

Abstract
Crk, the prototypical member of a class of SH2 and SH3 domain-containing proteins that controls the coordinated assembly of signaling complexes, is regulated by phosphorylation of Y221 in the linker region, which forms an intramolecular SH2-pY221 auto-clamp to interrupt SH2-SH3N signaling. Here, we show using LC-MS/MS and by generating phosphospecific antibodies that, iteratively with Y221, the Crk SH3C is routinely phosphorylated on Y239 and/or Y251 by several extracellular stimuli known to engage Crk. While phosphorylation at Y221 auto-inhibits the Crk SH2, phosphorylation of the SH3C generates an unconventional phosphoSH3C-SH3N unit in which the SH3N is fully functional to bind Polyproline Type II (PPII) ligands and the phosphoSH3C binds de novo to other SH2 domains. Using high throughput SH2 domain profiling, artificial neural network and position-specific scoring matrix based bio-informatics approaches, and unbiased MS, we found that the phosphoSH3C binds several SH2 domain-containing proteins, including specific non-receptor tyrosine kinases - Abl via pY251 and Csk via pY239. Functionally, we show that the phosphoSH3C modulates the Abl-mediated phenotypes of cell spreading and motility. Together, these studies describe a versatile mechanism wherein phosphorylation of Crk at Y221 is not an off switch but redirects signaling from the SH2-SH3N axis to a phosphoSH3C-SH3N axis, with the SH3N as a common denominator.
Keywords: Tyrosine phosphorylation, atypical Crk SH3 domain, non-canonical signaling
Introduction
The Src homology - 2 (SH2) and Src homology – 3 (SH3) domain containing Crk family of adaptor proteins assemble protein-protein complexes in a spatially and temporally regulated manner during cell adhesion, migration and efferocytosis (1–6). Crk signaling is mediated by the coordinated recruitment of specific tyrosine phosphorylated proteins, such as paxillin and p130cas, via the SH2 domain in the context of pY.X.X.P while the SH3N domain recruits specific guanine nucleotide exchange factors (GEFs) DOCK180 or C3G, by binding to conventional Polyproline Type II (PPII) motifs in the context of P.X.X.P.X. (K/R) (7–10). DOCK180 and C3G, in turn, activate the Rac1 and Rap1 GTPases, respectively (11, 12), which regulate cell migration or cell spreading. Signal transduction by Crk II and Crk L is negatively regulated by phosphorylation at Y221 (on CrkII) and Y207 (on CrkL) (13–15) to bring about an intramolecular association of the pY221 motif with the Crk SH2 domain thereby causing disassembly of Crk from proteins bound in trans to the SH2 domain (16).
The C-terminal SH3 domain (SH3C) of Crk is an atypical SH3 domain in that, unlike the N-terminal SH3 domain (SH3N), it does not bind conventional PPII motifs (17, 18). In contrast to the surface of the SH3N that has a hydrophobic ligand binding pocket lined by W169, Y186 and F141, the surface of the SH3C is lined by polar residues – Q244, H290 and Q274. Cis-trans isomerization about the G237 – P238 peptide bond in the chicken Crk II SH3N – SH3C unit has been shown to control accessibility of ligands to the SH3N where in the cis configuration, the SH3C engages the PPII binding pocket on the SH3N (19, 20). In human Crk II, the SH3N is negatively regulated by the SH3C and the inter-SH3 core region - residues 224–37 (22), which was shown to assemble CrkII into a structural state that resulted in reduced affinity for a PPII peptide derived from Sos1. These observations offer a molecular mechanism to explain why mutations or truncations in the SH3C activate the adaptor protein function of Crk. However, independent of its role in regulation of the SH3N, the physiological role of the SH3C in the context of Crk signaling is poorly understood.
Here, we found that both Y251 in the RT loop and Y239 at the SH3C boundary are iteratively and routinely phosphorylated with Y221, but at different stoichiometry with different extracellular stimuli. While phosphorylation at Y221 auto-inhibits the SH2 domain, it simultaneously generates a non-canonical phosphoSH3C-SH3N unit in Crk, with the SH3N as a common denominator. Our results define an affirmative role for the SH3C in signal transduction, and posit that phosphorylation at Y221 is not exclusively an off switch but redirects signaling by differential coupling of modular domains in Crk.
Historically, studies on Crk have impacted signal transduction by providing a paradigm for physical coupling by modular SH2 and SH3 domains. Here, we describe a novel paradigm whereby iterative tyrosine phosphorylation controls differential utilization of modular domains in Crk. Phosphorylation at Y221 functionally interrupts the SH2-SH3N axis while phosphorylation at Y239/Y251 iteratively with Y221 generates an unconventional phosphoSH3C-SH3N signaling unit. Our study presents a conceptual advance in the field by highlighting a novel role of tyrosine phosphorylation in regulating modular domain utilization in Crk. Future studies aimed to identify the repertoire of tyrosine kinases that control Y239 and Y251 phosphorylation, as well as identification of tumor types that dysregulate these phosphorylation events will greatly impact research on Crk biology.
Results
Identification of tyrosine phosphorylation sites on the Crk SH3C domain by LC-MS/MS
The Crk SH3C is an atypical SH3 domain that has distinct surface chemistry compared to conventional SH3 domains and does not bind conventional PPII motifs. Henceforth, unless otherwise specified, Crk II will be referred to as Crk, and ‘p’ denotes phosphotyrosine. By LC-MS/MS based phosphopeptide mapping of Crk following incubation with recombinant Abl kinase in vitro, three phosphorylation sites in the human Crk C-terminus were identified (i) Y251 in the extended RT-loop structure, (ii) Y239 at the boundary of the SH3C domain and (iii) Y221 previously identified as a motif that binds intramolecularly to the Crk SH2 domain to achieve auto-inhibition (Figs 1A, B and D).
