

Abstract
Peripheral insulin resistance is a key component of metabolic syndrome associated with obesity, dyslipidemia, hypertension, and type 2 diabetes. While the impact of insulin resistance is well recognized in the periphery, it is also becoming apparent in the brain. Recent studies suggest that insulin resistance may be a factor in brain aging and Alzheimer’s disease (AD) whereby intranasal insulin therapy, which delivers insulin to the brain, improves cognition and memory in AD patients. Here, we tested a clinically relevant delivery method to determine the impact of two forms of insulin, short-acting insulin lispro (Humalog) or long-acting insulin detemir (Levemir), on cognitive functions in aged F344 rats. We also explored insulin effects on the Ca2+-dependent hippocampal afterhyperpolarization (AHP), a well-characterized neurophysiological marker of aging which is increased in the aged, memory impaired animal. Low-dose intranasal insulin improved memory recall in aged animals such that their performance was similar to that seen in younger animals. Further, because ex vivo insulin also reduced the AHP, our results suggest that the AHP may be a novel cellular target of insulin in the brain, and improved cognitive performance following intranasal insulin therapy may be the result of insulin actions on the AHP.
Key Words: Calcium, Type 2 diabetes, Neuronal excitability, Memory, Afterhyperpolarization.
Results from preclinical settings argue that peripheral insulin resistance, a component of type 2 diabetes and/or the metabolic syndrome, may have a negative impact on cognitive function. Emerging studies report that in diabetic models, insulin responses are attenuated in the brain, possibly indicating the presence of insulin resistance in this tissue. Clinical and preclinical studies show that insulin and insulin-like growth factor-1 (IGF-1) receptors, their message and their function, are often reduced in the hippocampus during normal and pathological aging (1–6). Similarly, in animal models of aging and Alzheimer’s disease (AD), brain insulin receptor signaling is reduced, often in the presence of peripheral metabolic dysregulation (7–14). Importantly, several studies provide evidence that brain insulin therapy can greatly reduce cognitive decline with age and/or AD, and even improve memory in young adults [reviewed in ref. (15)].
In recent studies examining the effects of insulin on cognition, insulin has been administered via the intranasal route to bypass the blood brain barrier. Notably, Born and colleagues (16) have shown that intranasal insulin is capable of entering the human brain rapidly. However, the underlying mechanism(s) leading to cognitive improvement are not yet clear. Potential molecular/cellular targets affected by insulin in the brain and capable of offsetting cognitive decline include changes in neuronal firing (17), Ca2+-dependent K+ channels (18), GABA currents (19) and tau and Aβ metabolism (20–22). Insulin also alters synaptic structures (23,24), modifies synaptic plasticity (25–27) and increases delivery of NMDA receptors to the plasma membrane (28). Similar effects have been observed in response to IGF-1 replacement, showing increased AMPA-mediated excitatory postsynaptic potentials (29,30), and increased NMDA receptor trafficking (31), effects that appear capable of offseting age-dependent cognitive decline (32). Further, MRI data in men and women has provided evidence that intranasal insulin can have an impact on brain blood flow (33), and on whole-body insulin sensitivity via regulation of hypothalamic activity (34,35). Whether this later effect is purely mediated by central insulin effects or reflects permeation of small insulin amounts into the bloodstream is currently being investigated (36–38).
Another important candidate mechanism that may underlie the effects of intranasal insulin pertains to Ca2+ homeostasis. Ca2+ dysregulation is prominent in models of diabetes (39). In the dorsal root ganglion and the hippocampus of animals with STZ-induced diabetes (40,41), broader Ca2+ action potentials, larger Ca2+-dependent afterhyperpolarizations (AHPs), and aberrant intracellular Ca2+ release are seen (40,42,43). Impaired memory and long-term potentiation maintenance is also present (44). Clinical data also supports a role for Ca2+ dysregulation in peripheral insulin resistance as evidenced by the presence of abnormal and sustained adipocyte Ca2+ elevations in hyperinsulinemic, obese subjects (45). Similar evidence for an increase in Ca2+ levels in skin fibroblasts from type 2 diabetics has also been reported (46). In recent work, we have shown that the Ca2+-dependent AHP, which increases in amplitude and duration with aging (47–49), is sensitive to insulin (ex vivo), and that this sensitivity is reduced by a high fat diet (50).
While some studies in humans demonstrate the efficacy of intranasal insulin as a cognitive enhancer, studies in animal models are required to understand the potential mechanisms underlying the beneficial effects of intranasal insulin. However, only one prior study has measured the impact of intranasal insulin on brain function in the awake animal, focusing on olfactory discrimination in the young mouse (51). Because the greatest risk factor for AD is aging, research into how central nervous system insulin signaling affects brain aging may improve understanding of the biological conditions. Further, because of the sustained growth in the aging population, implementing strategies to reduce the impact of brain aging will likely benefit the aging population, and by extension, may reduce the incidence of age-related neurodegenerative diseases that share cellular mechanisms with brain aging (eg, AD).
We, therefore, assessed learning and memory performance in aged rats in response to two different insulin formulations (short-acting insulin Humalog and long-acting insulin Levemir), and quantified Ca2+ homeostasis. Insulin is available primarily in shorter- and longer-acting forms, and intranasal longer-acting insulin formulations (eg, Levemir) may potentially be superior to shorter-acting ones (eg, Humalog) especially in APOE-ε4 carriers (52). In addition, because insulin is often formulated with zinc, to more specifically address the impact of insulin, we studied a zinc-free insulin formulation (Apidra), as well as zinc alone on the AHP. Overall, our goals were to test whether intranasal insulin could reduce cognitive decline in aged animals, as well as redress the accompanying neuronal Ca2+ dysregulation seen in the hippocampus of aged animals.
Methods
Animals
Young male F344 (3 months old), aged male F344 (21 months old), and young male Sprague-Dawley rats (2–6 months old) were used for this study. Young F344 animals were not treated with insulin, and were used to help gauge the impact of intranasal insulin on memory function in aged F344. All animals were fed an 18% protein rodent diet (Harlan Teklad, diet #2018; Madison, WI).
In Vivo Insulin Delivery and Doses
Levemir doses were approximately 0.0715, 0.143, or 0.286 IU/day/rat delivered in two 5 µL applications applied to the tip of the right naris using a P10 Eppendorf pipette (Supplementary Figure 1) [adapted from (53)]. Humalog dose was chosen to deliver 0.0715 IU/day/rat for behavioral characterization. An intranasal 0.286 IU/rat dose was also administered once to aged animals (n = 3) to monitor the impact on peripheral glucose (Supplementary Table 1). Each application was separated by 1 minute, during which the animal was held supine and immobile in a DecapiCone (Braintree Scientific Inc., Braintree, MA). Each animal was acclimated to this procedure for 4 days prior to initiation of the Morris water maze protocol. All animal groups were exposed to the DecapiCone for the same duration, and no signs of stress from the bagging procedure were noted. The insulin doses were chosen to approximate several clinically relevant doses used in past studies ranging from 10 to 40 IU/day and were adjusted by brain weights. Assuming a 70-year old human brain weighs 1,360g (54), 10, 20, and 40 IU/day are equivalent to 0.0074, 0.0147, and 0.0294 IU/day/g of brain, respectively. Assuming a 21-month old F344 brain weighs 2.06g (55), our equivalent doses were 0.035, 0.069, and 0.138 IU/day/g of brain. It should be noted that these values may underestimate final insulin concentration given the greater olfactory epithelium surface in the nasal passages of the rat (50%), compared to man [8% (56)]. Insulin solutions were made fresh weekly from 100U/mL vials (Humalog, insulin lispro, Lilly; Levemir, insulin detemir, Novo Nordisk) and diluted into sterile 0.9% saline.
