

Abstract
Radiation hybrid (RH) mapping is limited by the inherent genomic instability of RH clones entailing both, limited DNA sample amounts and genomic heterogeneity of the clones. Here the instability of RH clones is quantified and the suitability of the multiple strand displacement whole genome amplification method (WGA) for radiation hybrid mapping is assessed. To quantify the instability of RH clones, eleven clones of a 10,000Rad rhesus macaque radiation hybrid panel were passaged ten times and analyzed by interspersed repeat sequence specific quantitative PCR and by genotyping of 46 macaque chromosome 5 STS markers. The quantitative PCR data indicate an average loss of 55% of the donor DNA over 10 passages. Over the same period, a dropout of 46.2% of the STS markers was observed. These data indicate a genome wide half-life of the donor DNA of 8.7 passages and of 10.6 passages for the chromosome 5 markers. The genotyping data of the genomic RH DNA were compared to three sets of WGA experiments: i) single wgaDNA amplifications, ii) six WGA replicates, and iii) secondary whole genome amplifications. The assays demonstrated concordance rates of 97.6%, 98%, and 99.3%, respectively, and indicated the marker specificity of some repeated WGA dropouts. The study confirms that WGA is suitable for RH mapping studies and WGA should enable the accurate analysis of almost an infinite numbers of markers. WGA will allow the analysis of earliest RH clone passages, thus limiting their heterogeneity and RH mapping artifacts.
INTRODUCTION
Gene mapping is reaching its pinnacle with the progression of whole genome sequence assemblies to the “finished” status. The assemblies will represent ultimate genetic maps in the form of complete, accurate, and annotated nucleotide sequences. The creation of such comprehensive genome sequence assemblies will however remain restricted to a few species in the near future since the recent breakthrough developments in DNA sequencing technology focus on short read lengths, which are more suitable for the efficient re-sequencing of genomes (Bentley, 2006; Wheeler et al., 2008). Thus the construction of high-resolution recombination maps and physical maps remains an active area of research (Matise et al., 2007; McKay et al., 2007; Shifman et al., 2006), providing chromosomal locations and inter-marker distances, landmarks for locating disease genes and species-specific frameworks for genome sequence assembly (Slonim et al., 1997). Radiation hybrid (RH) mapping has been exploited widely since 1990 (Cox et al., 1990) to construct physical maps. Since then, the technique has been refined and is used to construct high-resolution maps with high throughput techniques (McKay et al., 2007). High-resolution RH maps have proven to be useful for interrogating and amending genome sequence assemblies (Jann et al., 2006; Karere et al., 2008; McKay et al., 2007).
One of the limiting factors for RH map construction is the cell culture required to generate sufficient DNA for marker mapping and for sharing of the resource. The propagation of RH clones is especially difficult for non mammalian species like fishes (Senger et al., 2006) and plants (Wardrop et al., 2002) requiring some form of sample amplification (Wardrop et al., 2002). To generate RH clones, chromosomes of donor cells are fragmented by a lethal dose of X-rays. The resulting chromosome fragments are rescued by fusion to a recipient cell (mostly of rodent origin), deficient for a selection gene, for example TK or HRPT, and subsequent integration of the donor genome chromosomal fragments into the recipient karyotype. After culture in selective medium, individual RH clones are isolated and propagated in cell culture in increasing volumes. DNA from the resulting RH clones is genotyped for markers of interest. The random nature of both the X-ray fragmentation and of the integration into the recipient genome enables the generation of genome maps by linkage mapping approaches. However, since RH cell lines are unstable (Goodfellow, 1991; Graw et al., 1988; Walter and Goodfellow, 1995), results obtained with the same clone can only be combined if DNA originating from the same passage of cells has been tested. Thus, one of the major challenges in the construction of RH maps is the finite amount of DNA available for genotyping. The instability of the RH cell lines further induces strongly varying signal intensities during genotyping since certain donor DNA sequences might only be present in a fraction of the RH clone cells (Slonim et al., 1997). This problem necessitates duplicate genotyping for the verification of results and diminishes the precision of the RH mapping in cases of persisting ambiguity or induced artifacts. Ambiguous results are statistically very problematic and are often treated differently by the individual RH software packages (Agarwala et al., 2000). To meet the DNA quantity demands, the RH cell lines are typically passaged in multiple stages beginning with the establishment of a clone, passage into 24-well plates, and subsequent expansion in T25 flasks and roller bottles (Chowdhary et al., 2002). In cases where DNA stocks become inadequate, the hybrid cells have to been grown as a new passage. However, the extracted DNA then represents a different population of cells, usually necessitating re-typing of markers. The loss of unselected donor DNA fragments from somatic hybrid clones was already observed in early studies (Goss and Harris, 1977) and the process itself is not disputed. However, the data on the genomic instability are somewhat anecdotal since the published observations concern small genomic regions. Chromosome-wide or genome-wide data and quantifications are missing.
Since the 1990’s three major whole genome amplification (WGA) techniques including primer extension pre-amplification (PEP) (Zhang et al., 1992), degenerate oligonucleotide primed PCR (DOP) (Telenius et al., 1992), and multiple displacement amplification (MDA) (Dean et al., 2002) have been developed to address the problem of limited DNA samples. PEP and DOP are both PCR-based protocols, whereas MDA is an isothermal amplification employing the high-fidelity PHI (Φ) 29 phage DNA polymerase for DNA synthesis and strand displacement (Dean et al., 2002). While WGA techniques have been tested intensively on homogenous DNA samples for trait association studies (Pask et al., 2004), genetic disease research (Berthier-Schaad et al., 2007), and for the sequence analysis of DNA (Pinard et al., 2006), the efficiency and reliability of WGA techniques applied to heterogenous samples and their impact on the process of RH-mapping has not been analyzed in depth. Senger and colleagues (2006) did report a small-scale test and observed high genotype concordance (99%) between genomic and WGA DNA from two RH clones and between two independent reactions from a single clone.
