

Abstract
Infection with Plasmodium falciparum and vivax cause most cases of malaria. Emerging resistance to current antimalarial medications makes new drug development imperative. Ideally a new antimalarial drug should treat both falciparum and vivax malaria. Because malaria parasites are purine auxotrophic, they rely on purines imported from the host erythrocyte via Equilibrative Nucleoside Transporters (ENTs). Thus, the purine import transporters represent a potential target for antimalarial drug development. For falciparum parasites the primary purine transporter is the P. falciparum Equilibrative Nucleoside Transporter Type 1 (PfENT1). Recently we identified potent PfENT1 inhibitors with nanomolar IC50 values using a robust, yeast-based high throughput screening assay. In the current work we characterized the Plasmodium vivax ENT1 (PvENT1) homologue and its sensitivity to the PfENT1 inhibitors. We expressed a yeast codon-optimized PvENT1 gene in Saccharomyces cerevisiae. PvENT1-expressing yeast imported both purines ([3H]adenosine) and pyrimidines ([3H]uridine), whereas wild type (fui1Δ) yeast did not. Based on radiolabel substrate uptake inhibition experiments, inosine had the lowest IC50 (3.8 μM), compared to guanosine (14.9 μM) and adenosine (142 μM). For pyrimidines, thymidine had an IC50 of 183 μM (vs. cytidine and uridine; mM range). IC50 values were higher for nucleobases compared to the corresponding nucleosides; hypoxanthine had a 25-fold higher IC50 than inosine. The archetypal human ENT1 inhibitor 4-nitrobenzylthioinosine (NBMPR) had no effect on PvENT1, whereas dipyridamole inhibited PvENT1, albeit with a 40 μM IC50, a 1000-fold less sensitive than human ENT1 (hENT1). The PfENT1 inhibitors blocked transport activity of PvENT1 and the five known naturally occurring non-synonymous single nucleotide polymorphisms (SNPs) with similar IC50 values. Thus, the PfENT1 inhibitors also target PvENT1. This implies that development of novel antimalarial drugs that target both falciparum and vivax ENT1 may be feasible.
Keywords: Purines, Transporter, Malaria, Drug development, Nucleoside/nucleobase transport, Parasite, Plasmodium vivax, Single-nucleotide polymorphism (SNP)
Abbreviations: ACT, Artemisinin-based Combination Therapies; CQ, chloroquine; EC50, concentration causing 50% of maximal effect; ENT, equilibrative nucleoside transporter; EV, empty vector; hENT1, human ENT type 1; HTS, high throughput screen; IC50, concentration causing 50% inhibition; NBMPR, 4-nitrobenzylthioinosine; PfENT1, P. falciparum ENT type 1; PvENT1, P. vivax ENT type 1; SDM, synthetic defined media; SNP, single nucleotide polymorphism; WHO, World Health Organization; WT, wild type
Graphical abstract
Highlights
-
•
PvENT1 can be functionally expressed in Saccharomyces cerevisiae.
-
•
PvENT1 transports purine and pyrimidine nucleosides and nucleobases but does not transport nucleotides.
-
•
PvENT1 is inhibited by recently described PfENT1 inhibitors with similar potency.
-
•
Identified PvENT1 non-synonymous SNPs do not change PfENT1 inhibitor potency.
-
•
Plasmodium ENTs may be feasible target for development of novel antimalarial drugs.
1. Introduction
Malaria is a major global health problem and a socioeconomic burden in malaria endemic countries (Sachs and Malaney, 2002). According to the World Health Organization (WHO), in 2014 approximately 3.4 billion people were at risk for malaria infection (World Health Organization, 2014). Over 200 million clinical cases of malaria resulted in ∼600,000 deaths. Most deaths occurred in sub-Saharan Africa in young children and pregnant women (Snow et al., 2005, World Health Organization, 2014). Malaria is caused by infection with single-cell protozoan parasites from the genus Plasmodium. Five Plasmodium species infect humans (Plasmodium falciparum, vivax, malariae, ovale, and knowlesi). Ninety percent of clinical cases are due to infection with either P. falciparum or Plasmodium vivax (World Health Organization, 2014). P. falciparum is associated with the highest mortality (∼80% of all malaria-related deaths) but P. vivax infection is prevalent and associated with high morbidity (Rogerson and Carter, 2008, Anstey et al., 2009). The geographic overlap between P. falciparum and P. vivax endemic areas is significant, especially in tropical regions. Thus, new antimalarial drugs should target both species.
The development of resistance to antimalarial drugs has been a recurring problem. Chloroquine (CQ) was the mainstay of antimalarial chemotherapy until CQ resistance developed worldwide (Wellems and Plowe, 2001). In 2006, the WHO recommended Artemisinin-based Combination Therapies (ACT) as first-line treatment for P. falciparum infection. Unfortunately, resistance to current ACT regimens is expanding in Southeast Asia (Dondorp et al., 2011, Ariey et al., 2014, Hastings et al., 2015, Straimer et al., 2015). The fact that resistance to a three day ACT treatment course emerged in as little as a decade after the large scale introduction of ACTs as first line therapy underscores the importance of identifying new drug targets that take advantage of weaknesses in Plasmodium biology.
One potential target for the development of novel antimalarial drugs is the purine salvage pathway (Downie et al., 2008, Cassera et al., 2011, Frame et al., 2015a). Similar to other protozoa, Plasmodium species can perform de novo pyrimidine synthesis but are incapable of de novo purine synthesis (Manandhar and Van Dyke, 1975, Gero and O'Sullivan, 1990, Downie et al., 2008, Frame et al., 2015a). Therefore, Plasmodium parasites must import purines from the host cytoplasm. Imported purines are processed via the purine salvage pathway enzymes to form the purines required for RNA synthesis, DNA replication, and metabolism. Hence, the purine import and processing pathways are potential targets for antimalarial drug development (Downie et al., 2008, Ducati et al., 2013, Frame et al., 2015a).
Plasmodium parasites use equilibrative nucleoside transporters (ENT) to import purines (Landfear et al., 2004, Downie et al., 2008). Genomic sequence analysis of P. falciparum (3D7) and P. vivax (Sal I) (www.PlasmoDB.org) shows that both species possess four putative ENT homologues: PfENT1-4 and PvENT1-4 (Martin et al., 2005, Kirk and Lehane, 2014). P. falciparum ENTs have been studied more extensively. Multiple genetic, biochemical, and functional experiments show that PfENT1 is the principle route for purine uptake into the P. falciparum parasites. PfENT1 is localized to the parasite plasma membrane and transports both purine and pyrimidine substrates (Carter et al., 2000a, Parker et al., 2000, Rager et al., 2001, Riegelhaupt et al., 2010a). Genetic knockout of the PfENT1 gene (pfent1Δ) is lethal if the parasites are grown in concentrations of purines present in human blood, <10 μM (Traut, 1994, El Bissati et al., 2006, El Bissati et al., 2008, Frame et al., 2015b). However, PfENT1-knockout parasites survive when grown in culture with supra-physiologic purine concentrations (>100 μM) (El Bissati et al., 2006, El Bissati et al., 2008, Frame et al., 2015b). Thus, a secondary low affinity and/or low capacity purine transport pathway must be present, at least in the pfent1Δ parasites. The molecular basis for this secondary purine uptake pathway is unknown but may involve the AMP uptake pathway (Cassera et al., 2008) or possibly, PfENT4 (Frame et al., 2012).
To test whether chemical inhibition of PfENT1 would be lethal to P. falciparum parasites, we identified PfENT1 inhibitors using a yeast-based, high-throughput screen (HTS) (Frame et al., 2015b). We screened 64,500 compounds and identified 171 hits. Nine of the highest activity compounds that represent six distinct chemical scaffolds were characterized in depth. They blocked [3H]adenosine uptake into PfENT1-expressing yeast and into erythrocyte-free trophozoite stage parasites with 5–50 nM IC50 values and killed chloroquine-sensitive and -resistant P. falciparum parasites with 5–50 μM IC50 values (Frame et al., 2015b). These results provide strong support for the hypothesis that inhibition of purine uptake is a potential target for the development of novel antimalarial drugs.
Because of the extensive geographic overlap between vivax and falciparum malaria, an effective antimalarial drug should treat infection by both parasites. In the current work, we sought to characterize P. vivax ENT1 (PvENT1) functionally and determine whether the PfENT1 inhibitors also inhibit PvENT1. Based on its genomic sequence, PvENT1 is a 47.3 kDa, 416 amino acid protein. PvENT1 shares ∼75% amino acid sequence identity with PfENT1. However, unlike its P. falciparum homologue, the pvent1 gene is only moderately AT rich (57%; vs. 72% pfent1). Although no crystal structures are available for any known ENTs, glycosylation scanning and modeling algorithms support an 11-transmembrane segment topology with a cytoplasmic N-terminus and an extracellular C-terminus (Sundaram et al., 2001, Valdes et al., 2009). Hydropathy plots suggest that PvENT1 has a similar transmembrane topology to PfENT1. In the current work, we expressed PvENT1 in Saccharomyces cerevisiae. We showed that a similar group of purine and pyrimidine nucleobases and nucleosides competed with radioactive uridine or adenosine uptake into PvENT1-expressing yeast, but the measured IC50s were distinct from those previously reported for PfENT1-expressing yeast (Frame et al., 2015b). We determined the sensitivity of PvENT1 to the recently identified PfENT1 inhibitors. All nine PfENT1 inhibitors block PvENT1 with similar efficacy. Furthermore, the five known PvENT1 non-synonymous single nucleotide polymorphisms (SNP) have minimal impact on inhibitor potency.
