

Abstract
Previous studies have shown that female offspring are resistant to fetal stress-induced programming of ischemic-sensitive phenotype in the heart; however, the mechanisms responsible remain unclear. The present study tested the hypothesis that estrogen plays a role in protecting females in fetal programming of increased heart vulnerability. Pregnant rats were divided into normoxic and hypoxic (10.5% O2 from Day 15 to 21 of gestation) groups. Ovariectomy (OVX) and estrogen (E2) replacement were performed in 8-wk-old female offspring. Hearts of 4-mo-old females were subjected to ischemia and reperfusion injury in a Langendorff preparation. OVX significantly decreased postischemic recovery of left ventricular function and increased myocardial infarction, and no difference was observed between normoxic and hypoxic groups. The effect of OVX was rescued by E2 replacement. OVX decreased the binding of glucocorticoid receptor (GR) to glucocorticoid response elements at angiotensin II type 1 (Agtr1) and type 2 (Agtr2) receptor promoters, resulting in a decrease in Agtr1 and an increase in Agtr2 in the heart. Additionally, OVX decreased estrogen receptor (ER) expression in the heart and inhibited ER/GR interaction in binding to glucocorticoid response elements at the promoters. Consistent with the changes in Agtrs, OVX significantly decreased Prkce abundance in the heart. These OVX-induced changes were abrogated by E2 replacement. The results indicate that estrogen is not directly responsible for the sex dimorphism in fetal programming of heart ischemic vulnerability but suggest a novel mechanism of estrogen in regulating cardiac Agtr1/Agtr2 expression patterns and protecting female hearts against ischemia and reperfusion injury.
Keywords: angiotensin II receptor, estrogen, heart, ischemic injury
INTRODUCTION
Heart disease is a leading cause of death in the United States. Large epidemiological and animal studies have demonstrated that an adverse intrauterine environment leads to adaptive changes that may result in increased risk of ischemic heart disease in adulthood [1–4]. Hypoxia is a common intrauterine insult experienced by the developing fetus. Although a low oxygen tension is needed for proper heart formation and maturation during embryonic and fetal development, recent studies have shown that maternal chronic hypoxia results in promoter methylation and repression of cardioprotective genes, such as Prkce [5, 6]. This fetal stress-induced epigenetic reprogramming of gene expression patterns increased myocardial vulnerability to ischemia and reperfusion (I/R) injury in male offspring [6–9].
Of interest, female offspring were protected from the effect of prenatal hypoxia and showed no significant increase in myocardial vulnerability to I/R injury [9]. The question raised is whether postnatal ovarian hormones may be responsible for this gender-specific resistance in heart vulnerability to I/R injury observed in female offspring exposed to prenatal hypoxia. Observational studies have discovered that premenopausal women tend to be more resistant to cardiovascular disease compare to age-matched men. The risk of cardiovascular disease among women increases after the onset of menopause [10]. Although there exists controversy as to the role of estrogen replacement in cardiovascular protection in postmenopausal women, studies have shown that younger postmenopausal women given short courses of hormone replacement therapy gain added protection from cardiovascular disease [11]. This suggests that estrogens are a key factor in gender-specific cardioprotection, and their effects are dependent on duration and temporal exposure. Likewise, animal studies have shown that estrogens play a key role in maintaining vascular function and protecting against I/R injury [12–14]. Additionally, ovariectomized rats are more susceptible to injury from I/R insult but regain cardioprotection with estrogen replacement [15]. Estrogens exert their biological effects through the binding of two different estrogen receptors, estrogen receptor α (ERα) and estrogen receptor β (ERβ). ERα and ERβ are transcription factors that form dimers upon activation and regulate cell survival, growth, and metabolism through modulation of gene expression. Estrogens also activate an orphan G-protein-coupled receptor (GPR30), which is known to transiently activate signaling kinases that may play a role in cardioprotection [10, 16]. ERs may also cross talk with transcription factors, such as Sp1, which may result in direct or indirect binding to DNA for regulation of gene expression [17].
Another important factor in cardiovascular homeostasis and disease is angiotensin II (Ang II) hormonal regulation [18, 19]. Ang II acts on two main G-protein-coupled receptor subtypes, type 1 (Agtr1) and type 2 (Agtr2) Ang II receptors, which are expressed in cardiomyocytes and play a key role in myocardial response to I/R injury [20]. Previous studies have shown that acute stimulation of Agtr1 or blockade of Agtr2 in the heart protect the left ventricle (LV) from I/R injury [8, 21, 22]. The regulation of Agtr expression can be regulated by the glucocorticoid receptor (GR) when it binds to glucocorticoid response elements (GREs) within the promoter region of the Agtr promoter. Additionally, studies have shown that estrogen replacement regulates Agtr1 and Agtr2 expression in the kidney [23, 24]. However, the mechanisms underlying the estrogen-induced regulation of Agtr1 and Agtr2 expression in the heart are not well understood.
The role of postgestational estrogens in gender-specific fetal programming of cardiac disease is yet to be elucidated. Furthermore, the mechanisms of estrogens in protecting the heart from I/R injury are not fully understood. Herein, we present evidence that postnatal ovarian hormones are not responsible for the gender-specific resistance in cardiac I/R injury observed in female offspring exposed to antenatal hypoxia; however, estrogen protects the heart against cardiac I/R injury in both normoxic control and antenatal hypoxic female offspring. We further show that the cardioprotective effects of estrogen involve ER/GR cross talk at GREs located at Agtr1 and Agtr2 promoters, resulting in alterations in the Agtr1 to Agtr2 ratio in the heart.
MATERIALS AND METHODS
Experimental Animals
All the procedures and protocols were approved by the Institutional Animal Care and Use Committee of Loma Linda University and followed the guidelines by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Time-dated pregnant Sprague-Dawley rats were randomly divided into two groups: 1) normoxic control and 2) hypoxic treatment of 10.5% O2 from Days 15 to 21 of gestation as reported previously [9]. All of the dams were allowed to deliver at term, and the newborn pups were kept with their mothers until weaning. At weaning, only female offspring were kept for the present study.
Ovariectomy and Estrogen Replacement
Ovariectomy (OVX) and sham operation were performed in 8-wk-old female rats, as previously described [25–27]. Briefly, animals were anesthetized with intramuscular injection of 75 mg/kg ketamine and 5 mg/kg xylazine, and adequate anesthesia was determined by loss of the pedal withdrawal reflex. A ventral midline incision was made, the ovarian vessels were tied off, and the ovaries were removed (OVX group). The sham operation had a ventral midline incision, and the ovaries were visualized but not removed. Seven days after OVX, 17β-estradiol (E2) valerate minipellets (7.5 mg for 90-day release; Innovative Research of America) were implanted in half of animals for estrogen replacement as reported previously [25–27]. There were six groups of experimental animals: 1) normoxia-sham, 2) hypoxia-sham, 3) normoxia-OVX, 4) hypoxia-OVX, 5) normoxia-OVX+E2, and 6) hypoxia-OVX+E2. Our recent study [26] in the same animal model demonstrated that plasma estrogen levels were significantly lower in OVX rats (4 pg/ml) than those in sham rats (28 pg/ml), and E2 replacement restored plasma estrogen levels in OVX rats (32 pg/ml). In addition, plasma estrogen levels were not significantly different between antenatal hypoxic-treated and normoxic control animals [26].
