

Abstract
Multiplex ligand binding assays (LBAs) are increasingly being used to support many stages of drug development. The complexity of multiplex assays creates many unique challenges in comparison to single-plexed assays leading to various adjustments for validation and potentially during sample analysis to accommodate all of the analytes being measured. This often requires a compromise in decision making with respect to choosing final assay conditions and acceptance criteria of some key assay parameters, depending on the intended use of the assay. The critical parameters that are impacted due to the added challenges associated with multiplexing include the minimum required dilution (MRD), quality control samples that span the range of all analytes being measured, quantitative ranges which can be compromised for certain targets, achieving parallelism for all analytes of interest, cross-talk across assays, freeze-thaw stability across analytes, among many others. Thus, these challenges also increase the complexity of validating the performance of the assay for its intended use. This paper describes the challenges encountered with multiplex LBAs, discusses the underlying causes, and provides solutions to help overcome these challenges. Finally, we provide recommendations on how to perform a fit-for-purpose-based validation, emphasizing issues that are unique to multiplex kit assays.
KEY WORDS: biomarker, diagnostics, ligand binding assay, multiplex, validation
INTRODUCTION
Ligand binding assays (LBAs) are commonly used tools to measure many clinically relevant analytes, including biomarkers and new biologic-based drugs. Most LBAs rely on antibodies as critical reagents to capture and detect the analyte of interest in a biological matrix. Traditionally, these assays, which we refer to in this paper as single-plexed assays, are designed to detect the presence of a single analyte. Commonly used single-plexed assays rely on detection technologies that include enzyme-driven detection (ELISA), chemiluminescence (CL), radioactive isotopes, fluorescence (FL), and electrochemiluminescence (ECL). New technologies have been developed that allow multiple analytes to be measured simultaneously in a single sample, all in a single reaction vessel. Performing multiple assays together in this context is referred to as multiplexing. The demand for and usefulness of multiplex assays have resulted in a large number of premade commercially available multiplex assay kits offering a welcome increase in the number of tools that scientists have at their disposal besides reduced laboratory analysis time and sample volume requirements and cost savings. However, multiplex assays also present unique challenges not encountered with single-plexed assays. Unique challenges such as different detection ranges, cross-reactivity with multiplex assay reagents, increased specificity issues and matrix interference, cross-talk across assays, and changes in sensitivity, if not carefully addressed, can generate misleading results.
The development of new therapeutic drugs requires many years of preclinical and clinical studies in order to obtain drug approval. The importance of the data generated in these studies mandates the use of well-characterized assays, both in terms of analytical performance and reliability of the data. Thus, many of these assays are validated prior to use to ensure that the performance and data integrity are suitable for the intended use. There are many excellent publications that provide guidance on how to validate these assays and how to ensure that the analytical performance is fit for its intended purpose (1–3), as well as the recent draft guidance for industry on bioanalytical method validation (4). Despite these publications, some aspects of validation that are unique to multiplex assays are not covered in these publications.
In this publication, we first provide a brief overview of the available multiplexing technologies along with the benefits and challenges of each technology. Throughout the paper, we highlight some of the basic principles of LBA method validation for new users in the context of multiplexing. We then present an overview of the unique challenges with commercial multiplex assay kits and recommend solutions to help overcome these challenges. Finally, we provide recommendations on how to perform a fit-for-purpose validation of multiplex kit assays, with an emphasis on the unique aspects of multiplex assays.
OVERVIEW OF MULTIPLEXING TECHNOLOGIES
The emerging multiplex LBA technologies are based on either micro-bead or solid-phase planar array, requiring specialized instruments. The bead-based technologies typically distinguish LBAs from one another by assigning a unique bead set to each assay in contrast to solid-phase planar arrays which distinguish each LBA by a location on the solid-phase surface. There are differences in the mechanism used to detect and quantify the captured analyte for example, FL, CL, or ECL. There are also notable differences in the benefits and limitations for each of these technologies. The intended use should be the driving force on selecting the appropriate multiplex assay. In addition, several recent publications comprehensively review the latest multiplexing technologies (5–8).
Recommendations on How to Select a Suitable Multiplex Assay
Commercial multiplex kits are an economical way of measuring multiple analytes from a small volume of sample. They are generally designed to evaluate endogenous markers involved in a specific pathway or disease indication. A feasibility assessment of commercially available multiplex assay kit for the intended study involves the evaluation of a number of key parameters, including the range of detection; the availability of reliable and representative reference or purified sources of the analyte; and the performance of the multiplex in sample matrix in the presence of endogenous levels of analyte, specificity, sensitivity, vendor support, and other technology considerations. The use of commercial assay kits can expedite sample analysis and conserve resources. However, the trade-off is that these kits may not have been developed for the intended purpose of study of interest. In such circumstances, confirmation of acceptable performance must be demonstrated and may require feasibility testing. For example, methods commercialized to measure analytes in serum may be adapted to CSF collections. Likewise, vendor kits may require modification to meet regulatory requirements. Frequent issues include expanding the number/concentration of calibrators and quality controls. Currently available multiplex assay vendors are listed in Table I. The following section provides an outline of the recommended key parameters that will guide the user toward finding the best multiplex assay:
-
Quantitative range of detection and sensitivity
Most vendors provide calibration curve information with the multiplex assay product or kit insert for each analyte to determine whether the assay will provide sufficient sensitivity and range of detection. However, the range of detection reported by the vendor may not reflect the range of detection that will be encountered in a specific sample matrix of interest. If the quantitative range is unknown or questionable, the user should determine the range of detection for each analyte in both healthy and disease-state samples to ensure that the sample analyte levels fall within the quantitative range of the assay for all analytes. This can be done by working directly with the vendor or by performing additional development experiments in their own laboratory.
-
Performance in sample matrix
The majority of commercially available assays are developed and tested using purified proteins or protein mixtures made in assay buffer, occasionally tested in clinical matrix like serum or plasma. When matrix samples are used, they are often a combination of native and spiked analytes, due to variable levels of each analyte present in the samples. Commercially available multiplex kits are often manufactured to be used for several applications. As part of the feasibility assessment, for a specific study, it is important that the assay performance is assessed in its intended sample matrix. An increased number of samples may be needed to cover the ranges of all of the analytes in the multiplex. If the kit vendor or other reliable source cannot confirm performance of the assay in the intended sample matrix, the user will need to perform development experiments to test this parameter prior to kit selection for further use.
-
Specificity
The majority of multiplex assay vendors provide information on the specificity for each assay in the context of a multiplex environment. Ideally, the vendor should have performed both a single detection and single calibrator assay to ensure the specificity of each assay within the multiplex or looked at sample results of single-plexed assay compared to multiplex. This can be challenging for the scientist to perform due to a lack of availability of single-plexed reagents. Data from the vendor can give confidence in the specificity of the multiplex. However, depending on the intended use of the data, the user may need to perform additional specificity experiments on the assay to confirm the parameter.