Blood Glucose Levels
To ensure that the intranasal delivery did not result in reduced peripheral glucose levels, and to test whether stress from the acute restraint procedures (DecapiCone) could elevate blood glucose levels, we measured blood glucose (FreeStyle Lite glucometer; Abbott Laboratories, Abbott Park, IL) from dorsal tail veins in a subset of aged animals either exposed to intranasal Levemir (n = 5 from each treatment group) or Humalog (n = 3), both before and approximately 1 hour after dosing (Supplementary Table 1).
Animal Behavior, Treatment Groups and Morris Water Maze Procedures
Two cohorts of F344 animals were used for behavioral characterization (Supplementary Figure 1B). One cohort of animals (n = 50) was used to test the impact of long-acting insulin Levemir at three different doses (low, medium, and high; n = 10 per group) compared with an intranasal saline young group (3 months; n = 10) and an intranasal saline aged group (21 months old; n = 10). A second cohort of aged animals (n = 20) was used to test the impact of short-acting insulin Humalog against saline (n = 10 per group). For analysis, behavioral data from the two aged saline control groups in both cohorts were combined because memory performance on probe day was similar (path length to goal, p = .89 and cumulative search error, p = .913). One aged animal in the saline group died during acclimation prior to initiation of behavioral testing. The remaining 69 animals where trained on the Morris water maze (MWM). Animals were considered visually impaired or unable to learn the task if they could not find the platform on all three of the trials on the cue day (visual platform) and at least one of the remaining three training days. Based on these conditions, two aged Humalog animals and three aged low-dose Levemir animals were removed from the analysis. Behavioral results are therefore presented on 64 animals, 10 young saline, 19 aged saline, 8 aged low-dose humalog (10 IU/day), 7 aged low-dose Levemir (10 IU/day), 10 aged medium-dose Levemir (20 IU/day), and 10 aged high-dose Levemir (40 IU/day). On cue day, no significant differences were found across any of the groups on measures of path length (one-way ANOVA, p > .05) and swim speed across insulin dose (one-way ANOVA, post hoc p > .05), indicating motivation and motor activity were likely similar.
Insulin or saline treatment in both cohorts was initiated 4 days prior to the initiation of training (Supplementary Figure 1B) and was delivered 1–3 hours before testing on the MWM. The MWM protocol has been published elsewhere (50,57), but briefly, water temperature was maintained at 26°C –27°C, and black liquid tempera paint was used to make the water opaque and hide the submerged escape platform (~1.5cm below water surface). A Videomex-V acquisition system and water maze analysis software (version 4.64, Columbus Instruments, Columbus, OH) were used to track and measure animal movements. Animals were allowed 60 seconds to find the platform, after which they were guided to it. Each animal stayed on the platform for 30 seconds before returning to a heated holding chamber for about 2 minutes. On cue day, a white cup hanging above the partially submerged platform helped orient the animals. Animals were subjected to three trials per day (semi-random drop location for each trial) for cue and training days. Twenty-four hours after the last training day, a probe trial was initiated with the platform removed (60 seconds max swim time). Swim speed was derived from the distance travelled over time on the last trial of the third training day and on the first trial of the cue day. On all cue and training days the intertrial interval was approximately 150 seconds. All behavioral experiments were conducted with the experimenters blind to the treatment groups using color coding of the insulin and saline vials.
Electrophysiology
Hippocampal slices were used across two electrophysiological setups, one monitoring extracellular field potentials and, the other measuring intracellular potentials (eg, AHP). All data were acquired using pClamp 8.0 (Molecular Devices MDS, Toronto, Canada) through a Digidata 1320A A/D converter (Molecular Devices). Potentials (eg, amplitude and duration of AHPs, field EPSPs) were quantified off-line using Clampfit (Molecular Devices). Nineteen of the 20 behaviorally characterized aged animals (second cohort used to test the impact of short-acting insulin Humalog against saline) were used 2–5 weeks after the last day of behavioral characterization to determine the impact of single intranasal insulin dose in vivo. From those, a total of 30 slices used for extracellular field potential recordings (Figure 2D and E) and 13 cells used for intracellular recordings (Figure 2B and C) were used for statistical analyses and are presented using an n of 1 per cell/slice. In data presented in Figure 3, 16 Sprague-Dawley animals (2–6 months old) were used to monitor the impact of zinc and Humalog ex vivo both extracellularly and intracellularly (30 slices and 23 cells, respectively). Work presented in Figure 4 (intracellular physiology only) is compiled from a total of 17 F344 male rats split into young (n = 8; 3 months) and aged (n = 9; 22 months). This last group of animals yielded recordings from six cells for young artificial cerebro-spinal fluid (ACSF), eight cells from young Apidra, six cells for aged ACSF, and five cells for aged Apidra.
Figure 2.
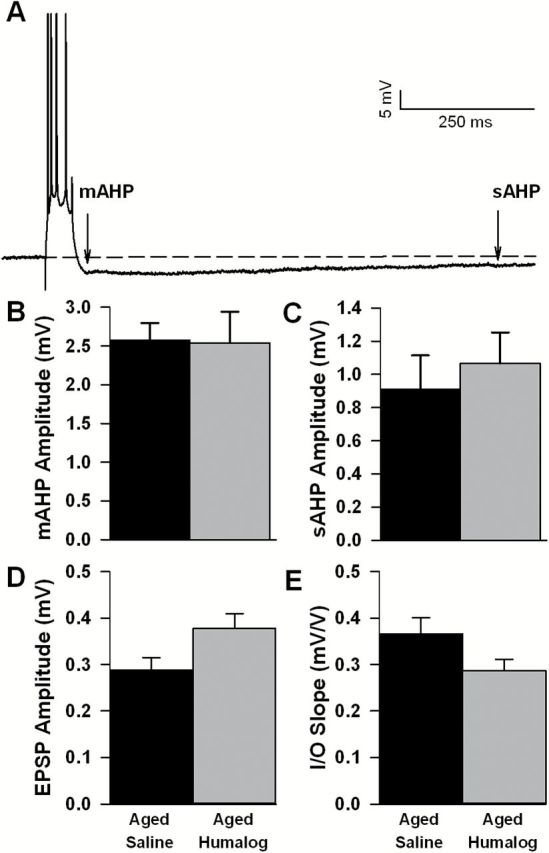
Impact of in vivo intranasal Humalog on intracellular and extracellular physiology. (A) Representative intracellular recording of the afterhyperpolarization (AHP) following a series of four action potentials (truncated to emphasize the AHP). The medium AHP (B) and slow AHP (C) amplitudes recorded 3–7 hours after intranasal Humalog in F344 rats were not significantly different. EPSP amplitudes (D) and I/O slopes (E) when recorded extracellularly 3–7 hours after intranasal Humalog also were not significantly altered. Mean ± SEM are shown.
Figure 3.