Hearts Subjected to I/R
Hearts of 4-mo-old offspring were isolated and retrogradely perfused via the aorta in a modified Langendorff apparatus as previously described [9]. After the baseline recording, hearts were subjected to 20 min of global ischemia by stopping the perfusion, followed by 30 min of reperfusion in a Langendorff preparation. LV-developed pressure, heart rate, change in pressure over time (i.e., dP/dtmax, dP/dtmin), and LV end diastolic pressure were continuously recorded. At the end of reperfusion, the LV was cut into four slices, incubated with 1% 2,3,5-triphenyl tetrazolium chloride solution for 15 min at 37°C, and immersed in formalin for 30 min. Each slice was then photographed separately, and areas of myocardial infarction in each slice were analyzed by computerized planimetry (Image J; National Institutes of Health), corrected for the tissue weight, summed for each heart, and expressed as a percentage of the total LV weight. Lactate dehydrogenase (LDH) activity was measured in coronary effluent collected at 30 sec before the onset of ischemia, and at 0, 1, 2, 3, 4, 5, 10, 15, 20, and 30 min of reperfusion. LDH activity was measured using a standard assay (TOX 7 kit; Sigma), following the manufacturer's directions.
Western Blot Analysis
LVs were homogenized in a lysis buffer containing 150 mM NaCl, 50 mM Tris HCl, 10 mM ethylenediaminetetraacetic acid, 0.1% Tween-20, 1% Triton, 0.1% β-mercaptoethanol, 0.1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, and 5 μg/ml aprotinin, pH 7.4, and allowed to incubate for 1 h on ice. Homogenates were then centrifuged at 4°C for 10 min at 10 000 × g and supernatants collected. Nuclear extracts were prepared from hearts using NXTRACT CelLytic Nuclear Extraction Kit (Sigma), based on the classical method of Dignam et al. [28]. This method is robust and has been tested by numerous studies. Although there is a paucity of nuclear compartment specific and unique protein markers and many, if not all, common nuclear proteins are also copresent in the cytosol, our previous studies showed the enrichment of transcription factors, including Sp1, Egr1, and ERα in the nuclear extract preparation. Protein concentrations were measured using a protein assay kit (Bio-Rad). Samples with equal amounts of protein were loaded onto 10% polyacrylamide gel with 0.1% SDS and separated by electrophoresis at 100 V for 90 min. Proteins were then transferred onto nitrocellulose membranes. Nonspecific binding sites was blocked for 1 h at room temperature in a Tris-buffered saline solution (20 mM Tris, 500 mM sodium chloride, pH 7.5) containing 5% dry-milk. The membranes were then probed with primary antibodies against Prkce (1:500 dilution), Prkcd (1:500 dilution), Agtr1 (1:100 dilution), Agtr2 (1:200 dilution), glucocorticoid receptor (GR, 1:2000 dilution), estrogen receptor α (ERα, 1:200 dilution), and ERβ (1:200 dilution) (Santa Cruz Biotechnology). To assure equal loading, band intensities were normalized to actin. After washing, membranes were incubated with secondary horseradish peroxidase-conjugated antibodies (Calbiochem). Proteins were visualized with enhanced chemiluminescence reagents, and blots were exposed to Hyperfilm. The results were analyzed with the Kodak ID image analysis software. The bands on the Western blots were quantified as pixel numbers.
Real-Time RT-PCR
RNA was extracted from LVs using TRIzol protocol (Invitrogen). Prkce, Prkcd, Agtr1a, Agtr1b, and Agtr2 mRNA abundance was determined by real-time RT-PCR using Icycler Thermal cycler (Bio-Rad), as described previously [8]. The primers used were: Prkce, 5′-GCTCAAGGGTAAGGATGAAGT-3′ (forward) and 5′-CTGGAAGCAGCAATAGAGTTG-3′ (reverse); Prkcd, 5′-ACAGAA GAAGCCCACCAT-3′ (forward) and 5′-GAACTCAGCCTTCCCGTT-3′ (reverse); Agtr1a, 5′-GGAGAGGATTCGTGGCTTGAG-3′ (forward) and 5′-CTTTCTGGGAGGGTTGTGTGAT-3′ (reverse); Agtr1b, 5′-ATGTCTCCAGTCCCCTCTCA-3′ (forward) and 5′-TGACCTCCCATCTCCTTTTG-3′ (reverse); and Agtr2, 5′-CAATCTGGCTGTGGCTGACTT-3′ (forward) and 5′-TGCACATCACAGGTCCAAAGA-3′ (reverse). Real-time RT-PCR was performed in a final volume of 25 μl. Each PCR reaction mixture consisted of primers, iScript reverse transcriptase (Bio-Rad), and iQ SYBR Green Supermix (Bio-Rad). We used the following RT-PCR protocol: 50°C for 10 min, 95°C for 5 min, followed by 40 cycles of 95°C for 10 sec, 56°C for 30sec, and 72°C for 20sec. GAPDH was used as an internal reference, and serial dilutions of the positive control was performed on each plate to create a standard curve. PCR was performed in triplicate, and threshold cycle numbers were averaged.
Electrophoretic Mobility Shift Assay
Nuclear extracts were collected from LVs of sham control and OVX and E2 replacement groups using NXTRACT CelLytic Nuclear Extraction Kit (Sigma). The oligonucleotide probes of GREs at rat Agtr1a and Agtr2 promoter region were labeled and subjected to gel shift assays using the Biotin 3′ end labeling kit and LightShift Chemiluminescent Electrophoretic Mobility Shift Assay (EMSA) kit (Pierce Biotechnology) as previously described [29]. Briefly, single-stranded oligos were incubated with terminal deoxynucleotidyl transferase (TdT) and biotin-11-dUTP in binding mixture for 30 min at 37°C. The TdT adds a biotin-labeled dUTP to the 3′-end of the oligonucleotides. The oligos were extracted using chloroform and isoamyl alcohol to remove the enzyme and unincorporated biotin-11-dUTP. Dot blots were performed to ensure the oligos were labeled equally. Combining sense and antisense oligos exposed to 95°C for 5 min was done to anneal complementary oligos. The labeled oligonucleotides were then incubated with or without nuclear extracts in the binding buffer (from the LightShift kit). Binding reactions were performed in 20 μl containing 50 fmol oligonucleotide probes, 1× binding buffer, 1 μg of poly (dI-dC), and 5 μg of nuclear extracts. For competition studies, increasing concentrations of nonlabeled homologous and heterologous oligonucleotides were added to the binding reactions. The samples were then run on a native 5% polyacrylamide gel. The contents of the gel were then transferred to a nylon membrane (Pierce) and crosslinked to the membrane using an ultraviolet crosslinker (125 mJoules/cm2). Membranes were blocked and then visualized using the reagents provided in the LightShift kit. The bands on EMSA blots were quantified as pixel numbers.