2. Materials and methods
2.1. Purine auxotrophic yeast (ade2Δ)
All genetically modified yeast were created using the S. cerevisiae BY4741strain that also had a deletion of the FUI1 gene (MATa; his3Δ 1; leu2Δ 0; met15Δ 0; ura3Δ 0; fui1Δ::KanMX4) (Winzeler et al., 1999). To disrupt de novo purine synthesis in S. cerevisiae, we replaced the yeast ADE2 gene (Chr. 15; phosphoribosyl-aminoimidazole carboxylase) with the hphNT1 (hygromycin B phosphotransferase) selectable marker using homologous recombination. Briefly, using two-step PCR, we created 45 nt 5′ and 3′ ade2 homology arms flanking the hphNT1 gene (which was amplified from the pFA6a-hphNT1 plasmid; gift from Dr. Ian Willis). The following sets of primers were used: 1st-stepF: 5′−GACAAAACAATCAAGTATGCGTACGCTGCAGGTCGACGGATCCCCG-3′, 1st stepR: 5′-GTATATCAATAAACTTATATATTAATCGATGAATTCGAGCTCG-3′; 2nd-stepF: 5′-AACAATCAAGAAAAACAAGAAAATCGGACAAAACAATCAAGTATG-3′, 2nd stepR: 5′-TTATAATTATTTGCTGTACAAGTATATCAATAAACTTATATATTA-3′. The underlined portion corresponds to the sequence of yeast chromosomal DNA flanking the ADE2 gene. Each 100 μL PCR reaction contained 50 ng DNA, 1x PfuUltraII Reaction Buffer, 250 μM of each dNTP (dATP, dCTP, dGTP, dTTP), 0.2 μM of each primer (F/R), 2% (v/v) PfuUltraII Fusion HS DNA Polymerase (Agilent). The PCR conditions were: 1 cycle—95 °C (2 min); 26 cycles—95 °C (30 s), 60 °C (30 s), 72 °C (30 s); 1 cycle—72 °C (2 min), 4 °C (hold). The PCR product was verified using ethidium bromide agarose gel electrophoresis, excised, and column purified. The amplicon was transformed into yeast (see below) and plated on YPD + 500 μM hygromycin B selection plates at 30 °C. The resulting yeast strain was MATa; his3Δ 1; leu2Δ 0; met15Δ 0; ura3Δ 0; fui1Δ::KanMX4, ade2Δ::hphNT1. Disruption of the ade2 gene was confirmed by PCR. The purine auxotrophic yeast was pigmented red and displayed a retarded growth phenotype under purine starved conditions (Kokina et al., 2014). Single colonies of the purine auxotrophic yeast were picked and expanded for transformation with the pCM189 constructs.
The purine auxotrophic yeast strain with the ADE2 gene deletion (ade2Δ::hphNT1) was used in all experiments in this paper and was either transformed with an episomal expression plasmid containing the PvENT1 gene or an empty vector (EV) lacking the PvENT1 gene. All subsequent references to “yeast” in the Results section refer to this purine auxotrophic strain.
2.2. PvENT1 DNA plasmid constructs
We synthesized a yeast codon-optimized version of the PvENT1 gene (Gene Designer® DNA2.0) based on the inferred amino acid sequence of PVX_083260 gene sequence in PlasmoDB (http://www.plasmodb.org/plasmo/) (DNA sequence available on request). The construct contained a 5′ BamHI restriction site, the 1251 bp codon-optimized gene, and a 3′ EcoRI restriction site. The construct was sub-cloned into the yeast pCM189 plasmid (Gari et al., 1997) using BamHI/EcoRI restriction enzymes and T4 DNA ligase. The endogenous EcoRI site upstream of the tetracycline regulatable element in pCM189 was deleted using site-directed mutagenesis prior to cloning steps. The plasmid is a low copy number, tetracycline regulatable yeast expression vector; selectable markers include an ampicillin resistance (AmpR; for maintenance in E. coli) and the orotodine 5′-phosphate decarboxylase (Ura3; for maintenance in S. cerevisiae). A strong synthetic promoter based on the CYC1 promoter drives PvENT1 expression. The final construct generated was pCM189tetOff-PvENT1 (Gari et al., 1997). The empty vector (EV) construct pCM189-EV lacked only the transporter gene. All sequences were verified by DNA sequencing (Genewiz, Inc.).
PvENT1 SNP mutant constructs were generated using the QuikChange site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's recommendations. Point mutations were verified by DNA sequencing.
2.3. Yeast transformation with PCR amplicon and pCM189 constructs
Yeast was transformed with plasmid or PCR DNA using the standard lithium-acetate/DMSO (8% v/v) method (Hill et al., 1991). For each transformation 10 mL of yeast culture was grown overnight in YPD media until the cell density reached ∼2 × 107 cells/mL. The culture was pelleted at 3500 × g, 5 min (room temperature; RT) and the yeast pellet was washed once with cold lithium acetate buffer (LiOAc: 100 mM LiOAc, 10 mM Tris pH 8; using 2 PBS vol/culture vol). The final pellet was resuspended with 100 μL of LiOAc to which 10 μL of salmon-sperm DNA (2 mg/mL) and 0.5–1 μg of DNA (PCR or construct) was added. The solution was incubated at RT for 5 min 280 μL of 50% (w/v) PEG (polyethylene glycol 3350) was added. The PEG-LiOAc suspension was incubated at 30 °C for 45 min. DMSO was added to the mixture and the sample was heat shocked at 42 °C for 15 min. Transformed samples were pelleted at 16,300 × g, 30 s (RT) and resuspended in appropriate media before being transferred onto selection plates. For the creation of ade2Δ::hphNT1 yeast, the final pellet was first resuspended in 10 mL YPD, allowed to recover for 3 h, pelleted again, and resuspended into a plating volume containing 500 μM hygromycin B. The final strains used in experiments had the following genotype: MATa; his3Δ 1; leu2Δ 0; met15Δ 0; ura3Δ 0; fui1Δ::KanMX4, ade2Δ::hphNT1 containing either pCM189-EV or pCM189-PvENT1 constructs.
2.4. Yeast growth media and determination of cell density
Yeast cells were grown in YPD media: 1% (w/v) yeast extract, 2% (w/v) peptone, and 2% (w/v) glucose. Purine auxotrophic yeast were maintained in synthetic defined media (SDM) containing 2% (w/v) dextrose, 0.5% (w/v) ammonium sulfate, 0.17% (w/v) yeast nitrogen base (US Biologicals, #Y2030), 0.02% (w/v) nutritional dropout mix (US Biologicals, cat# D9542; lacking uracil, adenine, histidine, and tryptophan), 40 mg/L tryptophan, and 40 mg/L histidine. Supplemental adenine (300 μM) or adenosine (1 mM) was added for culture maintenance of the EV and PvENT1 expressing yeast, respectively. Solid growth media contained the same SDM formulation supplemented with either 1 mM adenine or 10 mM adenosine in 2% (w/v) agar.
Yeast cell density was determined spectrophotometrically (Biorad Benchmark Plus) from 200 μL samples in 96-well clear, flat-bottom plates (Corning, #3596). OD600 = 0.018 correlated to ∼2 × 106 cells/mL using our experimental conditions.
2.5. Yeast growth and bioscreen assays
Purine auxotrophic yeast growth was assessed on both solid and liquid media where adenine or adenosine was the sole purine source. Briefly, the EV- and PvENT1-expressing yeast were grown in SDM-adenine media to mid-log phase, harvested at 3400 × g for 5 min (RT), and washed three times in sterile water. The final yeast cell pellet was diluted to 4 × 106 cells/mL in water. 96-well TC-treated plates were preloaded with 100 μL of purine serially diluted in 2× SDM lacking purine. 100 μL of cells were added to the plates and resuspended three times. The cells were incubated at 30 °C and evaluated for growth (OD600) after 17–21 h. Biological replicates (n ≥ 4) were done on different days. For growth on solid media, yeast were grown to a density of 106 cells/mL, serially diluted in water (1:5), inoculated onto SDM-agar plates (3000 cells/spot) and incubated at 30 °C for 2–3 days.
Bioscreen growth curve analysis for PvENT1 WT and SNP mutants was done using a Bioscreen C machine (Growth Curves USA, Piscataway, NJ). Briefly, cultures were grown in the appropriate yeast media to mid-log phase, washed as previously noted, and then diluted to ∼2 × 106 cells/mL (OD600 = 0.018) either in 1 mM adenine or 10 mM adenosine media. Three aliquots of 150 μL culture (per strain/per condition) were added to a 100-well plate. Growth at 30 °C was monitored every 15 min at OD600 for 72 h.