Figure 1. Identification of phosphorylation sites pY221, pY239 and pY251 on Crk by LC-MS/MS.

A, MS/MS spectrum of a quadruply-charged ion (m/z 1371.16) corresponding to a doubly phosphorylated peptide 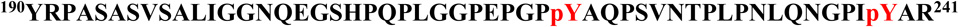 is shown. The phosphorylation sites are Y221 and Y239. B, MS/MS spectrum of a doubly-charged ion (m/z 1055.53) corresponding to the peptide sequence
is shown. The phosphorylation sites are Y221 and Y239. B, MS/MS spectrum of a doubly-charged ion (m/z 1055.53) corresponding to the peptide sequence 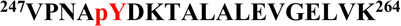 with a phosphorylation modification at Y251 is shown. The observed y- and b-ion series confirmed the peptide sequence and modification. C, GST-Crk was in vitro phosphorylated by immunoprecipitated Abl 1b in a kinase reaction and samples were analyzed by western blotting with anti-pY221 Crk (bottom), anti-pY239 Crk (middle) and anti-pY251 Crk (top) antibodies. D, Locations of Y239 and Y251 on the SH3C and Y221 on the inter-SH3 linker are depicted on the solution structure of Crk (PDB ID: 2EYZ).
with a phosphorylation modification at Y251 is shown. The observed y- and b-ion series confirmed the peptide sequence and modification. C, GST-Crk was in vitro phosphorylated by immunoprecipitated Abl 1b in a kinase reaction and samples were analyzed by western blotting with anti-pY221 Crk (bottom), anti-pY239 Crk (middle) and anti-pY251 Crk (top) antibodies. D, Locations of Y239 and Y251 on the SH3C and Y221 on the inter-SH3 linker are depicted on the solution structure of Crk (PDB ID: 2EYZ).
Specific Receptor Tyrosine Kinases induce distinct pY221/pY239/pY251 phosphorylation patterns on Crk
By generating affinity-purified phospho-specific antibodies towards the pY239 and pY251 phosphopeptide motifs, and using an available commercial anti-pY221 antibody, all three sites were found to be phosphorylated iteratively in vitro (Fig 1C) and in cells (Fig 2A) when Crk was co-expressed with Abl-1b (lane 6), consistent with the results of the LC-MS/MS analysis. Expression of individual point mutants of Crk shows the exquisite specificity of these antibodies (lanes 7–9), as no cross-reactivity was noted (Fig 2A).
Figure 2. RTKs show distinct preferences for phosphorylation of Crk at Y221/Y239/Y251.

A and C, 293T cells were transfected with the plasmids indicated and lysates were analyzed by western blotting with the antibodies shown on the left. B, MDA-MB-468 cells were stimulated with EGF (panel 1), MDA-MB-231 cells with HGF (panel 2) and NIH 3T3 cells with PDGF-BB (panel 3) for the times indicated or plated on Poly-D-Lysine or Fibronectin coated dishes for 20 minutes (panel 4). Samples were analyzed by western blotting with the antibodies indicated on the left. D, Summary of Crk phosphorylation patterns observed. Red, green and blue indicate positive for pY221, pY239 and pY251 respectively.
To examine whether tyrosine kinases other than Abl could induce pY239 and pY251, we analyzed endogenous pathways that have been implicated in Crk signaling. Towards this goal, we used (i) EGFR expressing MDA-MB-468 human breast cancer cells (ii) PDGFβR expressing mouse NIH 3T3 cells (iii) HGFR (c-Met) expressing MDA-MB-231 human breast cancer cells and (iv) NIH3T3 cells plated on fibronectin, which engages integrin mediated signaling via Crk (2, 23). EGFR, PDGFβR and HGFR have all been implicated in Crk signaling (24–28). We also examined Crk tyrosine phosphorylation downstream of the TAM family of tyrosine kinases, since Crk plays a pre-eminent role in TAM-mediated efferocytosis of apoptotic cells (6, 29).
Fig 2B shows experiments which involved no ectopic overexpression of growth factor receptor or Crk (see Fig S1B and table ST5 for quantification). Interestingly, EGF and fibronectin preferentially induced pY251 and pY221 (panel 1) while PDGF–BB preferentially induced pY239 and pY221 (panel 3) and HGF preferentially induced pY221 (panel 2), but not pY239 or pY251. Finally, we examined Crk phosphorylation downstream of constitutively active flag-tagged Fc-Tyro3 and Fc-Mer fusion proteins. Fc-Mer preferentially induced pY221 and pY239 (Fig. 2C, Fig. S1A), whereas Fc-Tyro3 induced pY221, pY239 and pY251. Taken together, these results suggest that EGFR, PDGFR, HGFR, Tyro3 and Mer, display distinct preferences for phosphorylation of Crk at Y221/Y239/Y251 (Fig 2D). Notably, in all cases examined to date, Y239 and Y251 on the SH3C are phosphorylated iteratively with Y221.
The Crk SH3N domain is accessible to ligands in SH2-autoinhibited (pY221-phosphorylated) Crk
By using high-resolution NMR to study the effect of tyrosine phosphorylation on structural rearrangements in Crk, we observed that the chemical shift differences (∆δ) between unphosphorylated (Crk) versus phosphorylated Crk (pCrk) revealed major shifts in the SH2 domain and the region around Y221 consistent with phosphorylation at Y221 and intramolecular pY221-Crk SH2 association (Fig 3A, stoichiometry of phosphorylation at Y221 was >95% by NMR). Notably, no drastic changes in the SH3N domain were observed, as exemplified by residues lining the SH3N-PPII binding pocket – F141, W169 and Y186 (Fig S2), suggesting that the Crk SH3N did not undergo a major change in accessibility upon phosphorylation at Y221 and intramolecular pY221-SH2 association. By comparing SH3N accessibility using a Crk SH3N-specific C3G PPII peptide with the sequence N-DNSPPPALPPKKRQSAPS-C (20), we observed that the SH3N was accessible to its PPII ligand in both Crk and pCrk as shown by shifts in the NMR peaks for F141, W169 and Y186 (Fig 3B). Consistently, binding affinities of Crk and pCrk for the C3G PPII peptide by ITC (Fig 3C) were similar (~ 2 uM) demonstrating that the Crk SH3N remains accessible to ligands upon phosphorylation at Y221 and intramolecular pY221-Crk SH2 association.
