

Graphical abstract

Abbreviations: HTS, high-throughput screen; SAR, structure–activity relationships; PRF, perforin; CTL, cytotoxic T lymphocytes; NK, natural killer cells; PAINS, pan-assay interference compounds; AgNO3, silver(I) nitrate; KF, potassium fluoride; PFP, pentafluorophenyl; DCC, N,N′-dicyclohexylcarbodiimide; HOBt, hydroxybenzotriazole; EDCI, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; DMF, dimethylformamide; KOAc, potassium acetate; DMSO, dimethyl sulfoxide; THF, tetrahydrofuran
Keywords: Perforin, Perforin inhibitor, Diarylthiophene, Bioisostere, Immunosuppressant
Abstract
Evolution from a furan-containing high-throughput screen (HTS) hit (1) resulted in isobenzofuran-1(3H)-one (2) as a potent inhibitor of the function of both isolated perforin protein and perforin delivered in situ by intact KHYG-1 NK cells. In the current study, structure–activity relationship (SAR) development towards a novel series of diarylthiophene analogues has continued through the use of substituted-benzene and -pyridyl moieties as bioisosteres for 2-thioxoimidazolidin-4-one (A) on a thiophene (B) -isobenzofuranone (C) scaffold. The resulting compounds were tested for their ability to inhibit perforin lytic activity in vitro. Carboxamide (23) shows a 4-fold increase over (2) in lytic activity against isolated perforin and provides good rationale for continued development within this class.
Perforin (PRF) is a calcium-dependent pore-forming glycoprotein found within granules of cytotoxic T lymphocytes (CTL) and natural killer (NK) cells.1, 2 These cytotoxic effector cells play an important role in immune-surveillance and homeostasis via a granule exocytosis pathway responsible for the elimination of neoplastic/virus-infected cells and intracellular pathogens.3 Numerous autoimmune diseases (e.g., insulin-dependent diabetes) and therapy-induced conditions (e.g., allograft transplantation rejection, graft-vs-host disease) adopt a synonymous biochemical pathway.4, 5 An integral part of this biological cascade is perforin oligomerisation in which monomeric perforin binds calcium upon entry into the immune synaptic cleft and forms highly ordered oligomers with 19–24 adjacent perforin monomers. These in turn form cylindrical-type pores which subsequently penetrate target cell membranes, allowing rapid delivery of cytotoxic granzymes into the cytosolic compartment, a process which culminates in apoptosis of the target cell. Highly specific inhibitors of perforin function are thus of interest as selective immunosuppressive drugs.
Isobenzofuran-1(3H)-one-based inhibitor 1 (Fig. 1) was a hit selected from a high-throughput screen of approximately 100,000 compounds sourced from commercial ‘lead discovery’ libraries.6 Subsequent SAR development led to the discovery of potent inhibitor 2 in which an ∼8-fold improvement in inhibition of isolated perforin protein function was achieved through replacement of a central furan ring with a 2,5-thiophene moiety.7 In more recently published work8 this template was elaborated further through variation of the isobenzofuranone C-subunit where an extensive SAR programme was conducted and the mechanism of perforin inhibition investigated using real-time microscopy.8, 9 The resulting isoindolinone-containing inhibitors were shown to have improved lytic activity against perforin delivered in situ by intact KHYG-1 NK cells and furthermore, were employed as probes to elucidate the role of perforin in the granule exocytosis pathway. This work resulted in the conclusion that this class of compounds target perforin after its release into the immune synapse.
Figure 1.

The development of new diarylthiophene inhibitors of perforin.
While substantial advances have been made in the development and understanding of the thioxoimidazolidinone-based series, more detailed in vitro and in vivo assessment7, 10 revealed several properties requiring further optimisation. Although well-tolerated in healthy mice, even those compounds deemed suitable candidates for further in vivo studies showed some toxicity toward whole NK cells.8 It is therefore likely that toxicity would also be observed in the immunocompromised mice required for an efficacy study. Also desirable would be the identification of new compounds with significantly improved potency to deliver a lower efficacious dose and better control of potential dose-related toxicity. Finally, while the thioxoimidazolidinone series passed various pan-assay interference compound (PAINS) filters,11 they still contain a potentially reactive Michael acceptor and exist as an inseparable (and interconverting) mixture of E- and Z-isomers.