-
Vendor support
A healthy business relationship with the vendor plays a critical role in assessing and evaluating the multiplex assays (9). Critical examples of communication from the vendor include manufacturing or reagent changes, supporting information on the kit performance, any information on, and availability of key reagents included in the kit and, most importantly, the availability of large lots of each kit (lot-to-lot variability is discussed in a later section). Ongoing timely technical support is essential to ensure that problems are quickly solved.
In summary, the selection criteria for the type of platform and its corresponding kits depend mainly on the above-mentioned four critical parameters. It is recommended that all of this information be utilized to make a decision to pursue a type of multiplexing platform. If these key parameters fail to provide scientifically sound information, or if some of these key assay attributes are missing, scientists are encouraged to strive for different technology platforms.
Table I.
Review of Some Current Multiplexing Platforms
| Technology | Surface | Detection | Benefits | Websites |
|---|---|---|---|---|
| Bioscale | Acoustic membrane particle technology | Sensor resonant frequency | Sensitivity | http://bioscale.com/vibe-workstation |
| Curiox | Magnetic beads (DropArray plates) | FL |
-Sensitivity -Automated -Kit menu -Custom assay development |
http://www.curiox.com/applications.html |
| Quanterix | Magnetic beads (SiMOA) | FL |
-Sensitivity -Automated -Kit menu -Custom assay development -Regulatory path from RUO to -IVD/CDx |
www.quanterix.com |
| Luminex | Fluorescent beads | FL |
-Sensitivity -Kit menu -Custom assay development -Regulatory path from RUO to -IVD/CDx -Flexibility to build your own assay |
http://www.luminexcorp.com |
| Cyto beads | Magnetic beads | FL |
-Sensitivity -Kit menu |
http://info.bio-rad.com |
| Meso Scale Discovery | Electrode carbon surface | ECL |
-Sensitivity -Kit menu -Custom assay development -Automation |
http://www.mesoscale.com |
| Aushon | Quartz | DCD |
-Sensitivity -Kit menu -Custom assay development |
http://www.aushon.com |
| Randox | Biochip | CL |
-Sensitivity -Kit menu -Custom assay development -Automation -Regulatory path from RUO to -IVD/CDx |
http://www.randox.com |
| Genalyte | Silicon photonic biosensors | Surface resonance units |
-Sensitivity -Kit menu -Custom assay development -Elimination of cross-talk |
http://genalyte.com |
F L fluorescence, RUO research uses only, IVD/CDx in vitro diagnostic/companion diagnostic, SiMOA single molecule array
MULTIPLEX ASSAY CHALLENGES AND SOLUTIONS
Multiplex assays inherently suffer from numerous analytical challenges during development, validation, and assay maintenance and throughout sample analysis. A few principal challenges to consider and their corresponding solutions are described below. Although this paper discusses mainly the challenges and solutions for commercial kit-based assays, certain key aspects of multiplex development are also included. When employing commercial kit-based assays, vendors typically provide supporting data that address these challenges through characterization work during their kit development; however, it is recommended that every scientist vigorously examine the data prior to validation and sample testing. Examples of solutions during various stages of multiplex validation, sample testing, and assay maintenance are described in Table II.
Table II.
Overview of Challenges and Solutions: Multiplex Validation, Sample Analysis, and Assay Maintenance
| Method stage | Challenge | Solution |
|---|---|---|
| Method validation | Data handling |
• Use built-in templates • Contract IT resources • Work with instrument/software vendors to assist with solutions |
| All pass/procedure if an analyte system fails? |
• Report data for analytes that pass, repeat run for analytes that do not pass (second run should mask results for passing analytes) • Analyte that does not meet validation acceptance criteria (if repeat analysis fails) should not be included in multiplex sample analysis. • Demonstrate that removal of capture/detect/analyte for failed assay does not change the multiplex assay performance |
|
| Lack of regulatory performance recommendations | Utilize approach based on bioanalytcial methods for biotherapeutics, implementing acceptance criteria prior to in-study analysis, and using fit-for-purpose approach based on intended use of the assay | |
| Sample analysis | Data handling (work list preparation, data management, assay approval, data reporting) |
• Possible use of macros, built-in templates • Contract IT resources • Work with instrument/software vendors to assist with solutions |
| All pass—if you must rerun one analyte, what do you do with data for other passing analytes from first run? | Report data for analytes that pass, repeat run for analytes that do not pass (second run should mask results for passing analytes). Analyte that does not meet sample analysis acceptance criteria (if repeat analysis fails) should not be included in final analysis. | |
| Identical curve fitting |
• Use best fit approach • It is acceptable to use different curve fits/weighting for different analytes; however, for a single analyte continue using same curve fitting after validation |
|
| Lack of regulatory performance recommendations |
• Utilize regulatory requirements that most closely meet the study requirements • FDA Draft Guidance on Bioanalytical Method Validation includes a discussion on biomarkers |
|
| Assay maintenance | Variability in reagent lots |
• Implement a defined process to investigate lot-to-lot variability • Apply a correction factor • Screen multiple lots • Acquire sufficient volumes of an original lot with expiration dating that allows completion of the program |
| Availability of critical reagents |
• Use surrogate molecules if needed • Initiate an analyte/antibody production program in anticipation starting the project • Contact vendors to assist with sourcing • Review research literature for possible academic sources |
Italicized points are unique to multiplexing
Challenges with Quantitative Ranges and Optimal Sample Dilution
In a multiplex assay, sample dilution of a particular analyte must take into account the concentrations of the other analytes present in the sample. There are times when a sample will have low concentrations of some of the analytes and high concentrations of others, making the decision to dilute the sample difficult. The challenge presented by this situation is to decide on the appropriate sample dilution factor that ensures that all the analytes in the sample fall into their respective quantitative range. The compromise in sample dilution may not be the optimal dilution for every analyte being measured. The following example further clarifies the situation.
In the development of a multiplex panel to measure apolipoprotein (Apo) profiles associated with cardiovascular disease (Fig. 1), it was noted that the optimal dilution (middle of the curve) for Apo AII was 1:200,000, whereas the optimal dilution for Apo B and Apo E was 1:4000 and 1:1000, respectively. A compromise was made for Apo B and Apo E at 1:2000, with most of the samples falling within a good range of the curve at that dilution. For Apo AII, a dilution of 1:2000 resulted in samples falling above the upper limit of quantitation (ULOQ); therefore, the assay was redesigned as a competitive assay, which decreased the assay’s sensitivity, however, brought the optimal sample dilution to 1:2000.
Fig. 1.