The present study addresses the questions of the loss of donor DNA between passages of RH clones and of the suitability of MDA amplified DNA to aid future RH mapping projects. The donor DNA loss was analyzed in two ways: For a specific chromosome, via STS marker typing, and on the genomic level by interspersed repeat sequence (IRS)-based quantitative PCR.
The suitability and sustainability of WGA RH cell DNA was tested on a larger sample size involving control experiments simulating the requirements of RH mapping studies. STS markers and RH cell clones previously characterized for the generation of a RH map of the rhesus macaque chromosome 5 (Karere et al., 2008) were re-analyzed in samples generated by WGA and in repeated cell culture passages. The results not only suggest that WGA can address the limitation of RH clone DNA samples and their sustainable distribution to the research community but further suggest improvements in the design of RH mapping studies.
MATERIALS AND METHODS
Cell line generation
Hybrid cell lines were generated from the fusion of male rhesus macaque and hamster fibroblast cell lines as previously described (Chowdhary et al., 2002; Womack et al., 1997). Briefly, the macaque cells were irradiated with an X-ray dosage 10,000Rad and fused with cells of a thymidine kinase-deficient (TK−) hamster cell line (A23). The HAT selected hybrid cells were grown in 24-well plates with HAT medium and passaged in T25 flasks to generate passage 5 cell lines that were cryopreserved in each into two aliquots. One set of the aliquots of these RH-clones were thawed and expanded previously to passage 6 (p6) for the generation of an RH map of rhesus macaque chromosome 5 (Karere et al., 2008).
Cell culture
For this study, a second set of passage 5 aliquots of eleven hybrid cell lines rich in macaque chromosome 5 material, as determined previously (Karere et al., 2008), were thawed and passaged once and then ten more times in a 1:3 ratio in T25 flasks to generate passage p6* and p16* cells (i.e.). Cells were harvested according to standard procedures and stored at −20°C until DNA extraction.
Quantitative PCR
The inter-Alu PCR protocol (Liu et al., 1993) was modified to allow discrimination of macaque and hamster genomic DNA samples and the application in real time quantitative PCR (qPCR) experiments. The Alu primers (Alu 1:GGATTACAGCYRTGAGCC; Alu 2: RCCAYTGCACTCCAGCCT) (Liu et al., 1993) were used to quantify the amount of macaque DNA in the 11 hybrid clones of the passages p6, p6* and P16*. The real-time PCR assay was standardized by performing duplicate serial dilutions (5-fold), consisting of 20ng, 4ng, 1.6ng, 0.8ng and 0.034ng of macaque:hamster DNA mix (1:5), to simulate realistic DNA ratios of RH clones. Quantitative PCR amplification of DNA was carried out in 25 μl reaction volumes containing 20ng of DNA, 2 μM of the primer pair and 1x SYBR green pre-mix solution (ABI, Applied Biosystems, Foster City, CA) as recommended by the manufacturer. A standard curve drawn from the quantified control sample was used to extrapolate the quantities of RH clone DNA. Hamster DNA (20ng) and water were used as negative controls. The following thermal profile was used: initial denaturation for 10 min at 95°C to activate the AmpliTaq Gold polymerase, followed by 40 cycles of denaturation at 95°C for 15 s and 70°C for 1 min to anneal and extend the primers. Quantitative PCR was conducted using an ABI 7300 System and analyzed by ABI SDS Software.
DNA isolation and whole genome amplification
Genomic DNA (gDNA) was isolated from the hybrid cells of passages p6* and p16* using the Puregene Core kit B (Qiagen, Valencia, CA). Whole genome amplifications were performed employing the MDA technique (Dean et al., 2002) with Repli-G minikits (Qiagen). To ensure optimal conditions for the amplification, each reaction used 75 ng template DNA. Three sets of whole genome amplifications were performed (i) amplifications of gDNA samples from eleven RH cell lines of passage p6*, (ii) gDNA samples from five of the eleven RH cell lines were additionally whole genome amplified in six replicate reactions, (iii) for all clones re-amplifications of the first wgaDNA samples were performed. Prior to DNA quantification and genotyping, wgaDNA, was purified using microcon centrifugal filters (Millipore Corporation, MA) to eliminate nonspecific primers and WGA reagents.
Genotyping
To investigate changes in the donor genome content between distinct cell line passages, 46 sequence-tagged site (STS) markers (Table S1) distributed evenly across macaque chromosome 5 (Mmul5) were genotyped on gDNA from both passage p6* and P16* of the eleven cell lines. The genotyping was performed in duplicate using standard PCR protocols as described (Karere et al., 2008). The reliability of the wgaDNA for RH typing was determined by typing markers on 30ng of wgaDNA sample, which was equivalent to a dilution of 1:40 of the WGA reaction in TLE buffer. Hamster and macaque gDNA samples were used as controls in both whole genome amplification reactions and PCRs. To test the quality of whole genome amplification, four hamster STS markers (Table 1) were typed on wgaDNA diluted at 1:40, 1:400, and 1:4000. Hamster STS PCRs were carried out using standard template DNA concentrations at an annealing temperature of 60°C. The PCR products were separated on 2% agarose gels, visualized by ethidium bromide staining, digitally recorded, and scored with GelScore software (http://www.wesbarris.com/GelScore). For the concordance calculations of the genotyping results, all STS markers that were consistently negative for a specific RH clone were excluded from any consideration to enhance the validity of the analysis.