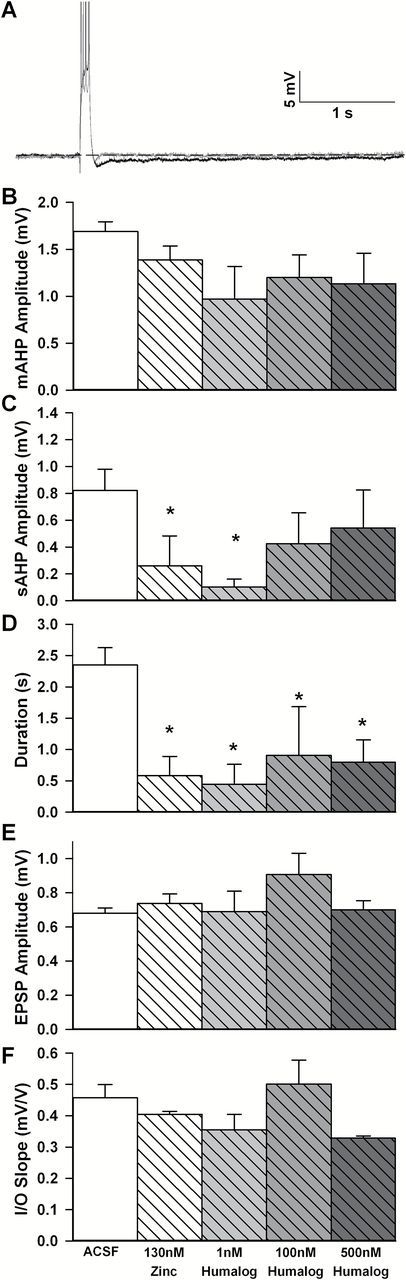
Impact of ex vivo Humalog and zinc on the afterhyperpolarization (AHP). (A) Representative voltage trace of the AHP recorded in a cell from an ACSF treated slice (black) and a Humalog treated slice (1nM, gray). Data are derived from young Sprague-Dawley animals. Traces are truncated to emphasize the AHP. Significant reductions in the slow AHP (sAHP) amplitude (C) and duration (D) were seen in response to Humalog as well as zinc. Note that zinc and low concentration Humalog seem to work more robustly on the sAHP [only a trend was noted for the medium AHP (B)]. No significant effect was detected during extracellular recording of field EPSP amplitudes (E) or slopes of the I/O curve (F). Mean ± SEM are shown.
Figure 4.
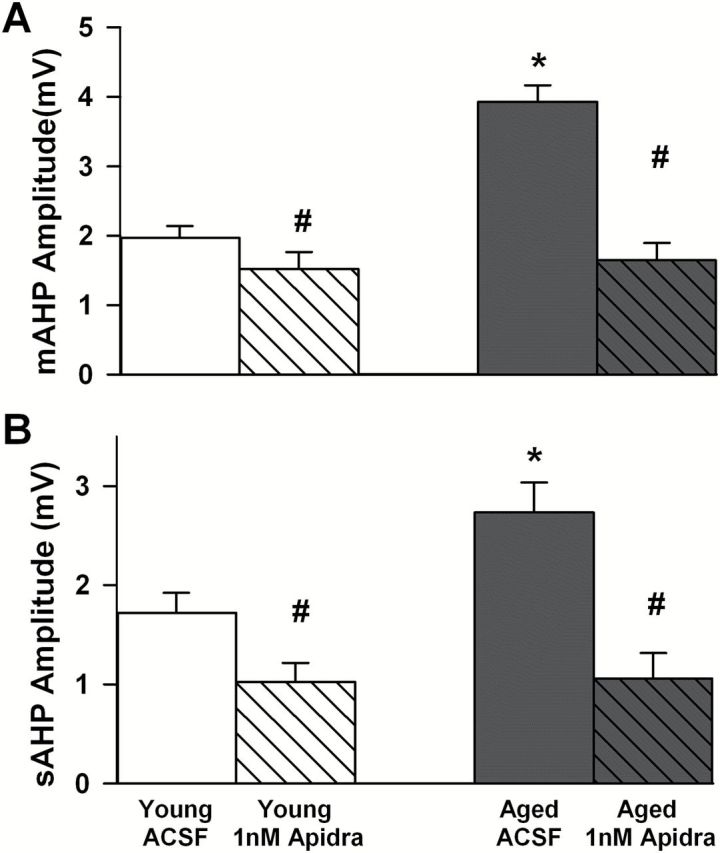
Zinc-free Apidra reduces the afterhyperpolarization (AHP) ex vivo. The medium AHP (mAHP) amplitude is shown in (A) and the slow AHP (sAHP) amplitude is shown in (B). Mean ± SEM are presented in response to 1nM Apidra. These data are taken from young and aged F344 rats. Asterisks indicate significant age-dependent increases in the mAHP and sAHP, and pound signs illustrate significant insulin-mediated reductions in the AHP. Again, the impact of insulin seems more robust on the sAHP compared to the mAHP. These recordings were obtained with 4.5mM glucose in the ACSF.
Field potentials
Rats were anesthetized with Isoflurane prior to decapitation. Brains were quickly removed and placed in oxygenated ACSF of the following composition (in mM): 114 NaCl, 3 KCl, 10 glucose, 1.25 KH2PO4, 26 NaHCO3, 8 MgCl2, and 0.1 CaCl2. Hippocampi were sectioned (coronal) with a Vibratome 3000 (Bannockburn, IL) set at 400 μm. Slices were transferred to an interface-type recording chamber (static) containing oxygenated recording ACSF of the following composition (in mM): 114 NaCl, 3 KCl, 10 glucose, 1.25 KH2PO4, 26 NaHCO3, 2.5 CaCl2, and 1.3 MgCl2. The recording chamber was kept at 32°C and warm moist 95% O2 and 5% CO2 was continually delivered to the chamber. Bipolar synaptic stimulation was delivered through a SD9K stimulator (Astro Med Inc., Grass Instr., Warwick, RI). Input/output curves (I/O) were recorded using a series of increasing voltage steps. The relationship between stimulus intensity and EPSP amplitudes (prior to dendritic spike contamination) was fit (linear, r 2 > .9) to yield a measure on the slope of the I/O. Data are also reported as the absolute amplitudes of the EPSPs measured as the maximum deflection from baseline. Insulin (1, 100, and 500nM Humalog) and/or zinc treatment (130 nM—see AHP-Intracellular Recordings section) under these conditions was initiated 1 hour after placing the slices in the chamber (compounds remained in the chamber for the duration of the experiment.
AHP-Intracellular Recordings
Hippocampal slices (350 µm) were transferred from the static chamber and placed in a perfusion chamber (RC22C, Warner Instruments, Co., Hamden, CT) and maintained in a continuous flow of oxygenated ACSF preheated at 32°C using a TC2Bip/HPRE2 in-line heating system (Cell Micro Controls, Northfolk, VA). The ACSF contained 2mM CaCl2 and 2mM MgCl2 and in some experiments (Figure 4 only), the traditional 10mM glucose concentration was replaced with 4.5mM glucose. This was done to test whether higher glucose concentrations altered insulin sensitivity. We chose 4.5mM to help maintain tissue health and physiology for extended hours (58), to approximate values in anesthetized preparations (59), and to consider fluctuations in brain glucose levels across a day (60). As previously described, cells were impaled with sharp microelectrodes filled with 2M KMeSO4 and 10mM HEPES, pH 7.4 [tip resistance 90–120 MΩ (47)]. Recordings of membrane input resistance (IR) were obtained in response to 800ms, 200 pA hyperpolarizing current injections using an Axoclamp 2B amplifier (Molecular Devices) while holding the cell at −70 mV. To generate an AHP cells were held at −65 mV (baseline) and depolarized with a 100ms current injection to generate four Na+ action potentials. The medium AHP (mAHP) was measured as the peak hyperpolarization immediately after the offset of the depolarizing current injection, the slow AHP (sAHP) was measured 800ms after the end of the current injection. The AHP duration was measured from the end of the depolarizing step until return to baseline.