To address these aforementioned issues the need for a 2-thioxoimidazolidin-4-one bioisosteric replacement was apparent and herein we report on the latest developments towards an exciting new class of nontoxic inhibitors of perforin function that show improved inhibition of the function of both isolated perforin protein and perforin delivered in situ by intact KHYG-1 NK cells.
Synthesis: Target compounds 6–23 (Scheme 1) were obtained in a linear four-step sequence starting with the conversion of commercially available 2-methyl-4-bromobenzoic acid to 5-bromoisobenzofuran-1(3H)-one 3.12 A sequential Finkelstein halogen exchange reaction13 gave 5-iodoisobenzofuran-1(3H)-one 4 in 87% yield. This can also be prepared in high yield through diazotization of the corresponding 5-aminoisobenzofuran-1(3H)-one using a literature procedure.14 Lactone derivative 4 was then utilised in a palladium-catalysed arylation reaction with 2-bromothiophene in the presence of a silver(I) nitrate/potassium fluoride (AgNO3/KF) activator system15 to yield key intermediate bromide 5. Subsequent Suzuki couplings with a variety of commercially available phenyl-boronates/boronic acids in the presence of catalytic PdCl2(dppf), afforded the respective diarylthiophene targets 6–23 in generally good yields. Isoindolinone-based versions of 23 (23a and 23b) were also prepared in exactly the same manner, using the isoindolin-1-one and 2-methylisoindolin-1-one-based analogues16 of key bromide 5 (5a and 5b; Scheme 1).
Scheme 1.

Reagents and conditions: (i) sealed tube, 1,4-dioxane, CuI (5.0 mol %), NaI, racemic trans-N,N′-dimethyl-1,2-cyclohexanediamine (10 mol %), 110 °C, N2, 18 h; (ii) 2-bromothiophene or 69, DMSO, KF, PdCl2(PPh3)2, AgNO3, 100 °C, N2, 2 h; (iii) toluene/EtOH, 2 M Na2CO3, PdCl2(dppf)·CH2Cl2, 85–90 °C, N2, 2–6 h; (iv) thiophene-2-boronic acid, toluene/EtOH, 2 M Na2CO3, PdCl2(dppf)·CH2Cl2, 85 °C, N2, 2 h.
Analogous methodology was adopted to generate secondary and tertiary carboxamide targets 29 and 30, by first reacting iodide 24 with 2-bromothiophene in the presence of AgNO3/KF (Scheme 2). The afforded bromide 25 was then converted to carboxamide intermediates 26 and 2717 through activation of the carboxylic acid to a pentafluorophenyl (PFP) ester followed by subsequent addition of the corresponding amine (methylamine or dimethylamine) in pyridine for 2–3 h at room temperature. Key boronate 28 was prepared in multi-gram quantities from bromide 318 (Scheme 1) and subsequently used in a Suzuki coupling with intermediates 26 and 27 to furnish carboxamide targets 29 (75%) and 30 (57%), respectively.
Scheme 2.

Reagents and conditions: (i) 2-bromothiophene, DMSO, KF, PdCl2(PPh3)2, AgNO3, 100 °C, N2, 2 h; (ii) (a) THF, pyridine, pentafluorophenyltrifluoroacetate, rt, 2 h; (b) THF, methylamine (40% soln.) or dimethylamine, rt, 2–3 h; (iii) toluene/EtOH, 2 M Na2CO3, PdCl2(dppf)·CH2Cl2, 85–90 °C, N2, 2–6 h.
Compounds 31 and 32, where the carboxamide side-chain was elaborated further by incorporating a series of basic side-chains, were accessed by applying the same chemistry as summarised in Scheme 1. A final step Suzuki coupling of key intermediate bromide 5 with the corresponding phenyl boronate (as according to the compounds of Table 1) gave 31 and 32 in good to modest yields, respectively, (61% and 42%).
Table 1.