Table 1.
Chinese hamster (Cricetulus griseus) STS primer sequences tested in the macaque RH clones.
| Marker | Forward (5′ – 3′) | Reverse (5′ – 3″) |
|---|---|---|
| LHB | GTTGCTATGGCTGCTGCTG | CTGGTGGTGAAGGTGATGC |
| ORC4 | CCAATGGAAAGAACTTCAGTCA | GAGGATGTTGCCCACTGC |
| SLC35C1 | GAGGATGTTGCCCACTGC | GTTGCTTGTCCACCACAGG |
| DNASE1L1 | CTTCAATGCTGACTGTGCATC | CACAATCCGATCGTAGGTACAG |
RESULTS
The instability of RH clones was assessed on the genomic level by IRS-based quantitative PCR and in detail for macaque chromosome 5 by STS marker genotyping. The qPCR data (Table 2, Fig. 1) display a high variation in the donor DNA content of the individual RH clone samples from different passages including losses of more than 90% of the donor DNA in p16*. Whereas a single clone, RH #147, showed an increase in donor DNA content, the p16* samples lost an average 55% of macaque IRS sequences present in p6*. The differences between the two independent cultures of passage 6 cells (p6 and p16*) were less pronounced. Forty-six markers, previously mapped on rhesus macaque chromosome 5 (Karere et al., 2008), were re-typed on gDNA of passages p6* and p16* of eleven RH clones (Table S1). The genotyping data show major discrepancies between cell culture passages p6* and p16* (Table 2, Fig. 2, Table S1). In agreement with the qPCR data, the call rates of genotypes generally declined from p6* to p16*. The average concordance rate between these two passages was 51.8%. Specifically, 46.2% of the genotypes showed a dropout in p16* and 5 of the possible 506 genotypes (2%) were positive in p16* while previously being absent in p6*. The latter “gained” p16* markers, D4S1582, HS3ST1, RAB28, PPBP, HS3ST1_1, were restricted to a single RH clone, RH #147. To examine the marker gain, additional experiments using 5-fold increased template DNA concentrations and higher numbers of PCR cycles, 60 instead of 35, were conducted. These amplifications revealed the presence of four out of five “gained” markers in the p6* using the modified PCR protocol geared towards highest sensitivity (data not shown). A comparison of the p6* data and the genotyping data of a previous thawing and culture of p6 cells (p6; data from (Karere et al., 2008) shows a concordance of 80.2%, with 9.7% of markers dropping out in p6, while 10.1% of markers were positive in p6 but not in p6* (Table 3, Table S1).
Table 2.
Marker Retention frequencies (%) and RT-PCR quantification of macaque DNA (%) present in 20ng of RH clone samples of passages p6, p6*, and p16*.
| 87 | 88 | 93 | 106 | 115 | 144 | 146 | 147 | 153 | 162 | 178 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p6 | qPCR | 2.69 | 5.17 | 8.13 | 2.40 | 9.60 | 3.71 | 1.68 | 2.51 | 1.88 | 0.81 | 2.33 |
| RF | 10.87 | 34.78 | 26.09 | 91.30 | 50.00 | 76.09 | 43.48 | 71.74 | 32.61 | 13.04 | 6.52 | |
| p6* | qPCR | 1.58 | 2.53 | 14.64 | 8.51 | 0.11 | 2.38 | 1.84 | 2.43 | 0.46 | 1.35 | 1.19 |
| RF | 10.87 | 30.43 | 28.26 | 80.43 | 50.00 | 76.09 | 58.70 | 71.74 | 17.39 | 13.04 | 6.52 | |
| p16* | qPCR | 0.27 | 2.31 | 3.74 | 1.77 | 2.11 | 1.35 | 0.49 | 4.25 | 0.23 | 0.14 | 0.19 |
| RF | 8.70 | 21.74 | 23.91 | 47.83 | 8.70 | 45.65 | 47.83 | 28.26 | 2.17 | 2.17 | 2.17 |
Figure 1.
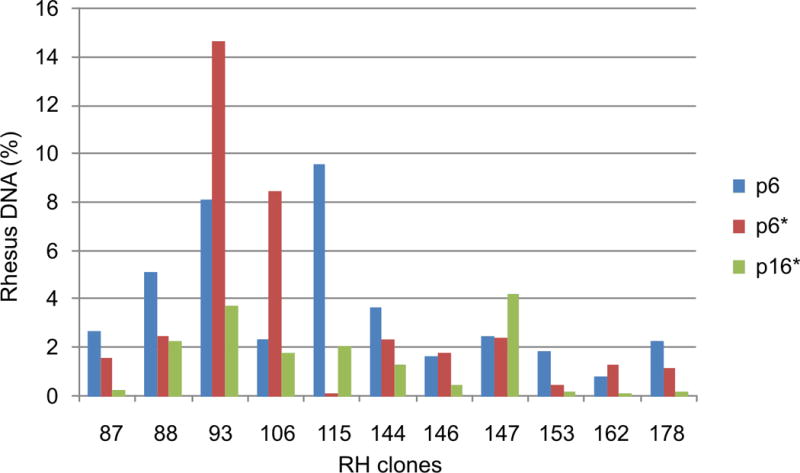
Real-Time PCR quantification of macaque DNA (%) present in 20 ng of RH clone samples of passages p6, p6* and p16*.
The qPCR assay employed inter-Alu-based fluorescence labeled primers to allow discrimination of macaque and hamster genomic DNA. The qPCR data indicate variation in the donor DNA content of the individual samples of different passages, with a substantial decline on average of 55% for the p16*.
Figure 2.