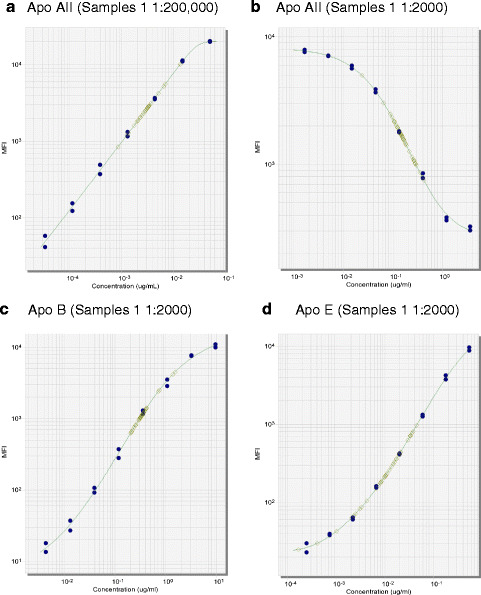
Multiplex curves and samples for apolipoproteins AII, B, and E. A multiplex assay was developed for serum apolipoproteins (Apo) on the Luminex platform. For the Apo AII assay (a), the optimal dilution was 1:200,000; however, the optimal dilution for Apo B (c) and Apo E (d) is closer to the 1:2000 range, with samples falling below the LLOQ at 1:200,000. Conversion of the Apo AII assay to the competitive format (b) decreased the assay sensitivity to bring the optimal dilution down to 1:2000. The calibrators are represented by the blue circles, and patient samples (n = 49) are represented by green squares
Another consideration for this quantitative range and sample dilution challenge is the matrix of the sample. LBAs typically perform differently in the samples taken from normal healthy subjects versus disease-state patients, and the levels of analytes in different sample matrices often substantially vary. Careful consideration of sample matrix should be evaluated as a possible solution to the challenge of finding an optimal sample dilution. Although MRD is determined based on fundamental principles of quantitative measurement of analyte, the issue is compounded by number of analytes in multiplex assay. Similar to sample dilution, MRD may also be impacted by matrix and level of analyte. Figure 2 illustrates the calculation of the minimum required dilution (MRD) using six serum samples, clarifying stepwise how the MRD can be determined. If an acceptable sample dilution cannot be achieved, the user should consider removing the problematic analytes from the multiplex panel and running them separately.
Fig. 2.
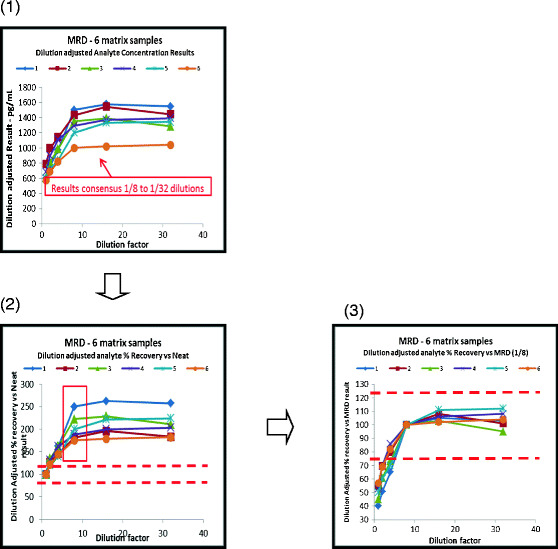
Calculation of optimal minimum required dilution (MRD): results of parallelism assessment for six individual matrix samples for a single analyte. In this figure, non-parallelism is apparent over part of the dilution range. Where there is parallelism across neat and diluted samples, no dilution will be required (1). The first chart shows test results adjusted for the dilution factor for all dilutions plotted against the actual dilution. In this example, the results increase as the dilution increases (due to matrix interference of some type) until it levels out. A consensus of results is observed once the interference effects have been sufficiently diluted out. In this chart, the dilutions from 1/8, 1/16, and 1/32 appear to have good consensus. This effectively indicates that an MRD of 1/8 is potentially needed (2). The second chart shows the same data calculated as % recovery using the neat sample result as the 100% target value. The red dashed lines are the acceptance limits for this particular case, and clearly, the results fall out with those, due to the matrix interference in the inadequately diluted samples (3). The next step to prove that the variance from 1/8 to 1/32 results is acceptable is to recalculate the % recovery but now using the 1/8 dilution results as the 100% target. Here, the variance of higher dilutions up to 1/32 meets the acceptance criteria and proves that the MRD is 1/8. It also shows that diluting samples up to 1/32 would achieve acceptable results. Parallelism is therefore shown between the 1/8 and the 1/32 dilutions. This example shows a single analyte across six individual matrix samples. It may be more appropriate to use a larger number of samples when conducting this test, although there is often difficulty in obtaining matrix with significant concentrations of the biomarker of interest to allow multiple dilutions to be assessed that result in levels within the analytical range. It is recommended that this experiment is conducted for every analyte in the multiplex panel. It is usually easier to assess data results in this way on an analyte-by-analyte basis rather than by presenting multiple analytes together from a single sample
Challenges with Cross-Reactivity (Specificity)
Cross-reactivity occurs when the capture or detection reagents in a LBA recognize similar epitopes on other analytes present in the sample. Epitopes that are located in conserved regions of related proteins, regions with similar secondary or tertiary structure, or similar amino acid sequences are often problematic. A multiplex assay creates an environment that is more susceptible to cross-reactivity issues than single-plexed assays. In the case of using commercial kits, manufacturers typically test for cross-reactivity in their multiplex assays. Due to the complexity and expense of reproducing this work, researchers should investigate the possibility of obtaining authenticated copies of raw data from the manufacturer, describing the results of their cross-reactivity experiments. It can be challenging to obtain this information from vendors. If it is obtained and depending upon the level of confidence in the data supplied, it may be possible to accept that information without having to repeat the tests separately. A second option is to compare multiplex to well-characterized single-plexed assays (10–12). Although the reagents provided in a single-plexed assay may or may not generate comparable results to the multiplex assay due to differences in reagents, this exercise could provide valuable information for assay performance. In the case where results are not comparable, a third approach to testing reagent cross-reactivity within a multiplexed assay is the “missing man” technique where all analytes except one are added to the assay. Changes in the performance of the multiplex assays (positive or negative) would suggest that cross-reactivity is occurring with the reagents being tested. Likewise, the assay for the omitted analyte should generate a signal at background level. An example of this is shown below (which may vary depending on the platform), whereby a series of plates are prepared, varying capture antibody target (C), analyte (A), and detection antibody (D), for a 10-plex:
(C, A, D) = (+10, +10, +10)
(+10, +9 , +10) = target cross-reactivity
(+10, +10, +9) = detection cross-reactivity
(+9, +10, +10) = capture cross-reactivity
As multiplex assays are far susceptible to cross-reactivity due to the complexity of multiple capture and detection reagents in a single format, we recommend that the scientist evaluate cross-reactivity if needed depending on the quality of the data available from the vendor.
Challenges of Cross-Talk
Cross-talk is different from cross-reactivity. Cross-reactivity deals specifically with chemical interference between antibody pairs and their analytes. Cross-talk in multiplex assays is any case in which a signal from one analyte in isolation creates an unwanted effect on another. Cross-talk is sometimes described as well-to-well or spot-to-spot “carryover,” “bleed-over,” or “leaching,” event which compromises the quantitation of each analyte. Multiplex ligand-binding assays that rely on “spots” within a well or solid surface use spot location to distinguish each individual analyte. It is generally typical that the vendor will provide the data supporting the lack of cross-talk. It is recommended that the feasibility test of cross-talk should be evaluated regardless of data availability from the vendor (13). One way to test for cross-talk is to vary the concentration of one analyte over the full dynamic range of the assay while keeping the other analytes in the multiplex at a low constant concentration. Blank samples should also be included in this test, which will reveal increased background signals caused by cross-talk. If preliminary experiments fail to confirm manufacturer’s claims, scientists are encouraged to assess alternative kits.