2.6. Inhibition of yeast growth with small molecule compounds
We evaluated the viability of purine auxotrophic yeast grown in the presence of various inhibitors. Briefly, 80 μL of exponentially growing PfENT1-expressing yeast (∼400,000 cells) were added to each well of a 384-well microplate (black, clear/flat-bottom, polystyrene; Corning #3712). The cells were grown in 1 mM adenosine—this concentration was picked based on growth that allowed maximum signal detection without saturating the culture/machine detection. Compound was serially diluted 3- or 4- fold in DMSO, and 0.8 μL was added to each well and resuspended three times. The final DMSO concentration was ∼1% (v/v). The plates were incubated at 30 °C for 15–17 h. Growth was evaluated at OD600, values were normalized to maximum growth with DMSO only and maximum death values for each compound. The normalized data was fit to a variable slope concentration-response model with no constraints to determine the concentration that gave half-maximal growth inhibition (IC50) using Prism 6.02 software (GraphPad). Biological replicates (n ≥ 4) were done on different days.
2.7. Radiolabel uptake time course experiments
PvENT1-expressing yeast were grown to mid-log phase in SDM +1 mM adenosine overnight at 30 °C. Cells were harvested by centrifugation at 3500× g for 1 min, RT. Cells were washed three times in yeast PBS (150 mM NaCl, 10 mM KH2PO4, 40 mM K2HPO4, 11 mM glucose, pH 7.4). The yeast pellet was resuspended in PBS to a final concentration of 2 × 108 cells/mL. 96-well microplate wells were loaded with 100 μl of 100 nM [3H]adenosine ([2,8-3H]adenosine; 35 Ci/mmol) or 500 nM [3H]uridine ([2,8-3H]uridine; 8.5 Ci/mmol). All radiolabels were purchased from Moravek Biochemicals. 100 μL of yeast cell suspension was added to the radiolabels at the appropriate times to generate the time-course: 0, 0.5, 1, 2.5, 5, 10, 15, 20, 30, 45, and 60 min. At the conclusion of the experiment, cells were harvested onto glass fiber filtermats (Filtermat A, GF/C; Perkin Elmer) using a TomTec 96-well cell harvester system (#96-3-469). Filtermats were dried, sealed in plastic bags containing Betaplate Scint LSC fluid (Perkin Elmer). Samples were counted using a microplate scintillation counter (1450 MicroBeta TriLux, Perkin Elmer). Total counts are represented as CPM/106 cells (mean ± SD) from n ≥ 3 independent experiments. The data was fit using a linear-regression model with no constraints (Prism 6.02).
2.8. Radiolabel-uptake inhibition by purines and pyrimidines
PvENT1-expressing yeast were grown and prepared as described above. To measure the concentration dependent inhibition of radiolabel uptake, 96-well plates were preloaded with 50 μL of 200 nM [3H]adenosine for pyrimidines or 1 μM [3H]uridine for purines in PBS. 50 μL of purine or pyrimidine (serially diluted 3- or 4- fold in buffer) was added to each well and resuspended. 100 μL yeast (2 × 108 cells/mL) were added to each well and incubated at RT for 15 min. At the end of each experiment, cells were harvested and radioactivity was counted. Biological replicates (n ≥ 3) were done on different days.
2.9. [3H]adenosine uptake inhibition by compounds
Compounds identified as PfENT1 inhibitors were purchased from Chembridge. Validation of purchased compounds by mass spectrometry and NMR were published previously (Frame et al., 2015b). To measure the concentration dependence of compound inhibition of [3H]adenosine uptake, 96-well plates were preloaded with 100 μL of 100 nM [3H]adenosine in PBS. 0.5 μL of compound (serially diluted 3- or 4- fold in DMSO, as described above) was added to each well and resuspended. 100 μL yeast (2 × 108 cells/mL) were added to each well and incubated at RT for 15 min. At the end of each experiment, cells were harvested and radioactivity was counted. Biological replicates (n ≥ 3) were done on different days.
3. Results
3.1. Expression of P. vivax ENT1 in yeast
The amino acid sequences of PvENT1 and PfENT1 are 75% identical. Since PfENT1 is a low affinity, broad-spectrum transporter of nucleosides and nucleobases, we hypothesized that PvENT1 would also recognize and transport these substrates. To study PvENT1, we expressed PvENT1 in S. cerevisiae yeast. We used a yeast codon-optimized version of the PvENT1 gene because species-specific differences in codon usage can affect gene expression levels (codon usage bias) (Hershberg and Petrov, 2009, Downie et al., 2010, Frame et al., 2012, Frame et al., 2015b). We used purine auxotrophic S. cerevisiae generated by deletion of the ADE2 gene, a critical enzyme in the yeast's de novo purine synthetic pathway. Purine auxotrophic ade2Δ yeast grow poorly when purine starved and display a characteristic red color phenotype (Dorfman, 1969, Kokina et al., 2014). Adenine can enter ade2Δ yeast cells via the endogenous FCY2 nucleobase transporter and rescue growth (Schmidt et al., 1984). Yeast do not encode an endogenous adenosine transporter. Thus, growth of the purine auxotrophic yeast with adenosine as the sole purine source can only occur if adenosine can enter the cell through the PvENT1 transporter.
Yeast transformed with either an empty vector (EV) or PvENT1 construct displayed similar concentration-dependent growth in liquid culture media containing adenine as the sole purine source (Fig. 1A) (EC50: 39 ± 2 μM EV vs. 27 ± 2 μM PvENT1). In contrast, PvENT1-expressing yeast grew in adenosine media (growth EC50: 394 ± 85 μM), while EV yeast failed to grow (Fig. 1A). Similar growth phenotypes were observed for the yeast cells grown on solid media (data not shown). Growth rescue with adenosine as the sole purine source demonstrated that the yeast cells produced functional PvENT1 protein. We assume that the PvENT1 protein trafficked to the yeast plasma membrane but we did not explicitly demonstrate this.
Fig. 1.

Function of PvENT1 in purine auxotrophic yeast. (A) The growth of purine auxotrophic yeast transformed with either empty vector (EV) or PvENT1 depends on the media concentration of adenine (Ade, empty symbols) or adenosine (Ado, filled symbols) when present as the sole purine source. Data points represent the mean ± SD of 3 technical replicates. EC50 values indicated in the text were measured from n ≥ 3 independent biological replicates. (B and C) PvENT1-mediated uptake of (B) 50 nM [3H]adenosine or (C) 250 nM [3H]uridine in purine auxotrophic yeast transformed with either empty vector or PvENT1. Data points in (B) and (C) represent counts (mean ± SD) over the course of 30 min from n ≥ 3 independent experiments.
Next, we examined whether PvENT1-expressing yeast could transport two radiolabeled substrates, adenosine and uridine. PvENT1-expressing yeast cells displayed time-dependent, linear accumulation of both [3H]adenosine and [3H]uridine (Fig. 1B and C, circles). As expected, EV transformed yeast not expressing PvENT1 did not accumulate either [3H]adenosine or [3H]uridine (Fig. 1B and C, squares). Since yeast lack an endogenous adenosine transporter and the endogenous FUI1 uridine transporter was deleted (fui1Δ), uptake of both radiolabeled substrates is only possible through PvENT1. The uptake of both purines and pyrimidines is consistent with the broad substrate specificity of the transporter.
3.2. PvENT1 mediated transport of purines and pyrimidines
We investigated whether other purines and pyrimidines could affect PvENT1-mediated uptake of radiolabeled adenosine or uridine. To avoid the potential for competition between the test substance and the radiolabeled substrate at cytoplasmic metabolic enzymes (Kirk et al., 2009, Riegelhaupt et al., 2010a), we used the inhibition of [3H]uridine uptake to characterize purine uptake and inhibition of [3H]adenosine to characterize pyrimidine uptake. Thus, the radiolabel is metabolized by different cytoplasmic enzymes than the competing cold substance. The only potential site of competition between the test substance and radioactive substrate is at the transporter. For these experiments, we assume that inhibition of radiolabel uptake is due to the competitive transport of the purine or pyrimidine test substance by the transporter. Unfortunately, due to financial constraints we did not have radiolabeled versions of all of the test substances to demonstrate that they were all transported into the yeast via PvENT1.
We quantified the observed effects as the IC50 of the test substance to inhibit the radiolabeled substrate uptake into the yeast. It should be noted that for these experiments, the ratio of the tritiated substrate concentration used in the uptake competition experiments to the test substance Km was between 10−3 and 10−4. Thus, by the Cheng–Prusoff equation the IC50 ≈ Ki, where Ki is the inhibition constant of the test substance for the transporter (Cheng and Prusoff, 1973).