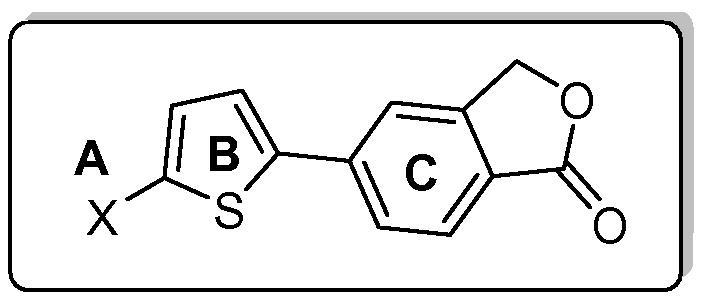
| Number | Scheme | Xa | Inhibition of Jurkat cell lysis IC50b (μM) |
|---|---|---|---|
| 6 | 1 | Ph | >20 |
| 7 | 1 | 2-Me Ph | >20 |
| 8 | 1 | 3-Me Ph | >20 |
| 9 | 1 | 4-Me Ph | >20 |
| 10 | 1 | 2-Cl Ph | >20 |
| 11 | 1 | 3-Cl Ph | >20 |
| 12 | 1 | 4-Cl Ph | >20 |
| 13 | 1 | 2-OMe Ph | >20 |
| 14 | 1 | 3-OMe Ph | >20 |
| 15 | 1 | 4-OMe Ph | 9.36 |
| 16 | 1 | 3-NH2 Ph | 13.97 |
| 17 | 1 | 4-NH2 Ph | 11.82 |
| 18 | 1 | 3-OH Ph | 5.89 |
| 19 | 1 | 4-OH Ph | >20 |
| 20 | 1 | 3-CN Ph | 20 |
| 21 | 1 | 4-CN Ph | 6.87 |
| 22 | 1 | 3-CONH2 Ph | 2.97 |
| 23 | 1 | 4-CONH2 Ph | 0.18 |
| 23ac | 1 | 4-CONH2 Ph | 6.04 |
| 23bd | 1 | 4-CONH2 Ph | >20 |
| 29 | 2 | 4-CONHMe Ph | 10.7 |
| 30 | 2 | 4-CONMe2 Ph | >20 |
| 31e | 1 | 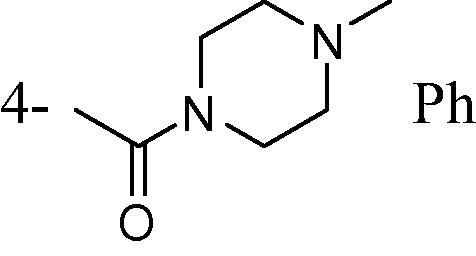 |
>20 |
| 32 | 1 | 4-CONH(CH2)3NMe2 Ph | >20 |
| 39 | 3 | 4-COOEt Ph | 10.97 |
| 40 | 3 | 3-OH, 4-COOEt Ph | >20 |
| 46 | 3 | 3-NH2, 4-COOH Ph | 5.84 |
| 48 | 3 | 3-OH, 4-CONH2 Ph | 0.67 |
| 49e | 3 | 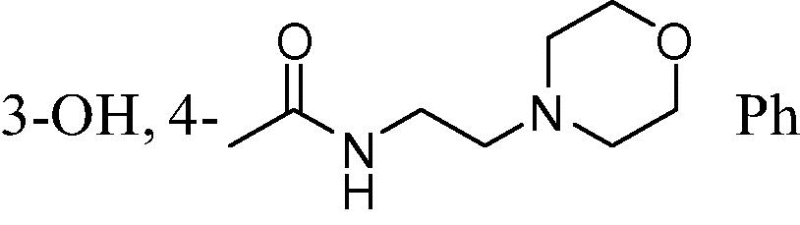 |
12.75 |
| 50e | 3 | 3-OH, 4-CONH(CH2)2NMe2 Ph | >20 |
| 51 | 3 | 3-OH, 4-CONHCH2CH2OH Ph | 3.31 |
| 52 | 3 | 3-OH, 4-CONHCH2CH(OH)CH3 Ph | >20 |
| 53 | 3 | 3-OH, 4-CONHCH2CONH2 Ph | 2.65 |
| 54 | 3 | 3-OH, 4-CONHCH2CH2CONH2 Ph | 7.12 |
| 55 | 3 | 3-NH2, 4-CONH2 Ph | 1.20 |
| 56e | 3 | 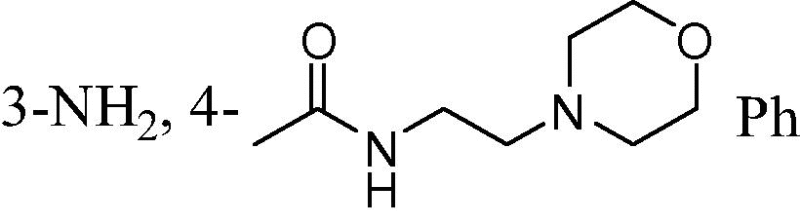 |
14.71 |
| 57e | 3 | 3-NH2, 4-CONH(CH2)2NMe2 Ph | >20 |
| 58 | 1 | 4-SMe Ph | >20 |
| 59 | 1 | 4-CH2OH Ph | 0.90 |
| 60 | 1 | 4-SO2Me Ph | >20 |
| 61 | 1 | 3-SO2NH2 Ph | 4.85 |
| 62 | 1 | 4-SO2NHtert-Bu Ph | 6.09 |
| 63 | 1 | 4-NHSO2Me Ph | 1.09 |
| 67 | 4 | 4-CONH2, 3-pyridyl | 0.92 |
| 70 | 1 | 4-Pyridyl | 4.53 |
| 71 | 1 | 5-Isobenzofuran-1(3H)-one | 6.24 |
Structure of the A-subunit.
Testing was carried out over a range of doses, with the IC50 being equal to the concentration at which 50% inhibition of the lysis of Jurkat cells by perforin was observed, as measured by 51Cr release. Values are the average of at least two independent IC50 determinations.
Isobenzofuran-1(3H)-one C-subunit replaced with an isoindolin-1-one.
Isobenzofuran-1(3H)-one C-subunit replaced with an 2-methylisoindolin-1-one.