Several concentrations and formulations of insulin were tested ex vivo by addition to the solution bathing the hippocampal slices. Humalog concentrations of 1, 100, and 500nM were used. A zinc (ZnCl2) concentration of 130nM was used to mirror levels of free zinc attained in 250nM Humalog. Apidra (insulin glulisine, Sanofi, a zinc-free insulin formulation) was used at 1nM and results were compared to ACSF. Under these experiments, Apidra was used to treat slices both in the static chamber and during recording of the AHP. Apidra was continuously perfused into the flow of the oxygenated recording ACSF using a precision perfusion pump (Orion Research, Model 341B, Boston, MA) and resulting in a final concentration of 1nM (diluted in sterile saline once a week from the 600 µM stock). Only neurons with IR > 25 MΩ, holding current < 500 pA and action potentials reaching above zero mV were included in the analysis.
Statistics
Statistical analyses testing for significance (p < .05) of main factors and interactions used a one-way or a two-way ANOVA as well as a t-test design (some electrophysiology). Fisher’s PLSD was used for post hoc comparisons. All tests were conducted using StatView statistical package (version 5, Cary, NC) or GraphPad Prism V5 (San Diego, CA). Together the behavioral and electrophysiological data presented here are derived from a total of 106 animals.
Results
Blood Analyses
Several studies show that intranasal insulin either has no effect, or a moderate lowering effect (within the euglycemic range) on peripheral blood glucose (36–38,53,61–63). A two-way RM ANOVA shows intranasal insulin did not alter peripheral blood glucose levels in a subset of the aged animals (n = 5/group) 30–90 minutes after intranasal insulin Levemir delivery (Supplementary Table 1). In another group of aged animals, 40 IU Humalog also was unable to alter blood glucose levels. This result is consistent with transport of insulin to the perivascular space around blood vessels, indicating the hormone may not have crossed into the blood stream (56,64).
Behavioral Analyses
All groups learned to find the platform in the MWM as demonstrated by a significant decrease in path length to goal across the first 3 days of training [F(2,116) = 27.2; p < .0001]. During this learning phase, path length (Figure 1A) however, showed no significant differences across groups with age or intranasal insulin dose (two-way RM ANOVA). As shown previously (57,65), analysis of swim speed revealed a significant group effect [F(5,58) = 12.5; p < .0001] with post hoc tests indicating aged animals swam at slower speeds compared to young animals, but no difference between the insulin treatment groups was noted (Figure 1D). Latency to target, as predicted from slower speeds in aged animals, was significant across 3 days of training [F(2,116) = 28.9; p < .0001], with increased latency seen in all aged animals compared with young saline animals (data not shown). Because of this decrease in swim speed we chose to assess search errors and quantify memory performance on the probe day (no platform) using a proximity measure analysis. This approach is less dependent on swim speed than latency and is tabulated using a cumulative search error derived from the subject’s distance to the platform summed over the time to reach the platform and is then subtracted from an ideal path (straight line); smaller values are indicative of better performance (66).
Figure 1.
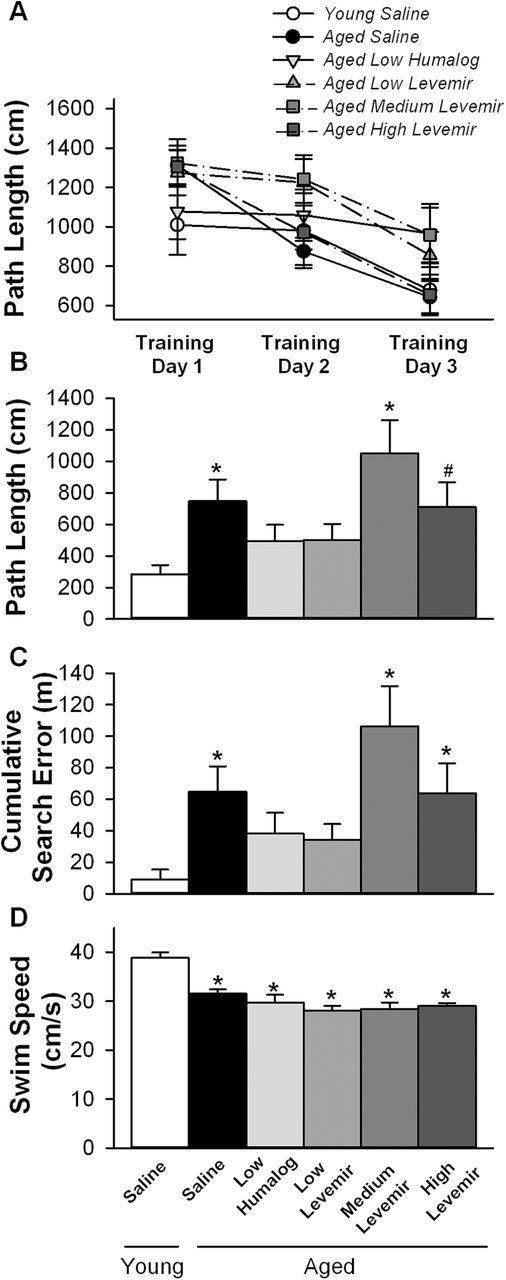
Morris water maze spatial learning and memory task. (A) Path length to submerged goal platform on the initial 3 days of training showed improved performance across all groups tested (RM ANOVA p < .0001), however, no group effect was seen. On probe day, with platform removed, animals receiving low doses of insulin (Humalog or Levemir) showed reduced path lengths to goal (B) and improved memory performance based on reductions in cumulative search error (C). Analysis of swim speed measured on the third training day shows aged F344 animals are slower [note no effect of insulin dose on speed (D)]. Mean ± SEM are shown. Asterisks indicate significant differences from the young saline group at p < .05; pound sign indicates a trend (p = .059) in comparison to the young saline group.
Compared to the learning component of the task, the 24-hour memory recall component measured during the probe trial (one-way ANOVA) revealed a significant group effect on path length to goal [F(5,58) = 2.9; p < .05) and on cumulative search error [F(5,58) = 3.3; p < .05). Post hoc analyses identified a significant increase on path length and cumulative search error in the aged saline (p < .05) and aged medium-dose Levemir (p < .005) groups compared with the young saline group (Figure 1B and C). A trend (p = .059) for improved performance was found for the high-dose Levemir animals on path length measures only. Notably, no significant differences were found between young saline treated and aged animals treated with either low-dose Humalog or low-dose Levemir on path length or cumulative proximal distance (Figure 1B and C), indicating that the lower dose intranasal insulin significantly reduced age-dependent memory loss. Additional post hoc analyses on memory performance in aged animals, comparing combined low dose insulins (Humalog and Levemir) with combined medium- and high-dose Levemir, revealed significant improvement in the low-dose insulin group on path length [F(1,33) = 5.25; p < .05] and cumulative search error [F(1,33) = 5.91; p < .05]. Thus, each of the different insulin formulations (Humalog and Levemir at low-doses) improved performance despite being administered to two separate cohorts of aged animals, indicating reproducibility of the insulin effect, and it appears lower, rather than higher doses of insulin are able to redress cognitive decline with aging.