We evaluated 11 purines for their ability to inhibit [3H]uridine uptake into PvENT1-expressing yeast. These included nucleobases, nucleosides, and nucleotides. Experiments were conducted at 15 min, which was within the linear phase of radiolabel uptake (Fig. 1C). Fig. 2A illustrates the experimental data for determination of IC50 values for the inhibition of radiolabel uptake by the purines inosine, hypoxanthine and xanthine. In this case, similar results were obtained whether the radiolabel substrate was a purine (adenosine) or a pyrimidine (uridine) implying that in both cases the inhibition of radiolabel uptake is due to competition at the PvENT1 transporter and not at a cytoplasmic metabolic enzyme. IC50 values for all purines tested are shown in Table 1. The nucleosides inosine and guanosine had the lowest IC50 values, in the low μM range, while their nucleobases were 50 and 300 times higher, respectively (Table 1). Adenosine had an IC50 value of 142 μM while the IC50 for its nucleobase adenine was one order of magnitude higher (Table 1).
Fig. 2.

Inhibition of uptake of [3H]adenosine or [3H]uridine into PvENT1-expressing yeast in the presence of selected purines and pyrimidines. (A) Uptake inhibition of radiolabel by select purine substrates after 15 min incubation period. (Urd) represents uptake of 250 nM [3H]uridine and (Ado) represents the same experiment done with 50 nM [3H]adenosine. (B) Uptake inhibition of 50 nM [3H]adenosine by select pyrimidine substrates in the uracil series after 15 min incubation. Single experiment traces are shown in (A) and (B). Inhibition of radiolabel uptake by purine/pyrimidine substrates were fit to a non-linear regression model, unless there was no observable effect (e.g., UMP).
Table 1.
IC50 values for inhibition of [3H]Uridine uptake by purines and [3H]Adenosine uptake by pyrimidines. Inhibition of uptake of [3H]uridine or [3H]adenosine by varying concentrations of purine/pyrimidine substrates evaluated in PvENT1-expressing yeast. IC50 values were obtained from concentration-response experiments similar to that presented in Fig. 2. Results are expressed as mean ± SD (μM) from n ≥ 3 independent experiments. Values in parenthesis are IC50 values determined using [3H]adenosine for purines or [3H]uridine for pyrimidines, (Urd = 250 nM [3H]uridine; Ado = 50 nM [3H]adenosine). NE signifies no observable effect on uptake of radiolabel at the concentrations tested.
| Purine | IC50 for inhibition of [3H]Uridine by indicated purine (μM) | Pyrimidine | IC50 for inhibition of [3H]Adenosine by indicated pyrimidine (μM) |
|---|---|---|---|
| Adenine | 1430 ± 736 (278 ± 62 Ado) | Thymine | 564 ± 145 |
| Adenosine | 142 ± 34 (74 ± 8 Ado) | Thymidine | 183 ± 53 (299 ± 77 Urd) |
| AMP | NE | Uracil | 3390 ± 1170 |
| Hypoxanthine | 99 ± 30 (81 ± 13 Ado) | Uridine | 1730 ± 809 |
| Inosine | 3.8 ± 1.4 (2.7 ± 0.7 Ado) | 5-Fluorouridine (5-FUrd) | 1770 ± 302 |
| Xanthine | 2550 ± 189 | UMP | NE |
| Xanthosine | 4310 ± 2870 | Cytosine | 6140 ± 1030 |
| Guanine | 6340 ± 2950 | Cytidine | NE |
| Guanosine | 14.9 ± 1.4 | 2-deoxycytidine | NE |
| 2-deoxyguanosine | 20.5 ± 4.8 | CMP | NE |
| GMP | NE | Orotic acid | NE |
Similarly, we evaluated 11 pyrimidines for their ability to inhibit the uptake of 50 nM [3H]adenosine. Fig. 2B illustrates the experimental data for determination of IC50 values for the inhibition of [3H]adenosine uptake into PvENT1-expressing yeast by members of the uracil series of pyrimidines. IC50 values for all pyrimidines tested are shown in Table 1. The nucleoside thymidine had the lowest IC50 of all pyrimidines tested (183 μM) followed by its nucleobase thymine (564 μM) (Table 1). The nucleobase cytosine caused slight inhibition of [3H]adenosine uptake while the remaining members of the family had negligible effects. The uracil family of pyrimidines had IC50 values in the low mM range (Fig. 2B, Table 1). 5-Fluorouridine (5-FUrd), a toxic uridine analog, had an IC50 value of 1.7 mM.
None of the purine or pyrimidine nucleotides tested inhibited the uptake of [3H]adenosine or [3H]uridine. This implies that nucleotides are not substrates for PvENT1. Overall, our data show that PvENT1 recognizes both purine and pyrimidine substrates in the form of nucleosides and nucleobases.
Substitution of choline chloride for NaCl in the [3H]adenosine uptake media had a minimal impact on 15 min [3H]adenosine uptake (data not shown). This indicated that purine uptake was not Na+ coupled in PvENT1.
3.3. Effect of hENT1 inhibitors and cytotoxic purines on PvENT1-mediated purine uptake
Mammalian ENTs are categorized based on their sensitivity to the inhibitor NBMPR (Griffiths et al., 1997, Baldwin et al., 2004). Human hENT1 is inhibited by sub-nanomolar NBMPR concentrations, whereas hENT2 requires μM-mM concentrations for complete inhibition. Similarly, hENT1 is inhibited by the coronary vasodilator dipyridamole in the 5–20 nM concentration range (Griffiths et al., 1997, Baldwin et al., 2004). NBMPR did not inhibit [3H]adenosine uptake into PvENT1-expressing yeast at concentrations up to 20 mM. Dipyridamole inhibited the transporter with an IC50 ∼ 40 μM, about three orders of magnitude higher than the IC50 for hENT1 inhibition (Table 2).
Table 2.
IC50 Values for inhibition of [3H]Adenosine uptake by human hENT1 inhibitors and cytotoxic purine derivatives. Effect of hENT1 inhibitors (NBMPR, nitrobenzylthioinosine; dipyridamole) and toxic purine analogs, 6-mercaptopurine and tubercidin (7-deaza-adenosine) on uptake inhibition of 50 nM [3H]adenosine into PvENT1-expressing yeast. Inhibition was evaluated over a range of concentrations up to 20 mM. IC50 values are represented as the mean ± SD from n = 3 separate experiments. NE signifies no observable effect.
| Compound | [3H]Adenosine uptake inhibition IC50 (μM) |
|---|---|
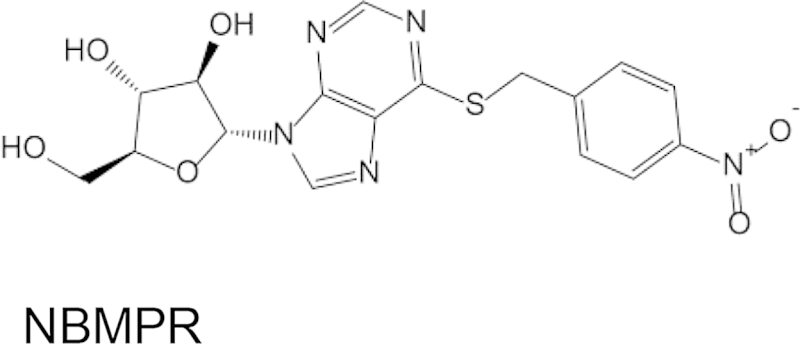 |
NE |
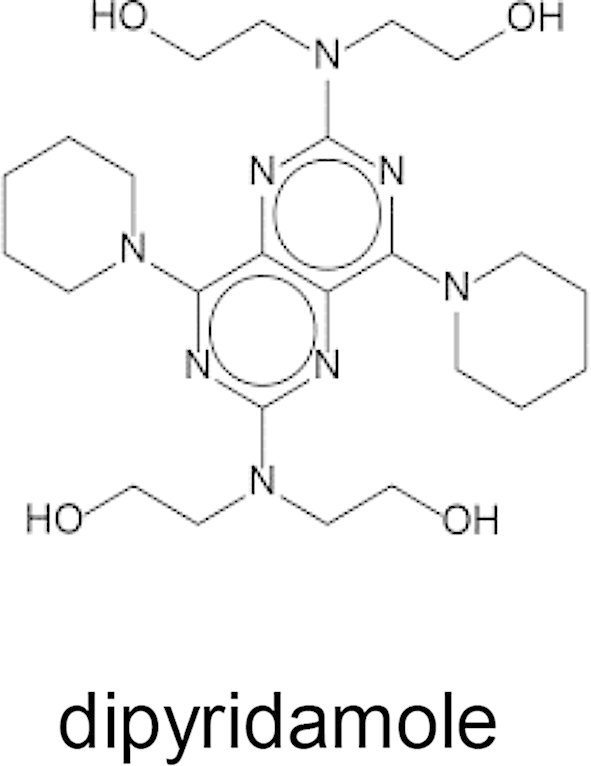 |
40 ± 21 |
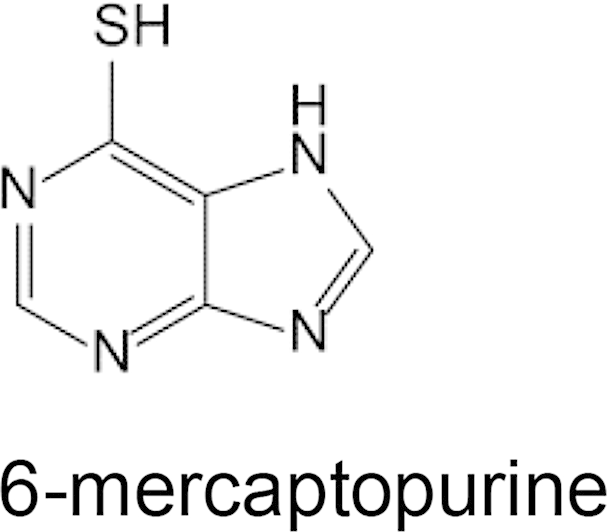 |
158 ± 31 |
 |
NE |
To explore determinants of purine recognition, we also examined the ability of two purine analogs to inhibit uptake of [3H]uridine. The immunosuppressive drug 6-mercaptopurine (6-MP) had an IC50 value of 160 μM. Surprisingly, tubercidin, a 7-deaza-adenosine derivative, failed to inhibit [3H]adenosine uptake (Table 2). Tubercidin is transported by PfENT1 (Riegelhaupt et al., 2010a). This suggests that the adenosine N7 position, which is absent in tubercidin but present in adenosine, is an important structural determinant for substrate recognition by PvENT1.