Detailed structures for compounds, 31, 49, 50, 56 and 57 can be found in the Supplementary information (Experimental Section).
With the chemistry now established for the mono-substituted A-ring targets shown in Scheme 1, Scheme 2, a series of di-substituted targets were investigated. Phenyl iodides 33–35 were prepared via esterification from their native carboxylic acids in the presence of EtOH and catalytic H2SO4, using conventional methodology (Scheme 3). The sequential step was carried out as for carboxylic acid analogue 25 (refer to Scheme 2) affording ethyl esters 36–38, these in turn were then coupled with key boronate ester 28 via a Suzuki reaction to give diarylthiophene esters 39–41 in good to excellent yields. Hydrolysis of the resulting esters with 2 M NaOH in MeOH under reflux conditions, gave bis-carboxylic acid intermediates 42–44, respectively. Subsequent dehydration to isobenzofuran-1(3H)-one precursors 45–47 was achieved quantitatively using a mixture of TFA/CH2Cl2 (1:1) at room temperature for approximately 3–4 h. Target compounds 48–57 were then synthesised utilising a variety of amide-coupling strategies (Scheme 3). Carboxamide 48 was prepared successfully in 57% yield going via the analogous PFP-active ester intermediate as employed for carboxamides 26 and 27 (refer to Scheme 2). Compounds 49–54, which contain various extended neutral and basic side-chains appended to the 4-carboxamide moiety, were accessed through treatment of the corresponding acid precursor with DCC and catalytic HOBt at elevated temperatures. Target compounds 55 (48%), 56 (80%), and 57 (38%), all of which contain a 3-substituted-phenyl primary amine, were accessed through an EDCI/HOBt-mediated amide-coupling in the presence of anhydrous DMF at 0 °C to room temperature over 1–2 h.
Scheme 3.

Reagents and conditions: (i) 2-bromothiophene, DMSO, KF, PdCl2(PPh3)2, AgNO3, 100 °C, N2, 2–4 h; (ii) toluene/EtOH, 2 M Na2CO3, PdCl2(dppf)·CH2Cl2, 85–90 °C, N2, 1–6 h; (iii) MeOH, 2 M NaOH, 100 °C, 2 h, HCl-workup; (iv) TFA–DCM (1:1), rt, 3–4 h; (v) THF, pyridine, pentafluorophenyltrifluoroacetate, rt, 2 h, R2-NH2, rt, 2 h, 48; or pyridine, DCC, HOBt, 75 °C, 5–6 h, 49–54; or anhydrous-DMF, EDCI·HCl, HOBt, rt, 2 h, R2-NH2, 0 °C → rt, 1–2 h, 55–57.
