

Abstract
The need for multiple vaccinations to enhance the immunogenicity of subunit vaccines may be reduced by delivering the vaccine over an extended period of time. Here, we report two novel injectable pentablock copolymer based thermoresponsive hydrogels made of polyethyleneglycol-polycaprolactone-polylactide-polycaprolactone-polyethyleneglycol (PEG-PCL-PLA-PCL-PEG) with varying ratios of polycaprolactone (PCL) and polylactide (PLA), as single shot sustained release vaccines. Pentablock copolymer hydrogels were loaded with vaccine-encapsulated poly lactic-co-glycolic acid nanoparticles (PLGA-NP) or with the soluble vaccine components. Incorporation of PLGA-NP into the thermoresponsive hydrogels increased the complex viscosity of the gels, lowered the gelation temperature, and minimized the burst release of antigen and adjuvants. The two pentablock hydrogels stimulated both cellular and humoral responses. The addition of PLGA-NP to the hydrogels sustained immune responses for up to 49 days. The polymer with a higher ratio of PCL to PLA formed a more rigid gel, induced stronger immune responses, and stimulated effective anti-tumor responses in a prophylactic melanoma tumor model.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-015-9843-4) contains supplementary material, which is available to authorized users.
KEY WORDS: nanoparticles, pentablock copolymers, sustained release, thermoresponsive hydrogels, vaccine delivery
INTRODUCTION
Subunit vaccines that utilize defined peptide, protein, or nucleic acid antigens are less reactogenic than whole-pathogen vaccines (1). However, use of subunit vaccines also results in poor immunogenicity due to lack of secondary signals (2). The weak immunogenicity of subunit vaccines may be overcome by delivering the vaccine multiple times or through using formulation strategies such as incorporating the antigen into sustained release formulations and including immune stimulatory adjuvants in the vaccine. The release of antigen in a sustained manner may increase the period of antigen interaction with the immune cells and thus lead to the induction of effective immune responses (3,4). Care must be taken with the development of such formulations as inappropriate release of vaccine antigen and adjuvant can induce cell sequestration and deletion (5).
Delivering subunit vaccines via particulate carriers to mimic pathogens is a strategy to enhance the immunogenicity of subunit vaccines (6). Additionally, particulate carriers may facilitate the synchronous presentation of vaccine antigens and adjuvants to dendritic cells, decreasing the potential for the development of tolerogenic immune response (7). Poly lactic-co-glycolic acid nanoparticles (PLGA-NP) have been extensively studied for vaccine delivery and have been reported to target dendritic cells naturally through phagocytosis with efficient delivery of the vaccine components (8). PLGA-NP can accommodate a wide range of actives (9) and in the present study were loaded with the model vaccine antigen ovalbumin and the adjuvants monophosphoryl lipid A (MPL) and Quil A (QA) before being combined with thermoresponsive hydrogels with the aim of creating a one-shot vaccine. MPL is a non-toxic, Toll-like receptor (TLR) 4 ligand isolated from the gram negative bacterium, Salmonella minnesota (10). MPL was the first TLR ligand to be approved for commercial use in humans and is reported to induce strong Th1 immune responses (11). Quil A is a non-TLR-dependent saponin adjuvant extracted from the bark of Quillaja saponaria (12). QA is capable of stimulating both cytotoxic T cell and humoral responses (13). Both MPL and QA can stimulate improved immune responses when administered individually, and recent studies suggest a combination of these adjuvants can induce stronger immune responses compared to each adjuvant delivered separately (14,15).
Thermoresponsive or in situ-forming hydrogels are temperature-sensitive injectable systems that are polymeric solutions at ambient temperature and that rapidly transform into sustained release gel depots upon administration into the body (16). Actives such as drugs and vaccines can be incorporated into hydrogels by simply mixing with the temperature-sensitive polymer solutions at ambient temperature (17). The triblock copolymer poloxamer 407 has been examined extensively as an in situ gelling system but the fast erosion of these gels in aqueous environments and the non-biodegradability of the polymer has limited its use as a sustained release system (18,19). Attempts to increase the stability of poloxamer through the addition of copolymers such as pluronic 25R4 (20), dextran (21), carrageenan (22), and methylcellulose (23) have had limited success. Biodegradable triblock copolymers made up of polycaprolactone (PCL) and polyethyleneglycol (PEG) (i.e., PEG-PCL-PEG, PCL-PEG-PCL) are reported to be stable in aqueous conditions but the crystallinity of the PCL blocks slowed degradation (24–26). In this study, two novel thermosensitive pentablock copolymers consisting of polyethyleneglycol-polycaprolactone-polylactide-polycaprolactone-polyethyleneglycol (PEG-PCL-PLA-PCL-PEG) were studied as injectable hydrogels, with the polylactide (PLA) blocks being added to accelerate the degradation of PCL (27,28). These tailor-made pentablock copolymers were developed for the sustained ocular delivery of drugs (29).
The overall aim of the current study was to develop a one-shot sustained release vaccine. Initial work in this study focussed on the preparation of pentablock copolymer hydrogels containing soluble and nanoparticle vaccine formulations. This was followed by rheological analysis, in vitro stability, and release studies to investigate the behavior of thermoresponsive hydrogels as sustained release formulations with and without PLGA-NP. Further in vivo vaccine and challenge studies were performed to examine the potential of these formulations in generating cellular and humoral responses.
MATERIALS AND METHODS
Materials
Poly(ethylene glycol), (PEG: 1 and 4 kDa), methoxy-PEG (550 Da), stannous octoate, -caprolactone, albumin from chicken egg (OVA, grade V, purity approximately 98%), fluorescein isothiocyanate (FITC, isomer 1), monophosphoryl lipid A from S. minnesota RE 595, and formic acid were acquired from Sigma-Aldrich (Missouri, USA). l-lactide and hexamethylenediamine (HMDI) were purchased from Acros organics (Morris Plains, NJ, USA). Purified saponin (Quil-A®) was obtained from Brenntag Biosector (Frederikssund, Denmark). PLGA copolymer with a 50:50 monomer ratio and inherent viscosity of 0.55 to 0.75 dL/g was acquired from Absorbable Polymers International (Pelham, USA). Polyvinyl alcohol (PVA), MW ∼ 25,000, was purchased from Polysciences, Inc. (Warrington, USA). Poloxamer 407 (Lutrol® F127) and Pluronic® 25R4 were obtained from BASF (Ludwigshafen, Germany). HPLC-grade chloroform, methanol, and acetonitrile were purchased form Merck KGaA (Darmstadt, Germany). Alum was obtained from SERVA Electrophoresis (Heidelberg, Germany). All the antibodies used for flow cytometry analysis were obtained from BD Pharmingen (San Diego, CA, USA). Water was purified using a Millipore® Purification System (USA) and was used during sample preparation and HPLC analysis. Nitrogen gas (oxygen free) was supplied by BOC Ltd., (Auckland, NZ).