Figure 3. Crk SH3N is accessible to ligands in pY221-Crk.


A, 1H-15N chemical shift differences between Crk and pCrk. ∆δ values are on y-axis and amino acid numbers are on the x-axis. Red bars reflect the residues with significant line broadening or for which the perturbation value cannot be computed. B, (Left) Overlay of 1H-15N HSQC of Crk (blue) and Crk + PPII ligand (green). (Right) Overlay of 1H-15N HSQC of pCrk (red) and pCrk + PPII ligand (yellow). C, ITC profiles of Crk (top) and pCrk (bottom) with the PPII peptide ligand. Kd values are displayed as inset. D, Pull downs with purified GST, unphosphorylated GST-Crk and in vitro phosphorylated GST-Crk from 293T cell lysates expressing Flag-paxillin. Samples were analyzed with anti-DOCK180, anti-Flag and anti-pY221 Crk antibodies, anti- pY239 Crk and anti-Crk antibodies. Quantification is shown with binding of DOCK180/flag-paxillin to GST control set to zero and binding to unphosphorylated Crk set to 1. E, 293T cells were transfected with the plasmids indicated and lysates were analyzed by western blotting with the antibodies shown on the left. S-S refers to the Crk SH3N-linker SH3C construct (amino acids 123–304).
Furthermore, we tested whether pY221-phosphorylated Crk can interact with a known physiological binding partner (Fig. 3D). Binding of Flag-Paxillin, an SH2 binding partner, to pCrk was greatly reduced compared to Crk consistent with near complete stoichiometry of phosphorylation at Y221. Concomitantly, GST-Crk was also phosphorylated at Y239 suggesting that the SH3C was accessible to phosphorylation and could form protein-protein interactions and possibly signaling complexes. In contrast, binding of DOCK180, an SH3N binding partner, to pCrk was not blocked suggesting that the Crk SH3N is accessible to its cognate protein ligands upon phosphorylation at Y221 and intramolecular pY221-Crk SH2 association. These results indicate that while phosphorylation at Y221 auto-inhibits the SH2 domain, it simultaneously generates a non-canonical phosphoSH3C-SH3N unit in Crk. To examine if this unit can sustain phosphorylation in cells, we expressed the Crk SH3N-linker SH3C unit (amino acids 123–304 with the Y221F mutation, referred to as S-S) with Abl in 293T cells (Fig 3E). While, phosphorylation at Y239 and Y251 was retained (although weaker at Y251 compared to WT), the W169K SH3N mutation (i.e. Abl binding site) drastically reduced phosphorylation by Abl suggesting that the SH3N could functionally engage PPII ligands (Abl in this case). These results suggest that in the non-canonical phosphoSH3C-SH3N unit, both phosphorylation on the SH3C and ligand binding to the SH3N, are maintained.
Distinct SH2 domain interaction landscapes for pY239 and pY251 on the phosphorylated Crk SH3C
Given that both Y251 and Y239 on the Crk SH3C were selectively tyrosine phosphorylated by distinct kinases, we hypothesized that each phosphotyrosine motif might also have the potential to recruit phosphotyrosine-specific recognition modules as part of a post-phosphorylation signaling mechanism. To identify binding targets, we used N-terminally biotinylated 16-mer phosphopeptides containing pY239 or pY251 to screen an SH2 domain library side-by-side (Fig. 4A, see methods for details). We identified two types of SH2 domain interactomes. The first group, represented by the Abl, Arg and Sck/ShcB SH2 domains, were common to both phospho-motifs (Fig 4B top panel, table ST1). The second group displayed differential selectivity to one or the other phospho-motifs (Fig 4B, middle and bottom panels). We then generated a profile of selectivity indices wherein selectivity index was defined as the ratio of the binding intensity of an SH2 domain obtained for pY239 to that obtained for pY251 or vice-versa. The maximum selectivity for either peptide in the first group of overlapping hits was not greater than ~ 2.4 (Fig 4C, top panel). Interestingly, in the second group, the pY239 specific hits displayed much higher selectivity (ranging from ~ 4 to 26) compared to the pY251 specific hits (ranging from ~ 1.8 to 2.7). Notably, the top hit for the pY239 peptide was the SH2 domain of C-terminal Src Kinase (Csk), which had the maximum selectivity index (~ 26) of all SH2 domains tested (Fig 4C, middle and bottom panels).
Figure 4. SH2 domain interaction landscapes of the pY239 and pY251 motifs.


A, Select hits from the rosette assay are shown for the pY239 and pY251 phosphopeptides. ‘Cont’ is a control unphosphorylated peptide. ‘Cont-P’ is a control phosphorylated peptide. N1-P is a non-specific phosphopeptide. B, Comparison of the top 10 hits of the pY239 phosphopeptide with those of the pY251 phosphopeptide. Top, Overlapping hits. Middle, pY239 specific hits. Bottom, pY251 specific hits. C, Selectivity indices of the overlapping hits (top), pY239 specific hits (middle) and pY251 specific hits (bottom). D, SH2 interaction landscapes of the pY239 (top) and pY251 motifs (bottom) obtained using NetPhorest. Y-axis indicates Probability Score. Top 10 hits are shown. E, Selectivity indices of the pY239 specific hits (top) and the pY251 specific hits (bottom). F, top, Human Crk SH3C (230–304) was phosphorylated in the TKB1 strain and used as bait for pull downs from MDA-MB-468 cell lysate. UI – Uninduced, IAA – Indoleacrylic Acid. Bottom, Phosphorylation status was analyzed by western blotting with the anti-pY251 and anti-pY239 Crk antibodies. G, Top, SYPRO RUBY stained gel of the samples indicated after pull downs with pGST, GST-SH3C or GST-pSH3C. Bottom, Western blot analysis with the anti-Csk antibody. H, Top, SYPRO Ruby stained gel of the samples obtained after pull downs with the pSH3C WT, Y239F, Y251F or Y239F/Y251F. Bottom, Western blot analysis with the anti-Csk antibody. I, 293T cells were transfected with the plasmids indicated and lysates were analyzed by western blotting with the antibodies shown on the left.