Selectivity Challenges
The ability of an assay to recognize only the analyte of interest in the presence of sample matrix is referred to as selectivity. Selectivity issues are amplified in multiplex assays due to the increased number of reagents and analytes being measured. Examples of interfering molecules that contribute to selectivity issues include soluble receptors, rheumatoid factor, and heterophilic antibodies (14). Similar to single-plexed assays, selectivity in a multiplex assay is typically tested by assessing recovery of the analyte from spiked samples containing the interference factor to be tested. Occasionally, selectivity issues may be present for some analytes but not others. Reproducibility of samples from one experiment to another may also be impacted. During study-specific feasibility experiments, it is recommended that a few individual samples, preferably target disease-state samples and the target matrix pool, are analyzed twice to evaluate whether the mean values for each experiments are within ≤30%. This exercise will help determine early on in the evaluation of a multiplex kit which analytes are likely to pass validation. In addition, reproducibility results will aid in establishing the MRD and will aid in setting up the parallelism experiments in validation. Solutions for selectivity include increasing the sample dilution or the addition of blocking agents, detergents, or heterophilic antibody blockers. This is a key challenge for multiplex assays, as changes in buffers needed for one analyte may negatively impact one or more of the other analytes in the panel. The scientist should evaluate whether selectivity issues are critical enough to address and then determine if the affected assay should be run as a single-plexed assay or if it is worthwhile to spend time and effort finding a solution that is amenable for the entire multiplex panel.
Challenges with Lot-to-Lot Variability
One of the most critical aspects of using commercial assays is lot-to-lot reproducibility (9). One strategy that is commonly used to overcome this limitation is the securing of a large number of kits prior to the study. This strategy can help overcome difficulties such as the halting of assay production in the middle of your study and preventing additional data variability that can be associated with lot changes. A second part of this strategy is to perform analysis on the samples in batches to minimize assay variability over time or with multiple kit lots. Finally, sparing a few kits from old lots enables the laboratory to bridge the old lot to new lots. More details on kit bridging are discussed later in this paper.
RECOMMENDED ADJUSTMENTS TO FIT-FOR-PURPOSE VALIDATION PRACTICES FOR MULTIPLEX KIT ASSAYS
There are several publications that describe how to perform a fit-for-purpose validation of commercial single-plexed LBAs (15,16). There are also several recent papers that have performed fit-for purpose validation of commercial multiplex kits (17–20). Many of the recommendations for single-plexed assays also apply for performing a fit-for-purpose validation of multiplex assays. The reader is encouraged to review these key publications for performing a fit-for-purpose validation on those aspects that are not covered here.
Biomarker Work Plan
A biomarker work plan (BWP) is a formal written document that establishes the study objectives for a bioanalytical project and provides general expectations for method performance (9). The BWP also defines the rigor of validation work necessary and addresses other considerations necessary for a successful outcome. While not a regulatory requirement, a BWP is good business practice, particularly for multiplex methodologies where method feasibility experiments, validation, and sample testing are more complex than single-plexed assays. A flexible strategy often helps overcome many issues generally observed during multiplex validation. Similar to what has been described in earlier publications for single-plexed assay validation (1,2), depending on the utility of the data from the multiplex method (and for each analyte in the multiplex), the scientist may choose to perform prevalidation experiments as recommended in this paper only or carry out a fit-for-purpose level of validation where the robustness is also assessed. An understanding of the importance (including the intended purpose) of each biomarker included in the panel prior to implementing validation experiments will help generate appropriate target acceptance criteria. Common target acceptance criteria for prevalidation and validation experiments are summarized in Table III.
Table III.
Parameters for Evaluation During Prevalidation and Validation Experiments, Including Target Acceptance Criteria
| Parameter to be measured | Number of plates for prevalidation | Number of plates for validation | Recommended target values (within quantitative range) |
|---|---|---|---|
| Standard curve accuracy and precision | Two plates | Six to ten plates |
Mean % CV of back-calculated concentrations ≤20% (inter-assay) Mean %RE of back-calculated concentrations ≤20%. Note that precision during validation is tested using validation samples prepared at three to five levels. |
| Validation samples | N/A | Three to five levels (LLOQ, low, mid, high, ULOQ) in chosen matrix | Test in duplicate over 3–5 days with multiple scientists. Results for each analyte should be accurate (%RE ≤30%) and precise (% CV ≤30%). |
| Limit of quantitation | N/A | N/A | Calculated using multiple runs from validation for each analyte |
| Parallelism or endogenous dilutional linearity (if endogenous levels are high enough) | Five individuals, disease-state individuals if available | Ten individuals, disease-state individuals if possible | Results for each dilution when recalculated for the dilution factor should be 100% original (neat) result ± (inter-assay CV% ×3). This is equivalent to accepting ±3SD of the expected result (approximate confidence limit 99.5%). This means that an acceptance limit is statistically significant to the specific method’s performance. |
| Matrix spike recovery | Five individual samples, unspiked and spiked at high QC and low QC levels, using disease-state individuals if available. | Ten individual samples, unspiked and spiked at high QC and low QC levels, using disease-state individuals if available. |
For both low and high spike: 3/5 during prevalidation and 7/10 during validation yield 70–130% of expected concentrations (sum of endogenous level plus spike level); % Recovery = Measured Concentration / Expected Concentration × 100%. |
| Dilutional linearity of high-spiked recombinant protein | Five individual disease-state samples (if available), spiking analyte at high QC levels, and diluted through assay range. | Five individual disease-state samples (if possible), spiking analyte at high QC levels, and diluted through assay range. | Results for each dilution when recalculated for the dilution factor should be within ±3SD of the original (neat) result. |
| Stability | N/A | If possible, three levels of endogenous samples. | ≥24 h at room temperature, ≥5 cycles of freeze/thaw. The mean values for stressed matrix should be within ±3SD of the mean values for unstressed matrix at each level. |
Quality controls are used during sample testing. It is recommended to test three levels chosen based on prevalidation experiments spanning the standard curve. It is expected that results for each analyte should be accurate (%RE ≤30%), and precise (% CV ≤30%) following general 4-6-30 rule; however, criteria may be flexible for certain analytes depending on the intended use, biological variability, etc.