Chromatin Immunoprecipitation
Chromatin extracts were prepared from LVs of sham control and OVX and E2 replacement groups. Chromatin immunoprecipitation (ChIP) assays were performed using the ChIP-IT kit (Active Motif) as previously described [6]. Briefly, hearts were exposed to 1% formaldehyde for 10 min to crosslink and maintain DNA/protein interactions. After the reactions were stopped with glycine, hearts were washed, chromatin isolated, and the DNA sheared into fragments (100–500 base pairs) using a sonicator. ChIP reactions were performed using GR antibody to precipitate the transcription factor/DNA complex. Crosslinking was then reversed using a salt solution and proteins digested with proteinase K. Primers flanking GRE1, GRE2, and GRE3 at the promoter of Agtr1a were used: 5′-TGGAACCAATGCTGCTTGTAA-3′ (forward) and 5′-CGAGTCCTCACTAGCAATTCA-3′(reverse); 5′-TTGCTAGTGAGGACTCGAATC-3′(forward) and 5′-GCATCGGGAGCCAGAATCA-3′ (reverse); and 5′-ATGCCATCTGTAATCCACAAC-3′ (forward) and 5′-GCCACATGAACTGACTCCAA-3′ (reverse), respectively. Primers flanking GRE4, GRE5, GRE6, GRE7, and GRE8 at the promoter of Agtr2 were used: 5′-GCAAGCAGGGTAGAGATTAAA-3′ (forward) and 5′-GACAGATTTTAAATAAATTCCC-3′(reverse); 5′-TGAGTAACTAATATCCCCATTT-3′(forward) and 5′-GCTTCACAAGCCACATCTCA-3′ (reverse); 5′-GCT GCTGCTGGCTGGTAT-3′(forward) and 5′-ACAAGTAA GGATGATTATG-3′(reverse); 5′-TTAATGTTTTGCAGCCAGAAA-3′(forward) and 5′-GGGGAGCCTTCAACCTACAT-3′(reverse); and 5′-CCAGAGGTCTGGTGCAGTTA-3′ (forward) and 5-ACTTACCTTAAAATGCAGGCT-3′(reverse), respectively. PCR amplification products were visualized on 2% agarose gel stained with ethidium bromide. To quantify PCR amplification, real-time PCR were carried out with 5 min initial denaturation followed by 45 cycles of 95°C for 10 sec, 56°C for 30 sec, and 72°C for 10 sec, using the iQ SYBR Green Supermix with iCycler real-time PCR system (Bio-Rad). To determine the cross talk of ER and GR at GREs, chromatin reimmunoprecipitation (Re-ChIP) assays were performed using the Re-ChIP-IT kit (Active Motif). Briefly, after the first ChIP reaction using GR antibody to precipitate the transcription factor/DNA complex, the second ChIP reaction was performed using ERα antibody to precipitate the transcription factor/DNA complex. Crosslinking was then reversed using a salt solution and proteins digested with proteinase K. PCR amplification products with the primers flanking GREs were visualized on 2% agarose gel stained with ethidium bromide. To ensure the proper immunoprecipitation in ChIP, normal immunoglobulin G (IgG) in lieu of specific antibodies was used in ChIP, followed by PCR amplification using primers flanking GREs. While specific amplicons were clearly visible with GR antibody, no specific amplicon was observed with normal IgG. In addition, non-GRE-site specific PCR primers were used to amplify DNA fragments pulled down with GR antibody, which showed no specific amplicon. For Re-ChIP assay, normal IgG was used as a negative control for the second ChIP reaction with ERα antibody. While the pull-down DNA fragments obtained by using ERα antibody were PCR amplified with GREs-specific PCR primers, normal IgG produced no specific amplicon in PCR.
Statistical Analysis
Data are expressed as mean ± SEM. Statistical significance (P < 0.05) was determined by analysis of variance (ANOVA).
RESULTS
Effects of OVX and E2 Replacement on Heart Susceptibility to I/R Injury
Preischemic values of LV function were not significantly different among all six groups (data not shown). In sham-operated female offspring, consistent with the previous finding [9], prenatal hypoxia did not significantly alter postischemic recovery of LV-developed pressure, LV end diastolic pressure, dP/dtmax, and dP/dtmin, nor myocardial infarct size and LDH release, as compared to the normoxic control (Fig. 1). OVX significantly decreased postischemic recovery of LV function and increased infarct size and LDH release in both normoxic control and hypoxic groups, which were abrogated by E2 replacement (Fig. 1). However, there were no significant differences in postischemic recovery of LV function and myocardial infarction between normoxic and fetal hypoxic groups in either groups of OVX or OVX with E2 replacement animals (Fig. 1).
FIG. 1.

Effect of ovariectomy (OVX) and estrogen replacement (OVX-E2) on postischemic recovery of LV function. Isolated hearts were studied in a Langendorff preparation in 4-mo-old females of normoxic control and antenatal hypoxic animals. Data are means ± SEM (n = 5). LV functional recovery data (A–C) were expressed as fold change of preischemic baseline values. Infarct size data (E) was expressed as a percentage of the total LV weight. Data of LDH release (F) were expressed as the area under curve (AUC) in coronary effluent collected from 30 sec before the onset of ischemia to 30 min of reperfusion. Data in A–D were analyzed by repeated measures two-way ANOVA with treatment as one factor and time as another, followed by Bonferroni posttests. Data in E and F were analyzed by two-way ANOVA with treatment as one factor and hypoxia as another, followed by Bonferroni posttests. *P < 0.05, OVX versus sham control or E2 replacement in both normoxic and hypoxic animals.
Effects of OVX and E2 Replacement on Agtr and Prkc Expression in the LV
OVX significantly decreased Agtr1 protein abundance but increased Agtr2 in the LV (Fig. 2A). This was associated with a down-regulation of Agtr1a mRNA and an up-regulation of Agtr2 mRNA without significant changes of Agtr1b mRNA (Fig. 2B). As shown in Figure 3, mRNA and protein abundance of Prkce, but not Prkcd, was significantly reduced in the LV in OVX as compared with sham animals. These OVX-induced changes in the heart were recovered by E2 replacement (Figs. 2 and 3).
FIG. 2.
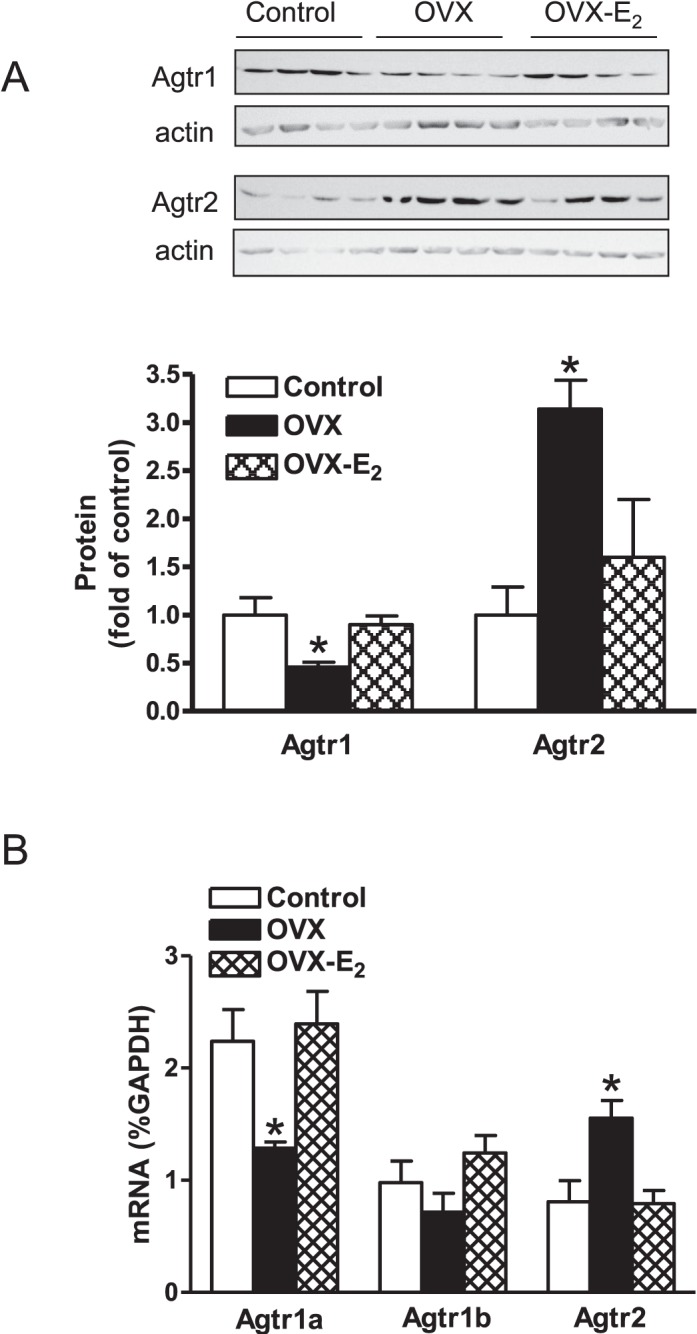
Effect of ovariectomy (OVX) and estrogen replacement (OVX-E2) on Agtr1 and Agtr2 protein and mRNA abundance in normoxic females. LVs were isolated from 4-mo-old normoxic females. A) Agtr1 and Agtr2 protein abundance was determined by Western blot analysis (n = 4). B) Agtr1a, Agtr1b, and Agtr2 mRNA abundance was determined by real-time RT-PCR (n = 5). Data are means ± SEM. Data were analyzed by one-way ANOVA followed by Neuman-Keuls posttests, within each receptor subtype. *P < 0.05, OVX versus sham control or E2 replacement.