Compounds 58–63 (Scheme 1) encompass a variety of A-ring moieties (such as an alcohol, a methanethiol and various sulfonamides). These were all accessed through a key Suzuki coupling step from key intermediate 5 with the corresponding substituted boronate, as according to the compounds of Table 1. Isobenzofuran-1(3H)-one dimer (71) was prepared in the same manner.
Bromide 64 was commercially available and was converted to iodide 65 via a copper-mediated halogen exchange reaction as adapted from literature procedure13 (Scheme 4). Boronate 66 was synthesised from corresponding bromide intermediate 5 in the presence of bis(pinacolato)diboron and KOAc and subsequently coupled with iodide 65 utilising Suzuki methodology to yield pyridyl carboxamide 67 in low yield.
Scheme 4.

Reagents and conditions: (i) sealed tube, 1,4-dioxane, CuI (5.0 mol %), NaI, racemic trans-N,N′-dimethyl-1,2-cyclohexanediamine (10 mol %), 110 °C, N2, 24 h; (ii) DMSO, bis(pinacolato)diboron, KOAc, PdCl2(dppf)·CH2Cl2, 90 °C, N2, 4 h; (iii) toluene/EtOH, 2 M Na2CO3, PdCl2(dppf)·CH2Cl2, 85–90 °C, N2, 1.5 h.
Finally, the preparation of 4-pyridyl-containing target 70 began with the Suzuki-coupling of 4-bromopyridine 68 with thiophene-2-boronic acid, as described by Effenberger et al.,19 to furnish 4-(2-thienyl)pyridine 69 in high yield. Isobenzofuran-1(3H)-one 4 was then installed through treatment with AgNO3 and KF in the presence of a palladium-complex, as previously discussed, to afford 4-pyridyl derivative 70 in 27% yield (Scheme 1).
Structure–activity relationships: Based on our previous studies described above, we hypothesised that selected substitutions on a benzene or pyridyl ring could potentially occupy the same space as the thioxoimidazolidinone pharmacophore, thereby mimicking the network of hydrogen bond donors/acceptors required for activity.8 This approach is illustrated in Figure 2 and highlights key objectives which include removal of the Michael acceptor and elimination of isomeric mixtures.
Figure 2.

Rationale for the development of new diarylthiophene inhibitors.
For the current study we elected to retain the isobenzofuranone C-subunit of 2 as this class is generally more potent than the corresponding isoindolinones8 which were originally introduced on the assumption they would be less susceptible to hydrolysis. However subsequent stability studies showed no clear advantage with both the isoindolinones and isobenzofuranones showing variable half-lives in vitro (microsome stability) and in vivo.11 Parent compound (6) was employed as a start point for the replacement of the 2-thioxoimidazolidin-4-one A-subunit, but due to its lack of potency (Jurkat IC50 >20 μM; Table 1) a series of simple substituted phenyl based derivatives were designed and investigated.
Compounds 7–15 (Table 1) follow an independent 2, 3, 4 substitution pattern on a benzene A-ring with derivative 15 (4-OMe) demonstrating activity against isolated perforin protein in our Jurkat assay (IC50 = 9.36 μM). The remaining methoxy isomers (13, 14) showed no detectable inhibition, suggesting that substitution para to the connecting thiophene B-subunit is essential for activity. Prompted by this encouraging result and with derivative 15 providing an H-bond acceptor moiety, we looked to introduce an NH2, OH and CN group at positions 3 and 4 accordingly. Isomers 16 (3-NH2) and 17 (4-NH2) both have comparable activity (IC50s = 13.97 and 11.82 μM, respectively), showing that substitution at the 3-position is tolerated in this particular case. This is confirmed further by 3-hydroxyl-containing compound 18 in which a 2-fold increase in potency is achieved (IC50 = 5.89 μM). Looking at 3-CN derivative 20 in comparison, activity is lost altogether suggesting that an H-bond donor may be required in this position for activity. In contrast, 4-OH containing compound 19 loses all activity whilst 4-CN containing compound 21 (IC50 = 6.87 μM) retains potency similar to that of 18. This reinforces the argument in that an H-bond acceptor at position 4 is more favourable and perhaps necessary for activity to exist in this series (15 and 21 vs 19).