Electrophysiology Analyses
Based on improved memory recall observed in aged animals treated with low-dose insulins, we next characterized the effects of in vivo intranasal insulin on hippocampal electrophysiology in a subset of the Humalog treated animals that were behaviorally characterized. Supplementary Table 2 shows that passive membrane properties were not different between groups and suggests cells with comparable baseline characteristics were analyzed. Intracellular recording of CA1 pyramidal neurons were obtained from animals treated with intranasal saline or Humalog (see Methods section). Approximately 3–7 hours following intranasal insulin delivery the Ca2+-dependent AHP was not significantly different between groups (Figure 2B and C; t-test). This was true for measures of AHP amplitude (medium and slow AHP) and duration (not shown). Extracellular synaptic potentials also showed no significant difference across groups (Figure 2D and E; t-test). EPSP amplitudes measured at 33% of the I/O were slightly larger in the Humalog treated animals and I/O slopes based on EPSP amplitude measures were somewhat reduced (Figure 2D and E, respectively, p < .1). These results are surprising given our prior report that insulin application to hippocampal slices reduces the AHP (50). In the prior study, however, hippocampal slices were perfused for 15 minutes with insulin and then studied electrophysiologically. The results obtained in the present study raise the possibility that the delay between intranasal insulin exposure and electrophysiological recording (3–7 hours) was too long and that the effect of insulin on the AHP may have dissipated. Thus, to confirm that insulin must be present in order to alter hippocampal physiology, we tested a range of ex vivo insulin concentrations and formulations on hippocampal slices from a separate group of animals.
We used young animals in the next series of experiments, and examined three different doses of Humalog, comparing Humalog’s actions to zinc and ACSF. In most insulin formulations, zinc is used to promote hexamer formation and prolong stability and duration of action in vivo. However, zinc is also known to modify ion flux across neuronal membranes, reducing activity at NMDA and GABAA receptors, ion channels associated with memory-related processes [reviewed in (67)]. Extracellular recordings in the presence of zinc or Humalog showed that neither EPSP amplitudes, nor I/O slopes were significantly altered when compared to ACSF controls (Figure 3E and F; one-way ANOVA). Similarly to data shown in Figure 3, these results indicate little if any, effects of insulin on extracellular postsynaptic potentials.
Intracellular recordings obtained from the same group of animals showed that the Ca2+-dependent AHP was sensitive to treatment with zinc or insulin. As previously reported, measures of postsynaptic excitability demonstrated the sensitivity of the AHP to both zinc (68) and insulin (50). A main effect of treatment on the slow AHP [F(4,18) = 3.34; p < .05] and its duration [F(4,18) = 4.49; p < .02] was seen across five groups of recorded cells (Figure 3C and D). Albeit being somewhat reduced, the mAHP was not significantly altered by insulin (Figure 3B). These results suggest a component of the insulin effect on the AHP could well be mediated by zinc found in this insulin formulation. To test the direct impact of ex vivo insulin on the AHP, we next investigated a zinc-free formulation of insulin (Apidra).
In this next series of experiments we obtained hippocampal slices from young (n = 8) and aged (n = 9) F344 rats and focused exclusively on intracellular potentials and the response to ex vivo Apidra exposure (1nM). Further, a recent study identified that 0.1–100nM exogenous insulin concentrations could reliably elicit dose-dependent activation of the insulin signaling pathway in human brain tissues (69), and approximated physiological levels near 1nM. For these reasons, we used 1nM Apidra. As previously reported (47–49,70) and shown here (Figure 4), mAHP and sAHP amplitudes recorded in CA1 pyramidal neurons were increased [F(1,24) = 17.6, p < .0005; F(1,24) = 4.7, p < .05, respectively] in aged, compared to young animals (two-way ANOVA). Treatment with the zinc-free Apidra insulin significantly reduced the AHP in slices from both young and aged rats as indicated by a main effect of treatment on the mAHP [F(1,24) = 32, p < .0001] and the sAHP [F(1,24) = 29.1, p < .0001). However, the Apidra-mediated AHP reduction was larger in aged animals compared to young animals (Figure 4A and B) as evidenced by a significant interaction term in the two-way ANOVA [F(1,24) = 10.9, p < .005 for the mAHP]. These results are consistent with our prior work using Humalog showing that the hippocampal AHP is sensitive to insulin (50). In the experiments presented here however, it is clear that insulin alone has a direct effect on the AHP, and that this effect persists even when lower, more physiological glucose levels are used in the ACSF (experiments in Figure 4 were conducted with 4.5 rather than the typical 10mM glucose concentration—see Methods section). Finally, it is interesting to note that Apidra did not alter holding current when compared to control cells (sup. Table 2), indicating insulin is neither activating nor inhibiting a tonic current near resting potential.
Discussion
Studies in humans have shown that intranasal insulin has promising cognition enhancing effects, alleviating, and perhaps even slowing the progression of age-related neurodegenerative disorders. However, the mechanisms underlying insulin’s effects in the brain are unclear. In order to identify potential mechanisms, we treated aged rodents with intranasal insulin, attempting to model aspects of the clinical trials reporting on the beneficial impact of intranasal insulin. Here, we describe physiological actions of insulin in the brain, highlighting a potential new mechanism of action of insulin on hippocampal function in both young and aged rats. This first analysis of intranasal insulin in aged animals demonstrates that intranasal insulin improved memory recall in aged F344 rats. We show that the effect on memory was observed in two separate cohorts of animals, using two different insulin formulations (Humalog and Levemir) delivered at relatively low dose (0.0715 IU/day/rat). Thus, these studies also demonstrate reproducibility of the effect of insulin on behavior. We also show that the Ca2+-dependent AHP, and in particular the slow AHP, a Ca2+-related biomarker of brain aging which increases with age and cognitive decline, is reduced by acute insulin exposure in aged animals and could well represent a novel target of brain insulin underlying the improvement seen in memory recall.
Long-acting insulin (Levemir) was as effective as short-acting insulin (Humalog) on memory recall in aged animals, inducing levels of performance indistinguishable from those seen in young animals. Levemir’s long duration of action is dependent on the presence of albumin in the periphery and a fatty acid modification of the insulin structure, increasing its’ affinity for albumin. Humalog’s shorter duration and faster onset are due to amino acid modifications in the β-chain of the insulin molecule. Because both versions of insulin were able to reverse cognitive decline to a similar degree in aged animals, our results suggest both insulins were able to gain access to the brain following intranasal administration despite structural differences. While results from clinical studies in memory-impaired older adults have reported improved word recall within 15-minute post-intranasal delivery (71), a prior clinical study on younger adults demonstrated the superiority of a faster acting insulin (Novolog) compared to regular insulin (Actrapid) on delayed word recall after 7 weeks of intranasal 160 IU/day Novolog (63). Using shorter delays (ie, 10 minutes) a recent study reveals 40 IU intranasal insulin delivery to young adult males improves odor-cued spatial memory (72). Clearly, the memory enhancing effects of insulin are depend on treatment regimen (acute vs subchronic), but also on the age and gender of the subject (73), as well as on the insulin formulation used (52,74). As mentioned above, a single, acute intranasal dose of insulin is capable of enhancing memory in humans and our electrophysiological studies showing an acute action of insulin on the AHP indicate that insulin can have rapid effects on neuronal processes critical to memory (see later). However, in the current study, it is unclear whether the effects on cognition observed were due to an acute action of insulin (ie, the dose received 1–3 hours prior to memory testing), or the result of the cumulative daily treatments received over the course of 8–11 days.