FIG. 3.
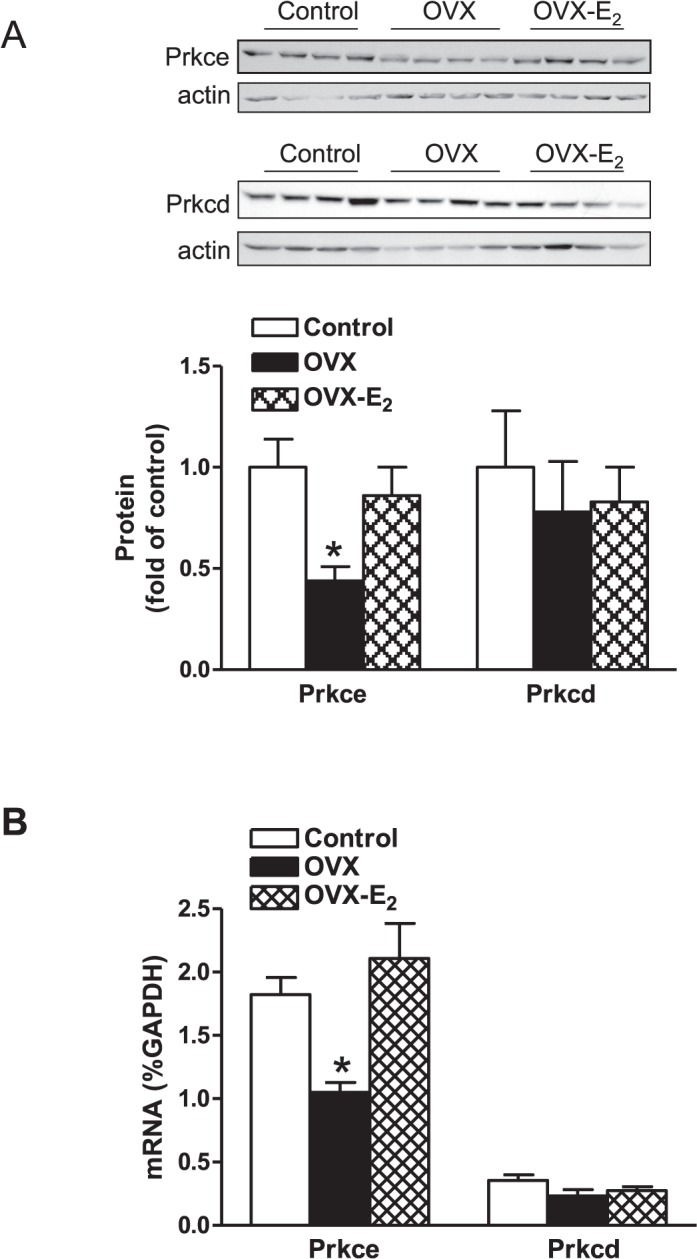
Effect of ovariectomy (OVX) and estrogen replacement (OVX-E2) on Prkce and Prkcd protein and mRNA abundance in normoxic females. LVs were isolated from 4-mo-old normoxic females. A) Protein abundance of Prkce and Prkcd, was determined by Western blot analysis (n = 4). B) Prkce and Prkcd mRNA abundance was determined by real-time RT-PCR (n = 5). Data are means ± SEM. Data were analyzed by one-way ANOVA followed by Neuman-Keuls posttests, within each isozyme. *P < 0.05, OVX versus sham control or E2 replacement.
Effects of OVX and E2 Replacement on GR Binding to GREs at Agtr1a and Agtr2 Promoters
Western blot analysis revealed that both OVX and E2 replacement had no significant effect on either total or nuclear GR protein abundance (data not shown). To determine the effect of OVX on GR binding to Agtr1a and Agtr2 promoters, competition EMSAs were performed in pooled nuclear extracts with the increasing ratio of unlabeled to labeled oligonucleotides encompassing GRE2 in the Agtr1a promoter and GRE4 in the Agtr2 promoter [8, 30]. As shown in Figure 4, OVX significantly decreased the binding affinity of nuclear extracts to both GRE sites at Agtr1a and Agtr2 promoters, which was recovered by E2 replacement. To further characterize the effect of OVX on GR binding to Agtr1a and Agtr2 promoters in vivo in the context of intact chromatin, ChIP assays were performed using a GR antibody. Figure 5 shows that OVX significantly decreased GR binding to GRE1, GRE2, and GRE3 sites at the Agtr1a promoter and GRE4, GRE5, GRE6, GRE7, and GRE8 sites at the Agtr2 promoter. Moreover, E2 replacement restored the binding of GR to the control levels (Fig. 5).
FIG. 4.
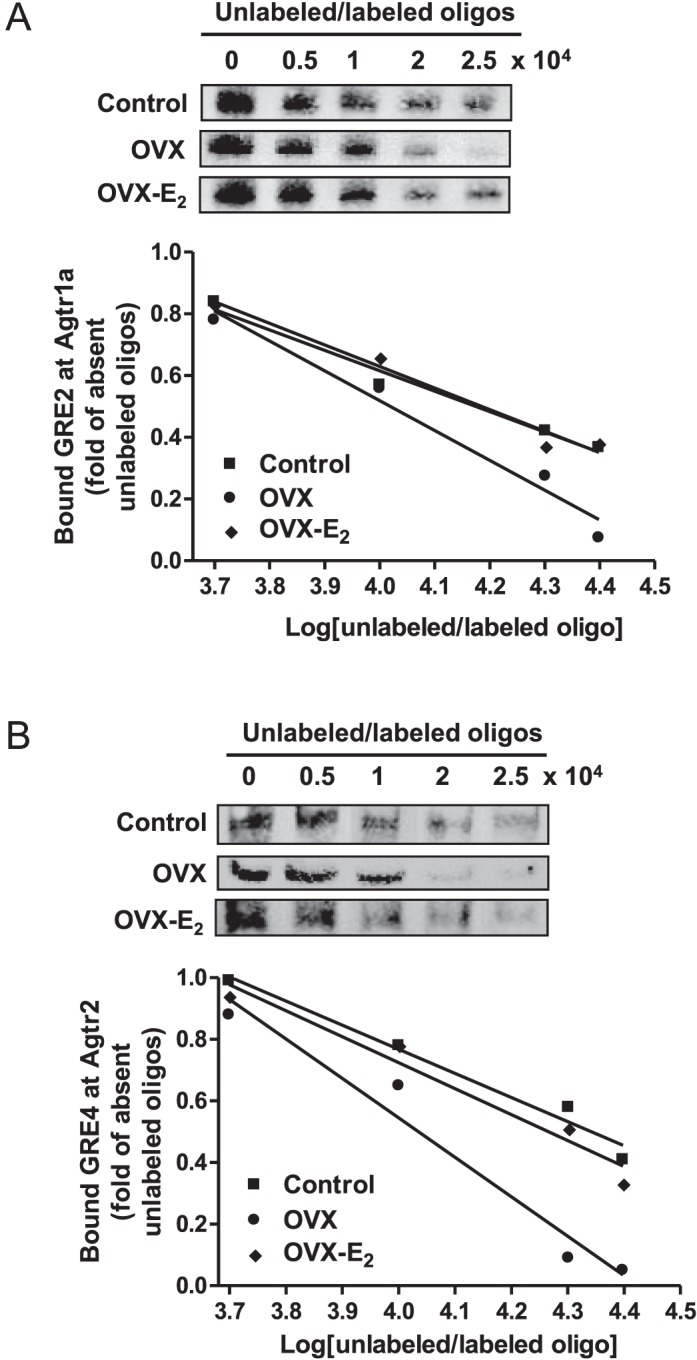
Effect of ovariectomy (OVX) and estrogen replacement (OVX-E2) on GR-binding affinity to GREs at Agtr1a and Agtr2 promoters in normoxic females. LVs were isolated from 4-mo-old normoxic females. The GR-binding affinity to GRE2 at Agtr1a promoter (A) and GRE4 at Agtr2 promoter (B) was determined in a competition EMSA performed in pooled nuclear extracts from LVs (n = 5 in each group) with increasing ratios of unlabeled to labeled oligonucleotides encompassing GRE2 at Agtr1a promoter or GRE4 at Agtr2 promoter.