Preparation of PLGA-NP
PLGA-NP were prepared by the double-emulsion method as previously reported (30,31). In the primary emulsion step, FITC-OVA (prepared as previously described (32)) or OVA (50 μL, 25 mg/mL in PBS) and QA (50 μL, 50 mg/mL in PBS) were added to the PLGA (300 μL, 10% w/v in chloroform) and MPL (100 μL, 25 mg/mL in chloroform). The resultant mixture was emulsified using an ultrasonic processor UP50H (Teltow, Germany) to form a primary emulsion. This primary emulsion was added to 2 mL of PVA solution (6% w/v PVA) and further emulsified by sonication to form a secondary emulsion. The secondary emulsion was then added drop wise into 8 mL of stirring 0.5% PVA solution at room temperature. Nanoparticles were collected after 3 h of stirring followed by centrifugation at 10,000×g (Eppendorf centrifuge, Hamburg, Germany) for 10 min. After centrifugation, nanoparticles were washed twice with cold water to remove the residual PVA and redispersed in PBS.
Particle-size distribution and zeta potential of PLGA-NP were determined by photon correlation spectroscopy (PCS) and electrophoretic mobility, using a Zetasizer Nano-ZS (Malvern Instruments, UK).
Synthesis, Preparation, and Loading of Thermosensitive Hydrogels
The two pentablock copolymers PEG550-PCL825-PLA550-PCL825-PEG550 (P1) and PEG550-PCL550-PLA1100-PCL550-PEG550 (P2) with different ratios of PCL and PLA blocks were synthesized as reported previously (29). Solutions (17% w/v) of the two pentablock copolymers in PBS (pH 7.4) were stirred overnight at 4°C. A triblock copolymer hydrogel (Tri), consisting of Poloxamer 407 and pluronic 25R4, was prepared as described previously (20). PLGA-NP containing OVA (or FITC-OVA when required), MPL and QA or soluble OVA (or FITC-OVA when required), MPL and QA were added to the thermosensitive polymer solutions which were then stirred overnight at 4°C to ensure homogenous mixing. All formulations contained 100 μg/mL OVA (or FITC-OVA when required), 100 μg/mL MPL, and 200 μg/mL QA.
Entrapment of FITC-OVA, MPL, and QA in PLGA-NP
An aliquot (20 mg) of PLGA-NP was added to 2 mL of 0.5 M sodium hydroxide and incubated overnight in dark with stirring to ensure complete dissolution of particles. Entrapment of FITC-OVA was determined by fluorescence spectroscopy (Shimadzu RF-540, Shimadzu Corporation, Kyoto, Japan) at excitation and emission wavelengths of 490 and 520 nm, respectively. The fluorescence of standard solutions of sodium hydroxide treated FITC-OVA was used to calculate the amount of FITC-OVA entrapped within PLGA-NP.
The loading of MPL and QA in PLGA-NP was quantified by a high-performance liquid chromatography/evaporative light scattering detection (HPLC-ELSD) method as previously described (33).
Rheological Measurements
Rheology measurements were performed using a cone and plate geometry on an Oscillatory HR-3 rheometer (TA Instruments). The rheometer was equipped with a temperature-controlled peltier plate and a 60-mm cone with an angle of 2°. All the measurements were performed in the linear viscoelastic region of the polymer. The sol–gel transition temperature was determined using temperature sweep measurements with 1°C intervals from 10 to 40°C. The sol–gel transition time and the angular frequency sweep measurements were determined at 37°C. All the samples were placed on the peltier plate at 5°C and a solvent trap was used to prevent evaporation of the sample.
In Vitro Gel Stability Studies
One milliliter of thermosensitive polymer solution was transferred into a vial and incubated at 37°C to induce gelation. The initial weight of the gel was noted (W1) and 1 mL of PBS prewarmed to 37°C was carefully layered on the gel surface. At predetermined time intervals, the PBS was removed, the weight of the remaining gel was recorded (W2), and the percentage gel weight loss was calculated.
Fluorescence Microscopy
FITC-OVA-loaded PLGA-NP, thermosensitive hydrogels containing soluble FITC-OVA, and FITC-OVA PLGA-NP were analyzed by fluorescence microscopy using an IX71/IX81 Olympus microscope (Olympus America, Inc.).
In Vitro Release Studies
In vitro release of FITC-OVA, MPL, and QA from PLGA-NP, PLGA-NP in hydrogels, and hydrogels was determined. A 20-mg aliquot of PLGA nanoparticles was transferred into Eppendorf tubes, and 1 mL of PBS was added. The samples were incubated at 37°C in the dark, and at predetermined time intervals the amount of FITC-OVA, QA, and MPL remaining in the nanoparticles was determined as described above.
A 1-mL aliquot of the thermosensitive polymer solutions containing vaccine-loaded PLGA-NP or soluble FITC-OVA, QA, and MPL was transferred into vials and incubated at 37°C to induce gelation. One milliliter of PBS prewarmed to 37°C was carefully layered over the surface of gels, and the vials were incubated at 37°C in the dark. At predetermined time intervals, 100 μL of release media was removed and replaced with an equal volume of PBS. FITC-OVA, QA, and MPL levels in the release media were then analyzed as described above and expressed as cumulative amount released (percent) versus time.
Mice
Male C57Bl/6 and OT-I and OT-II transgenic mice, 6–8 weeks old were bred and housed under specific pathogen-free conditions at the HTRU, Dunedin, New Zealand. T cell receptors specific for the CD8 (OVA257–264) and CD4 (OVA323–339) peptide epitopes of OVA are highly expressed in OT-I and OT-II transgenic mice, respectively (34,35). All the mice had free access to food and water during the entire period of experiments. All experiments were approved by the University of Otago Animal Ethics Committee.
Adoptive T Cell Transfer
OT-I and OT-II transgenic cells were transferred into C57Bl/6 mice on the day before immunization (4). In brief, spleens and lymph nodes were collected from OT-I and OT-II transgenic mice, a single-cell suspension at 1 × 106 cells/mL was prepared, and 200 μL was injected into the tail vein of C57Bl/6 mice the day before immunization.
Immunization Protocol
All the mice were injected with 200 μL of vaccine formulation subcutaneously in the dorsal skin fold on day 0 of experiment. Formulations used were OVA-MPL-QA entrapped PLGA-NP, thermosensitive polymer solutions loaded with OVA-MPL-QA entrapped PLGA-NP, and soluble OVA, MPL, and QA in thermosensitive polymer solutions. Each animal received a total of 20 μg of OVA, 20 μg of MPL, and 40 μg of QA (36). Alum containing 20 μg of OVA was given as the control formulation. Three days prior to sampling, all the mice were injected with 10 μg of OVA to maximize the detected immune response. Mice were sacrificed 21, 30, or 49 days after immunization and draining (axial and brachial) lymph nodes and blood were collected for further analysis.