The decrease in the AHP by insulin was observed in field CA1 of the hippocampus, a synaptic zone which plays a key role in memory processing. Decreasing the AHP in this structure would be expected to increase neuronal excitability and facilitate throughput during physiological activation. Consistent with this, exogenous insulin increases excitatory neurotransmitter receptor trafficking including NMDA and AMPA receptors (25–27,75), thus promoting network activation. Furthermore, pure insulin has been shown to activate an inward current and depolarize hypothalamic neurons via activation of transient receptor potential channels, providing supporting evidence that insulin raises excitability (76). However, recent results provide evidence that insulin or other therapies that raise insulin (exendin-4 and glucagon-like peptide-1), increase functional GABAA conductances in CA1 (19) as well as in CA3 pyramidal neurons (77). This observation is in line with prior reports of enhanced GABAA function in response to insulin, a resulting decrease in synaptic activity (17,18) and a long-lasting enhancement of inhibitory postsynaptic currents [IPSCs (78)]. The net result, therefore, of enhancing both steady-state hyperpolarizing (GABA-mediated) and short-term depolarizing forces (eg, reduced AHP or direct depolarization) could well increase synaptic signal-to-noise, thereby improving throughput and neuronal communication. Clearly, as seen in hypothalamic arcuate nucleus neurons, it is important to note that insulin’s modulatory influence on membrane potential/excitability, is insulin formulation dependent, as well as cell-type specific (76).
To extend slice health and allow for extended neuronal recordings, high glucose concentrations (10mM) are commonly used in the ex vivo hippocampal slice. However, this superphysiological glucose concentration may alter insulin sensitivity in the brain. For this reason, we used lower glucose levels in some experiments while maintaining tissue health (Table 2), and more closely approximating physiological glucose levels. Importantly, significant AHP reduction was seen irrespective of recording conditions (10 or 4.5mM glucose). Thus, the results presented here appear compatible with potential effects of insulin on hippocampal neurons in vivo.
The placebo group in almost all clinical trials studying the impact of intranasal insulin on memory function is generally exposed to either saline or HOE-31, a dilution buffer that does not contain zinc. Given that all insulin formulations except Apidra contain zinc, our results showing that Apidra is capable of reducing the AHP in hippocampal neurons ex vivo could have clinical significance. These data are consistent with insulin per se being able to alter hippocampal physiology, and suggest that insulin and not zinc may be responsible for the CNS effects of intranasal insulin on memory. Clearly further clinical trials with Apidra appear warranted.
Because the AHP is dependent on a Ca2+-activated K+ conductance, the insulin-mediated reductions in the AHP may be due to inhibition of Ca2+ influx (79) or inhibition of L-type Ca2+ channel currents as seen in pinealocytes (80). Taken together, these results suggest insulin acts through the insulin receptor to reduce calcium-mediated functions possibly through PI3 kinase- or tyrosine kinase-mediated phosphorylation of Ca2+ channel proteins. Indeed, insulin can enhance mitochondrial function and therefore, Ca2+ homeostasis through PI3 kinase mechanisms in the periphery (40). Insulin also increases sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) function in heart cells via an Akt (protein kinase B) pathway, and in neurons, insulin has been shown to reduce KV1.3 potassium channel function (81). Notably, all of these potential mechanisms are consistent with reductions in available Ca2+, and hence, reductions in the amplitude of Ca2+-dependent hyperpolarizing potentials.
Our data cannot address whether the beneficial actions of intranasal insulin on memory recall are mediated, in parts or in whole, by the effects of insulin on the AHP. While suggestive that this may be the case, we do not provide evidence to support this conclusion. Further, because young animals were not tested for the impact of intranasal insulin, future studies will be needed to test the hypothesis that the observed effect is age-selective, or generally nootropic. Finally, although a similar insulin dose yielded quantitatively similar enhancement on memory recall in two cohorts of aged animals, it is not clear that higher Humalog doses might also be ineffective, similarly to higher Levemir doses.
The present results shed new light on a previously unrecognized insulin mechanism in the brain. Reductions in the AHP could enhance neuronal communication and might represent a pathway through which low dose Humalog or Levemir improve memory recall in aged animals. We show that insulin targeted the AHP, a Ca2+-mediated conductance, and reduced AHP amplitude and duration, elevations of which are characteristic of brain aging. We propose that facilitating insulin signaling restores Ca2+ homeostasis in aged animals, resulting in optimal levels of membrane excitability and synaptic plasticity, in part by limiting the amplitude and duration of the AHP in CA1 neurons (Figures 4 and 5). During aging, reductions in brain insulin levels and/or insulin receptor function (see Introduction section) may help promote neuronal Ca2+ dysregulation, resulting in impaired membrane excitability and reduced synaptic plasticity (eg, larger AHP). Clearly, given the reductions in insulin receptor RNA and protein in the aged rodent (5,6), greater occupation of the leftover insulin receptors with insulin could well offset reductions in signaling through these receptors, or may even slow the age-related reduction in insulin receptor density. Collectively, these results indicate robust associations between brain insulin, Ca2+ homeostasis, and aging-related cognitive decline, and suggest the value of further developing “insulin-raising” strategies for treating cognitive decline in age and disease, perhaps curbing the development of AD.
Supplementary Material
Supplementary material can be found at: http://biomedgerontology.oxfordjournals.org/
Funding
This work was funded by the National Institutes of Health (R01 AG033649).
Supplementary Material
References
- 1. Talbot K, Wang HY. The nature, significance, and glucagon-like peptide-1 analog treatment of brain insulin resistance in Alzheimer’s disease. Alzheimers Dement. 2014;10(1 suppl):S12–S25. :10.1016/j.jalz.2013.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De Felice FG, Ferreira ST. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes. 2014; 632262–72. 10.2337/db13-1954 [DOI] [PubMed] [Google Scholar]
- 3. Deak F, Sonntag WE. Aging, synaptic dysfunction, and insulin-like growth factor (IGF)-1. J Gerontol A Biol Sci Med Sci. 2012;67:611–625. :10.1093/gerona/gls118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frölich L, et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm. 1998;105:423–438. [DOI] [PubMed] [Google Scholar]
- 5. Zaia A, Piantanelli L. Insulin receptors in the brain cortex of aging mice. Mech Ageing Dev. 2000;113:227–232. S0047637499001189 [pii]. [DOI] [PubMed] [Google Scholar]
- 6. Zhao WQ, et al. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol. 2004;490:71–81. :10.1016/j.ejphar.2004.02.045 [DOI] [PubMed] [Google Scholar]