N/A not applicable, % CV percent coefficient variation, %RE percent relative error, LLOQ lower limit of quantitation, ULOQ upper limit of quantitation
Number of Analytes in a Panel
Considerations for the number of total analytes in a multiplex panel include the intended use of the data and the required rigor of the validation acceptance criteria. In some cases, large multiplex panels with lower level of performance may be acceptable for discovery-type exploratory uses where the scientist is often only looking for patterns or relative differences between the analytes. Some of the analytes in a panel may not need as high a level of validation as other analytes in the panel, which should be stated in the validation plan. For example, higher analytical variability may be acceptable for certain analytes in a panel even in a clinical trial setting such as for pharmacodynamic (PD) biomarkers, if the planned study size provides adequate power to detect the effect/response of such analytes. However, if all analytes in the multiplex assay are to be used to support critical decisions, a high level of validation for all the analytes would be recommended. If there are cases where time is a critical factor, a smaller number of analytes in the panel is more practical, to balance between the quality of the data and the other benefits of multiplexing, such as savings on matrix volume, time, and cost. QC data from smaller panels are easier to interpret and are therefore enabling better decision making. In addition, the chances of failing the assay run acceptance criteria and having to retest samples increase considerably with the addition of more analytes in the panel.
Precision and Accuracy
In a multiplexing environment, the accuracy and precision of each assay will likely vary from the same assay run in single-plexed format. The user is encouraged to maintain a level of flexibility, based on the intended use, in setting up target prestudy and in-study validation acceptance criteria for each analyte that is part of a multiplex panel. Thus, this section focuses on the aspects of assay validation that are unique to multiplex assays. A set of validation samples (VS) for the analyte of interest at levels covering the target range of study samples are used to assess accuracy and precision. These VS may be used as the QCs when monitoring assay performance during sample analysis after validation is successfully completed. It is noted that some companies prefer to generate a separate set of QCs at different concentrations, within the range of quantitation. It is recommended that the decision is made based on quantity of samples and length of study, so that an uninterrupted supply of QCs is available to last through the study.
As with single-plexed assays, to monitor assay performance and precision in multiplex assay, endogenous QCs are recommended for as many analytes as possible. If an endogenous QC is identified early during development studies, it should be included in validation as it can provide greater confidence of assay performance during sample testing over time; however, unless there is an orthogonal method for determining concentration, the endogenous QC cannot be used to assess accuracy but only precision. However, in order to work with spiked QCs, the nominal value may need to be corrected for the endogenous concentration. In addition, if the manufacturers provide QCs to use with commercial kit assays, it can be most convenient source to prepare VS as well as in study QCs. It is recommended that QCs are as representative of “in-study” samples as possible and that they are produced in-house so that they help determine whether the kits are performing as expected by the manufacturer and to help bridge different lots of kits as necessary. This also helps to establish lab and method-specific analyte levels, as well as statistically relevant acceptable ranges. For some kits, in cases where the sample matrix has high endogenous levels of some of the analytes, the VS or dilutional linearity samples used to assess precision and accuracy may need to be made using a surrogate matrix. For in-study run acceptance criteria, the 4-6-30 rule may be acceptable for LBA quantitative methods. Conversely, we recommend that the acceptance criteria should be statistically aligned to method performance and be based on clinical understanding of the analyte, intended use, biological variability of patient population, and expected physiological changes.
Limits of Quantitation
Determining the limits of quantitation for multiplex assays is essentially the same process as used in single-plexed assays. Basically, each analyte should be prepared in target matrix at a range of concentrations. The accuracy and precision of each concentration should be calculated from the analysis of validation controls from multiple assay runs, and the lowest concentration that retains accuracy within ±30% and precision of arbitrary 30% CV is referred to as the lower limit of quantitation (LLOQ). However, the main issue with some analytes is the presence of endogenous analyte in the sample matrix. In these cases, either the endogenous level can be determined or a surrogate matrix may be used to determine the LLOQ. The highest concentration of the analyte that retains accuracy within ±30% and precision within arbitrary 30% is the ULOQ. Assessment of the ULOQ is typically easier to achieve given that sample matrix may be spiked with purified, recombinant analyte; thus, surrogate matrix is often not required, although it can be used with the QCs. It is important to understand that the LLOQ and ULOQ are measured in the presence of all the other assay reagents and analytes in the multiplex assay. The estimated LLOQ and ULOQ define the range of quantitation for each analyte in the multiplex assay.
Dilutional Linearity and Parallelism
Dilutional linearity should be assessed with at least five to ten samples that contain high levels of recombinant analyte. In the case of multiplex assays, this may require multiple sets of samples to ensure that all the analytes are evaluated. It may be possible to create a single set of dilution test samples. This could be done by spiking the samples with a purified, recombinant source of the analyte. Once a set(s) of dilutional linearity samples is obtained, the assessment of dilutional linearity is done in the same manner as for single-plexed assays except that information is generated on multiple analytes in lieu of one. Results for each dilution when recalculated for the dilution factor should be 100% original (neat) result ± 3SD (based on the inter-assay precision of each assay). The range of sample dilutions that meets these criteria dictates the acceptable dilution range of the assay and may be limited by analyte concentration in the samples tested. Equally, if there are consistent dilution-adjusted results that differ from the neat result, it is probable that the lowest dilution of the consistent results is defining the MRD required in the assay to overcome matrix interferences (Fig. 2).
The parallelism test determines whether the recombinant protein is appropriate for the measurement of the endogenous analyte. It is recommended that the link between reproducibility and parallelism is evaluated prevalidation as described earlier for each analyte of multiplex panel. This experiment cannot be done until the assay has been shown to have reliable performance with the kit standard. Parallelism is also assessed for as many analytes as possible using incurred individual subject samples that have high levels of the endogenous analytes or, if not available, commercial individual samples. The reproducibility experiments that are conducted during prevalidation indicate which analytes are likely to meet validation parallelism acceptance criteria and, therefore, which analytes will need to be taken out of the panel. Establishing parallelism for a multiplex assay follows the same guidelines as a single analyte assay. Assessing parallelism also assists in determining the minimum, and possibly maximum, sample dilution factor that can be used, as shown in Fig. 3 and as described above and in Fig. 2. If parallelism experiments can be completed with biological matrix containing sufficient endogenous concentrations of the analytes of interest, then these experiments prove the recovery of the true endogenous molecule. In circumstances where parallelism experiments are successful, spiked recovery and dilutional linearity experiments using exogenous molecules would add no value to the method performance details in relation to its ability to reproducibly quantify the endogenous biomarker.
Fig. 3.