To independently test the SH2 landscapes using a bioinformatics-based approach, we used published Artificial Neural Networks (ANN) and Position-Specific Scoring Matrices (PSSM) (30–32) to generate binding profiles for the pY239 and pY251 motifs (Fig 4D and table ST2). Not only were the binding profiles distinct but consistent with the experimental data shown above, Csk SH2 was the top hit for pY239, and displayed the highest selectivity index (Fig 4E). In contrast, the Abl/Arg SH2 domains were not among the top ten predicted hits for either peptide.
Complex formation and biological phenotypes influenced by phosphorylation of the Crk SH3C
To experimentally identify protein-protein assemblages mediated by the phosphorylated Crk SH3C domain in MDA-MB-468 cell lysate, we used recombinant, bacterially-expressed tyrosine phosphorylated CrkSH3C (herein referred to as pSH3C, see Fig 4F and methods). Analysis of pSH3C pull downs revealed a major 50 kD band, specific for pSH3C versus SH3C, which we identified as Csk by LC-MS/MS and confirmed by Western blotting (Fig 4G and table ST3). Further, consistent with the in vitro SH2 screen and bioinformatic analyses, the pSH3C- Csk complex was pY239 dependent and not pY251 dependent as the Y239F but not the Y251F mutation abrogated the interaction (Fig 4H). Since Csk is a negative regulator of Src-Family Kinases (SFKs), the pSH3C-Csk interaction could potentially activate a positive feed forward loop to sustain Src activation. Consistently, co-transfection of SFKs and Crk induced pY239 on Crk by Hck, Src, Lyn and Blk, which in turn were activated as evident by Western blotting with an anti-pY416 SFK antibody that recognizes activated SFKs (Fig 4I).
Despite the fact that both pY239 and pY251 peptides selected Abl and Arg SH2 domains as binding partners in vitro, analysis of the total pSH3C interactome by LC-MS/MS failed to detect Abl/Arg in complex with the pSH3C in the MDA-MB-468 cells (Table ST4) suggesting that unlike the pSH3C-Csk interaction, pSH3C-Abl/Arg is not a stand-alone interaction but might rather function within the Crk-Abl complex [already tethered via the CrkSH3N and Abl proline-rich domain (13, 15)] to affect Abl activation. To test this hypothesis both biochemically and functionally, we examined Abl activation and cell spreading on fibronectin since this is an Abl-modulated phenotype wherein activated Abl inhibits cell spreading (33, 34). Furthermore, adhesion to fibronectin induces pY251 (and pY221) on Crk. When Crk (−/−) MEFs stably reconstituted with EYFP, Crk WT or Crk Y251F were plated on fibronectin coated plates, Crk WT enhanced Abl activation while Crk Y251F attenuated Abl activation by ~25% (Fig 5A). Also, Crk Y251F accelerated the spreading kinetics of cells compared to Crk WT consistent with the role of activated Abl in inhibiting cell spreading (Fig 5D, top panel).
Figure 5. Phosphorylation of the Crk SH3C at Y251 inhibits cell spreading on fibronectin and promotes cell motility towards EGF.

A, Crk (−/−) MEFs stably reconstituted with EYFP, Crk WT or Crk Y251F were plated on fibronectin coated dishes for 20 minutes. Lysates were analyzed by western blotting with the antibodies shown. Quantification of pY245 on hyperactive Abl is shown on the right. All samples were run on the same gel and hence are comparable. However, the EYFP and WT lanes were not adjacent to the Y251F lane and hence have been demarcated by straight lines at the edges. B, MDA-MB-468 cells stably expressing EYFP, EYFP-Crk WT, EYFP-Crk Y251F were stimulated with EGF for 5 minutes and lysates were analyzed by western blotting with the antibodies shown. Quantification of pY245 Abl normalized to total Abl is shown on the right. C, Cells in B were analyzed for their motility towards EGF in a transwell Boyden chamber assay. ** indicates p<0.05. Representative images are shown on the left. D, Top, Cells in A were plated on fibronectin coated E-plates in triplicate and real-time cell spreading was recorded using xCelligence. Shown is a representative of two independent experiments each performed in triplicate. Quantification of cell index is shown as mean +/− SD. ‘**’ indicates p<0.05. Middle and Bottom, Cells in B in addition to MDA-MB-468 stably expressing Crk shRNA (Crk KD) were seeded into CIM-plates in quadruplicate in the middle panel and in duplicate in the bottom panel with an EGF gradient of 10 ng/ml and cell motility was recorded in real time using xCelligence. Middle panel is a representative of four independent experiments each performed in quadruplicate while the bottom panel is a representative of two independent experiments each performed in duplicate. Quantification of cell index is shown as mean +/− SD in the middle panel and as average of duplicates in the bottom panel. ’**’ indicates p<0.05.