Measurement of Cellular and Humoral Immune Responses
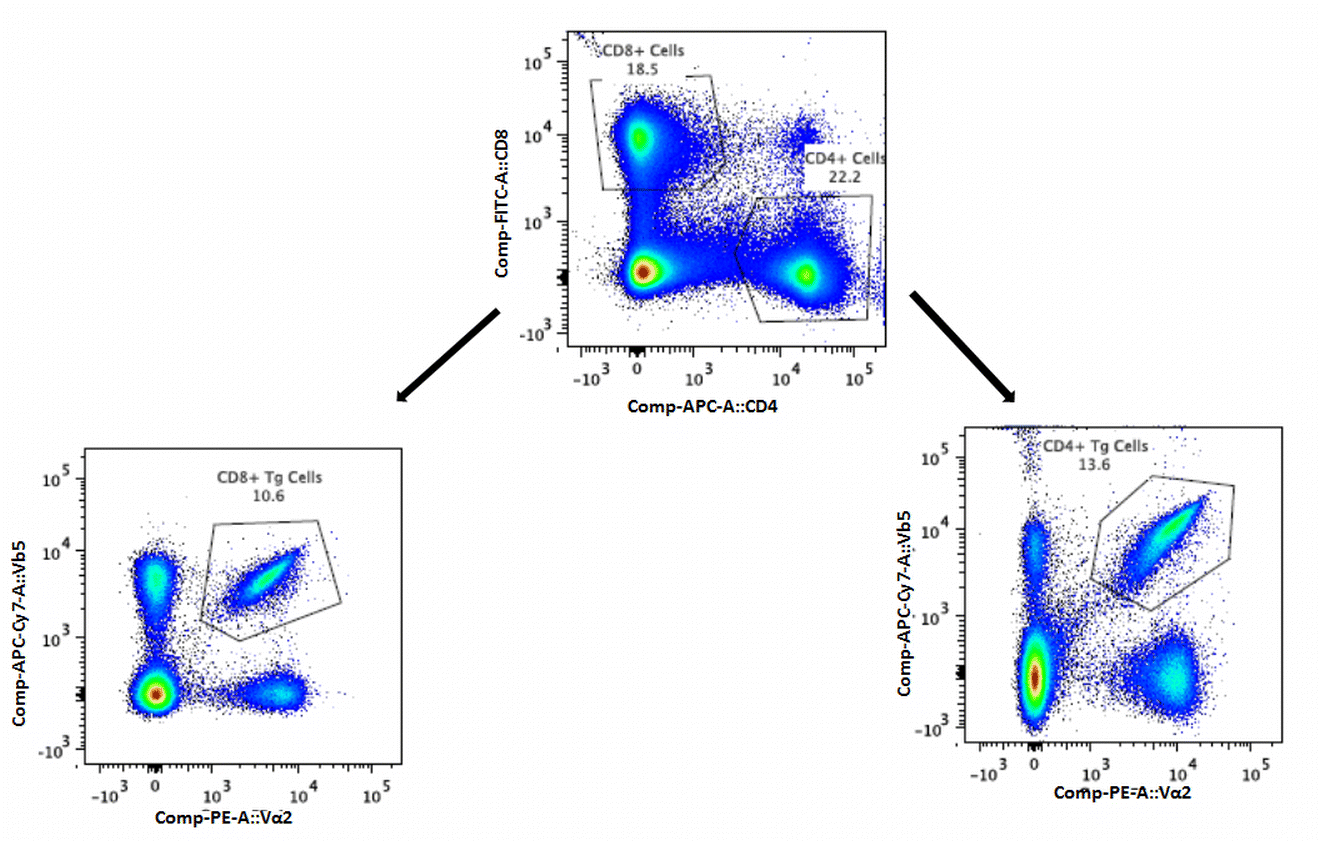
Single-cell suspensions were prepared from harvested lymph nodes, stained and analyzed by flow cytometry (BD FACS Canto II, BD Biosciences). Cells were first incubated with anti-CD16/CD32 antibody (2.4G2) and subsequently stained with an antibody mix consisting of anti-CD8 (53–6.7-FITC), anti-CD4 (APC), anti-Vα2 (B20.1-PE), and anti-Vβ5.1 (MR9-4-biotin). Secondary staining with SA-APC-Cy7 was then carried out. Data was analyzed using FlowJo software version 10.0.6 (Tree Star, Inc.) and representative flow cytometry plots are shown in supplementary Fig. 1. The total number of CD8 or CD4 transgenic cells was calculated by multiplying the percentage of antigen specific Vα2+ Vβ5+ CD8 T cells or Vα2+ Vβ5+ CD4 T cells by the total number of viable lymphocytes (37).
Serum was collected from centrifuged (2700×g for 10 min) blood samples and stored at −20°C till analysis. The serum samples were analyzed by ELISA for OVA-specific IgG antibody (38).
Tumor Experiments
Mice (eight per group) received either no treatment or were immunized subcutaneously in the dorsal skin fold with P1 hydrogels with blank PLGA-NP, P1 hydrogels with soluble vaccine, or P1 hydrogels with PLGA-NP vaccines as described above. Seven weeks later, all the mice were injected subcutaneously in the flank with B16. OVA melanoma cells (1 × 105) were acquired from 90% confluent cultures. The mice were examined every day and the tumor size was measured for every 2–3 days after appearance of first tumor using digital Vernier calipers. Mice with tumor sizes greater than 200 mm2 or meeting other predetermined humane endpoints were culled.
Statistical Analysis
Where applicable, results were analyzed for statistical significance with GraphPad Prism Version 6 using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison tests. Survival curves were compared using the Log-rank (Mantel-Cox) test.
RESULTS
Formulation Characterization
PLGA-NP containing the model antigen OVA and the adjuvants MPL and QA were successfully prepared by the double-emulsion method (Table I).
Table I.
Characterization of Blank PLGA-NP and FITC-OVA, MPL, and QA Loaded PLGA-NP. The Values Shown Are the Mean ± SD (n = 3)
| Formulation | Z-Average (nm) | PDI | Zeta potential (mV) | FITC-OVA EE (%) | MPL EE (%) |
QA EE (%) |
|---|---|---|---|---|---|---|
| Blank PLGA-NP | 250 ± 12.9 | 0.096 ± 0.069 | −34.83 ± 2.36 | – | – | – |
| FITC-OVA, MPL, QA PLGA-NP | 325 ± 15.1 | 0.257 ± 0.039 | −42.42 ± 3.59 | 77.5 ± 3.6 | 65.1 ± 3.1 | 57.3 ± 2.8 |
EE entrapment efficiency, PDI polydispersity index
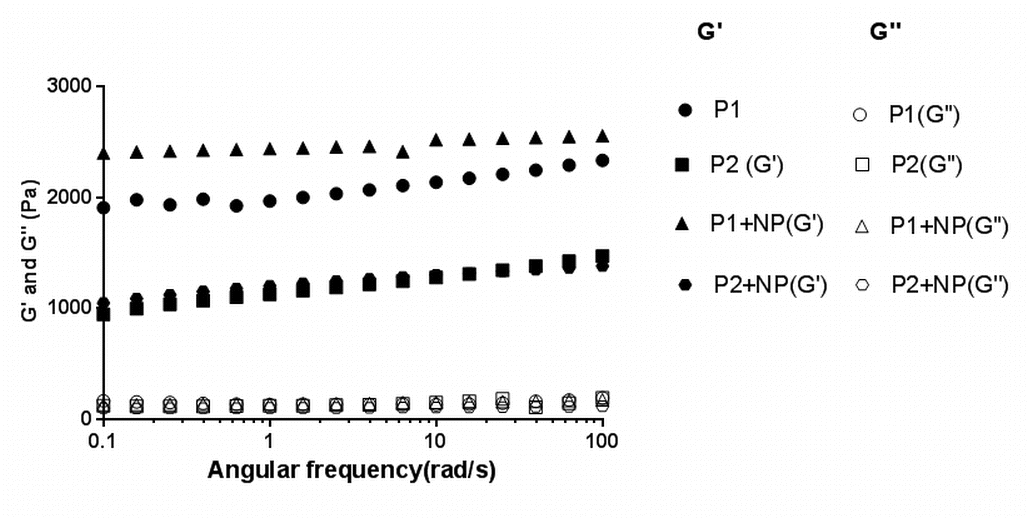
The sol–gel transition temperature and time were analyzed using oscillatory rheometry. A 17% (w/v) concentration of the polymer was chosen on the basis of preliminary studies (data not shown) examining the injectability of the hydrogels at room temperature. All the polymers analyzed were solutions at ambient temperature as indicated by the low complex viscosity values (Fig. 1a). With an increase in temperature, the thermoresponsive polymer solutions transformed into gels with a corresponding increase in complex viscosity. The sol–gel transition temperatures of P1 and P2 were determined to be 28 and 32°C, respectively. The addition of PLGA-NP to the polymer solutions resulted in a decrease in the sol–gel transition temperature and increase in the complex viscosity of the gel (Fig. 1a). The gelation temperatures of P1 and P2 hydrogels containing PLGA-NP were 26 and 28°C, respectively. All the pentablock copolymer formulations formed gels within 2 min at body temperature (Fig. 1b). The storage moduli (G’) and loss moduli (G”) for the systems with or without nanoparticles at body temperature are depicted in supplementary Fig. 2. For all the formulations, G’ is greater than G”, indicating the formation of rigid gels.