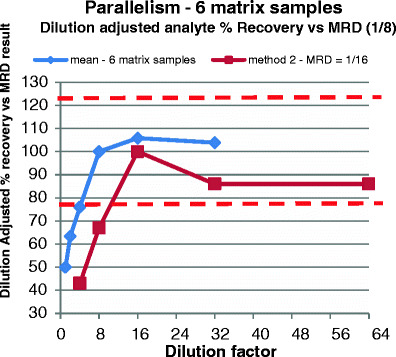
Determining parallelism when sufficient endogenous marker is present in matrix. Red dashed line indicates assay acceptance limits (≤23%—equivalent to ±3× inter-assay CV% for these particular methods). This is a parallelism experiment covering two different analytes. The lines represent mean % recovery of six different matrix samples. It demonstrates that there is a matrix interference effect when using a dilution of less than 1/8 (analyte #1—blue) or 1/16 (analyte #2—brown). Consensus is achieved over the dilution range of 1/8 to 1/32 and 1/16 to 1/64, respectively. Hence, different analytes may demonstrate different MRDs, and so for multiplexed assays, the analytical ranges and sensitivities of all the analytes combined may be compromised due to one or more analytes that require different MRDs. In this example, a dilution of 1/16 minimum must be used to capture valid results from both analytes. Larger multiplexes may give more differences, but overall, the largest MRD required will need to be used unless certain analytes are withdrawn from the panel. This could be because the resulting analytical range or sensitivity limits of the analytes are unsatisfactory due to the particular requirements of the project
Selectivity (Matrix Spike and Recovery)
Selectivity is the ability of a reagent or antibody to unequivocally recognize only the analyte of interest, typically recombinant, even in the presence of other components present in the matrix. For LBA multiplexing, it is important to note that the selected matrix or surrogate matrix may not be optimal for good recovery for all analytes, and therefore, the accurate measurement of each analyte spike may be compromised. For these experiments, ten individual samples, either normal or (and preferably) disease-state samples, are spiked with recombinant protein at high and low levels as predetermined for each analyte during prevalidation and are tested at the minimum assay dilution. It is recommended that 7/10 samples yield 70–130% of expected concentrations (sum of endogenous level plus spike level) calculated based on the formula: % Recovery = Measured Concentration / Expected Concentration × 100%. An alternative approach will be to use a subtraction method using the formula: Measured conc. minus endogenous level / Spiked level × 100%. This subtraction method has been supported by several publications (21–23). If an analyte fails to meet acceptance criteria, it should be removed from multiplex and tested as single assay. It should be noted here that if precision, parallelism, and dilutional linearity results prove to be acceptable, recovery is a less important component for relative quantitative assays; hence, the scientist is encouraged to make the decision regarding testing this parameter based on the risk level and considering the intended use of the multiplex assay. The spike recovery of a recombinant protein in different individual lots tests the effectiveness of the MRD to address non-specific interference as much as the potential for lack of reagent specificity due to cross-reactivity. In cases where literature or historical data point that the assay will not be measuring very high level samples or if there are sufficient endogenous levels available for each analyte, additional parallelism assessments would be more important to demonstrate the ability to quantify endogenous analyte—this is the intended purpose of the assay and would provide the needed confidence in the assay. While not a multiplex-specific issue, this is important to address in any biomarker relative quantitative assay.
Stability
The assessment of stability is the same for multiplex assays as it is for single-plexed assays. However, the challenge with multiplex samples is determining the conditions that are amenable for all the analytes. Suggested experiments are included in Table III. It is recommended and critical that freeze-thaw, bench-top, and long-term stability studies are initiated with endogenous analytes in target matrix, since endogenous analyte may behave differently from spiked recombinant analyte. Stability will be limited by the least stable analyte. In cases where one analyte is highly unstable than others in the multiplex, thus causing sample-handling issues for all the other analytes, the analyte may need to be removed from the multiplex panel. Stability assessments with recombinant material would be the same for multiplex as it is for single-plexed assays.
Solving Challenges with Lot-to-Lot Differences
A key consideration early in the selection of a multiplex kit is the availability of a large lot that is sufficient to avoid lot changes during the course of the study. This includes sufficient kits for prevalidation experiments, in-study validation, and sample analysis. Kit manufacturers usually have their own processes to monitor and control the kit manufacturing processes to limit variation between lots. However, when kits are used in longitudinal studies, variations in the performance of kits across multiple lots remain a genuine concern. This is especially true for multiplex assays given the complexity involved (analytes, capture and detection reagents, standards, etc.). The types of issues causing lot-to-lot variations from a single vendor are often related to critical reagents for one or more analytes, e.g., the quality of recombinant proteins in the kits, the coupling procedures for capture or detection reagents, and a general lack of information on critical reagent identity and characterization. A further concern is the unannounced changes in critical kit components, overall poor kit quality, and expiration dates on kits or individual component stability associated with kit lots during the study. Multiplex assays are sometimes manufactured by mixing some of the reagents from multiple single-plexed assays together, which makes lot-to-lot consistency difficult to control. Thus, for multiplex assays, an early assessment of the number of kits required to cover validation and sample analysis is highly recommended. If the intended use of the assay is exploratory, it may be sufficient to justify extending the expiration date based on an assessment of assay performance using precision and accuracy. For more definitive assays, evaluation of expiration dates should be more rigorous, and the methods discussed for assessing lot-to-lot variability may be applicable. In unforeseen circumstances when lots are changed, the bridging can be fulfilled using mainly three statistical approaches:
-
Show equivalence using ratios and limit of agreement
Differences in sample results from an old to a new lot can be assessed using the approach recommended for incurred sample reanalysis (24) and PK method cross-validation (25). We refer the readers to these papers for details and only briefly state the approach here. We recommend testing with both lots, side-by-side on the same day by the same scientist; 20–50 commercially purchased individual subject samples, with results that span the target range of study samples. Incurred clinical samples may be used with appropriate informed consent. For each sample, the ratio of new lot concentration to old lot concentration is calculated; from these, the mean ratio (MR), the 90% confidence interval of the mean ratio (MRL), and 67% limits of agreement (LA) are determined. The new lot is considered as similar to the old lot if MRL is within 0.8 to 1.25 and if LA is within 0.7 to 1.43. Even if the MRL is within 0.8 to 1.25 but does not contain the value 1, the bias between the old and new lots is considered as statistically significant. In this case, a correction factor based on the value of MR can be applied to the results from the new lot.
The importance of the use of a correction factor when lot-dependent performance changes are observed has also been discussed elsewhere, and the reader is referred to these papers (26,27). The use of a correction factor may be considered to compensate for content and proportional errors if experimental data have been generated and are available to support. This exercise will require careful and proper documentation during clinical sample testing. It is recommended that standard curves and controls be evaluated when results from multiple studies need to be linked, when the assay is transferred from one CRO to another, and when reagents from one kit to the next need to be bridged. As the PD data may be used for decision making, it is highly recommended that users of the biomarker data in the clinical team be kept informed of any performance variability. Due to the complexity of the multiplex assays, basing similarity on accuracy and precision performance may not be adequate. Application of a correction factor should be a risk-based approach: for example, the critical biomarker would require sufficiently accurate and precise data to determine dose-effective relationship while others may be additional confirmation of the biological effect. The scientist should remain aware that the application of a correction factor could compromise the data for critical biomarkers for decision making, while making corrections for non-critical exploratory markers could provide the additional information needed for path forward.
-
Correction factor using slope coefficient
An alternate statistical approach for determining a correction factor is to fit linear regressions for the QC responses versus nominal concentrations for both the old and new lots and determine if the two lines are parallel and super-imposable (28). If these conditions hold, the results for the two lots are considered similar and no correction factor is needed. If the slopes are similar, but there is a significant difference in intercepts, the ratio of responses at each QC concentration can be calculated and averaged across QC level, to provide a correction factor.