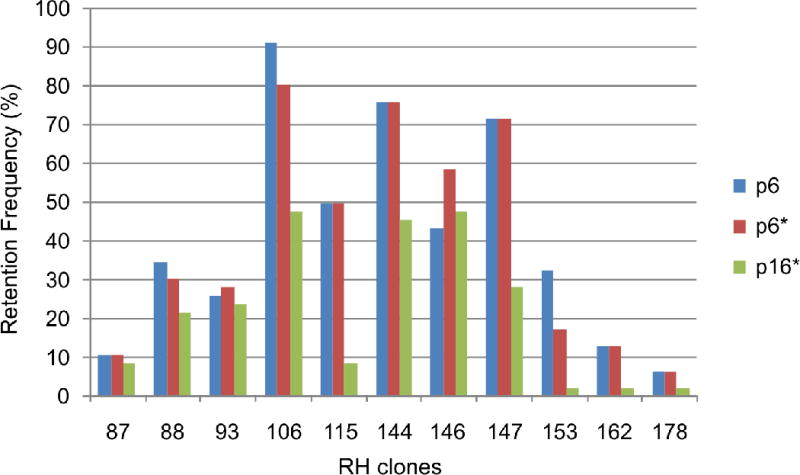
Retention frequencies (%) of markers amplified in RH clone samples.
The data show a general decline in marker retention frequencies between cell culture passages p6* and p16*, on average of 49.4% ranging from 87.5 to 15.4%.
Table 3.
Concordance of genotypes between RH clone passages p6, p6*, and p16*
| RH Clones | Comparison between p6* and p6 | Comparison between p6* and p16* | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| N1 | p6* drop-out (%) | p6 drop-out (%) | Concordance (%) | p6* drop-out (%) | p16* drop-out (%) | Concordance (%) | |
| MAEA | 4 | 0.0 | 25.0 | 75.0 | 0.0 | 25.0 | 75.0 |
| GRPEL1 | 4 | 0.0 | 0.0 | 100.0 | 0.0 | 25.0 | 75.0 |
| SORCS2 | 5 | 0.0 | 20.0 | 80.0 | 0.0 | 40.0 | 60.0 |
| MORF4 | 3 | 0.0 | 33.3 | 66.7 | 0.0 | 33.3 | 66.7 |
| MAN2B2 | 6 | 0.0 | 33.3 | 66.7 | 0.0 | 66.7 | 33.3 |
| STX18 | 3 | 0.0 | 0.0 | 100.0 | 0.0 | 33.3 | 66.7 |
| DRD5 | 4 | 25.0 | 25.0 | 50.0 | 0.0 | 66.7 | 33.3 |
| D4S1582 | 6 | 16.7 | 16.7 | 66.7 | 0.0 | 20.0 | 80.0 |
| HS3ST1 | 3 | 0.0 | 0.0 | 100.0 | 25.0 | 0.0 | 75.0 |
| HS3ST1_1 | 7 | 28.6 | 28.6 | 42.9 | 20.0 | 20.0 | 60.0 |
| RAB28 | 6 | 16.7 | 16.7 | 66.7 | 25.0 | 25.0 | 50.0 |
| CPEB2 | 6 | 0.0 | 16.7 | 83.3 | 0.0 | 66.7 | 33.3 |
| GBA3 | 4 | 0.0 | 0.0 | 100.0 | 0.0 | 50.0 | 50.0 |
| RBPSUH | 4 | 0.0 | 25.0 | 75.0 | 0.0 | 25.0 | 75.0 |
| STIM2 | 5 | 20.0 | 40.0 | 40.0 | 0.0 | 0.0 | 100.0 |
| PCDH7 | 5 | 20.0 | 0.0 | 80.0 | 0.0 | 25.0 | 75.0 |
| TBC1D1 | 4 | 25.0 | 0.0 | 75.0 | 0.0 | 0.0 | 100.0 |
| KIAA1239 | 7 | 28.6 | 0.0 | 71.4 | 0.0 | 50.0 | 50.0 |
| KLF3 | 6 | 0.0 | 0.0 | 100.0 | 0.0 | 100.0 | 0.0 |
| LIAS | 5 | 0.0 | 0.0 | 100.0 | 0.0 | 100.0 | 0.0 |
| RHOH | 3 | 0.0 | 0.0 | 100.0 | 0.0 | 33.3 | 66.7 |
| AB047909 | 5 | 0.0 | 0.0 | 100.0 | 0.0 | 60.0 | 40.0 |
| CCNI | 5 | 0.0 | 0.0 | 100.0 | 0.0 | 20.0 | 80.0 |
| SHROOM3 | 5 | 20.0 | 0.0 | 80.0 | 0.0 | 0.0 | 100.0 |
| ART3 | 5 | 0.0 | 0.0 | 100.0 | 0.0 | 20.0 | 80.0 |
| PPEF2 | 7 | 14.3 | 14.3 | 71.4 | 0.0 | 66.7 | 33.3 |
| RCHY1 | 4 | 0.0 | 0.0 | 100.0 | 0.0 | 25.0 | 75.0 |
| PPBP | 6 | 16.7 | 16.7 | 66.7 | 20.0 | 40.0 | 40.0 |
| EPHA5 | 6 | 0.0 | 50.0 | 50.0 | 0.0 | 66.7 | 33.3 |
| LPHN3 | 5 | 0.0 | 0.0 | 100.0 | 0.0 | 20.0 | 80.0 |
| IGFBP7 | 6 | 0.0 | 0.0 | 100.0 | 0.0 | 66.7 | 33.3 |
| REST | 7 | 0.0 | 0.0 | 100.0 | 0.0 | 42.9 | 57.1 |
| AB050266 | 5 | 20.0 | 0.0 | 80.0 | 0.0 | 50.0 | 50.0 |
| KDR | 4 | 0.0 | 0.0 | 100.0 | 0.0 | 25.0 | 75.0 |
| KIT | 6 | 16.7 | 0.0 | 83.3 | 0.0 | 60.0 | 40.0 |
| LNX | 6 | 16.7 | 0.0 | 83.3 | 0.0 | 80.0 | 20.0 |
| SPATA18 | 5 | 0.0 | 0.0 | 100.0 | 0.0 | 20.0 | 80.0 |
| DCUN1D4 | 7 | 0.0 | 0.0 | 100.0 | 0.0 | 50.0 | 50.0 |
| SOAT | 6 | 16.7 | 33.3 | 50.0 | 0.0 | 100.0 | 0.0 |
| KLHL8 | 4 | 25.0 | 0.0 | 75.0 | 0.0 | 100.0 | 0.0 |
| AB047948 | 5 | 40.0 | 0.0 | 60.0 | 0.0 | 100.0 | 0.0 |
| UCP1 | 4 | 25.0 | 25.0 | 50.0 | 0.0 | 100.0 | 0.0 |
| ARHGAP10 | 3 | 0.0 | 0.0 | 100.0 | 0.0 | 33.3 | 66.7 |
| CCNA2 | 7 | 71.4 | 14.3 | 14.3 | 0.0 | 50.0 | 50.0 |
| HSP90AA6P | 8 | 0.0 | 12.5 | 87.5 | 0.0 | 75.0 | 25.0 |
| ANP32C | 4 | 0.0 | 0.0 | 100.0 | 0.0 | 50.0 | 50.0 |
|
| |||||||
| Average | 10.1 | 9.7 | 80.2 | 2.0 | 46.2 | 51.8 | |
This table summarizes the concordances from Supplementary Table 1. N is the number of clones harboring the marker
Whole genome amplifications were successful in all genomic RH clone DNA samples tested as verified by control PCRs with four hamster STS markers (Table 1) on highly diluted WGA samples and by the concordance of the genotyping data to the gDNA samples. All WGA clone samples were positive for the four hamster markers in these experiments.