Fig. 1.
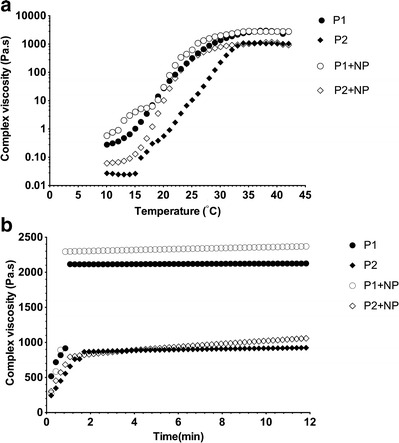
a Sol–gel transition temperature and b sol–gel transition time at body temperature of 17% (w/v) pentablock copolymers in the presence or absence of PLGA-NP loaded with OVA, MPL, and QA
In Vitro Gel Stability and Release Studies
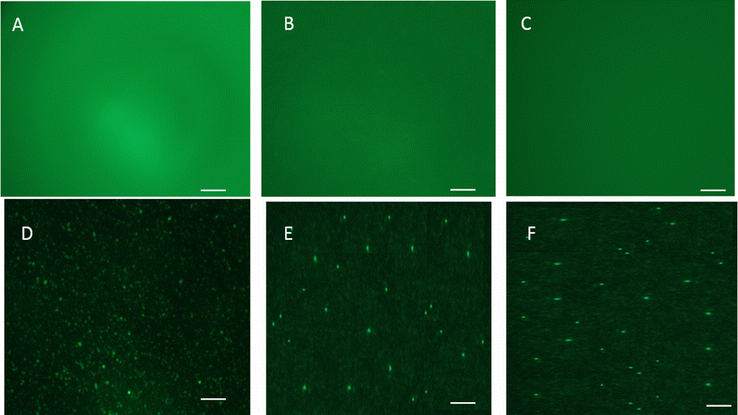
Both pentablock copolymers were stable in aqueous environments, unlike the triblock copolymers (Fig. 2), and by the end of 30 days, only 43% of the P1 gel and 23% of P2 gel remained intact (Fig. 2). The dissolution of P1 gel was slightly slower than that of the P2 gel at all time points. The stability of the gels was increased in systems containing nanoparticles when compared to systems without nanoparticles. After 30 days, 65% of the P1 gel with nanoparticles and 57% of the P2 formulation with nanoparticles remained intact. NP stability in the hydrogels was confirmed by fluorescence microscopy (supplementary Fig. 3).
Fig. 2.
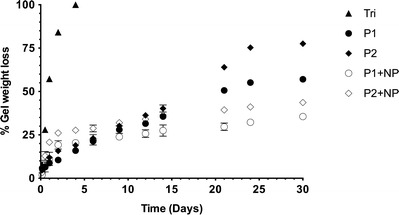
In vitro gel stability of modified triblock copolymer (Tri) and pentablock copolymer hydrogels (P1 and P2) with and without PLGA-NP (NP) at 37°C in PBS buffer (pH 7.4). The values shown are the mean ± SD (n = 3)
The in vitro release of FITC-OVA, MPL, and QA from PLGA-NP, thermoresponsive pentablock copolymer hydrogels, and PLGA-NP-loaded thermoresponsive hydrogels was evaluated over 30 days (Fig. 3). For the hydrophilic actives (FITC-OVA and QA), release was fastest and more complete from the NP (>85% release at 30 days). Release from the hydrogels was slower and less complete (60–70% release at 30 days) and the inclusion of NP into the hydrogels further slowed release, with less than 40% of FITC-OVA being released after 30 days. The inclusion of vaccine-loaded NP into hydrogels also reduced the initial burst release of the hydrophilic actives. The release of the hydrophobic active MPL was less affected by the formulation; however, the same trends were present. Interestingly, release of actives from the P1 gels was slower than from the P2 gels.
Fig. 3.
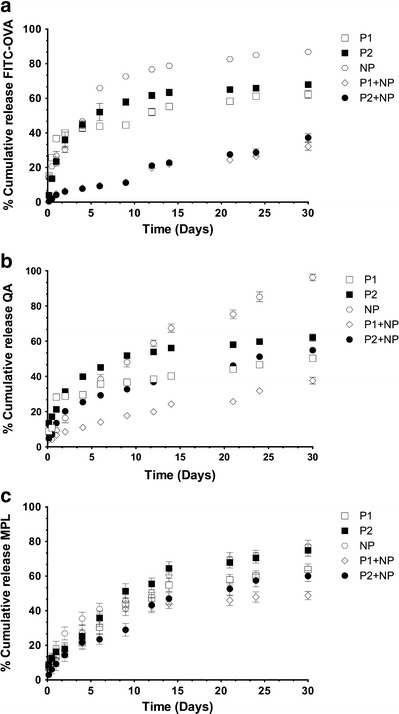
In vitro release profiles of a FITC-OVA, b QA, and c MPL from PLGA-NP (NP) and pentablock copolymer hydrogels (P1 and P2) with and without PLGA-NP. Data shown are the mean ± SD (n = 3)
Pentablock Copolymer Hydrogels Induced Long-Lasting Antigen-Specific Immune Responses
The cellular immune responses induced by triblock and pentablock copolymer hydrogels were investigated 21, 30, and 49 days after immunization (Fig. 4), and a number of interesting trends were apparent. Pentablock copolymer hydrogels induced significantly greater CD8+ T cell expansion than did the triblock hydrogels, the PLGA-NP and alum. Interestingly, the development of immune responses to the nanoparticle in hydrogel vaccines were delayed but after 49 days, P1 hydrogels containing nanoparticles stimulated a significantly greater expansion of CD8+ T cells than did the alum vaccine (p < 0.001). Mice immunized with P1 formulations had a higher number of CD8+ T cells compared to mice immunized with P2 formulations, both when loaded with soluble actives or with vaccine in PLGA-NP. The CD4+ responses were not as clear cut but similar trends in responses were observed with the response to the PLGA-NP-loaded gels being delayed and the P1 vaccines being superior to the P2 vaccines. Mice immunized with a single dose of PLGA-NP or an alum-adjuvanted vaccine developed only weak CD4+ responses.
