. Biophys. Res. Commun. 246, 7–12 [DOI] [PubMed] [Google Scholar]
- 27.Miyaura C., Kusano K., Masuzawa T., Chaki O., Onoe Y., Aoyagi M., Sasaki T., Tamura T., Koishihara Y., and Ohsugi Y. (1995) Endogenous bone-resorbing factors in estrogen deficiency: cooperative effects of IL-1 and IL-6. J. Bone Miner. Res. 10, 1365–1373 [DOI] [PubMed] [Google Scholar]
- 28.Li H. J., Reinhardt F., Herschman H. R., and Weinberg R. A. (2012) Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2, 840–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wicki A., and Rochlitz C. (2012) Targeted therapies in breast cancer. Swiss Med. Wkly. 142, w13550. [DOI] [PubMed] [Google Scholar]
- 30.Alcolea S., Antón R., Camacho M., Soler M., Alfranca A., Avilés-Jurado F. X., Redondo J. M., Quer M., León X., and Vila L. (2012) Interaction between head and neck squamous cell carcinoma cells and fibroblasts in the biosynthesis of PGE2. J. Lipid Res. 53, 630–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brandner J. M., and Haass N. K. (2013) Melanoma's connections to the tumour microenvironment. Pathology 45, 443–452 [DOI] [PubMed] [Google Scholar]
- 32.Plotkin L. I., Speacht T. L., and Donahue H. J. (2015) Cx43 and mechanotransduction in bone. Curr. Osteoporos. Rep. 13, 67–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elander N., Ungerbäck J., Olsson H., Uematsu S., Akira S., and Söderkvist P. (2008) Genetic deletion of mPGES-1 accelerates intestinal tumorigenesis in APC (Min/+) mice. Biochem. Biophys. Res. Commun. 372, 249–253 [DOI] [PubMed] [Google Scholar]
- 34.Nakanishi M., Montrose D. C., Clark P., Nambiar P. R., Belinsky G. S., Claffey K. P., Xu D., and Rosenberg D. W. (2008) Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 68, 3251–3259 [DOI] [PubMed] [Google Scholar]
- 35.Hanaka H., Pawelzik S. C., Johnsen J. I., Rakonjac M., Terawaki K., Rasmuson A., Sveinbjörnsson B., Schumacher M. C., Hamberg M., Samuelsson B., Jakobsson P. J., Kogner P., and Rådmark O. (2009) Microsomal prostaglandin E synthase 1 determines tumor growth in vivo of prostate and lung cancer cells. Proc. Natl. Acad. Sci. U.S.A. 106, 18757–18762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang S. F., Chen M. K., Hsieh Y. S., Chung T. T., Hsieh Y. H., Lin C. W., Su J. L., Tsai M. H., and Tang C. H. (2010) Prostaglandin E2/EP1 signaling pathway enhances intercellular adhesion molecule 1 (ICAM-1) expression and cell motility in oral cancer cells. J. Biol. Chem. 285, 29808–29816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J. I., Lakshmikanthan V., Frilot N., and Daaka Y. (2010) Prostaglandin E2 promotes lung cancer cell migration via EP4-bArrestin1-c-Src signalsome. Mol. Cancer Res. 8, 569–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain S., Chakraborty G., Raja R., Kale S., and Kundu G. C. (2008) Prostaglandin E2 regulates tumor angiogenesis in prostate cancer. Cancer Res. 68, 7750–7759 [DOI] [PubMed] [Google Scholar]
- 39.Yang L., Huang Y., Porta R., Yanagisawa K., Gonzalez A., Segi E., Johnson D. H., Narumiya S., and Carbone D. P. (2006) Host and direct antitumor effects and profound reduction in tumor metastasis with selective EP4 receptor antagonism. Cancer Res. 66, 9665–9672 [DOI] [PubMed] [Google Scholar]
- 40.Brown J. E., and Coleman R. E. (2012) Denosumab in patients with cancer-a surgical strike against the osteoclast. Nat. Rev. Clin. Oncol. 9, 110–118 [DOI] [PubMed] [Google Scholar]
- 41.Hatazawa R., Tanigami M., Izumi N., Kamei K., Tanaka A., and Takeuchi K. (2007) Prostaglandin E2 stimulates VEGF expression in primary rat gastric fibroblasts through EP4 receptors. Inflammopharmacology 15, 214–217 [DOI] [PubMed] [Google Scholar]
- 42.Guo X., Oshima H., Kitmura T., Taketo M. M., and Oshima M. (2008) Stromal fibroblasts activated by tumor cells promote angiogenesis in mouse gastric cancer. J. Biol. Chem. 283, 19864–19871 [DOI] [PubMed] [Google Scholar]
- 43.Relf M., LeJeune S., Scott P. A., Fox S., Smith K., Leek R., Moghaddam A., Whitehouse R., Bicknell R., and Harris A. L. (1997) Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor β-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res. 57, 963–969 [PubMed] [Google Scholar]
- 44.Berger W., Setinek U., Mohr T., Kindas-Mügge I., Vetterlein M., Dekan G., Eckersberger F., Caldas C., and Micksche M. (1999) Evidence for a role of FGF-2 and FGF receptors in the proliferation of non-small cell lung cancer cells. Int. J. Cancer. 83, 415–423 [DOI] [PubMed] [Google Scholar]
- 45.Giri D., Ropiquet F., and Ittmann M. (1999) Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin. Cancer Res. 5, 1063–1071 [PubMed] [Google Scholar]
- 46.Polnaszek N., Kwabi-Addo B., Peterson L. E., Ozen M., Greenberg N. M., Ortega S., Basilico C., and Ittmann M. (2003) Fibroblast growth factor 2 promotes tumor progression in an autochthonous mouse model of prostate cancer. Cancer Res. 63, 5754–5760 [PubMed] [Google Scholar]
- 47.Finetti F., Solito R., Morbidelli L., Giachetti A., Ziche M., and Donnini S. (2008) Prostaglandin E2 regulates angiogenesis via activation of fibroblast growth factor receptor-1. J. Biol. Chem. 283, 2139–2146 [DOI] [PubMed] [Google Scholar]
- 48.Rao R., Redha R., Macias-Perez I., Su Y., Hao C., Zent R., Breyer M. D., and Pozzi A. (2007) Prostaglandin E2-EP4 receptor promotes endothelial cell migration via ERK activation and angiogenesis in vivo. J. Biol. Chem. 282, 16959–16968 [DOI] [PubMed] [Google Scholar]