3.4. Effect of small molecule PfENT1 inhibitors on PvENT1
Using a yeast-based high throughput screen (HTS), we recently identified small molecule inhibitors of PfENT1 (Frame et al., 2015b). Of the 171 hits identified, we validated the efficacy of nine of the highest activity compounds, representing six different chemotypes (Fig. 3A) using a series of yeast- and parasite-based assays. In the current work, we explored the ability of these nine inhibitors to block PvENT1 using two different assays: 1) inhibition of [3H]adenosine uptake into PvENT1-expressing yeast, and 2) inhibition of the growth of PvENT1-expressing purine auxotrophic yeast with adenosine as the sole purine source. All nine compounds produced concentration-dependent inhibition of [3H]adenosine uptake with IC50 values in the 2–40 nM range (Fig. 3B, Table 3). We assume that these compounds that inhibit radiolabeled substrate uptake in the nanomolar concentration range were blocking the transporter and were not transported. However, we do not have experimental evidence to support this assumption (Frame et al., 2015b). For comparison purposes, Table 3 includes the IC50 values obtained in the comparable experiment with PfENT1-expressing yeast (Frame et al., 2015b). The IC50 values for inhibition of [3H]adenosine uptake by PvENT1-and PfENT1-expressing yeast are within a factor of five of each other. Because the compounds have similar efficacy in the yeast based inhibition of [3H]adenosine uptake assays with PfENT1 and PvENT1 and all nine compounds inhibited P. falciparum proliferation in culture (Frame et al., 2015b), we hypothesize that they will also inhibit P. vivax proliferation. However, due to the difficulty of growing P. vivax in culture we cannot currently test this.
Fig. 3.

Structures of nine PfENT1 inhibitors and the concentration–response relationships for the effects on PvENT1. (A) PfENT1 inhibitors identified in a yeast-based HTS and characterized in yeast and parasite based assays as described previously (Frame et al., 2015b). Structures of the nine compounds are labeled by their Rank Order # from the initial screen. The nine compounds represent six distinct chemotypes. (B) Concentration–response relationships for the effects of the nine PfENT1 inhibitors on [3H]adenosine uptake into PvENT1-expressing yeast. CPM, counts per minute. Results are from one representative experiment.
Table 3.
Efficacy of PfENT1 inhibitors against PvENT1 using two assays of PvENT1 function. IC50 values for inhibition of [3H]adenosine uptake into or growth of PvENT1-expressing yeast by nine PfENT1 inhibitors. IC50 values are represented as the mean ± SD from n ≥ 3 separate experiments. Compound numbers based on Rank Order# in HTS (Frame et al., 2015b). Compound structures are in Fig. 3A. Data for IC50 values for compound efficacy against PfENT1 in the [3H]adenosine uptake assay are shown for comparison.
| Compound# | PvENT1 IC50 in [3H]Adenosine uptake assay (nM) | PfENT1 IC50 in [3H]Adenosine uptake assay (nM)a | PvENT1 IC50 for growth inhibition of purine auxotrophic yeast in adenosine containing media (nM) |
|---|---|---|---|
| 1 | 1.8 ± 0.8 | 3.0 ± 1.3 | 63 ± 26 |
| 2 | 22.4 ± 11.8 | 10 ± 8.4 | 703 ± 178 |
| 3 | 2.6 ± 0.9 | 2.4 ± 1.5 | 77 ± 8 |
| 4 | 32.3 ± 19.9 | 13.7 ± 7.5 | 1170 ± 188 |
| 5 | 31.7 ± 7.6 | 22.7 ± 7.2 | 785 ± 390 |
| 6 | 9.5 ± 3.7 | 9.6 ± 7.4 | 396 ± 50 |
| 7 | 5.2 ± 2.6 | 10.9 ± 6.8 | 155 ± 26 |
| 13 | 7.5 ± 4.0 | 3.9 ± 1.6 | 159 ± 50 |
| 19 | 8.3 ± 0.9 | 38.4 ± 16.5 | 234 ± 41 |
PfENT1 data from (Frame et al., 2015b).
Furthermore, all nine compounds inhibited the growth of PvENT1-expressing purine auxotrophic yeast with adenosine as the sole purine source in a concentration-dependent manner with IC50 values ranging from 60 nM to 1.1 μM (Table 3). A linear regression fit between the IC50 values in the two assays had a slope = 30 (95% confidence interval = 23 to 38; R2 = 0.93). Thus, on average, the compound IC50 values were ∼30-fold higher in the growth assay compared to the radiolabel uptake experiments (Table 3).
3.5. Effect of PvENT1 non-synonymous SNPs on purine IC50 values and sensitivity to inhibitors
The substrate and inhibitor profiles described above are for the PvENT1 amino acid sequence encoded by the gene from the El Salvador I isolate (Carlton et al., 2008). To date, genomic sequencing of 170 P. vivax field isolates has identified six unique non-synonymous SNPs in the PvENT1 gene (PVX_083260). Two of the SNPs, M99I and Q367K, always appear together (http://www.plasmodb.org/plasmo/showRecord.do?name=GeneRecordClasses.GeneRecordClass&project_id=PlasmoDB&source_id=PVX_083260, accessed 6/30/15). The SNPs, strain and protein locations are: D23E (Thailand VKTS-52, N-terminus), M99I/Q367K double mutant (Brazil strain; TM 3 and TM10-11 loop), Q178K (Columbia_30103103280, TM5-6 loop), L188M (PNG58 and others, TM6), and N329S (N. Korea strain; TM 9). We evaluated the functional significance of the PvENT1 polymorphisms using the yeast expression system.
Yeast expressing WT and each of the SNP mutant PvENT1s grew at a similar rate with adenine as the sole purine source (Fig. 4A left panel and B). This was expected because the yeast express an endogenous adenine transporter. With adenosine as the sole purine source, the M99I/Q367K double mutant PvENT1-expressing yeast grew at rates comparable to WT (Fig. 4A right panel and C). The D23E, Q178K, and L188M mutants also had comparable growth rates (data not shown). This implied that these yeast all expressed PvENT1 to a similar extent and with similar transport capacity. The Q178K and L188M mutations increased the hypoxanthine IC50 values (Table 4). D23E, Q178K and L188M increased the inosine IC50 values by 6- to 26-fold. In contrast, none of the adenosine IC50 values were significantly different than WT, although the Q178K IC50 value was 3 times higher (Table 4).
Fig. 4.

Analysis of PvENT1-dependent growth of SNP mutations expressed in yeast. (A) Equal number of mid-log phase PvENT1 WT and SNP mutant strain yeast cells were serially diluted 5-fold and spotted onto adenine (1 mM; left) or adenosine (10 mM; right) agar media plates. (B and C) Bioscreen analysis growth curves for WT and PvENT1 SNP mutant strains grown in liquid culture media containing (B) 1 mM adenine and (C) 10 mM adenosine as the sole purine source. OD600 measurements were obtained at 15-min intervals (see Materials and Methods). Note in some places the traces for the different mutants are overlapping.
Table 4.