Fig. 4.
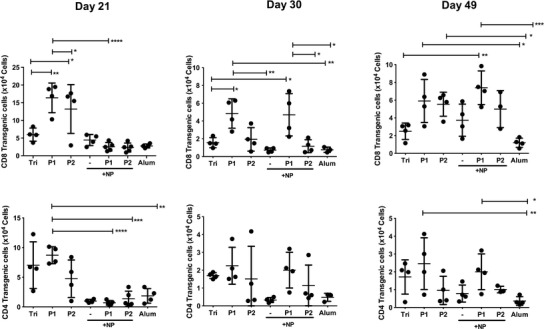
Expansion of OVA-specific CD8 and CD4 transgenic T cells in lymph nodes 21, 30, and 49 days post immunization with the triblock copolymer (Tri), the pentablock copolymers (P1 and P2) with or without PLGA-NP (NP) or alum. Data presented are the individual mice and the group mean ± SD (n = 3–5 mice, *p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001)
Ova-specific IgG responses were determined by ELISA (Fig. 5). The alum-adjuvanted vaccines stimulated high antibody titres as expected while the triblock copolymer and the PLGA-NP vaccines stimulated modest antibody titres. In agreement with previous results, the pentablock copolymers containing soluble vaccines stimulated high antibody titres earlier than pentablock copolymers containing PLGA-NP vaccines. Antibody responses were long lived and detectable out to 49 days post immunization.
Fig. 5.
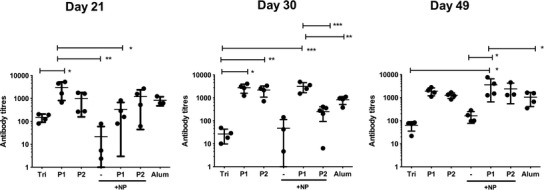
Ova-specific IgG antibody titres in serum of mice after 21, 30, and 49 days post immunization with the triblock copolymer (Tri), the pentablock copolymers (P1 and P2) with or without PLGA-NP (NP) or alum. Data presented are the individual mice and the group mean ± SD (n = 3–5 mice, *p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001)
Pentablock Copolymer Hydrogels Induced Long-Lived Cytotoxic Immune Responses
The next step in evaluating the potential of these vaccine formulations was to use a functional assay. As these vaccines stimulated long-lasting cellular responses, the ability of the vaccines to stimulate memory cytotoxic immune responses able to prevent the establishment of tumors was investigated. From the previous experiments, the pentablock copolymer with the higher PCL to PLA ratio (P1) appeared to stimulate stronger immune responses than P2 and was therefore utilized in the tumor challenge experiment. Mice were immunized with P1 pentablock hydrogels loaded with either the soluble vaccine or the PLGA-NP vaccine or with a control vaccine with no antigen and 49 days later were injected subcutaneously with 1 × 105 B16.OVA melanoma cells (Fig. 6 and supplementary Fig. 4). Mice immunized with P1 hydrogels containing blank nanoparticles and the no treatment group had measureable tumors day 12 after tumor injection. After 25 days, 90% of the mice in the blank nanoparticle and no treatment group were euthanized after reaching predetermined clinical endpoints. After 40 days, 87% survival of mice immunized with the P1 hydrogel PLGA nanoparticle vaccine and 75% survival of mice immunized with the P1 hydrogels containing soluble vaccine were observed. The surviving mice were rechallenged on the other flank with 1 × 105 B16.OVA melanoma cells 60 days after the first challenge (109 days after immunization). After rechallenge, mice immunized with P1 hydrogels with PLGA-NP vaccine had 75% survival, whereas 50% survival of mice immunized with the P1 hydrogels containing soluble vaccine was found. Mice immunized with the P1 hydrogels containing either soluble or NP vaccine had significantly greater survival than mice immunized with the P1 hydrogels containing blank NP (p < 0.0001). However, there was no significant difference in survival of mice immunized with the P1 hydrogels containing soluble or NP vaccine (p = 0.1611).
Fig. 6.
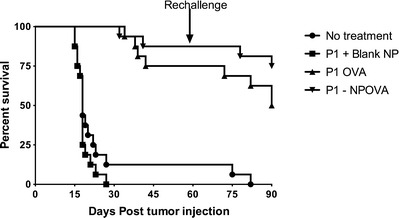
Prophylactic anti-tumor response. Survival analysis of mice (n = 16, from two independent experiments) injected subcutaneously with 1 × 105 B16.OVA melanoma cells 49 days post immunization with the P1 pentablock gel loaded with blank PLGA-NP (P1 + blank NP), with vaccine-loaded PLGA-NP (P1-NPOVA) or with soluble vaccine (P1 OVA). Unimmunized mice were included as a control (no treatment). Surviving mice were rechallenged 60 days after the initial tumor challenge in the opposite flank with 1 × 105 B16.OVA melanoma cells
DISCUSSION
During a naturally occurring infection, antigens are released from the site of infection as the pathogen replicates until such time as the pathogens are destroyed by the immune system. This is the situation we wish to recreate with our vaccine formulation and the sustained release of antigens from depot formulations is one such strategy for achieving this (6). Modified triblock copolymer based thermoresponsive gels previously tested in our lab lacked stability (36); therefore in this study, we utilized pentablock copolymers. The two novel pentablock copolymers used in the study had the same block arrangement but differed in the ratio of PCL and PLA, having either a 3:1 (P1) or 1:1 (P2) ratio of PCL to PLA. The presence of PEG blocks made both polymers hydrophilic and able to be easily solubilized in aqueous systems. The aqueous polymer solutions consist of self-assembled polymeric micelles and upon an increase in temperature the micelles aggregate to form a gel (29). While both P1 and P2 transformed from solutions to gels at physiological temperature, P1 formed more rigid gels because of increased hydrophobic interactions between the micelles. The transition time of sol to gel for the polymers was less than 2 min, which is advantageous as it reduces the likelihood of dose dumping and the potential for the gel to be diluted with physiological fluids at the site of injection (39).
A particulate carrier loaded with the vaccine antigen and adjuvants was incorporated into the formulation in order to mimic the presence of pathogens and to facilitate synchronous uptake of the vaccine components by APCs (7,8). PLGA-NP were chosen due to their biocompatibility, stability, and versatility. The addition of PLGA-NP to the gels decreased the sol–gel transition time and temperature, likely due to the increased hydrophobic interactions between the PLGA-NP and the gels.