We also tested an EGF-inducible system wherein Crk is phosphorylated at pY251 and pY221. MDA-MB-468 cells stably expressing Crk WT displayed enhanced Abl activation which was attenuated by Crk Y251F (Fig 5B). Furthermore, Crk Y251F expressing cells displayed attenuated cell motility towards EGF compared to Crk WT (Fig 5C, Fig 5D middle panel and Fig S3A). Imatinib treated Crk WT cells displayed an overlapping migration profile to that of Y251F cells suggesting that Crk pY251 promotes cell motility in an Abl kinase dependent manner (Fig 5D, bottom panel). These results suggest that although Crk pY251-Abl SH2 may not be a stand-alone interaction, it influences Abl-modulated biological phenotypes by promoting Abl transactivation within the Crk-Abl complex. Consistent with these results, biochemically, Crk pY251 has been shown to promote transactivation of Abl by Crk (35). Furthermore, co-expression of the Crk SH3N-linker-SH3C with Abl in 293T cells was sufficient to transactivate Abl and was in turn phosphorylated at Y239 and Y251 (Figs. S3B and 3E). Taken together, these data suggest that the Crk pSH3C-SH3N unit is a functional signaling axis.
Discussion
Despite a high degree of evolutionary conservation, the role of the atypical Crk SH3C domain has remained elusive. Previous studies suggest that phosphorylation of Crk at Y221 functions as an off switch as it induces SH2 domain auto-inhibition. In this study, we show that iterative tyrosine phosphorylation of Y221 in the linker region and within the SH3C (at Y239 and/or Y251) changes the domain utilization in Crk from a SH2-SH3N axis to a non-canonical phosphoSH3C-SH3N axis (Fig. 6). Although the repertoire of tyrosine kinases that phosphorylate SH3C is not fully realized, such differential domain utilization rationalizes a new level of complexity and versatility in terms of the adaptor protein function of Crk.
Figure 6. Model depicting a non-canonical signaling scheme for Crk mediated by pY239 and pY251.

Our data predicts that pY221 would turn off the SH2-SH3N axis while iteratively phosphorylated pY239/pY251 would turn on the pSH3C – SH3N signaling axis predicting a dynamic turnover and/or relocalization of protein complexes. In light of our findings and other independent reports, atleast two modes of auto-inhibition may exist - pY221 alone in the absence of pY239/pY251 on the SH3C or alternate auto-inhibitory domain organization that blocks SH3N accessibility (leftmost two scenarios).
An important consequence of the above findings is that phosphorylation at Y221 will not be an endpoint for signaling by Crk when it occurs concomitantly with Y239 and/or Y251 phosphorylation on the SH3C. In support of this model, we observed by NMR, ITC and pull down experiments that Crk can exist in a conformation in which the SH3N remains accessible to ligands after pY221 phosphorylation, whereby the SH2 is occupied by the intramolecular auto-clamp. Plausibly, this permits Crk to retain adaptor protein function in conjunction with binding partners engaged by the pY239/pY251 motifs on the SH3C. However, in contrast to our results, Crk has been shown to also adopt a conformation wherein accessibility to the SH3N is drastically reduced upon phosphorylation at Y221 and intramolecular pY221-SH2 association (22). Given the conformational versatility inherent in Crk, we do not exclude the possibility that multiple modes of auto-inhibition may exist. This could be achieved by selective phosphorylation at Y221 alone in the absence of SH3C phosphorylation, alternate SH3N-inhibitory domain arrangements in Crk or SH3N post-translational modifications, an example of which would be phosphorylation at Y186 on the Crk SH3N (a conventional SH3 domain) which forms a part of its hydrophobic ligand binding pocket. Phosphorylation of this conserved tyrosine has been shown to reduce the affinity of SH3 domains for ligands in Btk, Abl and p130Cas/BCAR1(36–39). However, in our LC-MS/MS analysis, we did not observe phosphorylation of Crk at Y186 (Fig S4).
As alluded to above, the possibility of retention of adaptor function upon SH3N/PPII engagement in conjunction with complexes formed by pY239/pY251 on the Crk SH3C formed the rationale for identifying SH2 domain containing proteins that bound the pY239/pY251 motifs. The identification of Csk that selectively binds pY239 and not pY251 on the SH3C predicts a specific feed-forward loop whereby activation of Src Family Kinases (SFKs) could be sustained upon phosphorylation at Y239 on the Crk SH3C. Upon being iteratively phosphorylated with Y221, pY239 on the SH3C would be expected to bind and spatially separate Csk from locations where SFK activity is induced. This suggests a mechanism whereby the kinetics of SFK activation can be fine-tuned to impact relevant biological phenotypes.
Although both the pY239 and pY251 phosphopeptides were selective for the Abl and Arg SH2 domains, the pSH3C-Abl/Arg interaction could not be recapitulated from cells suggesting that while the pSH3C-Csk complex can form de novo, the pSH3C-Abl interaction likely plays an accessory role within the assembled Crk-Abl complex to impinge on Abl activation by SH2 domain displacement. Consistently, in scenarios where pY251 was induced (iteratively with pY221), the Y251F mutation partially inhibited Abl activation and also influenced the Abl-modulated phenotypes of cell spreading and motility, suggesting that modulation of Abl activity by Crk pY251 is functionally important. Considering the high selectivity observed of pY239 for Csk, we expect that pY251 rather than pY239 would impact Abl activation. Importantly, these findings suggest how signaling downstream of RTKs can be dissected by Crk to impinge on specific non-RTKs – Csk or Abl. This lends new mechanistic insight into how RTKs can specifically induce Csk/Src or Abl-dependent pathways via the non-canonical (pY239/pY251) SH3C-SH3N module in Crk. Since we found distinct patterns of Crk pY221/pY239/pY251 phosphorylation induced by different factors and kinases, this immediately suggests the possibility to achieve different signaling outcomes via differential Crk phosphorylation. While we do not exclude the possibility of differentially phosphorylated forms of Crk, we have not observed phosphorylation of Y239 or Y251 in the absence of Y221 phosphorylation under physiological conditions so far. An important future goal will be to identify the full repertoire of tyrosine kinases that impinge on the Crk SH3C and biological contexts in which they segregate to Csk/Src or Abl/Arg pathways.