Purine IC50 values for inhibition of 3H-substrate uptake by yeast expressing PvENT1 containing mutations identified in P. vivax field isolates with non-synonymous SNPs in the pvent1 gene. [3H]uridine uptake was used to assess PvENT1 activity. #Indicates that the radiolabel used was [3H]adenosine, not [3H]uridine. *Significantly different than WT (p < 0.05), unpaired Student t-test unequal variance. Values are mean ± SD.
| IC50 values for inhibition of [3H]substrate uptake by the indicated purine (μM) | |||
|---|---|---|---|
| SNP | Hypoxanthine | Inosine | Adenosine |
| WT | 141 ± 24# | 5 ± 4# | 167 ± 52 |
| D23E | 150 ± 164 | 123 ± 113 | 94 ± 93 |
| M99I/Q367K | 107 ± 13# | 6 ± 2# | 206 ± 28 |
| Q178K | 530 ± 110* | 32 ± 5* | 600 ± 270 |
| L188M | 1200 ± 780 | 130 ± 23* | 190 ± 120 |
| N329S | 158 ± 18# | 10 ± 4# | 205 ± 31 |
The growth behavior of yeast expressing the N329S mutation was distinctly different from WT and the other SNPs. The N329S yeast grew on adenine as the sole purine source implying that the purine salvage pathway was intact. In contrast, with adenosine as the sole purine source, N329S-PvENT1-expressing yeast behaved similar to the EV-transformed yeast—the growth rate was minimal even in the presence of 10 mM adenosine (Fig. 4C). This phenotype was observed with N329S-PvENT1-expressing yeast from several independently transformed colonies and from two independent transformations. However, if the N329S-expressing yeast were grown in media with adenine as the purine source, we could measure [3H]adenosine uptake. While the IC50 values for inhibition of [3H]adenosine uptake by three purines were similar to WT PvENT1 (Table 4) the amount of uptake at 15 min was 5–10-fold lower than the amount of uptake into WT-expressing yeast. Thus, the N329S mutant transported [3H]adenosine, but either the rate of transport per transporter or the number of N329S transporters in the plasma membrane was lower than for WT. Differences in the media pH and composition in the uptake and growth assays did not affect N329S function. The [3H]adenosine uptake experiments were done at pH 7.4 whereas the growth media had a pH ∼4.2. [3H]adenosine uptake in 15 min for both WT- and N329S-expressing yeast was comparable in PBS at pH 4.2 and 7.4, the absolute amounts were different for WT and N329S (n = 5, data not shown). Furthermore, the amount of [3H]adenosine taken up in 15 min was comparable whether the buffer was PBS or SDM media (n = 4, data not shown). Thus, neither a difference in the effect of media pH on the N329S mutant or the media composition explains the failure of N329S-expressing yeast to grow with adenosine as the sole purine source.
We determined whether the PvENT1 non-synonymous SNP mutations altered the sensitivity to the PfENT1 inhibitor compounds (Fig. 3A). For most of the compounds, the PvENT1 SNPs had IC50 values comparable to WT PvENT1 in the [3H]adenosine uptake inhibition assays. For the D23E mutant, the IC50 values for compounds 3, 6 and 7 were ∼3.5 fold higher than WT (p < 0.05, data not shown), but the mutant was more sensitive than WT to compounds 1 and 4. The N329S mutant induced a 2.6 fold increase in the IC50 of compound 2 relative to WT. No SNP caused a similar effect across all nine compounds. The results demonstrated that the SNP mutations do not confer resistance to the majority of the PfENT1 inhibitors identified and characterized in our initial screen.
4. Discussion
Multidrug-resistant malaria parasites have become more common. Thus it is imperative to develop new antimalarial compounds that target novel aspects of parasite biology. Inhibition of the purine import pathway(s) is one potential target for the development of novel antimalarial drugs (Carter et al., 2001, Downie et al., 2006, El Bissati et al., 2006, Riegelhaupt et al., 2010a, Frame et al., 2015b). However, a major hurdle has been the difficulty in developing HTS assays to identify inhibitors of equilibrative nucleoside transporters. Recently, we described a yeast-based growth assay that we used to screen a 64,500 compound library. We identified inhibitors of PfENT1, the primary purine import pathway in P. falciparum parasites (Frame et al., 2015b). Nine of the highest affinity PfENT1 inhibitors, comprising six distinct chemotypes, kill P. falciparum parasites in culture (Frame et al., 2015b). Because of the considerable geographical overlap in the distributions of P. falciparum and P. vivax, to be effective, novel drugs should ideally target both species. In the present work we have characterized the P. vivax equilibrative nucleoside transporter homolog, PvENT1, in a yeast expression system. We show that it has a similar, but distinct, substrate profile compared to PfENT1. Furthermore, the PfENT1 inhibitors that we identified previously (Frame et al., 2015b) are highly efficacious inhibitors of PvENT1. This implies that it may be feasible to develop inhibitors of the primary malaria purine import transporter as potential novel antimalarial drugs that will be effective against both falciparum and vivax malaria.
Despite 75% amino acid sequence identity between PfENT1 and PvENT1, PvENT1 displays functional differences from PfENT1 in terms of substrate interaction profiles. PvENT1 is more selective for purines than for pyrimidines. Compared to PfENT1, the IC50 values for inosine and guanosine were lower than for adenosine, a profile similar to the Leishmania donovani LdNT2 transporter (Carter et al., 2000b). Inosine had the lowest IC50 for inhibition of [3H]uridine uptake, followed by guanosine and adenosine/hypoxanthine. Hypoxanthine and adenosine are endogenous purine substrates for PfENT1 (and for isolated P. falciparum parasites). PvENT1 had 4–5 fold lower IC50 values for hypoxanthine and adenosine, ∼100 μM and ∼140 μM, respectively, than PfENT1, ∼480 μM for hypoxanthine and ∼650 μM for adenosine (Riegelhaupt et al., 2010a). These differences may relate to the fact that P. vivax parasites only infect reticulocytes, immature erythrocytes, whereas P. falciparum parasites infect mature erythrocytes as well as reticulocytes. The cytoplasmic environment and purine composition is different in immature vs mature erythrocytes (Srivastava et al., 2015). It remains to be determined if the difference is a consequence of their respective preference for immature versus mature erythrocytes. Furthermore, it is unknown whether the differences in purine IC50 values for PvENT1 versus PfENT1 are physiologically significant.
One of the important findings from this study was that the PfENT1 inhibitors that we identified previously are also potent inhibitors of PvENT1 with IC50 values in the low nM range. In the present study, we showed that the nine compounds inhibited [3H]adenosine uptake into PvENT1-expressing yeast and inhibited adenosine-dependent growth of PvENT1-expressing purine auxotrophic yeast. We previously showed that these compounds are not toxic to yeast at concentrations up to hundreds of micromolar (Frame et al., 2015b). Thus, at the sub-micromolar concentrations where the compounds inhibit adenosine-dependent growth, it is likely due to inhibition of PvENT1-dependent adenosine uptake. The efficacy of the nine compounds in both assays is consistent with their site of action being inhibition of PvENT1. The differences in the IC50 values measured in the two assays is a function of the fundamental differences in the assays and does not represent a difference in the strength of the inhibitor's interaction with PvENT1. The IC50 values in the growth experiments depend on the level of PvENT1 expression and thus, the amount of excess purine transport capacity relative to the purine requirements for yeast cell growth and proliferation. Thus, the IC50 values in the growth experiments do not directly reflect the affinity of the inhibitors for PvENT1, but rather the fraction of expressed PvENT1 that must be inhibited to make purine uptake limit yeast cell growth. In the inhibition of [3H]adenosine uptake assay, the measured IC50 values are a direct measure of interaction between the inhibitor and PvENT1.
Alternatively, the adenosine concentration is 1 mM in the growth assay and only 50 nM in the [3H]adenosine uptake assay, in the former case, ∼10 times the adenosine IC50 and in the latter ∼2000-fold lower. The difference may represent competition between adenosine and the inhibitor for an overlapping binding site. Overall, our results provide strong evidence that the PfENT1 small molecule inhibitors also block PvENT1, in many cases with higher potency (Frame et al., 2015b). The ability of these nine compounds to inhibit both PfENT1 and PvENT1 provides further support for the hypothesis that malaria purine transporters are a reasonable target for the development of novel antimalarial drugs.
In contrast to the efficacy of the PfENT1 inhibitors against PvENT1, hENT1 inhibitors had little or no efficacy against PvENT1. PvENT1 was not sensitive to NBMPR, an inosine analog where the C6 oxygen is replaced with the more bulky S-nitrobenzyl moiety. This result is in agreement with the NBMPR-insensitivity observed for other protozoan ENTs (de Koning et al., 2005). Dipyridamole did inhibit PvENT1 with a 40 μM IC50 but this is over 1000-fold higher than its efficacy against hENT1. The Plasmodium homologs are only 17% sequence identical to hENT1 so this result was not unexpected. Since both hENT1 and the Plasmodium ENTs recognize purines with similar affinity, this suggests that dipyridamole binding affinity probably depends on residues in the transporter besides those involved in purine recognition.
We investigated the impact of known non-synonymous PvENT1 SNPs identified in sequenced field isolates. Growth of yeast expressing five of the six SNPs with adenosine as the sole purine source was similar to growth rates of yeast expressing WT PvENT1. We were surprised that yeast expressing the N329S-PvENT1 variant were unable to grow in adenosine, although they grew normally with adenine as the sole purine source, because the IC50 values for adenosine, inosine, and hypoxanthine inhibition of [3H]uridine uptake for N329S-expressing yeast were similar to the IC50 values obtained with WT-expressing yeast. We ruled out differences in the media pH or composition as potential effects. It is most likely due to a difference in N329S function or level of expression relative to WT. Further experiments will be necessary to explain these results.