A key requirement for the gels was stability and sustained release of antigen. Both gels were highly stable with only minor effects on stability resulting from the variation in the ratio of PCL to PLA. Increased hydrophobic interactions of PLGA-NP with the gels further increased the aqueous stability of particle-loaded formulations, consistent with the rheology measurements. The release of FITC-OVA, QA, and MPL from PLGA-NP exhibited an initial burst phase due to surface adsorption; however, when the nanoparticles were incorporated into the hydrogel, the burst effect was reduced due to the presence of the additional diffusion layer. The release of antigen and adjuvant from this combination formulation was overall slower as compared to formulations where antigen and adjuvant were loaded directly into the hydrogels, again due to the presence of two diffusion barriers—the particles and then the gel. The release of antigen and adjuvant in vitro was incomplete over the 30-day examination period, and visual inspection of the mice culled at day 49 revealed the continued presence of a depot at the site of immunization. This type of release resembles a chronic rather than an acute infection, which may be detrimental to the immune response generated as chronic exposure to antigen can result in cell deletion or exhaustion (40). However, this will depend on how the antigen and adjuvant are being released over this extended period of time, for example if antigen continues to be released by itself after adjuvant release is complete or if release stays at high levels or reduces down to low levels. Low-level release of antigen in an immunogenic form may enrich memory cell populations (41) rather than exhausting effector cells.
The immunogenicity of these promising formulations was then examined. Immune responses to acute infections peak around day 10, therefore antigen release for at least 10–15 days would be desirable for the stimulation of robust immune responses (42). Modified triblock copolymer hydrogels do not stimulate sustained immune responses due to poor stability and fast erosion of the gel (around 3 days) (36) and were included here as a control along with alum, a gold standard adjuvant widely used in the clinic. The pentablock copolymer hydrogels with their increased stability and sustained release of antigen and immunomodulators stimulated strong, long-lasting T cell and antibody responses. These results are in agreement with the recent investigations on chitosan thermoresponsive hydrogels as sustained vaccine delivery systems; however, those systems had less than optimal formulation characteristics (36,43). Interestingly, the inclusion of PLGA-NP in the hydrogels delayed the induction of both cellular and humoral immune responses, possibly due to the reduced burst release and overall slower release of vaccine components. Recent literature suggests that multiple administrations of nanoparticle vaccines are required to stimulate effective cellular and humoral responses (44,45). Consistent with these studies, our results demonstrated that a single dose of PLGA-NP vaccine is not sufficient to stimulate potent cellular or humoral immune responses.
The choice of adjuvant plays an important role in the formulation design of vaccines. Kamath et al. showed that the uptake of antigen by APCs before adjuvant reduces uptake and inhibits immunity, but that adjuvant uptake by APCs before antigen may not (7). Therefore, NPs were included in the formulation to ensure synchronous exposure of APC to both antigen and adjuvant. However, with the formulation used here, this did not seem necessary as the hydrogels loaded with the individual vaccine components also stimulated robust immune responses. This is likely related to the choice and combination of adjuvants used in this vaccine where a hydrophilic TLR-independent and a hydrophobic TLR-dependent adjuvant were used. The release of the adjuvants was similar or slightly faster than that of the protein antigen, which would result in APC being exposed to them either at the same time or to adjuvant before antigen, while the depot formulation would ensure exposure at the same place. The importance of the depot in creating a focus for the developing immune response was demonstrated by the much weaker immune responses stimulated by the NP vaccine.
P1 hydrogels were selected for the functional studies of immunity as these more rigid gels stimulated slightly stronger immune responses. As the vaccines stimulated strong CD8+ T cells responses, which are more difficult to generate as they require antigen cross presentation (46), a tumor challenge model was chosen to assess functional cytotoxicity (47). The tumor challenge was administered 49 days after vaccination, and remarkably a single dose of the hydrogel vaccine, with or without NP, was able to stimulate long-lived immunity able to mediate killing of the tumor cells in the majority of mice. Over half the mice were able to reject a further challenge 109 days after the initial immunization. These are impressive results for a single dose of a protein vaccine which are notorious for requiring booster immunizations and for not being strong stimulators of cytotoxic immune responses (48).
CONCLUSION
In this study, thermoresponsive injectable hydrogel vaccines were developed with the aim of delivering a subunit vaccine for a sustained period of time and to stimulate robust long-lived immune responses. This was achieved using two novel PEG-PCL-PLA-PCL-PEG pentablock copolymers. With the combination of adjuvants used here, the loading of the vaccine into a nanoparticle, to ensure synchronous uptake of both antigen and adjuvant by APC, was not required. However, this is likely a result of the physical and immunological properties of the adjuvants used and will not apply to all vaccines. Release of the vaccine in vitro from these formulations was incomplete over 30 days, which may be not optimal. Further studies should be carried out to accurately assess release in vivo and to develop a formulation which releases antigen over a shorter period of time (10–15 days). These points notwithstanding, the vaccines developed here were able, after a single dose, to stimulate robust cellular and humoral immune responses able to reject tumor cells given a month and a half post immunization.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Gating strategy for transgenic CD4+ and CD8+ T cells. CD4+ and CD8+ T cells were gated for CD4+ Vα2+ Vβ5+ or CD8+ Vα2+ Vβ5+ transgenic cells. Colors are indicative of relative cell density in a particular area, with relative density decreasing in the order of red>yellow>green>light blue>dark blue. (GIF 229 kb)
{kind=link}
The storage and loss moduli of 17% polymer 1 and polymer 2 in the presence or absence of PLGA nanoparticles at body temperature. (GIF 54 kb)
{kind=link}
Fluorescence microscopy images of FITC-OVA in a PBS, b P1 gel, and c P2 gel respectively, and FITC-OVA PLGA-NP in d PBS, e, P1 gel, and f P2 gel, respectively. Scale bar is 200 μm. (GIF 143 kb)
{kind=link}
Prophylactic anti-tumor response. Tumor volume in individual mice (n = 16, from two independent experiments) that developed tumors following subcutaneous injection of 1 × 105 B16.OVA melanoma cells 49 days post immunization with the P1 pentablock gel loaded with blank PLGA NP (P1 + blank NP), with vaccine-loaded PLGA NP (P1-NPOVA) or with soluble vaccine (P1 OVA). Unimmunized mice were included as a control (no treatment). Surviving mice were rechallenged 60 days after the initial tumor challenge in the opposite flank with 1 × 105 B16.OVA melanoma cells. (GIF 90 kb)
Acknowledgments
SB is supported by University of Otago Postgraduate Scholarship.