Finally, in a comparison between Crk and CrkL (Fig S5), the residue on CrkL corresponding to Crk-Y239 is phenylalanine suggesting that this pY239-mediated signaling mechanism is specific for Crk. Further, the sequence around Y251 diverges from PNAYDKTALALE on Crk to PCAYDKTALALE on CrkL. This may result in differential binding to PTB domains which recognize residues N-terminal to the phosphorylated tyrosine residue. The Y221/Y207 negative regulatory sites and the Y251 motifs appear to be well conserved across humans, mice, rats, xenopus and chicken while the Crk-Y239 motif is conserved across humans, mice, rats and chicken suggesting conservation of function through evolution. Therefore, apart from domain organization differences (40), post-translational modification by phosphorylation at Y239 and/or Y251 likely determines differences in downstream signaling initiated by Crk and CrkL.
Materials and Methods
Phospho-specific antibodies
Phospho-specific antibodies to pY239 and pY251 were generated by immunizing rabbits with synthetic 14-mer phosphopeptides corresponding to residues surrounding Y239 and Y251 on Crk respectively. Antibodies were purified by positive selection on a phosphopeptide bound column followed by negative selection on a unphosphorylated peptide bound column.
Cell culture and growth factor stimulation
For stimulation, cells were serum-starved in 0.5% serum for atleast 12 hours followed by stimulation with growth factor at 100ng/ml or held in suspension for 30 minutes and then plated on dishes coated with either 50 or 100 ug/ml of poly-D-lysine or fibronectin. Adherent cells were harvested after 20 minutes.
In vitro phosphorylation
For LC-MS/MS analysis of phosphorylation sites on Crk, bacterially expressed and purified GST- CrkII was incubated with immunoprecipitated Abl 1b (from 293T cells) in 50mM Tris, 150mM NaCl, 10mM MgCl2, 5mM ATP and 1mM DTT at pH 7.5 for 12 hours. The reactions were boiled in SDS Sample buffer, resolved by SDS-PAGE and stained with SYPRO RUBY. The bands corresponding to GST-CrkII were excised and analyzed by LC-MS/MS on an Orbitrap instrument.
Isothermal Titration Calorimetry and NMR Analysis
For NMR, all samples were dialyzed in NMR buffer 50 mM potassium phosphate (pH 6.8), 140 mM NaCl and 1 mM β-mercaptoethanol). NMR experiments were performed on Varian 800- and 600-MHz and Bruker 700- and 600-MHz spectrometers. Abl kinase domain (Abl KD) was expressed and purified as described before (41, 42). Purified and Tev-processed Crk II was phosphorylated by the addition of catalytic amounts of Abl KD in buffer (50mM KPi (pH 6.8), 150mM NaCl, 1mM β-mercaptoethanol) supplemented with 5 mM ATP and 10 mM MgCl2. The reaction was carried out at room temperature. Stoichiometry of phosphorylation at Y221 was >95% by NMR.
All calorimetric titrations were performed on an iTC200 microcalorimeter (GE). Unphosphorylated or phosphorylated CrkII was at 40 µM in the sample cell and the PPII ligand was at 400 µM in the injection syringe. Ligand was dissolved in the flow through of the last buffer exchange. Data for the preliminary injection, which are affected by diffusion of the solution from and into the injection syringe during the initial equilibration period, were discarded. The data were fitted with Origin 7.0.
GST – pull down experiments
For generation of tyrosine phosphorylated Crk SH3C, the E-coli TKB1 strain was used (Stratagene). The Crk SH3C (230 – 304) was cloned into pET42a under a LacZ promoter and then transformed into the parental E.coli BL-21 strain or the TKB1 strain which harbors a plasmid encoding the EphB4 tyrosine kinase domain under a TrpE promoter. IPTG was used to induce synthesis of the GST-SH3C in both BL-21 and TKB1 strains. Addition of Indoleacrylic Acid (IAA) to TKB1 induced synthesis of the EphB4 kinase that phosphorylated GST-SH3C at both Y239 and Y251 to yield pSH3C (Fig 4F). GST-fusion proteins (bait) were then bound to a glutathione-agarose column, washed and incubated with lysates of unstimulated MDA-MB-468 cells. After further washing, bound proteins were eluted with glutathione.
For analysis of SH3N accessibility in full length CrkII, 1uM of purified GST-CrkII was phosphorylated by incubating with 10 uM purified Abl KD in 50 mM Tris, 150mM NaCl, 10mM MgCl2, 5mM ATP and 1mM DTT at pH 7.5 overnight. GST-CrkII was then captured on glutathione-agarose beads and then incubated with lysates over-expressing flag-tagged paxillin for 1 hour at 4 C. Beads were then spun down and washed twice in HNTG buffer (0.1% Triton-X-100) and then boiled in SDS sample buffer.
High-throughput SH2 domain screens and bioinformatic analysis
The Crk pY239 and pY251 phosphopeptides were spotted on a gelatin-coated nitrocellulose membrane in register with the wells of a 96-well plate. Followed by incubation with soluble human GST-SH2 domains as probes (one in each well), HRP-conjugated GSH was used to detect and quantify binding by chemiluminescence as described before (43). The list of SH2 domain probes and clone information can also be found at (http://www.addgene.org/Bruce_Mayer/). For bioinformatic analyses, predictions of SH2 interaction landscapes were made based on ANNs and PSSMs through NetPhorest (http://netphorest.info/index.php).