FIG. 5.
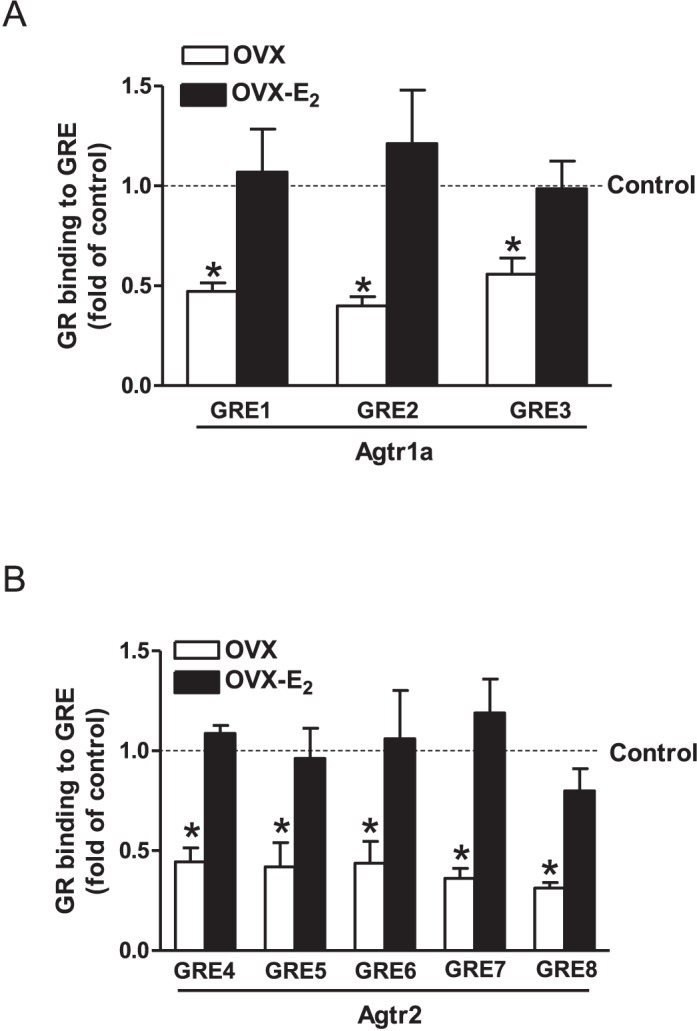
Effect of ovariectomy (OVX) and estrogen replacement (OVX-E2) on GR binding to GREs at Agtr1a and Agtr2 promoters in normoxic females. LVs were isolated from 4-mo-old normoxic females. GR binding to GREs at Agtr1a promoter (A) and GREs at Agtr2 promoter (B) was determined with ChIP assay using a GR antibody. Data are means ± SEM (n = 5 in each group). Data were analyzed by one-way ANOVA followed by Neuman-Keuls posttests, within each GRE site. *P < 0.05, OVX versus sham control or E2 replacement.
Effects of OVX and E2 Replacement on ER Abundance and ER/GR Cross Talk at GREs in Agtr1a and Agtr2 Promoters
OVX significantly decreased both ERα and ERβ protein abundance in the LV, which was rescued by E2 replacement (Fig. 6). ER may regulate gene expression by cross talking with several transcription factors, such as Sp1, Ap1, and NF-κB. To determine whether ER cross talks with GR in binding to GREs at Agtr1a and Agtr2 promoters, Re-ChIP assays were performed using both GR and ERα antibodies. Colocalization of ERα with GR was demonstrated at all GREs examined, including GRE1, GRE2, and GRE3 sites at the Agtr1a promoter and GRE4, GRE5, GRE6, GRE7, and GRE8 sites at the Agtr2 promoter (Fig. 7).
FIG. 6.
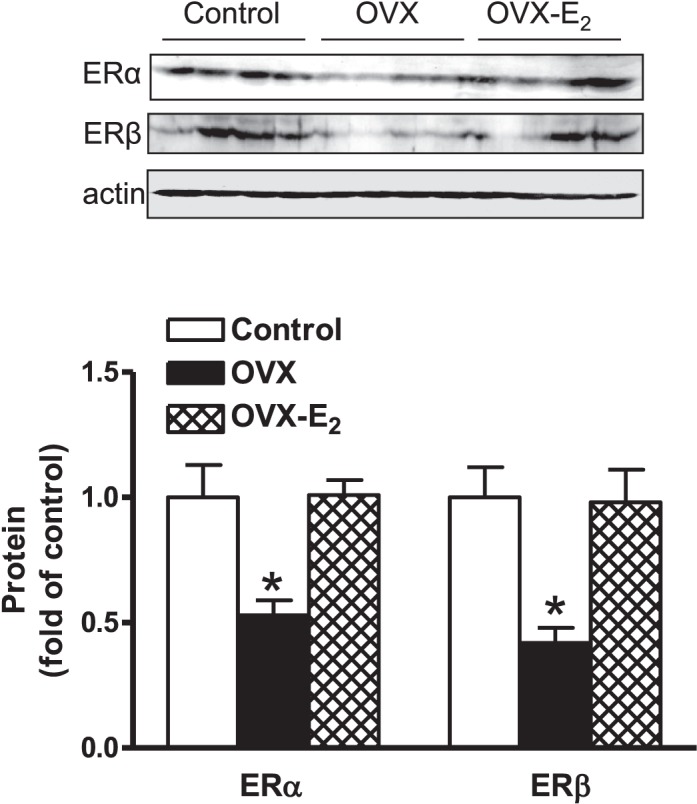
Effect of ovariectomy (OVX) and estrogen replacement (OVX-E2) on ERα and ERβ protein abundance in normoxic females. LVs were isolated from 4-mo-old normoxic females. Protein abundance of ERα and ERβ was determined by Western blot analysis. Data are means ± SEM (n = 4). Data were analyzed by one-way ANOVA followed by Neuman-Keuls posttests, within each receptor subtype. *P < 0.05, OVX versus sham control or E2 replacement.
FIG. 7.
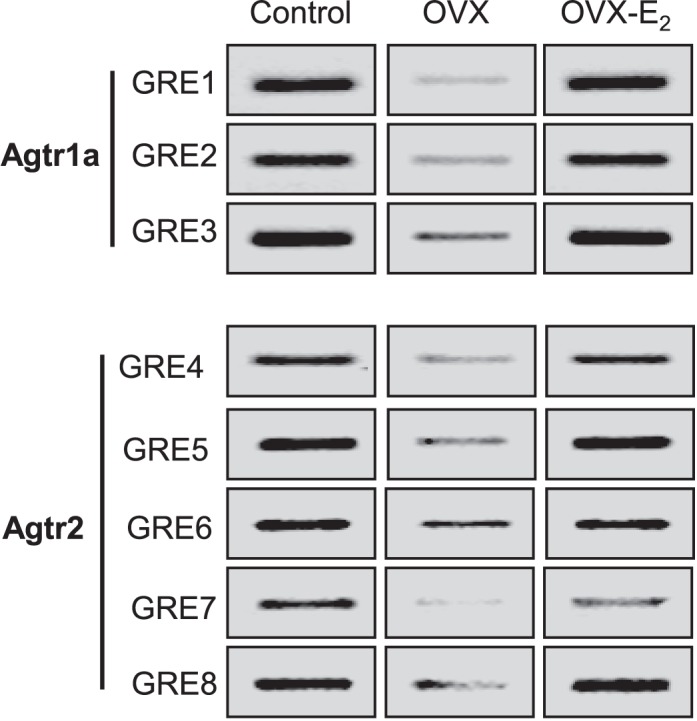
Effect of ovariectomy (OVX) and estrogen replacement (OVX-E2) on ER/GR cross talk in binding to GREs at Agtr1a and Agtr2 promoters in normoxic females. LVs were isolated from 4-mo-old normoxic females. Cross talk binding of ER and GR to GREs at Agtr1a and Agtr2 promoters was determined by Re-ChIP assay with first GR pull-down and second ERα pull-down in pooled samples of LVs (n = 5 in each group). The blots were run on one membrane for each GRE. To be consistent with other figures, the presentation of the blot was reordered.