- 7. Rowe WB, et al. Hippocampal expression analyses reveal selective association of immediate-early, neuroenergetic, and myelinogenic pathways with cognitive impairment in aged rats. J Neurosci. 2007;27:3098–3110. :10.1523/JNEUROSCI.4163-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stranahan AM, et al. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci. 2008;11:309–317. :10.1038/nn2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pedersen WA, Flynn ER. Insulin resistance contributes to aberrant stress responses in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol Dis. 2004;17:500–506. :10.1016/j.nbd.2004.08.003 [DOI] [PubMed] [Google Scholar]
- 10. Thibault O, et al. Hippocampal calcium dysregulation at the nexus of diabetes and brain aging. Eur J Pharmacol. 2013;71934–43. 10.1016/j.ejphar.2013.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao W, et al. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem. 1999;274:34893–34902. [DOI] [PubMed] [Google Scholar]
- 12. Zhao WQ, et al. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008;22:246–260. doi:10.1096/fj.06-7703com [DOI] [PubMed] [Google Scholar]
- 13. McNay EC, Recknagel AK. Brain insulin signaling: a key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. Neurobiol Learn Mem. 2011;96:432–442. :10.1016/j.nlm.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blalock EM, et al. Aging-related gene expression in hippocampus proper compared with dentate gyrus is selectively associated with metabolic syndrome variables in rhesus monkeys. J Neurosci. 2010;30:6058–6071. :10.1523/JNEUROSCI.3956-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Freiherr J, et al. Intranasal insulin as a treatment for Alzheimer’s disease: a review of basic research and clinical evidence. CNS Drugs. 2013;27:505–514. :10.1007/s40263-013-0076-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Born J, et al. Sniffing neuropeptides: a transnasal approach to the human brain. Nat Neurosci. 2002;5:514–516. :10.1038/nn849 [DOI] [PubMed] [Google Scholar]
- 17. Palovcik RA, et al. Insulin inhibits pyramidal neurons in hippocampal slices. Brain Res. 1984;309:187–191. 0006-8993(84)91028-X [pii]. [DOI] [PubMed] [Google Scholar]
- 18. O’Malley D, Shanley LJ, Harvey J. Insulin inhibits rat hippocampal neurones via activation of ATP-sensitive K+ and large conductance Ca2+-activated K+ channels. Neuropharmacology. 2003;44:855–863. S0028390803000819 [pii]. [DOI] [PubMed] [Google Scholar]
- 19. Jin Z, et al. Insulin reduces neuronal excitability by turning on GABA(A) channels that generate tonic current. PLoS One. 2011;6:e16188. 10.1371/journal.pone.0016188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao L, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci. 2004;24:11120–11126. :10.1523/JNEUROSCI.2860-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Planel E, et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci. 2007;27:13635–13648. :10.1523/JNEUROSCI.2860-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salkovic-Petrisic M, et al. Modeling sporadic Alzheimer’s disease: the insulin resistant brain state generates multiple long-term morphobiological abnormalities including hyperphosphorylated tau protein and amyloid-beta. J Alzheimers Dis. 2009;18:729–750. :10.3233/JAD-2009-1184 [DOI] [PubMed] [Google Scholar]
- 23. Chiu SL, Cline HT. Insulin receptor signaling in the development of neuronal structure and function. Neural Dev. 2010;5:7. :10.1186/1749-8104-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mielke JG, Wang YT. Insulin, synaptic function, and opportunities for neuroprotection. Prog Mol Biol Transl Sci. 2011;98:133–186. 10.1016/B978-0-12-385506-0.00004-1 [DOI] [PubMed] [Google Scholar]
- 25. van der Heide LP, et al. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-D-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J Neurochem. 2005;94:1158–1166. :10.1111/j.1471-4159.2005.03269.x [DOI] [PubMed] [Google Scholar]
- 26. Man HY, et al. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25:649–662. S0896-6273(00)81067-3 [pii]. [DOI] [PubMed] [Google Scholar]
- 27. Huang CC, Lee CC, Hsu KS. An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. J Neurochem. 2004;89:217–231. :10.1111/j.1471-4159.2003.02307.x [DOI] [PubMed] [Google Scholar]
- 28. Skeberdis VA, et al. Insulin promotes rapid delivery of N-methyl-D-aspartate receptors to the cell surface by exocytosis. Proc Natl Acad Sci U S A. 2001;98:3561–3566. 10.1073/pnas.051634698 98/6/3561 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramsey MM, et al. Functional characterization of des-IGF-1 action at excitatory synapses in the CA1 region of rat hippocampus. J Neurophysiol. 2005;94:247–254. :10.1152/jn.00768.2004 [DOI] [PubMed] [Google Scholar]
- 30. Molina DP, et al. Growth hormone and insulin-like growth factor-I alter hippocampal excitatory synaptic transmission in young and old rats. Age (Dordr). 2012;351575–87. 10.1007/s11357-012-9460-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen BS, Roche KW. Growth factor-dependent trafficking of cerebellar NMDA receptors via protein kinase B/Akt phosphorylation of NR2C. Neuron. 2009;62:471–478. :10.1016/j.neuron.2009.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sonntag WE, Ramsey M, Carter CS. Growth hormone and insulin-like growth factor-1 (IGF-1) and their influence on cognitive aging. Ageing Res Rev. 2005;4:195–212. [DOI] [PubMed] [Google Scholar]
- 33. Novak V, Milberg W, Hao Y, et al. Enhancement of vasoreactivity and cognition by intranasal insulin in type 2 diabetes. Diabetes Care. 2014;37:751–759. :10.2337/dc13-1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heni M, et al. Central insulin administration improves whole-body insulin sensitivity via hypothalamus and parasympathetic outputs in men. Diabetes. 2014;634083–8. 10.2337/db14-0477 [DOI] [PubMed] [Google Scholar]
- 35. Kullmann S, et al. Intranasal insulin modulates intrinsic reward and prefrontal circuitry of the human brain in lean women. Neuroendocrinology. 2013;97:176–182. :10.1159/000341406 [DOI] [PubMed] [Google Scholar]
- 36. Ott V, et al. Central nervous insulin administration does not potentiate the acute glucoregulatory impact of concurrent mild hyperinsulinemia. Diabetes. 2014. :10.2337/db14-0931 [DOI] [PubMed] [Google Scholar]
- 37. Dash S, et al. Intranasal insulin suppresses endogenous glucose production in humans compared to placebo, in the presence of similar venous insulin concentration. Diabetes, 2014. :10.2337/db14-0685 [DOI] [PubMed] [Google Scholar]
- 38. Ferreira de Sa DS, et al. Cortisol, but not intranasal insulin, affects the central processing of visual food cues. Psychoneuroendocrinology. 2014;50C:311–320. :10.1016/j.psyneuen.2014.09.006 [DOI] [PubMed] [Google Scholar]
- 39. Verkhratsky A, Fernyhough P. Calcium signalling in sensory neurones and peripheral glia in the context of diabetic neuropathies. Cell Calcium. 2014;56362–371. 10.1016/j.ceca.2014.07.005 [DOI] [PubMed] [Google Scholar]
- 40. Huang TJ, et al. Diabetes-induced alterations in calcium homeostasis in sensory neurones of streptozotocin-diabetic rats are restricted to lumbar ganglia and are prevented by neurotrophin-3. Diabetologia. 2002;45:560–570. 10.1007/s00125-002-0785-x [DOI] [PubMed] [Google Scholar]
- 41. Huang TJ, et al. Neurotrophin-3 prevents mitochondrial dysfunction in sensory neurons of streptozotocin-diabetic rats. Exp Neurol. 2005;194:279–283. :10.1016/j.expneurol.2005.03.001 [DOI] [PubMed] [Google Scholar]
- 42. Kamal A, et al. Increased spike broadening and slow afterhyperpolarization in CA1 pyramidal cells of streptozotocin-induced diabetic rats. Neuroscience. 2003;118:577–583. S0306452202008746 [pii]. [DOI] [PubMed] [Google Scholar]
- 43. Kruglikov I, et al. Diabetes-induced abnormalities in ER calcium mobilization in primary and secondary nociceptive neurons. Pflugers Arch. 2004;448:395–401. :10.1007/s00424-004-1263-8 [DOI] [PubMed] [Google Scholar]