Using incurred or purchased samples that cover the target range (n = 25–50), predicted concentrations for samples assayed with the new lot (y-axis) regressed on the concentrations from the old lot (x-axis) should show strong agreement with the “identity line” (old lot = new lot; slope = 1 and intercept = 0) if there are no lot differences. If the 95% confidence interval for the slope includes “1” and the 95% confidence interval for the intercept includes “0,” then there is strong agreement between lot results (results fall close to the identity line) and no correction is needed. If the intercept is not significantly different from 0 but the slope is significantly different than 1, the slope coefficient is the correction factor. The slope multiplied by the new-lot-predicted concentration provides the appropriate adjustment.
-
Correction factor using regression equation based on predicted values
Another approach is to prepare two aliquots of each sample to be assayed in two different experiments. In the first experiment, the samples are assayed with both lots, and the concentrations for the samples assayed with the new lot are regressed against those from the old lot. Using the regression equation, new predicted values for these are generated from the regression equation. The second set of samples is then assayed with the new lot, and the actual concentrations obtained for these samples are compared to the values predicted from the regression calculations. The acceptance of the two assay lots would be based on achieving results for the second set of data that were within the expected analytical performance of the original method. Interested scientists are encouraged to read Clinical Laboratory Improvement Amendments (CLIA) guidelines (29) to understand the basics of using correction factor.
In conclusion, various statistical tools and experimental approaches are available in the field to compare two lots of kits. Based on the limited experience available to date with multiplex assays, we recommend a two-tiered approach to allow for a full assessment. Lots can be compared using option 1 based on the ratio calculation since this can be achieved using an Excel spreadsheet. If it is determined that the lots are equivalent using option 1, no correction factor is needed. If, however, the two lots are determined to be different, option 2 provides a better description of lots differences. Parallel slopes with a shift of intercepts suggest that a correction factor can be applied as described for option 2. Unparallel slopes indicate that a correction factor cannot be applied since lot differences change with changes in concentration. The approaches taken must be applied to each of the analytes in the multiplex assay. It should be noted that in multiplex assays, often the scientists face the situation where correction is required for some analytes and not for the others. A scientific decision should govern the path forward, e.g., either excluding analytes completely or continue using analytes without correction factor. Correction factors can be applied to individual analytes as needed.
Considerations for Transitioning Multiplex Assays into Diagnostic Assays
Diagnostic assays are performed in CLIA-certified laboratories to assist health care providers in identifying disease, health risk factors, and guiding medical treatment. Homebrew or research-use-only bioanalytical methods may evolve to laboratory developed tests—performed as diagnostics assays in a single laboratory under the authority of Center for Medicare and Medicaid Services (30,31) and eventually to FDA-approved in vitro diagnostic assays (IVDs). Companion diagnostics are IVDs that provide information that is essential for the safe and effective use of a corresponding therapeutic. There is an increased use of companion diagnostic assays to improve the design of clinical trials and the efficiency of patient selection for specific therapeutic interventions. The advantages of incorporating a multiplexed companion diagnostic assay into the development of a pharmaceutical program are similar to those previously discussed—including the ability to simultaneously screen multiple analytes from a small sample size. Commercially available diagnostic kits are available for indications such as autoimmune, oncology, and infectious disease.
Companion diagnostics must formally be approved by the FDA (CDRH) as in vitro diagnostics as category 3 high complexity devices (42 CFR 493.17). As such, companion diagnostics must undergo both an analytical and clinical validation (21 CFR 809.3). On August 2014, FDA released the final Guidance on In Vitro Companion Diagnostic Devices (32). Transforming a LBA into a companion diagnostic assay requires a tremendous amount of work and knowledge of diagnostic assays. It is out of the scope of this paper to describe the detailed parameters for LDTs and other diagnostic assays. Some key areas that need to be addressed for an assay to be converted and marketed as a companion diagnostic include ensuring that the proper diagnostic rights on all key reagents have been obtained, approval of the instrumentation and software, thorough review of the relevant patents, and an evaluation of already approved assays. Multiplex assays must meet all the requirements just as single-plexed assays. However, one of the biggest challenges would be the GMP manufacturing of the multiplex assay, which would have to meet rigorous testing and ruggedness criteria. The manufacturing of the reagents, calibration standards, and reference standards would also have to be defined and characterized. An additional challenge with multiplex assays would be to define the contribution of each aspect of the multiplex panel, either individually or in combination. All these factors taken together will also impact “intended use”, which is the basis of the approval of the multiplex assay. Thus, the transition of multiplex assays to a diagnostic assay would require a significant amount of investment in both time and money. The recommendation would be to form a close relationship with a partner that has the needed expertise to transition the assay into an approved diagnostic assay.
CONCLUSIONS
Multiplex assays are very powerful analytical tools that are becoming more widely used in the drug development process. There are many unique challenges associated with multiplex biomarker assays that are different than those encountered with single-plexed assays. We have identified these unique challenges and provided recommendations to overcome them. The challenges include the selection, characterization, and validation of multiplex assays within the context of fit-for-purpose biomarker assay development. Specifically unique are the challenges with different detection ranges, more complex specificity issues and matrix interference, cross-talk, and cross-reactivity between reagents. The guidelines that we have provided mainly apply to assays that are for research uses only (RUO). It is up to the discretion of the scientist to choose the appropriate level of characterization depending on the intended use of the multiplex. In these recommendations, we have specified acceptance criteria for assessing assay parameters during validation and solutions to challenges associated with lot-to-lot variability with multiplex kits, in particular a statistical approach to applying a correction factor between lots if necessary. We also have highlighted how to handle data when one or more of the analytes fail during validation or during in-study sample analysis.
Acknowledgments
Authors thank FDA members for their critical review of the manuscript. We also thank the members of The Ligand Binding Assay Bioanalytical Focus Group (LBAFG) of the BIOTEC Section, American Association of Pharmaceutical Scientists (AAPS) organization, for critical review of the manuscript.
Disclaimer
The contents of this article reflect the personal opinions of the authors and may not represent the official perspectives of their affiliated organizations.
Conflict of Interest
Andreas Jeromin is an advisor to Quanterix Corporation and owns stock options.
Footnotes
This manuscript is the result of a collaborative effort from the AAPS Biomarker Discussion group, part of the Ligand Binding Assay Bioanalytical Focus Group.
References
- 1.Lee JW, Weiner RS, Sailstad JM, Bowsher RR, Knuth DW, O’Brien PJ, et al. Method validation and measurement of biomarkers in nonclinical and clinical samples in drug development: a conference report. Pharm Res. 2005;22(4):499–511. doi: 10.1007/s11095-005-2495-9. [DOI] [PubMed] [Google Scholar]
- 2.Lee JW, Devanarayan V, Barrett YC, Weiner R, Allinson J, Fountain S, et al. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm Res. 2006;23:312–328. doi: 10.1007/s11095-005-9045-3. [DOI] [PubMed] [Google Scholar]
- 3.Chau C, Rixie O, McLeod H and W Figg, Validation of analytic method for biomarkers used in drug development, Clinical Cancer Research, 2008 14(19). [DOI] [PMC free article] [PubMed]
- 4.Draft guidance for industry: bioanalytical method validation. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM), September 2013.