DISCUSSION
The novelty of the present study is two-fold. First, postnatal ovarian hormones are not directly responsible for the sex dimorphism in fetal programming of heart ischemic vulnerability in female offspring exposed to prenatal hypoxia; however, estrogen protects against cardiac I/R injury in both normoxic control and antenatal hypoxic female offspring. Second, the mechanisms by which estrogen protects female hearts from I/R injury involve changes in the Agtr2 to Agtr1 ratio in the heart. This is mediated by enhanced GR binding to Agtr2 and Agtr1a promoters through increased ER/GR cross talk, resulting in up-regulation of Agtr1 and down-regulation of Agtr2 expression, as well as increased Prkce abundance in the heart.
In the present study, we demonstrated that OVX increased cardiac I/R injury in female offspring in both normoxic control and prenatally hypoxic groups and that E2 replacement ameliorated these effects. This is consistent with previous studies in rats, showing that OVX increased cardiac LDH and CK release and decreased viable cardiomyocytes when exposed to I/R insult [15, 31, 32]. Estrogen replacement reversed these effects, suggesting that estrogen-dependent pathways are vital in gender-specific cardioprotection against I/R injury. Additionally, previous studies demonstrated that chronic estrogen withdrawal in ovariectomized rats abolished the cardioprotective effects of ischemia preconditioning through the reduction of Prkce activity [33]. In addition to the chronic effect, acute administration of estradiol prior to I/R insult was found cardioprotective in both rabbit [34] and canine [35] models.
Previously, we demonstrated that maternal chronic hypoxia reduced myocardial recovery from I/R insult in adult male but not female offspring [7, 9]. This sex-dependent vulnerability of fetal programming has also been observed in other studies [36, 37]. Of importance, the present study demonstrated that the lack of effect in female offspring in response to fetal stress-induced programming of heart ischemic vulnerability was maintained in OVX animals. This indicates that postnatal estrogens may not confer an added advantage in protecting females from fetal programming of cardiac dysfunction and suggests a novel mechanism occurred in the early developmental stage. Consistent with this notion, neurodevelopmental studies suggest that sex chromosomes may play a role in gender-specific ischemic sensitivity in cerebral vascular accidence [38]. Determining the mechanisms through which female offspring negate the programming effects of fetal stress is an intriguing area of study that deserves further investigation.
The finding that OVX altered Agtr1 and Agtr2 expression patterns in the heart provides a possible mechanism through which estrogen protects myocardial tissue. Our data suggest that estrogen increases Agtr1 but decreases Agtr2 expression in the heart. Although there has been some controversy about the specificity of commercial Agtr1 antibodies in mouse tissues [39], the present finding that OVX and estrogen-mediated changes in Agtr1 protein abundance were consistent with the changes in Agtr1 mRNA levels suggests estrogen-mediated up-regulation of Agtr1 expression in the heart. Similar findings were obtained in dexamethasone-induced changes in Agtr1 protein abundance determined by the Agtr1 antibody, which corresponded with the changes in Agtr1 mRNA in the heart [40]. In agreement with our findings, it was previously reported that E2 increased Agtr1 in Wistar rats [24, 41]. This is intriguing given that previous studies have found the direct cardioprotective effect of Agtr1 and the opposite effect of Agtr2 in the acute setting of I/R injury [8]. Hence, the ratio of Agtr1 to Agtr2 in the heart plays an important role in sensitizing the myocardium for recovery or dysfunction from I/R injury. Rodents have two types of Agtr1: Agtr1a and Agtr1b. Although the expression of Agtr1a and Agtr1b genes in rodents are under different promoter controls, they are functionally and pharmacologically indistinguishable at the protein level. In the present study, we have shown that OVX-mediated decrease in Agtr1 protein in the heart is mainly due to down-regulation of Agtr1a mRNA. Unlike rodents, large mammals, including humans, have only one type of Agtr1, which is equivalent to Agtr1a in rodents. There is no human equivalent Agtr1 to the rodent Agtr1b. Interestingly, Agtr1a is up-regulated by binding of glucocorticoids to GREs in the heart [30]. Conversely, modulation of GREs by glucocorticoids at the Agtr2 promoter exerts inhibitory effects on Agtr2 gene expression [8]. In the present study, competition EMSAs demonstrated that OVX significantly decreased the GR-binding affinity to GREs at Agtr1a and Agtr2 promoters, which was recovered by E2 replacement. This finding was further demonstrated by ChIP assays. Although the ChIP assay does not quantify protein-binding affinity to DNA, it shows that the protein binds to a specific region of DNA in vivo in the context of intact chromatin. While caution should be observed in the interpretation of reliable quantification in the ChIP assay, it has been widely used to estimate the interaction of protein to DNA. Taken together, these findings suggest that E2 replacement abrogated OVX-induced reduction of Agtr1 and elevation of Agtr2 expression via rescuing GR binding to GREs at both Agtr1a and Agtr2 promoters in the heart. Nonetheless, other studies have found that E2 replacement in OVX rats reduced cardiac Agtr1 but enhanced Agtr2 in 1-yr-old OVX rats [42], suggesting that differential regulation of estrogens on Agtr that may be age dependent.
The present study demonstrated that OVX significantly decreased both ERα and ERβ protein abundance in the heart, which was recovered by E2 replacement. This is consistent with the premise that ovarian steroid estrogen maintains and regulates the expression of their own receptors ERα and ERβ, and is largely in agreement with the findings of exogenous estrogen treatments in vivo on steroid receptor expression in the heart and vasculature [41, 43]. ERs have been implicated in cardioprotection. The acute activation of ERα is known to stimulate eNOS with cardioprotection against I/R [44], whereas ERα knockout also blocks cardioprotection in female offspring [45]. Likewise, ERβ-knockout females are at greater risk of injury from I/R insults [14]. The finding that OVX decreased ER expression and reduced ERα/GR cross talk in binding to GREs located in the promoters of both Agtr1a and Agtr2 genes is of great interest and suggests an intriguing mechanism that estrogens may regulate Agtr1a and Agtr2 expression in the heart. Previous studies have shown that ER/GR cross talk at AP-1 response elements [46]. Additionally, pulmonary inflammatory models have found a cross talk between ER/GR in gene regulation [47]. Perhaps ER/GR exerts tissue-specific effects. It is likely that the ER/GR interaction may mediate cardioprotective effects of estrogens by enhancing GR binding to the GREs at Agtr1a and Agtr2 promoters and altering the Agtr1 and Agtr2 ratio, leading to a cardioprotective phenotype. While a focus of the present study was ERs, we cannot rule out the involvement of additional mechanisms such as the signaling kinase PI3K or GPR30, which are regulated by estrogens. These pathways may work independent or synergistically with ERs to modulate the Agtr1 to Agtr2 ratio for cardioprotection.