Some naturally occurring PfENT1 SNPs have functional effects on purine uptake (e.g., F394L; ref. (Riegelhaupt et al., 2010b)) (Table 4). Given the small numbers of identified PvENT1 non-synonymous SNPs and that these SNPs have similar affinity for the identified PfENT1 inhibitors, currently existing SNPs do not represent a potential source of pre-existing resistance to the inhibitors.
Most protozoan species have de novo pyrimidine synthesis pathways and do not incorporate host pyrimidines into DNA/RNA. Nevertheless, our experiments show that PvENT1 can bind to and probably transport various pyrimidine compounds. In the case of uridine, we showed that it is transported by PvENT1. Knowledge of the PvENT1 pyrimidine binding profile may be useful in designing drugs that target the de novo pyrimidine biosynthetic pathway and might use PvENT1 as a transport pathway for drug entry.
5. Conclusions
The parasite purine salvage pathway has been a target of various potent antimalarial compounds (e.g., deoxycoformycin, immucillins) (Webster et al., 1984, Kicska et al., 2002, Ting et al., 2005). In this body of work, we have characterized the functional properties of PvENT1, a potential antimalarial target. While PfENT1 and PvENT1 share a high degree of amino acid sequence identity, differences in amino acids in PvENT1 result in altered affinities for purine substrates. Nonetheless, PvENT1 (and the known non-synonymous SNP variants) are inhibited by a new class of small molecule inhibitors that target the P. falciparum purine importer PfENT1. Development of these inhibitors as novel antimalarial drugs may provide an additional arsenal of antimalarial compounds that have no overlap with current chemotherapeutics.
Conflicts of interest
The authors declare the following potential competing financial interest(s): A patent application is pending on the yeast-based assays as a method to identify nucleoside transporter inhibitors. A second pending patent covers the use of the 171 compounds as PfENT1 inhibitors and as novel antimalarial drugs. The patents have not been licensed to date and no negotiations are currently in progress.
Author contributions
RD, IJF and MHA designed the experiments. RD and YS performed the experiments. All authors analyzed data and participated in writing and/or editing the manuscript.
Acknowledgments
We thank David Pierce for expert technical assistance and Andrea Lopez and Adi Berman for contributions to the project while working in the Einstein Summer Undergraduate Research Program. RD and IJF were supported in part by the NIGMS Medical Scientist Training Program grant T32-GM007288. This work was supported by funds from the Albert Einstein College of Medicine and from NIH R01AI116665 to MHA. The funders had no role in the study design, collection or analysis of data or in writing and submission of this manuscript.
References
- Anstey N.M., Russell B., Yeo T.W., Price R.N. The pathophysiology of vivax malaria. Trends Parasitol. 2009;25:220–227. doi: 10.1016/j.pt.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Ariey F., Witkowski B., Amaratunga C., Beghain J., Langlois A.C., Khim N., Kim S., Duru V., Bouchier C., Ma L., Lim P., Leang R., Duong S., Sreng S., Suon S., Chuor C.M., Bout D.M., Menard S., Rogers W.O., Genton B., Fandeur T., Miotto O., Ringwald P., Le Bras J., Berry A., Barale J.C., Fairhurst R.M., Benoit-Vical F., Mercereau-Puijalon O., Menard D. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin S.A., Beal P.R., Yao S.Y., King A.E., Cass C.E., Young J.D. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 2004;447:735–743. doi: 10.1007/s00424-003-1103-2. [DOI] [PubMed] [Google Scholar]
- Carlton J.M., Adams J.H., Silva J.C., Bidwell S.L., Lorenzi H., Caler E., Crabtree J., Angiuoli S.V., Merino E.F., Amedeo P., Cheng Q., Coulson R.M., Crabb B.S., Del Portillo H.A., Essien K., Feldblyum T.V., Fernandez-Becerra C., Gilson P.R., Gueye A.H., Guo X., Kang'a S., Kooij T.W., Korsinczky M., Meyer E.V., Nene V., Paulsen I., White O., Ralph S.A., Ren Q., Sargeant T.J., Salzberg S.L., Stoeckert C.J., Sullivan S.A., Yamamoto M.M., Hoffman S.L., Wortman J.R., Gardner M.J., Galinski M.R., Barnwell J.W., Fraser-Liggett C.M. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455:757–763. doi: 10.1038/nature07327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter N.S., Ben Mamoun C., Liu W., Silva E.O., Landfear S.M., Goldberg D.E., Ullman B. Isolation and functional characterization of the PfNT1 nucleoside transporter gene from Plasmodium falciparum. J. Biol. Chem. 2000;275:10683–10691. doi: 10.1074/jbc.275.14.10683. [DOI] [PubMed] [Google Scholar]
- Carter N.S., Drew M.E., Sanchez M., Vasudevan G., Landfear S.M., Ullman B. Cloning of a novel inosine-guanosine transporter gene from Leishmania donovani by functional rescue of a transport-deficient mutant. J. Biol. Chem. 2000;275:20935–20941. doi: 10.1074/jbc.M002418200. [DOI] [PubMed] [Google Scholar]
- Carter N.S., Landfear S.M., Ullman B. Nucleoside transporters of parasitic protozoa. Trends Parasitol. 2001;17:142–145. doi: 10.1016/s1471-4922(00)01806-7. [DOI] [PubMed] [Google Scholar]
- Cassera M.B., Hazleton K.Z., Riegelhaupt P.M., Merino E.F., Luo M., Akabas M.H., Schramm V.L. Erythrocytic adenosine monophosphate as an alternative purine source in Plasmodium falciparum. J. Biol. Chem. 2008;283:32889–32899. doi: 10.1074/jbc.M804497200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassera M.B., Zhang Y., Hazleton K.Z., Schramm V.L. Purine and pyrimidine pathways as targets in Plasmodium falciparum. Curr. Top. Med. Chem. 2011;11:2103–2115. doi: 10.2174/156802611796575948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Prusoff W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- de Koning H.P., Bridges D.J., Burchmore R.J. Purine and pyrimidine transport in pathogenic protozoa: from biology to therapy. FEMS Microbiol. Rev. 2005;29:987–1020. doi: 10.1016/j.femsre.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Dondorp A.M., Fairhurst R.M., Slutsker L., Macarthur J.R., Breman J.G., Guerin P.J., Wellems T.E., Ringwald P., Newman R.D., Plowe C.V. The threat of artemisinin-resistant malaria. N. Engl. J. Med. 2011;365:1073–1075. doi: 10.1056/NEJMp1108322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfman B.Z. The isolation of adenylosuccinate synthetase mutants in yeast by selection for constitutive behavior in pigmented strains. Genetics. 1969;61:377–389. doi: 10.1093/genetics/61.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downie M.J., El Bissati K., Bobenchik A.M., Nic Lochlainn L., Amerik A., Zufferey R., Kirk K., Ben Mamoun C. PfNT2, a permease of the equilibrative nucleoside transporter family in the endoplasmic reticulum of Plasmodium falciparum. J. Biol. Chem. 2010;285:20827–20833. doi: 10.1074/jbc.M110.118489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downie M.J., Kirk K., Mamoun C.B. Purine salvage pathways in the intraerythrocytic malaria parasite Plasmodium falciparum. Eukaryot. Cell. 2008;7:1231–1237. doi: 10.1128/EC.00159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downie M.J., Saliba K.J., Howitt S.M., Broer S., Kirk K. Transport of nucleosides across the Plasmodium falciparum parasite plasma membrane has characteristics of PfENT1. Mol. Microbiol. 2006;60:738–748. doi: 10.1111/j.1365-2958.2006.05125.x. [DOI] [PubMed] [Google Scholar]
- Ducati R.G., Namanja-Magliano H.A., Schramm V.L. Transition-state inhibitors of purine salvage and other prospective enzyme targets in malaria. Future Med. Chem. 2013;5:1341–1360. doi: 10.4155/fmc.13.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bissati K., Downie M.J., Kim S.K., Horowitz M., Carter N., Ullman B., Ben Mamoun C. Genetic evidence for the essential role of PfNT1 in the transport and utilization of xanthine, guanine, guanosine and adenine by Plasmodium falciparum. Mol. Biochem. Parasitol. 2008;161:130–139. doi: 10.1016/j.molbiopara.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bissati K., Zufferey R., Witola W.H., Carter N.S., Ullman B., Ben Mamoun C. The plasma membrane permease PfNT1 is essential for purine salvage in the human malaria parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A. 2006;103:9286–9291. doi: 10.1073/pnas.0602590103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame I.J., Deniskin R., Arora A., Akabas M.H. Purine import into malaria parasites as a target for antimalarial drug development. Ann. N. Y. Acad. Sci. 2015;1342:19–28. doi: 10.1111/nyas.12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame I.J., Deniskin R., Rinderspacher A., Katz F., Deng S.X., Moir R.D., Adjalley S.H., Coburn-Flynn O., Fidock D.A., Willis I.M., Landry D.W., Akabas M.H. Yeast-based high-throughput screen identifies Plasmodium falciparum equilibrative nucleoside transporter 1 inhibitors that kill malaria parasites. ACS Chem. Biol. 2015;10:775–783. doi: 10.1021/cb500981y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame I.J., Merino E.F., Schramm V.L., Cassera M.B., Akabas M.H. Malaria parasite type 4 equilibrative nucleoside transporters (ENT4) are purine transporters with distinct substrate specificity. Biochem. J. 2012;446:179–190. doi: 10.1042/BJ20112220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari E., Piedrafita L., Aldea M., Herrero E. A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast. 1997;13:837–848. doi: 10.1002/(SICI)1097-0061(199707)13:9<837::AID-YEA145>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Gero A.M., O'Sullivan W.J. Purines and pyrimidines in malarial parasites. Blood Cells. 1990;16 467–484; discussion 485–498. [PubMed] [Google Scholar]