Compounds 22 and 23 were designed with a carboxamide moiety installed at positions 3 and 4, respectively. Substitution at position 3 was well tolerated giving rise to an IC50 = 2.97 μM—2-fold greater than our previously most active compound 18. However when the primary amide is moved to position 4, a dramatic increase in the ability to inhibit perforin lytic activity is seen. Benzene-4-carboxamide 23 has a potency approximately 4-fold greater than lead thioxoimidazolidinone 2 (Figure 1, Figure 2) against isolated perforin (0.18 μM vs 0.78 μM, respectively). Furthermore, the corresponding pyridine-4-carboxamide 67 is also one of our most potent compounds (IC50 = 0.92 μM). Although at the outset a decision was made to retain the more potent isobenzofuranone C-subunit, for completeness the analogous isoindolin-1-one (23a) and 2-methylisoindolin-1-one (23b) derivatives were also prepared. As expected this modification resulted in a loss of activity. Conversion of the primary amide of 23 to a secondary derivative (29) results in a loss of efficacy, while the presence of a tertiary amide (30, 31) abolishes activity completely. Ester 39 showed limited activity (10.97 μM), while compounds 40, 46, 48 and 55 were designed in an effort to combine the outstanding potency of 23 with the preferred 3-OH of 18 or 3-NH2 of 16, thereby introducing an H-bond donor at the 3-position and adding an ionisable centre to assist solubility. While this approach did generate potent (and slightly more soluble—see Supplementary data) compounds, none were an improvement on 23. In an extension of this strategy, solubilising sidechains were introduced in examples 32, 48–54 and 56, 57. Disappointingly, compounds with strongly or weakly basic sidechains (32, 49, 50, 56, 57) displayed poor activity or were inactive, while neutral compounds 51 and 53 showed only moderate activity (IC50s 3.31 and 2.65 μM), respectively.
In order to further increase diversity and enrich the SAR of the diarylthiophene series, a range of commercially available boronates were deployed in Suzuki reactions with key intermediate 5 (Scheme 1), generating analogues 58–63. Results were mixed, with the methyl alcohol 59 and methyl sulfonamide 63 (IC50s 0.92 and 1.09 μM, respectively) the best of this set. Finally, given similarities in SAR between the A- and C-subunits7 the symmetrical analogue 71 was synthesised but proved only moderately potent as well as extremely insoluble.
Biological activity and stability: The five most potent inhibitors of isolated recombinant perforin (23, 48, 59, 63, and 67 from Table 1) were then subjected to more advanced assessment. Preliminary stability studies were carried out by incubation in human or mouse plasma at 37 °C with the percentage parent remaining measured at 24 h (Table 2). All five inhibitors were significantly more stable in mouse plasma compared to human. This result is not unexpected, as the anticancer agent camptothecin which also contains a lactone moiety has been shown to co-exist in both the closed and ring-opened form and that this equilibrium is distinctly different for human and mouse plasma. In mouse plasma the ratio of open to closed is 50:50%, while in human plasma this shifts to 90:10 due to the strong affinity of human serum albumin for the ring-opened form.20 It is likely that the same phenomenon is operating in the present case and while not necessarily an issue for mouse studies, will need to be addressed in future work.
Table 2.
Capacity of selected compounds to inhibit perforin delivered by KHYG-1 NK cells
| Number | Jurkat IC50a (μM) | KHYG-1 inhibitionb (% at 20 μM) | KHYG-1 viabilityc (%) | Plasma stabilityd (% at 24 h) |
|
|---|---|---|---|---|---|
| Mouse | Human | ||||
| 23 | 0.18 | 63 | 90 | 75 | 46 |
| 48 | 0.67 | 20 | 92 | 75 | 49 |
| 59 | 0.90 | 13 | 79 | 87 | 73 |
| 63 | 1.09 | No activity | — | 80 | 70 |
| 67 | 0.92 | 80 | 82 | 66 | 39 |
Data given for five most potent compounds as determined by the Jurkat assay.
Inhibition by compound (20 μM) of the perforin-induced lysis of K562 target cells when co-incubated with KHYG-1 human NK cells (see Supplementary data). Percent inhibition calculated compared to untreated control.