References
- 1.Arnon R, Ben-Yedidia T. Old and new vaccine approaches. Int Immunopharmacol. 2003;3(8):1195–204. doi: 10.1016/S1567-5769(03)00016-X. [DOI] [PubMed] [Google Scholar]
- 2.Kersten GFA, Crommelin DJA. Liposomes and ISCOMs. Vaccine. 2003;21(9–10):915–20. doi: 10.1016/S0264-410X(02)00540-6. [DOI] [PubMed] [Google Scholar]
- 3.Zinkernagel RM, Ehl S, Aichele P, Oehen S, Kundig T, Hengartner H. Antigen localisation regulates immune responses in a dose- and time-dependent fashion: a geographical view of immune reactivity. Immunol Rev. 1997;156:199–209. doi: 10.1111/j.1600-065X.1997.tb00969.x. [DOI] [PubMed] [Google Scholar]
- 4.Myschik J, McBurney WT, Hennessy T, Phipps-Green A, Rades T, Hook S. Immunostimulatory biodegradable implants containing the adjuvant Quil-A-Part II: in vivo evaluation. J Drug Target. 2008;16(3):224–32. doi: 10.1080/10611860701848886. [DOI] [PubMed] [Google Scholar]
- 5.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang X-F, et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat Med. 2013;19(4):465–72. doi: 10.1038/nm.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gordon S, Teichmann E, Young K, Finnie K, Rades T, Hook S. In vitro and in vivo investigation of thermosensitive chitosan hydrogels containing silica nanoparticles for vaccine delivery. Eur J Pharm Sci. 2010;41(2):360–8. doi: 10.1016/j.ejps.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Kamath AT, Mastelic B, Christensen D, Rochat AF, Agger EM, Pinschewer DD, et al. Synchronization of dendritic cell activation and antigen exposure is required for the induction of Th1/Th17 responses. J Immunol. 2012;188(10):4828–37. doi: 10.4049/jimmunol.1103183. [DOI] [PubMed] [Google Scholar]
- 8.Foged C, Sundblad A, Hovgaard L. Targeting vaccines to dendritic cells. Pharm Res. 2002;19(3):229–38. doi: 10.1023/A:1014474414097. [DOI] [PubMed] [Google Scholar]
- 9.Gamvrellis A, Leong D, Hanley JC, Xiang SD, Mottram P, Plebanski M. Vaccines that facilitate antigen entry into dendritic cells. Immunol Cell Biol. 2004;82(5):506–16. doi: 10.1111/j.0818-9641.2004.01271.x. [DOI] [PubMed] [Google Scholar]
- 10.Baldridge JR, Crane RT. Monophosphoryl lipid A (MPL) formulations for the next generation of vaccines. Methods. 1999;19(1):103–7. doi: 10.1006/meth.1999.0834. [DOI] [PubMed] [Google Scholar]
- 11.Baldridge JR, McGowan P, Evans JT, Cluff C, Mossman S, Johnson D, et al. Taking a toll on human disease: toll-like receptor 4 agonists as vaccine adjuvants and monotherapeutic agents. Expert Opin Biol Ther. 2004;4(7):1129–38. doi: 10.1517/14712598.4.7.1129. [DOI] [PubMed] [Google Scholar]
- 12.Kensil C, Wu J-Y, Soltysik S. Structural and immunological characterization of the vaccine adjuvant QS-21. In: Powell M, Newman M, editors. Vaccine design. Pharmaceutical biotechnology. US: Springer; 1995. pp. 525–41. [DOI] [PubMed] [Google Scholar]
- 13.Demana PH, Fehske C, White K, Rades T, Hook S. Effect of incorporation of the adjuvant Quil A on structure and immune stimulatory capacity of liposomes. Immunol Cell Biol. 2004;82(5):547–54. doi: 10.1111/j.0818-9641.2004.01276.x. [DOI] [PubMed] [Google Scholar]
- 14.Mikloska Z, Rückholdt M, Ghadiminejad I, Dunckley H, Denis M, Cunningham AL. Monophosphoryl lipid A and QS21 increase CD8 T lymphocyte cytotoxicity to herpes simplex virus-2 infected cell proteins 4 and 27 through IFN-γ and IL-12 production. J Immunol. 2000;164(10):5167–76. doi: 10.4049/jimmunol.164.10.5167. [DOI] [PubMed] [Google Scholar]
- 15.Rattanapak T, Young K, Rades T, Hook S. Comparative study of liposomes, transfersomes, ethosomes and cubosomes for transcutaneous immunisation: characterisation and in vitro skin penetration. J Pharm Pharmacol. 2012;64(11):1560–9. doi: 10.1111/j.2042-7158.2012.01535.x. [DOI] [PubMed] [Google Scholar]
- 16.Ruel-Gariepy E, Leroux JC. In situ-forming hydrogels—review of temperature-sensitive systems. Eur J Pharm Biopharm. 2004;58(2):409–26. doi: 10.1016/j.ejpb.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 17.Jeong B, Kim SW, Bae YH. Thermosensitive sol–gel reversible hydrogels. Adv Drug Deliv Rev. 2002;54(1):37–51. doi: 10.1016/S0169-409X(01)00242-3. [DOI] [PubMed] [Google Scholar]
- 18.Dumortier G, Grossiord JL, Agnely F, Chaumeil JC. A review of poloxamer 407 pharmaceutical and pharmacological characteristics. Pharm Res. 2006;23(12):2709–28. doi: 10.1007/s11095-006-9104-4. [DOI] [PubMed] [Google Scholar]
- 19.Kabanov AV, Batrakova EV, Alakhov VY. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Release. 2002;82(2–3):189–212. doi: 10.1016/S0168-3659(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 20.Kojarunchitt T, Hook S, Rizwan S, Rades T, Baldursdottir S. Development and characterisation of modified poloxamer 407 thermoresponsive depot systems containing cubosomes. Int J Pharm. 2011;408(1–2):20–6. doi: 10.1016/j.ijpharm.2011.01.037. [DOI] [PubMed] [Google Scholar]
- 21.Paavola A, Yliruusi J, Rosenberg P. Controlled release and dura mater permeability of lidocaine and ibuprofen from injectable poloxamer-based gels. J Control Release. 1998;52(1–2):169–78. doi: 10.1016/S0168-3659(97)00206-X. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Zhu Y-Y, Wei G, Lu W-Y. Effect of carrageenan on poloxamer-based in situ gel for vaginal use: improved in vitro and in vivo sustained-release properties. Eur J Pharm Sci. 2009;37(3–4):306–12. doi: 10.1016/j.ejps.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 23.El-Kamel AH. In vitro and in vivo evaluation of Pluronic F127-based ocular delivery system for timolol maleate. Int J Pharm. 2002;241(1):47–55. doi: 10.1016/S0378-5173(02)00234-X. [DOI] [PubMed] [Google Scholar]
- 24.Gong C, Shi S, Dong P, Kan B, Gou M, Wang X, et al. Synthesis and characterization of PEG-PCL-PEG thermosensitive hydrogel. Int J Pharm. 2009;365(1–2):89–99. doi: 10.1016/j.ijpharm.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 25.Ma G, Miao B, Song C. Thermosensitive PCL-PEG-PCL hydrogels: synthesis, characterization, and delivery of proteins. J Appl Polym Sci. 2010;116(4):1985–93. [Google Scholar]
- 26.Gou M, Zheng L, Peng X, Men K, Zheng X, Zeng S, et al. Poly(epsilon-caprolactone)-poly(ethylene glycol)-poly(epsilon-caprolactone) (PCL-PEG-PCL) nanoparticles for honokiol delivery in vitro. Int J Pharm. 2009;375(1–2):170–6. doi: 10.1016/j.ijpharm.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 27.Chen CC, Chueh JY, Tseng H, Huang HM, Lee SY. Preparation and characterization of biodegradable PLA polymeric blends. Biomaterials. 2003;24(7):1167–73. doi: 10.1016/S0142-9612(02)00466-0. [DOI] [PubMed] [Google Scholar]