The WGAs yielded an average of 10 μg of wgaDNA ranging from 9.3 to 10.7 μg, indicating consistency of the reactions. The amplification of the gDNA was on average 133-fold after purification. A high level of concordance, 97.6%, was seen between the STS marker (N = 46) typing data of the genomic, gDNAp6*, and the wgaDNAP6* samples (Table S1). The differences are due exclusively to dropouts in the WGA samples. For 11 markers that had dropouts in the eleven samples, three dropped out more than once and a single marker, LNX, was absent in all WGA samples. The six independent WGA replicates from each of 5 RH clones displayed an even higher concordance of 99.3% for the 10 markers typed, indicating a profound consistency of wgaDNA for genotyping. The secondary WGA products (WGA2), re-amplified from primary wgaDNA samples, displayed only a single marker, LPHN3, dropout, implying 98% concordance (Table S1).
DISCUSSION
RH mapping is the predominant method for generating high-resolution genomic maps and has recently seen tremendous progress by the introduction of high-throughput genotyping methods (McKay et al., 2007). The remaining limitations for RH mapping, limited DNA samples and heterogeneity of DNA samples, are both related to the instability of the RH clone cell lines. The instability has been described previously (Goodfellow, 1991; Goss and Harris, 1977), but so far has not been quantified. As an example of a mammalian RH panel suitable for high-resolution RH mapping, we did examine clones of our 10,000Rad rhesus macaque RH panel by IRS-based quantitative PCR and by STS marker genotyping. The variation of the donor DNA content was studied over 10 low split-ratio cell culture passages to increase the number of observations and their representativeness. This does not imply that such an extended cell culture is applied in typical RH mapping projects.
The inter-ALU PCR quantifies the dispersed repetitive sequences as a representative of the macaque genomic DNA. The method might slightly underestimate the total amount of macaque DNA in the RH clone samples since some of the targets can be expected to have been interrupted by the irradiation. However the sizes of the inter-ALU PCR products, ranging from 100bp to 4kb (Liu et al., 1993), the majority being less than 2kb, can be estimated to be at least 100 times smaller than the fragments produced by the 10,000Rad irradiation of the macaque cells. For comparison, a 32,000Rad irradiation of genomic DNA produced fragments of about 150kb (Konfortov et al., 2000). Due to the size range differences, the interruption of IRS-PCR targets should cause only minor changes in the quantification of the donor DNA. The qPCR is used here to analyze the changes between different passages of RH cells, thus the influence of such artifacts should be negligible. The qPCR data demonstrate an average loss of 55% of the macaque donor DNA over ten passages of RH clone cell culture equaling a rate of 5.5% loss per passage. Since a uniform decay is not typical for biological processes a “natural decay” with a half-life of 8.7 passages is likely a better model for the donor DNA loss process. The increase of the donor DNA in a single RH clone could be attributed to amplifying mutations of the donor DNA but, as discussed below, could also be a consequence of the heterogeneity of the RH clone cultures and the varying growth rates of cell lineages. An indicator of the heterogeneity of the RH cell cultures are the low amounts of donor DNA. For example, the p6 samples display an average content of 3.7% macaque DNA; about half the estimated value derived from the genotyping data; Table 2 and (Karere et al., 2008). The presence of multiple cell populations with varying donor DNA content seems to be the best explanation for these differences.