- 5.Tighe P, Negm O, Todd I, Fairclough L. Utility, reliability and reproducibility of immunoassay multiplex kits. Methods. 2013;61:23–29. doi: 10.1016/j.ymeth.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Ling MM, Ricks C, Lea P. Multiplexing molecular diagnostics and immunoassays using emerging microarray technologies. Expert Rev Mol Diagn. 2007;7(1):87–98. doi: 10.1586/14737159.7.1.87. [DOI] [PubMed] [Google Scholar]
- 7.Hsu HY, Joos To and Koga H. Multiplex microsphere-based flow cytometric platforms for protein analysis and their application in clinical proteomics—from assays to results. Electrophoresis 30(23) 4008-19. [DOI] [PubMed]
- 8.Marquette CA, Corgier BP, Blum LJ. Recent advances in multiplex immunoassays. Bioanalysis. 2012;4(8):927–936. doi: 10.4155/bio.12.56. [DOI] [PubMed] [Google Scholar]
- 9.Khan M, Bowsher RR, Cameron M, Devanarayan V, Keller S, King L, et al. Application of commercial kits for quantitative biomarker determinations in drug development: recommendations for method adaptation and validation. Bioanalysis. 2014;7(2):229–242. doi: 10.4155/bio.14.274. [DOI] [PubMed] [Google Scholar]
- 10.Salvante KG, Brindle E, McConnell D, O’Connor K, Nepomnaschy PA. Validation of a new multiplex assay against individual immunoassays for the quantification of reproductive, stress, and energetic metabolism biomarkers in urine specimens. Am J Hum Biol. 2012;24(1):81–86. doi: 10.1002/ajhb.21229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bastarache JA, et al. Validation of a multiplex electrochemiluminescent immunoassay platform in human and mouse samples. J Immunol Methods. 2014;408:13–23. doi: 10.1016/j.jim.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Backen The combination of circulating Ang1 and Tie2 levels predicts progression-free survival advantage in bevacizumab-treated patients with ovarian cancer. Clin Cancer Res. 2014;20(17):4549–58. doi: 10.1158/1078-0432.CCR-13-3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marchese RD, Puchalski D, Miller P, Antonello J, Hammond O, Green T, et al. Optimization and validation of a multiplex, electrochemiluminescence-based detection assay for the quantitation of immunoglobulin G serotype-specific antipneumococcal antibodies in human serum. Clin Vaccine Immunol. 2009;16(3):387–396. doi: 10.1128/CVI.00415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Todd DJ, Knowlton N, Amato M, Frank MB, Schur PH, Izmailova ES, et al. Erroneous augmentation of multiplex assay measurements in patients with rheumatoid arthritis due to heterophilic binding by serum rheumatoid factor. Arthritis Rheum. 2011;63(4):894–903. doi: 10.1002/art.30213. [DOI] [PubMed] [Google Scholar]
- 15.Fraser S, Soderstrom C. Due diligence in the characterization of matrix effects in a total IL-13 Singulex™ method. Bioanalysis. 2014;6(8):1123–1129. doi: 10.4155/bio.14.42. [DOI] [PubMed] [Google Scholar]
- 16.Bowsher RR, Sailstad JM. Insights in the application of research-grade diagnostic kits for biomarker assessments in support of clinical drug development: bioanalysis of circulating concentrations of soluble receptor activator of nuclear factor ligand. J Pharm Biomed Anal. 2008;48:1282–1289. doi: 10.1016/j.jpba.2008.09.026. [DOI] [PubMed] [Google Scholar]
- 17.Belabani, et al. A condensed performance-validation strategy for multiplex detection kits used in studies of human clinical samples. J Immunol Methods. 2013;387:1–10. doi: 10.1016/j.jim.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 18.Backen AC, et al. ‘Fit-for-purpose’ validation of SearchLight multiplex ELISAs of angiogenesis for clinical trial use. J Immunol Methods. 2009;342:106–114. doi: 10.1016/j.jim.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Chowdhury F, et al. Validation and comparison of two multiplex technologies, Luminex® and Mesoscale Discovery, for human cytokine profiling. J Immunol Methods. 2009;340:55–64. doi: 10.1016/j.jim.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 20.Li, et al. Validation of a multiplex immunoassay for serum angiogenic factors as biomarkers for aggressive prostate cancer. Clin Chim Acta. 2012;413:1506–1511. doi: 10.1016/j.cca.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stevenson L, Kelley M, Gorovits B, et al. Large molecule specific assay operations: recommendations for best practices and harmonization from the Global Bioanalysis Consortium Harmonization Team. AAPS J. 2014;16(1):83–88. doi: 10.1208/s12248-013-9542-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bastarache JA, Koyama T, Wickersham NE, Mitchell DB, Mernaugh RL, Ware LB. Accuracy and reproducibility of a multiplex immunoassay platform: a validation study. J Immunol Methods. 2011;367:33–39. doi: 10.1016/j.jim.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marcelletti JF, Evans CL, Saxena M, Lopez AE. Calculations for adjusting endogenous biomarker levels during analytical recovery assessments for ligand-binding assay bioanalytical method validation. AAPS J. 2015 doi: 10.1208/s12248-015-9756-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rocci ML, Devanarayan V, Haughey DB, Jardieu P. Confirmatory reanalysis of incurred bioanalytical samples, AAPS J 9(3) Article 40, 336-343 (2007). [DOI] [PMC free article] [PubMed]
- 25.Thway TM, Ma M, Lee J, Sloey B, Yu S, Wang YM, et al. Experimental and statistical approaches in method cross-validation to support pharmacokinetic decisions. J Pharm Biomed Anal. 2009;49(3):613–8. doi: 10.1016/j.jpba.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Ezzelle J, Rodriguez-Chavez IR, et al. Guidelines on good clinical laboratory practice: bridging operations between research and clinical research laboratories. J Pharm Biomed Anal. 2008;46(1):18–29. doi: 10.1016/j.jpba.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.King LE, Farley E, Imazato M, et al. Ligand binding assay critical reagents and their stability: recommendations and best practices from the Global Bioanalysis Consortium Harmonization team. AAPS J. 2014;16(3):504–15. doi: 10.1208/s12248-014-9583-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weisberg S. Applied linear regression, 3rd Ed. published by Wiley/Interscience (2005).
- 29.CLSI Guidelines: EP9A2IR: method comparison and bias estimation using patient samples: approved guideline 2nd edition: Interim Revision 2011.
- 30.FDA Draft Guidance. In vitro companion diagnostic devices draft guidance for industry, food and drug administration staff, and clinical laboratories: framework for regulatory oversight of laboratory developed tests (LDTs), (October 2014).
- 31.FDA Final Guidance. Draft guidance for industry, food and drug administration staff, and clinical laboratories: FDA notification and medical device reporting for laboratory developed tests (LDTs) (October 2014).
- 32.FDA Final Guidance. Guidance for industry and food and drug administration staff: in vitro companion diagnostic devices (August 2014).