- Griffiths M., Beaumont N., Yao S.Y., Sundaram M., Boumah C.E., Davies A., Kwong F.Y., Coe I., Cass C.E., Young J.D., Baldwin S.A. Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat. Med. 1997;3:89–93. doi: 10.1038/nm0197-89. [DOI] [PubMed] [Google Scholar]
- Hastings I.M., Kay K., Hodel E.M. How robust are malaria parasite clearance rates as indicators of drug effectiveness and resistance? Antimicrob. Agents Chemother. 2015;59:6428–6436. doi: 10.1128/AAC.00481-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R., Petrov D.A. General rules for optimal codon choice. PLoS Genet. 2009;5:e1000556. doi: 10.1371/journal.pgen.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J., Donald K.A., Griffiths D.E. DMSO-enhanced whole cell yeast transformation. Nucleic Acids Res. 1991;19:5791. doi: 10.1093/nar/19.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kicska G.A., Tyler P.C., Evans G.B., Furneaux R.H., Schramm V.L., Kim K. Purine-less death in Plasmodium falciparum induced by immucillin-H, a transition state analogue of purine nucleoside phosphorylase. J. Biol. Chem. 2002;277:3226–3231. doi: 10.1074/jbc.M105906200. [DOI] [PubMed] [Google Scholar]
- Kirk K., Howitt S.M., Broer S., Saliba K.J., Downie M.J. Purine uptake in Plasmodium: transport versus metabolism. Trends Parasitol. 2009;25:246–249. doi: 10.1016/j.pt.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Kirk K., Lehane A.M. Membrane transport in the malaria parasite and its host erythrocyte. Biochem. J. 2014;457:1–18. doi: 10.1042/BJ20131007. [DOI] [PubMed] [Google Scholar]
- Kokina A., Kibilds J., Liepins J. Adenine auxotrophy–be aware: some effects of adenine auxotrophy in Saccharomyces cerevisiae strain W303-1A. FEMS Yeast Res. 2014;14:697–707. doi: 10.1111/1567-1364.12154. [DOI] [PubMed] [Google Scholar]
- Landfear S.M., Ullman B., Carter N.S., Sanchez M.A. Nucleoside and nucleobase transporters in parasitic protozoa. Eukaryot. Cell. 2004;3:245–254. doi: 10.1128/EC.3.2.245-254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manandhar M.S., Van Dyke K. Detailed purine salvage metabolism in and outside the free malarial parasite. Exp. Parasitol. 1975;37:138–146. doi: 10.1016/0014-4894(75)90064-8. [DOI] [PubMed] [Google Scholar]
- Martin R.E., Henry R.I., Abbey J.L., Clements J.D., Kirk K. The ‘permeome’ of the malaria parasite: an overview of the membrane transport proteins of Plasmodium falciparum. Genome Biol. 2005;6:R26. doi: 10.1186/gb-2005-6-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker M.D., Hyde R.J., Yao S.Y., McRobert L., Cass C.E., Young J.D., McConkey G.A., Baldwin S.A. Identification of a nucleoside/nucleobase transporter from Plasmodium falciparum, a novel target for anti-malarial chemotherapy. Biochem. J. 2000;349:67–75. doi: 10.1042/0264-6021:3490067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rager N., Mamoun C.B., Carter N.S., Goldberg D.E., Ullman B. Localization of the Plasmodium falciparum PfNT1 nucleoside transporter to the parasite plasma membrane. J. Biol. Chem. 2001;276:41095–41099. doi: 10.1074/jbc.M107037200. [DOI] [PubMed] [Google Scholar]
- Riegelhaupt P.M., Cassera M.B., Frohlich R.F., Hazleton K.Z., Hefter J.J., Schramm V.L., Akabas M.H. Transport of purines and purine salvage pathway inhibitors by the Plasmodium falciparum equilibrative nucleoside transporter PfENT1. Mol. Biochem. Parasitol. 2010;169:40–49. doi: 10.1016/j.molbiopara.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegelhaupt P.M., Frame I.J., Akabas M.H. Transmembrane segment 11 appears to line the purine permeation pathway of the Plasmodium falciparum equilibrative nucleoside transporter 1 (PfENT1) J. Biol. Chem. 2010;285:17001–17010. doi: 10.1074/jbc.M110.115758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogerson S.J., Carter R. Severe vivax malaria: newly recognised or rediscovered. PLoS Med. 2008;5:e136. doi: 10.1371/journal.pmed.0050136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs J., Malaney P. The economic and social burden of malaria. Nature. 2002;415:680–685. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- Schmidt R., Manolson M.F., Chevallier M.R. Photoaffinity labeling and characterization of the cloned purine-cytosine transport system in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 1984;81:6276–6280. doi: 10.1073/pnas.81.20.6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow R.W., Guerra C.A., Noor A.M., Myint H.Y., Hay S.I. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A., Creek D.J., Evans K.J., De Souza D., Schofield L., Muller S., Barrett M.P., McConville M.J., Waters A.P. Host reticulocytes provide metabolic reservoirs that can be exploited by malaria parasites. PLoS Pathog. 2015;11:e1004882. doi: 10.1371/journal.ppat.1004882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straimer J., Gnadig N.F., Witkowski B., Amaratunga C., Duru V., Ramadani A.P., Dacheux M., Khim N., Zhang L., Lam S., Gregory P.D., Urnov F.D., Mercereau-Puijalon O., Benoit-Vical F., Fairhurst R.M., Menard D., Fidock D.A. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science. 2015;347:428–431. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram M., Yao S.Y., Ingram J.C., Berry Z.A., Abidi F., Cass C.E., Baldwin S.A., Young J.D. Topology of a human equilibrative, nitrobenzylthioinosine (NBMPR)-sensitive nucleoside transporter (hENT1) implicated in the cellular uptake of adenosine and anti-cancer drugs. J. Biol. Chem. 2001;276:45270–45275. doi: 10.1074/jbc.M107169200. [DOI] [PubMed] [Google Scholar]
- Ting L.M., Shi W., Lewandowicz A., Singh V., Mwakingwe A., Birck M.R., Ringia E.A., Bench G., Madrid D.C., Tyler P.C., Evans G.B., Furneaux R.H., Schramm V.L., Kim K. Targeting a novel Plasmodium falciparum purine recycling pathway with specific immucillins. J. Biol. Chem. 2005;280:9547–9554. doi: 10.1074/jbc.M412693200. [DOI] [PubMed] [Google Scholar]
- Traut T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- Valdes R., Arastu-Kapur S., Landfear S.M., Shinde U. An ab Initio structural model of a nucleoside permease predicts functionally important residues. J. Biol. Chem. 2009;284:19067–19076. doi: 10.1074/jbc.M109.017947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster H.K., Wiesmann W.P., Pavia C.S. Adenosine deaminase in malaria infection: effect of 2′-deoxycoformycin in vivo. Adv. Exp. Med. Biol. 1984;165(Pt A):225–229. doi: 10.1007/978-1-4684-4553-4_44. [DOI] [PubMed] [Google Scholar]
- Wellems T.E., Plowe C.V. Chloroquine-resistant malaria. J. Infect. Dis. 2001;184:770–776. doi: 10.1086/322858. [DOI] [PubMed] [Google Scholar]
- Winzeler E.A., Shoemaker D.D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J.D., Bussey H., Chu A.M., Connelly C., Davis K., Dietrich F., Dow S.W., El Bakkoury M., Foury F., Friend S.H., Gentalen E., Giaever G., Hegemann J.H., Jones T., Laub M., Liao H., Liebundguth N., Lockhart D.J., Lucau-Danila A., Lussier M., M'Rabet N., Menard P., Mittmann M., Pai C., Rebischung C., Revuelta J.L., Riles L., Roberts C.J., Ross-MacDonald P., Scherens B., Snyder M., Sookhai-Mahadeo S., Storms R.K., Veronneau S., Voet M., Volckaert G., Ward T.R., Wysocki R., Yen G.S., Yu K., Zimmermann K., Philippsen P., Johnston M., Davis R.W. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- World Health Organization . World Health Organization; Switzerland: 2014. WHO Global Malaria Programme: World Malaria Report 2014. [Google Scholar]