The retention of macaque chromosome 5 markers over ten passages was analyzed in greater detail. Chromosome 5 is not under selection of the TK gene, is of medium size, and thus should be a good representative of the genome-wide average. Over ten passages, p6* to p16*, a high discordance, 48.2%, was observed in positive genotype calls, indicating an average discordance rate of about 5% per passage, in agreement with the qPCR data. Since the predominant change is the dropout of markers (Table 2), this indicates a half-life of the chromosome 5 markers of 10.6 passages. Unexpectedly a single p16*clone sample also displayed the presence of five markers not detected in the earlier p6* sample using the standard genotyping protocol. This “gain of loci” is likely an indirect consequence of the RH clone instability. The observed high instability of RH clones necessarily induces a fast loss of the clonality of the culture, resulting in multiple lineages of RH cells in the same culture (Slonim et al., 1997). Since the integrated donor DNA can have growth inhibiting effects (Senger et al., 2006), as readily observed by the strongly varying growth rates of RH clones, the unstable lineages inside a RH clone culture are bound to experience varying selective advantages. Thus, the seeming “gain of loci” in the advanced passage is likely due to the expansion of a previously underrepresented lineage, experiencing a growth promoting mutation. This hypothesis is supported by the genotyping experiments featuring high template DNA concentrations and PCR cycle numbers, which were able to detect the presence of 4 out of 5 markers “gained” in p16* also in the p6* samples. It should be noted that the RH clone experiencing the marker gain, RH #147, also displayed a donor DNA gain in the qPCR experiments.
The instability of RH clones is further apparent when comparing the STS data of p6 and p6*, two sets of samples resulting from independent cultures of twin passage 5 aliquots. The p6 and p6* sample sets showed a discordance of 14.4%. This observation is in agreement with the process of natural decay, suggesting that earlier passages should experience higher marker dropout rates than the 5% average observed over ten passages.
WGA of earliest RH clone passages could be an approach to avoid the high genomic heterogeneity of typical RH clone samples described above. Thus, the suitability of Φ 29 phage DNA polymerase based MDA for RH mapping was investigated through three sets of experiments simulating the requirements of an RH mapping project. The reliability of wgaDNA genotyping data from RH clone samples, measured as concordance rate with data from gDNA, was high at 97.6%. This rate is similar to the concordance rates observed for SNP BeadArray genotyping (Pask et al., 2004; Tzvetkov et al., 2005). The even higher concordance rate (99.3%) of the six sets of replicate wgaDNA experiments concurs with previous results of a smaller scale experiment involving two independent whole genome amplifications from a single clone (Senger et al., 2006). Together these studies confirm a very high reproducibility of the MDA technique with RH clone DNA and show that concerns about the application of MDA on irradiated samples (Bergen et al., 2005) do not apply for the RH protocol where the cells are cultured after irradiation. Our data show further that the dropout of some markers in WGA experiments is reproducible (Pugh et al., 2008) and thus marker specific. For example, one marker failed to amplify by WGA in all clones in which it was present (five out of five samples; Table S1). Two further markers showed repeated WGA failure. This observation is in agreement with previous SNP genotyping studies that showed limited marker dropouts specifically in very GC-rich and telomere near genome regions (Berthier-Schaad et al., 2007). The reproducibility of WGA-related marker dropouts will enable the identification of susceptible markers in control MDA experiments with genomic DNA of the donor species and thus their elimination from RH studies. It can be expected that, after the exclusion of WGA-failure-susceptible markers, the concordance between gDNA and wgaDNA genotypes will be close to the 99.3% rate observed for the replicate MDA experiments described above. Thus, the rate of possibly MDA induced errors should be similar to replicate STS marker genotyping experiments. The present data show that MDA is a reliable genome amplification method for RH clone samples. The 99% concordance of second-generation WGA data to the gDNA data demonstrates further its sustainability in repeated amplifications. The testing of the highly diluted samples of the whole genome amplification products with hamster STS marker positive controls is suggested as a quality control of the amplification reaction to make sure that the experiment is not affected by WGA artifacts.
In the present study, consistent 133-fold amplifications of RH-clone DNA were achieved by MDA. The enhanced yield of sample DNA is in stark contrast to the three-fold gain achieved by cell culture passaging, which was accompanied by a 5% marker dropout rate. The MDA amplification rate can certainly be further improved by larger scale experiments employing maxi-sized WGA-kits instead of the mini-kits that were used for this study. Because of its reliability and high amplification rate, MDA will enable considerable improvements of the experimental design in RH mapping. Not only is the sample DNA limitation surmounted, but MDA will also allow the analysis of very early RH clone passages. The current need for a expansion of RH clone cells to generate enough sample DNA for large scale RH mapping experiments goes along with the two drawbacks of significant loci losses and increasing heterogeneity of the cell population, both caused by the inherent instability of RH clones. Theoretical calculations did conclude previously that RH mapping would be most efficient when RH clone panels with retention frequencies of 40 to 50% are employed (Boehnke et al., 1991; Jones, 1996; Lunetta et al., 1995). For comparison, the macaque RH clone panel studied previously (Karere et al., 2008), displays an average retention frequency of 19%, which is representative for 10,000Rad panels. The analysis of passages as early as the second passage will create RH panels with highly elevated marker retention frequencies. It will further reduce the number of subpopulations in the samples and thus minimize the occurrence of ambiguous genotyping data and of selective dropouts of weakly performing STS primers. A single T25 flask per RH clone would provide enough starting material for multiple whole genome amplifications and thus whole genome mapping.