A possible mechanism in Agtr1/Agtr2-mediated cardioprotection involves Prkce [48, 49]. The present study demonstrated that OVX inhibited the Agtr1 to Agtr2 ratio, which was associated with a decrease in Prkce expression in the heart, which was restored by estrogen replacement. This is in agreement with the previous study showing that estrogen replacement restored Prkce in OVX rats [33]. Prkce plays a pivotal role of cardioprotection in heart I/R [6, 50, 51]. The study in a Prkce-knockout mouse model has demonstrated that Prkce expression is not required for cardiac function under normal physiological conditions, but it is necessary for acute cardioprotection during cardiac I/R [52]. Future studies will be needed to determine whether the Prkce activity is altered by estrogen-mediated changes in the Agtr1 to Agtr2 ratio in the heart.
Sex differences are demonstrated in fetal stress-induced developmental programming of cardiovascular dysfunction, and females tend to be more resistant. Although the mechanisms remain largely elusive, the present study demonstrates that the resistance of female offspring to fetal programming of ischemic-sensitive phenotype in the heart involves mechanisms mediated by factors other than postnatal sex steroid hormones, suggesting early developmental pathways that may protect female offspring later in life. In addition, other factors contributing to the increased sensitivity in male offspring may lead to sex difference in susceptibility to I/R. Of importance, the present investigation provides evidence of a novel mechanism by which estrogen protects female hearts against I/R injury via a cross talk between ER/GR in regulating endogenous protective mechanisms in the heart. These findings are important because they provide a mechanistic understanding that contributes to our knowledge of sex steroid hormones in gender-specific protection of cardiovascular function in women. It should be noted that although reactive oxygen species is formed following I/R, estrogen does not increase the antioxidant superoxide dismutase in the heart as has been suggested by some [53].
Footnotes
This work was supported by National Institutes of Health Grants HL118861 (L.Z.), DA032510 (D.X.), and by the Regents of the University of California Tobacco Related Disease Research Program (TRDRP) grant 22XT-0022 (D.X.).
REFERENCES
- Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077–1081. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- Bateson P, Barker D, Clutton-Brock T, Deb D, D'Udine B, Foley RA, Gluckman P, Godfrey K, Kirkwood T, Lahr MM, McNamara J, Metcalfe NB, et al. Developmental plasticity and human health. Nature. 2004;430:419–421. doi: 10.1038/nature02725. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- Chen M, Xiong F, Zhang L. Promoter methylation of Egr-1 site contributes to fetal hypoxia-mediated PKCepsilon gene repression in the developing heart. Am J Physiol Regul Integr Comp Physiol. 2013;304:R683–689. doi: 10.1152/ajpregu.00461.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson AJ, Chen M, Xue Q, Xiao D, Zhang L. Chronic prenatal hypoxia induces epigenetic programming of PKCepsilon gene repression in rat hearts. Circ Res. 2010;107:365–373. doi: 10.1161/CIRCRESAHA.110.221259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Xiao Y, Estrella JL, Ducsay CA, Gilbert RD, Zhang L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J Soc Gynecol Investig. 2003;10:265–274. doi: 10.1016/s1071-5576(03)00074-1. [DOI] [PubMed] [Google Scholar]
- Xue Q, Dasgupta C, Chen M, Zhang L. Foetal hypoxia increases cardiac AT(2)R expression and subsequent vulnerability to adult ischaemic injury. Cardiovasc Res. 2011;89:300–308. doi: 10.1093/cvr/cvq303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Q, Zhang L. Prenatal hypoxia causes a sex-dependent increase in heart susceptibility to ischemia and reperfusion injury in adult male offspring: role of protein kinase C epsilon. J Pharmacol Exp Ther. 2009;330:624–632. doi: 10.1124/jpet.109.153239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–1811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- Yang XP, Reckelhoff JF. Estrogen, hormonal replacement therapy and cardiovascular disease. Curr Opin Nephrol Hyperten. 2011;20:133–138. doi: 10.1097/MNH.0b013e3283431921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z, Carlson SH, Chen YF, Oparil S, Wyss JM. Estrogen depletion induces NaCl-sensitive hypertension in female spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1934–R1939. doi: 10.1152/ajpregu.2001.281.6.R1934. [DOI] [PubMed] [Google Scholar]
- Oparil S, Chen SJ, Chen YF, Durand JN, Allen L, Thompson JA. Estrogen attenuates the adventitial contribution to neointima formation in injured rat carotid arteries. Cardiovasc Res. 1999;44:608–614. doi: 10.1016/s0008-6363(99)00240-0. [DOI] [PubMed] [Google Scholar]
- Wang M, Crisostomo PR, Markel T, Wang Y, Lillemoe KD, Meldrum DR. Estrogen receptor beta mediates acute myocardial protection following ischemia. Surgery. 2008;144:233–238. doi: 10.1016/j.surg.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Zhai P, Eurell TE, Cotthaus R, Jeffery EH, Bahr JM, Gross DR. Effect of estrogen on global myocardial ischemia-reperfusion injury in female rats. Am J Physiol Heart Circ Physiol. 2000;279:H2766–H2775. doi: 10.1152/ajpheart.2000.279.6.H2766. [DOI] [PubMed] [Google Scholar]
- Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Sanda N, Miyawaki Y, Fujimori Y, Yamada T. Takagi A, Murate T, Saito H, Kojima T. Down-regulation of PROS1 gene expression by 17beta-estradiol via estrogen receptor alpha (ERalpha)-Sp1 interaction recruiting receptor-interacting protein 140 and the corepressor-HDAC3 complex. J Biol Chem. 2010;285:13444–13453. doi: 10.1074/jbc.M109.062430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadoke PW, Lindsay RS, Seckl JR, Walker BR, Kenyon CJ. Altered vascular contractility in adult female rats with hypertension programmed by prenatal glucocorticoid exposure. J Endocrinol. 2006;188:435–442. doi: 10.1677/joe.1.06506. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Sherman RC, Welham SJ, Nwagwu MO, Gardner DS, Jackson AA. Intrauterine programming of hypertension: the role of the renin-angiotensin system. Biochem Soc Trans. 1999;27:88–93. doi: 10.1042/bst0270088. [DOI] [PubMed] [Google Scholar]
- Schneider MD, Lorell BH. AT(2), judgment day: which angiotensin receptor is the culprit in cardiac hypertrophy? Circulation. 2001;104:247–248. doi: 10.1161/01.cir.104.3.247. [DOI] [PubMed] [Google Scholar]
- Ford WR, Clanachan AS, Jugdutt BI. Opposite effects of angiotensin AT1 and AT2 receptor antagonists on recovery of mechanical function after ischemia-reperfusion in isolated working rat hearts. Circulation. 1996;94:3087–3089. doi: 10.1161/01.cir.94.12.3087. [DOI] [PubMed] [Google Scholar]
- Ford WR, Khan MI, Jugdutt BI. Effect of the novel angiotensin II type 1 receptor antagonist L-158,809 on acute infarct expansion and acute anterior myocardial infarction in the dog. Can J Cardiol. 1998;14:73–80. [PubMed] [Google Scholar]
- Baiardi G, Macova M, Armando I, Ando H, Tyurmin D, Saavedra JM. Estrogen upregulates renal angiotensin II AT1 and AT2 receptors in the rat. Regul Pept. 2005;124:7–17. doi: 10.1016/j.regpep.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Krishnamurthi K, Verbalis JG, Zheng W, Wu Z, Clerch LB, Sandberg K. Estrogen regulates angiotensin AT1 receptor expression via cytosolic proteins that bind to the 5′ leader sequence of the receptor mRNA. Endocrinology. 1999;140:5435–5438. doi: 10.1210/endo.140.11.7242. [DOI] [PubMed] [Google Scholar]