Cell spreading and cell motility using xCelligence
For cell spreading, 10,000 viable cells were seeded into each well of fibronectin coated E-plates (Acea BioSciences Inc.) after background measurement. Impedance to current through the electrodes is recorded every 30 seconds to generate cell index, a parameter directly proportional to the cell area in contact with the surface electrodes. For cell motility, 40,000 viable cells were seeded into each well of the upper chamber of CIM-plates, the undersides of which were coated with 10 ug/ml Type I collagen. The lower chamber contained 10 or 25 ng/ml EGF as the chemoattractant. Cell index was recorded every 15 minutes to generate real time migration profiles.
Statistical analysis
Where indicated, statistical analysis was performed using SPSS (IBM Inc) software. For Figs 5C and Fig S3A, ANOVA was performed without assuming equal variances across groups. Tamhane’s T2 test was employed for post-hoc analysis at a significance threshold of p=0.05. For Fig. 5D top and middle panels, two-factor ANOVA was performed followed by Tukey’s HSD for post-hoc analysis at a significance threshold of p=0.05.
Supplementary Material
Acknowledgements
This work was supported by the NIH grant CA165077 to RBB and Rutgers Foundation grant to RBB and in part by NIH grant to CGK and RBB GM080308. The LC-MS/MS work was funded in part by an NIH grant NS046593, for the support of the Rutgers Neuroproteomics Core Facility.
We thank Chingiz Underbayev, WenI Tsou and Anita Antes for technical support; Sushil Kumar and Stanley Kimani for discussion.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Klemke RL, et al. CAS/Crk coupling serves as a "molecular switch" for induction of cell migration. The Journal of cell biology. 1998;140(4):961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vuori K, Hirai H, Aizawa S, Ruoslahti E. Introduction of p130cas signaling complex formation upon integrin-mediated cell adhesion: a role for Src family kinases. Molecular and cellular biology. 1996;16(6):2606–2613. doi: 10.1128/mcb.16.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332(6161):272–275. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- 4.Hedgecock EM, Sulston JE, Thomson JN. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science. 1983;220(4603):1277–1279. doi: 10.1126/science.6857247. [DOI] [PubMed] [Google Scholar]
- 5.Tosello-Trampont AC, Brugnera E, Ravichandran KS. Evidence for a conserved role for CRKII and Rac in engulfment of apoptotic cells. J Biol Chem. 2001;276(17):13797–13802. doi: 10.1074/jbc.M011238200. [DOI] [PubMed] [Google Scholar]
- 6.Albert ML, Kim JI, Birge RB. alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat Cell Biol. 2000;2(12):899–905. doi: 10.1038/35046549. [DOI] [PubMed] [Google Scholar]
- 7.Knudsen BS, Feller SM, Hanafusa H. Four proline-rich sequences of the guanine-nucleotide exchange factor C3G bind with unique specificity to the first Src homology 3 domain of Crk. The Journal of biological chemistry. 1994;269(52):32781–32787. [PubMed] [Google Scholar]
- 8.Matsuda M, et al. CRK protein binds to two guanine nucleotide-releasing proteins for the Ras family and modulates nerve growth factor-induced activation of Ras in PC12 cells. Mol Cell Biol. 1994;14(8):5495–5500. doi: 10.1128/mcb.14.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasegawa H, et al. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol. 1996;16(4):1770–1776. doi: 10.1128/mcb.16.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birge RB, et al. Identification and characterization of a high-affinity interaction between v-Crk and tyrosine-phosphorylated paxillin in CT10-transformed fibroblasts. Molecular and cellular biology. 1993;13(8):4648–4656. doi: 10.1128/mcb.13.8.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gotoh T, et al. Identification of Rap1 as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol Cell Biol. 1995;15(12):6746–6753. doi: 10.1128/mcb.15.12.6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiyokawa E, et al. Activation of Rac1 by a Crk SH3-binding protein, DOCK180. Genes Dev. 1998;12(21):3331–3336. doi: 10.1101/gad.12.21.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feller SM, Knudsen B, Hanafusa H. c-Abl kinase regulates the protein binding activity of c-Crk. The EMBO journal. 1994;13(10):2341–2351. doi: 10.1002/j.1460-2075.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Jong R, ten Hoeve J, Heisterkamp N, Groffen J. Tyrosine 207 in CRKL is the BCR/ABL phosphorylation site. Oncogene. 1997;14(5):507–513. doi: 10.1038/sj.onc.1200885. [DOI] [PubMed] [Google Scholar]
- 15.Ren R, Ye ZS, Baltimore D. Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes & development. 1994;8(7):783–795. doi: 10.1101/gad.8.7.783. [DOI] [PubMed] [Google Scholar]
- 16.Rosen MK, et al. Direct demonstration of an intramolecular SH2-phosphotyrosine interaction in the Crk protein. Nature. 1995;374(6521):477–479. doi: 10.1038/374477a0. [DOI] [PubMed] [Google Scholar]
- 17.Muralidharan V, et al. Solution structure and folding characteristics of the C-terminal SH3 domain of c-Crk-II. Biochemistry. 2006;45(29):8874–8884. doi: 10.1021/bi060590z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reichman C, et al. Transactivation of Abl by the Crk II adapter protein requires a PNAY sequence in the Crk C-terminal SH3 domain. Oncogene. 2005;24(55):8187–8199. doi: 10.1038/sj.onc.1208988. [DOI] [PubMed] [Google Scholar]