Conclusion
MDA can resolve the problem of limited DNA samples in RH mapping, allowing for the analysis of an almost infinite number of markers and facilitating the liberal distribution of RH clone samples among the research community. Although with improving genotyping technologies (Steemers and Gunderson, 2007) smaller DNA amounts are required, the technique is not currently widely applied as the DNA microarrays are only available for a few selected species. The new data on marker dropouts in cell culture and reproducibility of MDA suggest that an analysis of the earliest passage RH clone DNAs would allow for RH mapping experiments with two significant advantages: (i) higher efficiency due to the higher retention frequencies achievable (fewer RH clones needed), and (ii) increased precision due to the higher uniformity of the cell populations representing individual RH clones. MDA seems especially appropriate for systems where the growth of RH clones is difficult, e.g. for non mammalian species like fishes (Sarropoulou et al., 2007) and plants (Wardrop et al., 2002). The use of early passage whole genome amplified DNA samples, combined with the developing microarray systems for high-throughput genotyping (McKay et al., 2007; Prasad et al., 2007), should elevate the efficiency of RH mapping significantly.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health, National Center for Research Resources [R24 RR017584]. We thank Dr. James Womack and Elaine Owens (Texas A&M University) and Lee Millon (UC Davis, Veterinary Genetics Laboratory) for the generation of the rhesus macaque radiation hybrid cells.
Footnotes
Supplementary Information
Genotype comparisons between passages p6* and p6, p16*, or, p6*wga. The table presents the genotyping raw data of the PCR analyses (‘1’ represents amplification of the particular marker in the cell line. ‘0’ indicates no amplification of the particular marker in the cell line).
References
- Agarwala R, Applegate DL, Maglott D, Schuler GD, Schaffer AA. A fast and scalable radiation hybrid map construction and integration strategy. Genome Res. 2000;10:350–64. doi: 10.1101/gr.10.3.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006;16:545–52. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Bergen AW, Qi Y, Haque KA, Welch RA, Garcia-Closas M, Chanock SJ, Vaught J, Castle PE. Effects of electron-beam irradiation on whole genome amplification. Cancer Epidemiol Biomarkers Prev. 2005;14:1016–9. doi: 10.1158/1055-9965.EPI-04-0686. [DOI] [PubMed] [Google Scholar]
- Berthier-Schaad Y, Kao WH, Coresh J, Zhang L, Ingersoll RG, Stephens R, Smith MW. Reliability of high-throughput genotyping of whole genome amplified DNA in SNP genotyping studies. Electrophoresis. 2007;28:2812–7. doi: 10.1002/elps.200600674. [DOI] [PubMed] [Google Scholar]
- Boehnke M, Lange K, Cox DR. Statistical methods for multipoint radiation hybrid mapping. Am J Hum Genet. 1991;49:1174–88. [PMC free article] [PubMed] [Google Scholar]
- Chowdhary BP, Raudsepp T, Honeycutt D, Owens EK, Piumi F, Guerin G, Matise TC, Kata SR, Womack JE, Skow LC. Construction of a 5000(rad) whole-genome radiation hybrid panel in the horse and generation of a comprehensive and comparative map for ECA11. Mamm Genome. 2002;13:89–94. doi: 10.1007/s00335-001-2089-8. [DOI] [PubMed] [Google Scholar]
- Cox DR, Burmeister M, Price ER, Kim S, Myers RM. Radiation hybrid mapping: a somatic cell genetic method for constructing high-resolution maps of mammalian chromosomes. Science. 1990;250:245–50. doi: 10.1126/science.2218528. [DOI] [PubMed] [Google Scholar]
- Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, Bray-Ward P, Sun Z, Zong Q, Du Y, Du J, Driscoll M, Song W, Kingsmore SF, Egholm M, Lasken RS. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99:5261–6. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow PN. Irradiation and fusiongene transfer. In: MURRAY EJ, editor. Methods in Molecular Biology. Clifton: Humana Press; 1991. [Google Scholar]
- Goss SJ, Harris H. Gene transfer by means of cell fusion I. Statistical mapping of the human X-chromosome by analysis of radiation-induced gene segregation. J Cell Sci. 1977;25:17–37. doi: 10.1242/jcs.25.1.17. [DOI] [PubMed] [Google Scholar]
- Graw S, Davidson J, Gusella J, Watkins P, Tanzi R, Neve R, Patterson D. Irradiation-reduced human chromosome 21 hybrids. Somat Cell Mol Genet. 1988;14:233–42. doi: 10.1007/BF01534584. [DOI] [PubMed] [Google Scholar]
- Jann OC, Aerts J, Jones M, Hastings N, Law A, Mckay S, Marques E, Prasad A, Yu J, Moore SS, Floriot S, Mahe MF, Eggen A, Silveri L, Negrini R, Milanesi E, Ajmone-Marsan P, Valentini A, Marchitelli C, Savarese MC, Janitz M, Herwig R, Hennig S, Gorni C, Connor EE, Sonstegard TS, Smith T, Drogemuller C, Williams JL. A second generation radiation hybrid map to aid the assembly of the bovine genome sequence. BMC Genomics. 2006;7:283. doi: 10.1186/1471-2164-7-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HB. Hybrid selection as a method of increasing mapping power for radiation hybrids. Genome Res. 1996;6:761–9. doi: 10.1101/gr.6.8.761. [DOI] [PubMed] [Google Scholar]