- Ojeda NB, Grigore D, Robertson EB, Alexander BT. Estrogen protects against increased blood pressure in postpubertal female growth restricted offspring. Hypertension. 2007;50:679–685. doi: 10.1161/HYPERTENSIONAHA.107.091785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Huang X, Xue Q, Zhang L. Antenatal hypoxia induces programming of reduced arterial blood pressure response in female rat offspring: role of ovarian function. PLoS One. 2014;9:e98743. doi: 10.1371/journal.pone.0098743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Huang X, Yang S, Zhang L. Estrogen normalizes perinatal nicotine-induced hypertensive response in adult female rat offspring. Hypertension. 2013;61:1246–1254. doi: 10.1161/HYPERTENSIONAHA.113.01152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Levovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Research. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Zhang H, Zhang L. Direct effect of cocaine on epigenetic regulation of PKCepsilon gene repression in the fetal rat heart. J Mol Cell Cardiol. 2009;47:504–511. doi: 10.1016/j.yjmcc.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo DF, Uno S, Inagami T. Steroid hormones upregulate rat angiotensin II type 1A receptor gene: role of glucocorticoid responsive elements in rat angiotensin II type 1A promoter. J Steroid Biochem Mol Biol. 1995;53:69–73. doi: 10.1016/0960-0760(95)00023-s. [DOI] [PubMed] [Google Scholar]
- Wu Q, Zhao Z, Sun H, Zhang Y, Hao YL, Sun YW. The cardioprotection of estrogen on solated global myocardial ischemia/reperfusion injury in ovariectomized rats [in Chinese] Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2009;25:355–360. [PubMed] [Google Scholar]
- Xu Y, Arenas IA, Armstrong SJ, Plahta WC, Xu H, Davidge ST. Estrogen improves cardiac recovery after ischemia/reperfusion by decreasing tumor necrosis factor-alpha. Cardiovasc Res. 2006;69:836–844. doi: 10.1016/j.cardiores.2005.11.031. [DOI] [PubMed] [Google Scholar]
- Shinmura K, Nagai M, Tamaki K, Bolli R. Loss of ischaemic preconditioning in ovariectomized rat hearts: possible involvement of impaired protein kinase C epsilon phosphorylation. Cardiovasc Res. 2008;79:387–394. doi: 10.1093/cvr/cvn086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale SL, Birnbaum Y, Kloner RA. Estradiol, administered acutely, protects ischemic myocardium in both female and male rabbits. J Cardiovasc Pharmacol Ther. 1997;2:47–52. doi: 10.1177/107424849700200106. [DOI] [PubMed] [Google Scholar]
- Lee TM, Su SF, Tsai CC, Lee YT, Tsai CH. Cardioprotective effects of 17 beta-estradiol produced by activation of mitochondrial ATP-sensitive K(+)Channels in canine hearts. J Mol Cell Cardiol. 2000;32:1147–1158. doi: 10.1006/jmcc.2000.1167. [DOI] [PubMed] [Google Scholar]
- Netuka ISO, Maly J, Besik J, Neckar J, Kolar F, Ostadalova I, Pirk J, Ostadal B. Effect of perinatal hypoxia on cardiac tolerance to acute ischaemia in adult male and female rats. Clin Exp Pharmacol Physiol. 2006;33:714–719. doi: 10.1111/j.1440-1681.2006.04423.x. [DOI] [PubMed] [Google Scholar]
- Hinojosa-Laborde C, Mifflin SW. Sex differences in blood pressure response to intermittent hypoxia in rats. Hypertension. 2005;46:1016–1021. doi: 10.1161/01.HYP.0000175477.33816.f3. [DOI] [PubMed] [Google Scholar]
- Lentini E, Kasahara M, Arver S, Savic I. Sex differences in the human brain and the impact of sex chromosomes and sex hormones. Cerebral Cortex. 2013;23:2322–2336. doi: 10.1093/cercor/bhs222. [DOI] [PubMed] [Google Scholar]
- Herrera M, Sparks MA, Alfonso-Pecchio AR, Harrison-Bernard LM, Coffman TM. Lack of specificity of commercial antibodies leads to misidentification of angiotensin type 1 receptor protein. Hypertension. 2013;61:253–258. doi: 10.1161/HYPERTENSIONAHA.112.203679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Q, Patterson AJ, Xiao D, Zhang L. Glucocorticoid modulates angiotensin II receptor expression patterns and protects the heart from ischemia and reperfusion injury. PLoS One. 2014;9:e106827. doi: 10.1371/journal.pone.0106827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricchiuti V, Lian CG, Oestreicher EM, Tran L, Stone JR, Yao T, Seely EW, Williams GH, Adler GK. Estradiol increases angiotensin II type 1 receptor in hearts of ovariectomized rats. J Endocrinol. 2009;200:75–84. doi: 10.1677/JOE-08-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Arenas IA, Armstrong SJ, Davidge ST. Estrogen modulation of left ventricular remodeling in the aged heart. Cardiovasc Res. 2003;57:388–394. doi: 10.1016/s0008-6363(02)00705-8. [DOI] [PubMed] [Google Scholar]
- Byers MJ, Zangl A, Phernetton TM, Lopez G, Chen DB, Magness RR. Endothelial vasodilator production by ovine uterine and systemic arteries: ovarian steroid and pregnancy control of ERα and ERβ levels. J Physiol. 2005;565:85–99. doi: 10.1113/jphysiol.2005.085753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103:401–406. doi: 10.1172/JCI5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Crisostomo P, Wairiuko GM, Meldrum DR. Estrogen receptor-alpha mediates acute myocardial protection in females. Am J Physiol Heart Circ Physiol. 2006;290:H2204–2209. doi: 10.1152/ajpheart.01219.2005. [DOI] [PubMed] [Google Scholar]
- Uht RM, Anderson CM, Webb P, Kushner PJ. Transcriptional activities of estrogen and glucocorticoid receptors are functionally integrated at the AP-1 response element. Endocrinology. 1997;138:2900–2908. doi: 10.1210/endo.138.7.5244. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Bruscoli S, Crisafulli C, Mazzon E, Agostini M. Muia C, Esposito E, Di Virgilio R, Meli R, Vegeto E, Maggi A, Riccardi C. Estrogen receptor antagonist fulvestrant (ICI 182,780) inhibits the anti-inflammatory effect of glucocorticoids. Mol Pharmacol. 2007;71:132–144. doi: 10.1124/mol.106.029629. [DOI] [PubMed] [Google Scholar]
- Diaz RJ, Wilson GJ. Selective blockade of AT1 angiotensin II receptors abolishes ischemic preconditioning in isolated rabbit hearts. J Mol Cell Cardiol. 1997;29:129–139. doi: 10.1006/jmcc.1996.0258. [DOI] [PubMed] [Google Scholar]
- Xu Y, Arenas IA, Armstrong SJ, Plahta WC, Xu H, Davidge ST. Estrogen improves cardiac recovery after ischemia/reperfusion by decreasing tumor necrosis factor-alpha. Cardiovasc Res. 2006;69:836–844. doi: 10.1016/j.cardiores.2005.11.031. [DOI] [PubMed] [Google Scholar]
- Chen L, Hahn H, Wu G, Chen CH, Liron T. Schechtman D, Cavallaro G, Banci L, Guo Y, Bolli R, Dorn GW II, Mochly-Rosen D. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci U S A. 2001;98:11114–11119. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping P, Song C, Zhang J, Guo Y, Cao X. Li RC, Wu W, Vondriska TM, Pass JM, Tang XL, Pierce WM, Bolli R. Formation of protein kinase C(epsilon)-Lck signaling modules confers cardioprotection. J Clin Invest. 2002;109:499–507. doi: 10.1172/JCI13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C epsilon. J Biol Chem. 2004;279:3596–3604. doi: 10.1074/jbc.M311459200. [DOI] [PubMed] [Google Scholar]
- Furuya K, Chaudhuri G. Estradiol does not influence myocardial superoxide dismutase activity in rabbits. J Cardiovasc Pharmacol. 1993;22:65–68. doi: 10.1097/00005344-199307000-00011. [DOI] [PubMed] [Google Scholar]