Viability of KHYG-1 NK cells after 24 h by Trypan blue exclusion assay (see Supplementary data).
Data given as % parent measured at 24 h in mouse and human plasma (see Supplementary data for further details).
The compounds were then evaluated for their ability to block the effect of perforin delivered by whole KHYG-1 NK cells (Table 2). Employing whole NK cells is a more rigorous model of conditions in vivo than the use of isolated recombinant protein since effector cell identification of the labelled target cell and formation of an immune synapse are required for perforin release. Any putative inhibitor must also possess the ability to access perforin released into the synaptic cleft. After co-incubation of inhibitor with KHYG-1 NK cells in medium for 30 min at room temperature, 51Cr-labelled K562 leukaemia target cells were added and cell lysis evaluated after 4 h incubation at 37 °C by measuring 51Cr release. The viability of the NK cells in the presence of inhibitor was also assessed 24 h. later to confirm that any inhibitory activity exhibited by the compounds was due to blocking the action of perforin and not nonspecific killing of the effector cells. Viable and dead cells were counted and percent viability calculated based on total cell count (for further details see Supplementary data). Of the five compounds selected for further testing, three showed poor or no activity; the 3-OH, 4-CONH2 compound 48, 4-methyl alcohol 59 and methanesulfonamide 63. Gratifyingly, the most potent compound against isolated perforin (23) showed comparable activity (60% inhibition of lytic activity at 20 μM concentration) to many of the active compounds contained in our previous report on the thioxoimidazolidinone-based series.8 In addition, pyridinecarboxamide 67 was essentially equivalent to our previously most potent in vivo candidates (80% inhibition of lysis). Most importantly, this activity was observed in the absence of toxicity toward the effector cells (90% and 82% viability, respectively). For the purposes of this work, compounds are classified as toxic if NK cell viability falls below 70%. This is due to the importance of CTLs and NK cells in the overall immunological response which means that it is essential that any pharmacological intervention allows rapid recovery of these cytotoxic effector cells for normal immunological function.
In summary a new series of benzene- and pyridine-carboxamides have been designed as isosteric replacements for our previous thioxoimidazolidinone-based series. Several of these compounds showed improved inhibition of perforin-mediated lysis, with benzenecarboxamide 23 demonstrating a 4-fold increase (0.18 μM) over the corresponding thioxoimidazolidinone inhibitor 2 (0.78 μM). Compound 23 and its pyridine analogue 67 also showed excellent activity against the lytic action of whole human NK cells, at a level comparable to our previously identified in vivo candidates.8 Use of an isostere in place of the thioxoimidazolidinone A-subunit has also enabled removal of a potentially reactive Michael acceptor moiety and elimination of E- and Z-isomeric mixtures. Finally, these compounds do not appear to possess the marginal toxicity associated with many of the thioxoimidazolidinones.
We can conclude that we have identified viable replacements for the thioxoimidazolidinone subunit while improving potency. We have also overcome several key concerns in relation to this historic series and now seek to expand on these findings to identify a suitable in vivo candidate. The mechanism of action of these inhibitors is still under investigation, however given our previous conclusion that this class of compounds target perforin after its release into the immune synapse it is most likely that inhibition occurs by either (1) blocking the monomers from association with the target cell membrane, (2) counteracting the oligomerisation process that forms the pre-pore, or (3) by blocking of the conformational changes required for pore formation. This work is currently ongoing and further developments will be reported in the future.
Acknowledgments
This work was supported by the Wellcome Trust (Grant 097767) and the Auckland Division of the Cancer Society of New Zealand. K.M.H. thanks the Academy of Finland (Grant 135439) and the Orion-Farmos Research Foundation for financial support. We also thank Sisira Kumara and Karin Tan for HPLC studies, and Maruta Boyd and Shannon Black for NMR studies.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2015.12.003.
Supplementary data
Experimental procedures.