- 28.Ge H, Hu Y, Yang S, Jiang X, Yang C. Preparation, characterization, and drug release behaviors of drug-loaded ε-caprolactone/L-lactide copolymer nanoparticles. J Appl Polym Sci. 2000;75(7):874–82. doi: 10.1002/(SICI)1097-4628(20000214)75:7<874::AID-APP3>3.0.CO;2-G. [DOI] [Google Scholar]
- 29.Patel SP, Vaishya R, Mishra GP, Tamboli V, Pal D, Mitra AK. Tailor-made pentablock copolymer based formulation for sustained ocular delivery of protein therapeutics. J Drug Deliv. 2014;2014:401747. doi: 10.1155/2014/401747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silva JM, Videira M, Gaspar R, Preat V, Florindo HF. Immune system targeting by biodegradable nanoparticles for cancer vaccines. J Control Release. 2013;168(2):179–99. doi: 10.1016/j.jconrel.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 31.Hamdy S, Haddadi A, Somayaji V, Ruan D, Samuel J. Pharmaceutical analysis of synthetic lipid A-based vaccine adjuvants in poly (D, L-lactic-co-glycolic acid) nanoparticle formulations. J Pharm Biomed. 2007;44(4):914–23. doi: 10.1016/j.jpba.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Könnings S, Copland MJ, Davies NM, Rades T. A method for the incorporation of ovalbumin into immune stimulating complexes prepared by the hydration method. Int J Pharm. 2002;241(2):385–9. doi: 10.1016/S0378-5173(02)00270-3. [DOI] [PubMed] [Google Scholar]
- 33.Bobbala S, McDowell A, Hook S. Quantitation of the immunological adjuvants, monophosphoryl lipid A and Quil A in poly (lactic-co-glycolic acid) nanoparticles using high performance liquid chromatography with evaporative light scattering detection. J Chromatogr B. 2015;975:45–51. doi: 10.1016/j.jchromb.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based a- and b-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76(1):34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 35.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76(1):17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 36.Kojarunchitt T, Baldursdottir S, Dong Y-D, Boyd BJ, Rades T, Hook S. Modified thermoresponsive poloxamer 407 and chitosan sol–gels as potential sustained-release vaccine delivery systems. Eur J Pharm Biopharm. 2015;89:74–81. doi: 10.1016/j.ejpb.2014.11.026. [DOI] [PubMed] [Google Scholar]
- 37.Filatenkov AA, Jacovetty EL, Fischer UB, Curtsinger JM, Mescher MF, Ingulli E. CD4 T cell-dependent conditioning of dendritic cells to produce IL-12 results in CD8-mediated graft rejection and avoidance of tolerance. J Immunol. 2005;174(11):6909–17. doi: 10.4049/jimmunol.174.11.6909. [DOI] [PubMed] [Google Scholar]
- 38.McBurney WT, Lendemans DG, Myschik J, Hennessy T, Rades T, Hook S. In vivo activity of cationic immune stimulating complexes (PLUSCOMs) Vaccine. 2008;26(35):4549–56. doi: 10.1016/j.vaccine.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 39.Charrueau C, Tuleu C, Astre V, Grossiord JL, Chaumeil JC. Poloxamer 407 as a thermogelling and adhesive polymer for rectal administration of short-chain fatty acids. Drug Dev Ind Pharm. 2001;27(4):351–7. doi: 10.1081/DDC-100103735. [DOI] [PubMed] [Google Scholar]
- 40.Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2009;106(21):8623–8. doi: 10.1073/pnas.0809818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim TS, Hufford MM, Sun J, Fu Y-X, Braciale TJ. Antigen persistence and the control of local T cell memory by migrant respiratory dendritic cells after acute virus infection. J Exp Med. 2010;207(6):1161–72. doi: 10.1084/jem.20092017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whitmire JK, Murali-Krishna K, Altman J, Ahmed R. Antiviral CD4 and CD8 T-cell memory: differences in the size of the response and activation requirements. Philos Trans R Soc Lond B Biol Sci. 2000;355(1395):373–9. doi: 10.1098/rstb.2000.0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Highton AJ, Kojarunchitt T, Girardin A, Hook S, Kemp RA. Chitosan hydrogel vaccine generates protective CD8 T cell memory against mouse melanoma. Immunol Cell Biol. 2015;93(7):634–40. doi: 10.1038/icb.2015.14. [DOI] [PubMed] [Google Scholar]
- 44.Varypataki E, van der Maaden K, Bouwstra J, Ossendorp F, Jiskoot W. Cationic liposomes loaded with a synthetic long peptide and poly(I:C): a defined adjuvanted vaccine for induction of antigen-specific T cell cytotoxicity. AAPS J. 2015;17(1):216–26. doi: 10.1208/s12248-014-9686-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joshi V, Geary S, Salem A. Biodegradable particles as vaccine delivery systems: size matters. AAPS J. 2013;15(1):85–94. doi: 10.1208/s12248-012-9418-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12(8):557–69. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 47.Gilboa E. The promise of cancer vaccines. Nat Rev Cancer. 2004;4(5):401–11. doi: 10.1038/nrc1359. [DOI] [PubMed] [Google Scholar]
- 48.Atanackovic D, Altorki NK, Cao Y, Ritter E, Ferrara CA, Ritter G, et al. Booster vaccination of cancer patients with MAGE-A3 protein reveals long-term immunological memory or tolerance depending on priming. Proc Natl Acad Sci U S A. 2008;105(5):1650–5. doi: 10.1073/pnas.0707140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gating strategy for transgenic CD4+ and CD8+ T cells. CD4+ and CD8+ T cells were gated for CD4+ Vα2+ Vβ5+ or CD8+ Vα2+ Vβ5+ transgenic cells. Colors are indicative of relative cell density in a particular area, with relative density decreasing in the order of red>yellow>green>light blue>dark blue. (GIF 229 kb)
The storage and loss moduli of 17% polymer 1 and polymer 2 in the presence or absence of PLGA nanoparticles at body temperature. (GIF 54 kb)
Fluorescence microscopy images of FITC-OVA in a PBS, b P1 gel, and c P2 gel respectively, and FITC-OVA PLGA-NP in d PBS, e, P1 gel, and f P2 gel, respectively. Scale bar is 200 μm. (GIF 143 kb)
Prophylactic anti-tumor response. Tumor volume in individual mice (n = 16, from two independent experiments) that developed tumors following subcutaneous injection of 1 × 105 B16.OVA melanoma cells 49 days post immunization with the P1 pentablock gel loaded with blank PLGA NP (P1 + blank NP), with vaccine-loaded PLGA NP (P1-NPOVA) or with soluble vaccine (P1 OVA). Unimmunized mice were included as a control (no treatment). Surviving mice were rechallenged 60 days after the initial tumor challenge in the opposite flank with 1 × 105 B16.OVA melanoma cells. (GIF 90 kb)