- 19.Sarkar P, Saleh T, Tzeng SR, Birge RB, Kalodimos CG. Structural basis for regulation of the Crk signaling protein by a proline switch. Nat Chem Biol. 2011;7(1):51–57. doi: 10.1038/nchembio.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarkar P, Reichman C, Saleh T, Birge RB, Kalodimos CG. Proline cis-trans isomerization controls autoinhibition of a signaling protein. Mol Cell. 2007;25(3):413–426. doi: 10.1016/j.molcel.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Q, Leiva MC, Fischkoff SA, Handschumacher RE, Lyttle CR. Leukocyte chemotactic activity of cyclophilin. The Journal of biological chemistry. 1992;267(17):11968–11971. [PubMed] [Google Scholar]
- 22.Kobashigawa Y, et al. Structural basis for the transforming activity of human cancer-related signaling adaptor protein CRK. Nat Struct Mol Biol. 2007;14(6):503–510. doi: 10.1038/nsmb1241. [DOI] [PubMed] [Google Scholar]
- 23.Nojima Y, et al. Integrin-mediated cell adhesion promotes tyrosine phosphorylation of p130Cas, a Src homology 3-containing molecule having multiple Src homology 2-binding motifs. The Journal of biological chemistry. 1995;270(25):15398–15402. doi: 10.1074/jbc.270.25.15398. [DOI] [PubMed] [Google Scholar]
- 24.Birge RB, Fajardo JE, Mayer BJ, Hanafusa H. Tyrosine-phosphorylated epidermal growth factor receptor and cellular p130 provide high affinity binding substrates to analyze Crk-phosphotyrosine-dependent interactions in vitro. J Biol Chem. 1992;267(15):10588–10595. [PubMed] [Google Scholar]
- 25.Hashimoto Y, et al. Phosphorylation of CrkII adaptor protein at tyrosine 221 by epidermal growth factor receptor. J Biol Chem. 1998;273(27):17186–17191. doi: 10.1074/jbc.273.27.17186. [DOI] [PubMed] [Google Scholar]
- 26.Antoku S & Mayer BJ. Distinct roles for Crk adaptor isoforms in actin reorganization induced by extracellular signals. Journal of cell science. 2009;122(Pt 22):4228–4238. doi: 10.1242/jcs.054627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamorte L, Rodrigues S, Naujokas M, Park M. Crk synergizes with epidermal growth factor for epithelial invasion and morphogenesis and is required for the met morphogenic program. J Biol Chem. 2002;277(40):37904–37911. doi: 10.1074/jbc.M201743200. [DOI] [PubMed] [Google Scholar]
- 28.Lamorte L, Royal I, Naujokas M, Park M. Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol Biol Cell. 2002;13(5):1449–1461. doi: 10.1091/mbc.01-10-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci. 2005;118(Pt 3):539–553. doi: 10.1242/jcs.01632. [DOI] [PubMed] [Google Scholar]
- 30.Tinti M, et al. The SH2 domain interaction landscape. Cell reports. 2013;3(4):1293–1305. doi: 10.1016/j.celrep.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang H, et al. Defining the specificity space of the human SRC homology 2 domain. Molecular & cellular proteomics : MCP. 2008;7(4):768–784. doi: 10.1074/mcp.M700312-MCP200. [DOI] [PubMed] [Google Scholar]
- 32.Li L, et al. Prediction of phosphotyrosine signaling networks using a scoring matrix-assisted ligand identification approach. Nucleic acids research. 2008;36(10):3263–3273. doi: 10.1093/nar/gkn161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antoku S, Saksela K, Rivera GM, Mayer BJ. A crucial role in cell spreading for the interaction of Abl PxxP motifs with Crk and Nck adaptors. Journal of cell science. 2008;121(Pt 18):3071–3082. doi: 10.1242/jcs.031575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin H & Wang JY. Abl tyrosine kinase promotes dorsal ruffles but restrains lamellipodia extension during cell spreading on fibronectin. Molecular biology of the cell. 2007;18(10):4143–4154. doi: 10.1091/mbc.E07-01-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kardinal C, et al. Rational development of cell-penetrating high affinity SH3 domain binding peptides that selectively disrupt the signal transduction of Crk family adapters. Amgen Peptide Technology Group. Annals of the New York Academy of Sciences. 1999;886:289–292. doi: 10.1111/j.1749-6632.1999.tb09439.x. [DOI] [PubMed] [Google Scholar]
- 36.Park H, et al. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity. 1996;4(5):515–525. doi: 10.1016/s1074-7613(00)80417-3. [DOI] [PubMed] [Google Scholar]
- 37.Morrogh LM, Hinshelwood S, Costello P, Cory GO, Kinnon C. The SH3 domain of Bruton's tyrosine kinase displays altered ligand binding properties when auto-phosphorylated in vitro. Eur J Immunol. 1999;29(7):2269–2279. doi: 10.1002/(SICI)1521-4141(199907)29:07<2269::AID-IMMU2269>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 38.Chen S, O'Reilly LP, Smithgall TE, Engen JR. Tyrosine phosphorylation in the SH3 domain disrupts negative regulatory interactions within the c-Abl kinase core. J Mol Biol. 2008;383(2):414–423. doi: 10.1016/j.jmb.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janostiak R, et al. Tyrosine phosphorylation within the SH3 domain regulates CAS subcellular localization, cell migration, and invasiveness. Mol Biol Cell. 2011;22(22):4256–4267. doi: 10.1091/mbc.E11-03-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jankowski W, et al. Domain organization differences explain Bcr-Abl's preference for CrkL over CrkII. Nat Chem Biol. 2012;8(6):590–596. doi: 10.1038/nchembio.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seeliger MA, et al. High yield bacterial expression of active c-Abl and c-Src tyrosine kinases. Protein science : a publication of the Protein Society. 2005;14(12):3135–3139. doi: 10.1110/ps.051750905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobashigawa Y, Naito M, Inagaki F. An efficient method for protein phosphorylation using the artificially introduced of cognate-binding modules into kinases and substrates. Journal of biotechnology. 2007;131(4):458–465. doi: 10.1016/j.jbiotec.2007.07.956. [DOI] [PubMed] [Google Scholar]
- 43.Machida K, et al. High-throughput phosphotyrosine profiling using SH2 domains. Mol Cell. 2007;26(6):899–915. doi: 10.1016/j.molcel.2007.05.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.