- 44. Biessels GJ, et al. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res. 1998;800:125–135. S0006-8993(98)00510-1 [pii]. [DOI] [PubMed] [Google Scholar]
- 45. Draznin B, et al. Possible role of cytosolic free calcium concentrations in mediating insulin resistance of obesity and hyperinsulinemia. J Clin Invest. 1988;82:1848–1852. :10.1172/JCI113801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Duner E, et al. Intracellular free calcium abnormalities in fibroblasts from non-insulin-dependent diabetic patients with and without arterial hypertension. Hypertension. 1997;29:1007–1013. :10.1523/JNEUROSCI.4171-05.2006 [DOI] [PubMed] [Google Scholar]
- 47. Gant JC, et al. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci. 2006;26:3482–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oh MM, Oliveira FA, Disterhoft JF. Learning and aging related changes in intrinsic neuronal excitability. Front Aging Neurosci. 2010;2:2. :10.3389/neuro.24.002.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tombaugh GC, Rowe WB, Rose GM. The slow afterhyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. J Neurosci. 2005;25:2609–2616. :10.1523/JNEUROSCI.5023-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pancani T, et al. Effect of high-fat diet on metabolic indices, cognition, and neuronal physiology in aging F344 rats. Neurobiol Aging. 2013;34:1977–1987. :10.1016/j.neurobiolaging.2013.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Marks DR, et al. Awake intranasal insulin delivery modifies protein complexes and alters memory, anxiety, and olfactory behaviors. J Neurosci. 2009;29:6734–6751. :10.1523/JNEUROSCI.1350-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Claxton A, et al. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J Alzheimers Dis. 2014. :10.3233/JAD-141791 [DOI] [PubMed] [Google Scholar]
- 53. Pillion DJ, Fyrberg MD, Meezan E. Nasal absorption of mixtures of fast-acting and long-acting insulins. Int J Pharm. 2010;388:202–208. :10.1016/j.ijpharm.2010.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dekaban AS. Changes in brain weights during the span of human life: relation of brain weights to body heights and body weights. Ann Neurol. 1978;4:345–356. :10.1002/ana.410040410 [DOI] [PubMed] [Google Scholar]
- 55. Schoeffner DJ, et al. Organ weights and fat volume in rats as a function of strain and age. J Toxicol Environ Health A. 1999;56:449–462. :10.1080/009841099157917 [DOI] [PubMed] [Google Scholar]
- 56. Lochhead JJ, Thorne RG. Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev. 2012;64:614–628. :10.1016/j.addr.2011.11.002 [DOI] [PubMed] [Google Scholar]
- 57. Blalock EM, et al. Effects of long-term pioglitazone treatment on peripheral and central markers of aging. PLoS One. 2010;5:e10405. :10.1371/journal.pone.0010405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dong WQ, et al. The rat hippocampal slice preparation as an in vitro model of ischemia. Stroke. 1988;19:498–502. [DOI] [PubMed] [Google Scholar]
- 59. McNay EC, Gold PE. Extracellular glucose concentrations in the rat hippocampus measured by zero-net-flux: effects of microdialysis flow rate, strain, and age. J Neurochem. 1999;72:785–790. [DOI] [PubMed] [Google Scholar]
- 60. Levin BE, Dunn-Meynell AA, Routh VH. CNS sensing and regulation of peripheral glucose levels. Int Rev Neurobiol. 2002;51:219–258. [DOI] [PubMed] [Google Scholar]
- 61. Benedict C, et al. Intranasal insulin improves memory in humans. Psychoneuroendocrinology. 2004;29:1326–1334. :10.1016/j.psyneuen.2004.04.003 [DOI] [PubMed] [Google Scholar]
- 62. Benedict C, et al. Intranasal insulin enhances postprandial thermogenesis and lowers postprandial serum insulin levels in healthy men. Diabetes. 2011;60:114–118. :10.2337/db10-0329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Benedict C, et al. Intranasal insulin improves memory in humans: superiority of insulin aspart. Neuropsychopharmacology. 2007;32:239–243. :10.1038/sj.npp.1301193 [DOI] [PubMed] [Google Scholar]
- 64. Hanson LR, Frey WH., II Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008;9(suppl 3):S5. :10.1186/1471-2202-9-S3-S5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Searcy JL, et al. Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer’s disease. J Alzheimers Dis. 2012;30:943–961. :10.3233/JAD-2012-111661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gallagher M, Burwell R, Burchinal M. Severity of spatial learning impairment in aging: development of a learning index for performance in the Morris water maze. Behav Neurosci. 1993;107:618–626. [DOI] [PubMed] [Google Scholar]
- 67. Sensi SL, et al. The neurophysiology and pathology of brain zinc. J Neurosci. 2011;31:16076–16085. :10.1523/JNEUROSCI.3454-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mayer ML, Vyklicky L., Jr The action of zinc on synaptic transmission and neuronal excitability in cultures of mouse hippocampus. J Physiol. 1989;415:351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Talbot K, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–1338. :10.1172/JCI59903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Disterhoft JF, Oh MM. Alterations in intrinsic neuronal excitability during normal aging. Aging Cell. 2007;6:327–336. :10.1111/j.1474-9726.2007.00297.x [DOI] [PubMed] [Google Scholar]
- 71. Reger MA, et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis. 2008;13:323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Brunner YF, et al. Central insulin administration improves odor-cued reactivation of spatial memory in young men. J Clin Endocrinol Metab. 2014. :10.1210/jc.2014-3018 [DOI] [PubMed] [Google Scholar]
- 73. Benedict C, et al. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J Clin Endocrinol Metab. 2008;93:1339–1344. :10.1210/jc.2007-2606 [DOI] [PubMed] [Google Scholar]
- 74. Rosenbloom MH, et al. A single-dose pilot trial of intranasal rapid-acting insulin in apolipoprotein e4 carriers with mild-moderate Alzheimer’s disease. CNS Drugs. 2014. :10.1007/s40263-014-0214-y [DOI] [PubMed] [Google Scholar]
- 75. Liu L, Brown JC, 3rd, Webster WW, Morrisett RA, Monaghan DT. Insulin potentiates N-methyl-D-aspartate receptor activity in Xenopus oocytes and rat hippocampus. Neurosci Lett. 1995;192:5–8. 030439409511593L [pii]. [DOI] [PubMed] [Google Scholar]
- 76. Qiu J, et al. Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab. 2014;19:682–693. :10.1016/j.cmet.2014.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Korol SV, et al. Glucagon-like peptide-1 (GLP-1) and exendin-4 transiently enhance GABAA receptor-mediated synaptic and tonic currents in rat hippocampal CA3 pyramidal neurons. Diabetes. 2014. :10.2337/db14-0668 [DOI] [PubMed] [Google Scholar]
- 78. Vetiska SM, et al. GABAA receptor-associated phosphoinositide 3-kinase is required for insulin-induced recruitment of postsynaptic GABAA receptors. Neuropharmacology. 2007;52:146–155. :10.1016/j.neuropharm.2006.06.023 [DOI] [PubMed] [Google Scholar]
- 79. O’Malley D, Harvey J. MAPK-dependent actin cytoskeletal reorganization underlies BK channel activation by insulin. Eur J Neurosci. 2007;25:673–682. :10.1111/j.1460-9568.2007.05347.x [DOI] [PubMed] [Google Scholar]
- 80. Chik CL, et al. Insulin and insulin-like growth factor-I inhibit the L-type calcium channel current in rat pinealocytes. Endocrinology. 1997;138:2033–2042. :10.1210/endo.138.5.5129 [DOI] [PubMed] [Google Scholar]
- 81. Fadool DA, et al. Tyrosine phosphorylation modulates current amplitude and kinetics of a neuronal voltage-gated potassium channel. J Neurophysiol. 1997;78:1563–1573. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.