- Karere GM, Froenicke L, Millon L, Womack JE, Lyons LA. A high-resolution radiation hybrid map of rhesus macaque chromosome 5 identifies rearrangements in the genome assembly. Genomics. 2008 doi: 10.1016/j.ygeno.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konfortov BA, Cohen HM, Bankier AT, Dear PH. A high-resolution HAPPY map of Dictyostelium discoideum chromosome 6. Genome Res. 2000;10:1737–42. doi: 10.1101/gr.141700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Siciliano J, Seong D, Craig J, Zhao Y, De Jong PJ, Siciliano MJ. Dual Alu polymerase chain reaction primers and conditions for isolation of human chromosome painting probes from hybrid cells. Cancer Genet Cytogenet. 1993;65:93–9. doi: 10.1016/0165-4608(93)90213-6. [DOI] [PubMed] [Google Scholar]
- Lunetta KL, Boehnke M, Lange K, Cox DR. Experimental design and error detection for polyploid radiation hybrid mapping. Genome Res. 1995;5:151–63. doi: 10.1101/gr.5.2.151. [DOI] [PubMed] [Google Scholar]
- Matise TC, Chen F, Chen W, De La Vega FM, Hansen M, He C, Hyland FC, Kennedy GC, Kong X, Murray SS, Ziegle JS, Stewart WC, Buyske S. A second-generation combined linkage physical map of the human genome. Genome Res. 2007;17:1783–6. doi: 10.1101/gr.7156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckay SD, Schnabel RD, Murdoch BM, Aerts J, Gill CA, Gao C, Li C, Matukumalli LK, Stothard P, Wang Z, Van Tassell CP, Williams JL, Taylor JF, Moore SS. Construction of bovine whole-genome radiation hybrid and linkage maps using high-throughput genotyping. Anim Genet. 2007;38:120–5. doi: 10.1111/j.1365-2052.2006.01564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pask R, Rance HE, Barratt BJ, Nutland S, Smyth DJ, Sebastian M, Twells RC, Smith A, Lam AC, Smink LJ, Walker NM, Todd JA. Investigating the utility of combining phi29 whole genome amplification and highly multiplexed single nucleotide polymorphism BeadArray genotyping. BMC Biotechnol. 2004;4:15. doi: 10.1186/1472-6750-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinard R, De Winter A, Sarkis GJ, Gerstein MB, Tartaro KR, Plant RN, Egholm M, Rothberg JM, Leamon JH. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 2006;7:216. doi: 10.1186/1471-2164-7-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A, Schiex T, Mckay S, Murdoch B, Wang Z, Womack JE, Stothard P, Moore SS. High resolution radiation hybrid maps of bovine chromosomes 19 and 29: comparison with the bovine genome sequence assembly. BMC Genomics. 2007;8:310. doi: 10.1186/1471-2164-8-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh TJ, Delaney AD, Farnoud N, Flibotte S, Griffith M, Li HI, Qian H, Farinha P, Gascoyne RD, Marra MA. Impact of whole genome amplification on analysis of copy number variants. Nucleic Acids Res. 2008;36:e80. doi: 10.1093/nar/gkn378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarropoulou E, Franch R, Louro B, Power DM, Bargelloni L, Magoulas A, Senger F, Tsalavouta M, Patarnello T, Galibert F, Kotoulas G, Geisler R. A gene-based radiation hybrid map of the gilthead sea bream Sparus aurata refines and exploits conserved synteny with Tetraodon nigroviridis. BMC Genomics. 2007;8:44. doi: 10.1186/1471-2164-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger F, Priat C, Hitte C, Sarropoulou E, Franch R, Geisler R, Bargelloni L, Power D, Galibert F. The first radiation hybrid map of a perch-like fish: the gilthead seabream (Sparus aurata L) Genomics. 2006;87:793–800. doi: 10.1016/j.ygeno.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Shifman S, Bell JT, Copley RR, Taylor MS, Williams RW, Mott R, Flint J. A high-resolution single nucleotide polymorphism genetic map of the mouse genome. PLoS Biol. 2006;4:e395. doi: 10.1371/journal.pbio.0040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slonim D, Kruglyak L, Stein L, Lander E. Building human genome maps with radiation hybrids. J Comput Biol. 1997;4:487–504. doi: 10.1089/cmb.1997.4.487. [DOI] [PubMed] [Google Scholar]
- Steemers FJ, Gunderson KL. Whole genome genotyping technologies on the BeadArray platform. Biotechnol J. 2007;2:41–9. doi: 10.1002/biot.200600213. [DOI] [PubMed] [Google Scholar]
- Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, Tunnacliffe A. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 1992;13:718–25. doi: 10.1016/0888-7543(92)90147-k. [DOI] [PubMed] [Google Scholar]
- Tzvetkov MV, Becker C, Kulle B, Nurnberg P, Brockmoller J, Wojnowski L. Genome-wide single-nucleotide polymorphism arrays demonstrate high fidelity of multiple displacement-based whole-genome amplification. Electrophoresis. 2005;26:710–5. doi: 10.1002/elps.200410121. [DOI] [PubMed] [Google Scholar]
- Walter MA, Goodfellow PN. Irradiation and fusion gene transfer. Mol Biotechnol. 1995;3:117–28. doi: 10.1007/BF02789107. [DOI] [PubMed] [Google Scholar]
- Wardrop J, Snape J, Powell W, Machray GC. Constructing plant radiation hybrid panels. Plant J. 2002;31:223–8. doi: 10.1046/j.1365-313x.2002.01351.x. [DOI] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, Mcguire A, He W, Chen YJ, Makhijani V, Roth GT, Gomes X, Tartaro K, Niazi F, Turcotte CL, Irzyk GP, Lupski JR, Chinault C, Song XZ, Liu Y, Yuan Y, Nazareth L, Qin X, Muzny DM, Margulies M, Weinstock GM, Gibbs RA, Rothberg JM. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–6. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- Womack JE, Johnson JS, Owens EK, Rexroad CE, 3rd, Schlapfer J, Yang YP. A whole-genome radiation hybrid panel for bovine gene mapping. Mamm Genome. 1997;8:854–6. doi: 10.1007/s003359900593. [DOI] [PubMed] [Google Scholar]
- Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N. Whole genome amplification from a single cell: implications for genetic analysis. Proc Natl Acad Sci U S A. 1992;89:5847–51. doi: 10.1073/pnas.89.13.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.