References and notes
- 1.Lopez J.A., Brennan A.J., Whisstock J.C., Voskoboinik I., Trapani J.A. Trends Immunol. 2012;33:406. doi: 10.1016/j.it.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Law R.H.P., Lukoyanova N., Voskoboinik I., Caradoc-Davies T.T., Baran K., Dunstone M.A., D’Angelo M.E., Orlova E.V., Coulibaly F., Verschoor S., Browne K.A., Ciccone A., Kuiper M.J., Bird P.I., Trapani J.A., Saibil H.R., Whisstock J.C. Nature. 2010;468:447. doi: 10.1038/nature09518. [DOI] [PubMed] [Google Scholar]
- 3.Voskoboinik I., Smyth M.J., Trapani J.A. Nat. Rev. Immunol. 2006;6:940. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 4.Barry M., Bleackley R.C. Nat. Rev. Immunol. 2002;2:401. doi: 10.1038/nri819. [DOI] [PubMed] [Google Scholar]
- 5.Veale J.L., Liang L.W., Zhang Q., Gjertson D.W., Du Z., Bloomquist E.W., Jia J., Qian L., Wilkinson A.H., Danovitch G.M., Pham P.-T.T., Rosenthal J.T., Lassman C.R., Braun J., Reed E.F., Gritsch H.A. Hum. Immunol. 2006;67:777. doi: 10.1016/j.humimm.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Trapani, J. A.; Smyth, M. J. WO 2005083098 A1, 1 March, 2005.
- 7.Spicer J.A., Huttunen K.M., Miller C.K., Denny W.A., Ciccone A., Browne K.A., Trapani J.A. Bioorg. Med. Chem. 2012;20:1319. doi: 10.1016/j.bmc.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 8.Spicer J.A., Lena G., Lyons D.M., Huttunen K.M., Miller C.K., O’Connor P.D., Bull M., Helsby N.A., Jamieson S.M.F., Denny W.A., Ciccone A., Browne K.A., Lopez J.A., Rudd-Schmidt J., Voskoboinik I., Trapani J. J. Med. Chem. 2013;56:9542. doi: 10.1021/jm401604x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopez J.A., Jenkins M.R., Rudd-Schmidt J.A., Brennan A.J., Danne J.C., Mannering S.I., Trapani J.A., Voskoboinik I. J. Immunol. 2013;191:2328. doi: 10.4049/jimmunol.1301205. [DOI] [PubMed] [Google Scholar]
- 10.Bull M.R., Spicer J.A., Huttunen K.M., Denny W.A., Ciccone A., Browne K.A., Trapani J.A., Helsby N.A. Eur. J. Drug Metab. Pharmacokinet. 2015;40:417. doi: 10.1007/s13318-014-0220-y. [DOI] [PubMed] [Google Scholar]
- 11.Baell J.B., Holloway G.A. J. Med. Chem. 2010;53:2719. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 12.Hayat S., Atta-ur-Rahman, Choudary M.I., Khan K.M., Bayer E. Tetrahedron Lett. 2001;42:1647. [Google Scholar]
- 13.Klapars A., Buchwald S.L. J. Am. Chem. Soc. 2002;124:14844. doi: 10.1021/ja028865v. [DOI] [PubMed] [Google Scholar]
- 14.Kumar, N.; Nayyer, S.; Mitu, G. Patent WO 2006103550 A1, 5 October, 2006.
- 15.Kobayashi K., Sugie A., Takahashi M., Masui K., Mori A. Org. Lett. 2005;7:5083. doi: 10.1021/ol052063y. [DOI] [PubMed] [Google Scholar]
- 16.Spicer, J. A.; Denny, W. A.; Miller, C. K.; O’Connor, P. D.; Huttunen, K.; Trapani, J. A.; Hill, G.; Alexander, K. Patent WO 2014028968 A1, 27 February, 2014.
- 17.Ernst, G.; Frietze, W.; Jacobs, R.; Phillips, E. Patent WO 2005061510 A1, 7 July, 2005.
- 18.Dally, R. D.; Dodge, J. A.; Hummel, C. W.; Jones, S. A.; Shepherd, T. A.; Wallace, O. B.; Weber, W. W. Patent WO 2005073205 A1, 11 August, 2005.
- 19.Effenberger F., Endtner J.M., Miehlich B., Münter J.S.R., Vollmer M.S. Synthesis. 2000;9:1229. [Google Scholar]
- 20.Giovanella B.C., Harris N., Mendoza J., Cao Z., Liehr J., Stehlin J.S. Ann. N.Y. Acad. Sci. 2000;922:27. doi: 10.1111/j.1749-6632.2000.tb07022.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures.