

Abstract
FOXP3 functions not only as the master regulator in regulatory T cells but also as an X-linked tumor suppressor. The tumor suppressive activity of FOXP3 has been observed in tumor initiation, but its role during tumor progression remains controversial. Moreover, the mechanism of FOXP3-mediated tumor suppressive activity remains largely unknown. Using chromatin immunoprecipitation sequencing, we identified a series of potential FOXP3-targeted microRNAs (miRs) in MCF7 cells. Notably, FOXP3 significantly induced the expression of miR-146a/b. In vitro, FOXP3-induced miR-146a/b prevented tumor cell proliferation and enhanced apoptosis. Functional analyses in vitro and in vivo revealed that FOXP3-induced miR-146a/b negatively regulate NF-κB activation by inhibiting the expression of IRAK1 and TRAF6. In chromatin immunoprecipitation assays, FOXP3 directly bound the promoter region of miR-146a but not of miR-146b, and FOXP3 interacted directly with NF-κB p65 to regulate an miR-146-NF-κB negative feedback regulation loop in normal breast epithelial and tumor cells, as demonstrated with luciferase reporter assays. Although FOXP3 significantly inhibited breast tumor growth and migration in vitro and metastasis in vivo, FOXP3-induced miR-146a/b contributed only to the inhibition of breast tumor growth. These data suggest that miR-146a/b contribute to FOXP3-mediated tumor suppression during tumor growth by triggering apoptosis. The identification of a FOXP3-miR-146-NF-κB axis provides an underlying mechanism for disruption of miR-146 family member expression and constitutive NF-κB activation in breast cancer cells. Linking the tumor suppressor function of FOXP3 to NF-κB activation reveals a potential therapeutic approach for cancers with FOXP3 defects.
Keywords: FOXP3, microRNA, NF-κB, breast cancer
Introduction
MicroRNA-146a (miR-146a) is overexpressed in Foxp3+ Tregs and is critical for Treg function in the immune system (1), suggesting a link between FOXP3 and miR-146a. The miR-146 family includes miR-146a/b in humans, but only miR-146a in mice. Many of the predicted target genes are common to both miR-146a/b, but each miR may have a different post-transcriptional processing mechanism due to genomic location (miR-146a/b on human chromosome 5 and 10, respectively). Notably, NF-κB directly induces miR-146a in a dose-dependent manner (2), whereas miR-146a negatively regulates NF-κB activation through targeted inhibition of the NF-κB inducers TRAF6 and IRAK1 in breast cancer cells, suggesting an miR-146a-NF-κB negative feedback regulation loop (3). Accumulating data also demonstrate that miR-146a/b inhibit cell proliferation, invasion, and metastasis in human cancers, including breast cancer (3, 4). Although dispute about the expression levels of miR-146a in human cancers remains, most breast cancer cell lines have low expression levels of miR-146a compared with the normal breast epithelial cell line MCF10A (4, 5). Importantly, miR-146a-knockout mice develop spontaneous myeloid sarcomas and lymphomas at a high rate (6, 7), and depletion of miR-146a has been implicated in human myeloid malignancies (8). Thus, there is consistent evidence that miR-146a functions as a tumor suppressor.
Studies have also identified additional miR-146a targets involved in cell proliferation, differentiation, and migration of cancer cells, including EGFR (9, 10), CXCR4 (11), NOTCH1 (12), ROCK1 (13), PRKCE (14), and others, but these targets require further validation. Additionally, the regulatory mechanisms controlling miR-146a/b are largely unknown. NF-κB, breast cancer metastasis suppressor 1 (4), p53-binding protein-1 (15), and tumor necrosis factor-related apoptosis-inducing ligand (11) were identified as transactivators of miR-146a/b in breast cancer cells. These proteins induce miR-146a to suppress either NF-κB-dependent tumor growth or chemokine (C-X-C motif) receptor 4-mediated tumor metastasis in breast cancer cells. However, the mechanism through which miR-146a controls tumor development and/or metastasis remains debated. Given the critical roles for miR-146a/b and FOXP3 in cancer biology (6, 7, 16-18), we tested whether miR-146a/b are involved in FOXP3-mediated tumor suppression in breast cancer cells.
Materials and Methods
Cell lines, antibodies, DNA constructs, and reagents
Breast cancer cell lines MCF7, T47D, BT474, MDA-MB-468, and MDA-MB231 and the pre-neoplastic breast epithelial cell line MCF10A were obtained from the American Type Culture Collection (Manassas, VA). Cell lines were authenticated by examination of morphology and growth characteristics and confirmed to be mycoplasma-free. Cells were maintained in DMEM supplemented with 10% FBS (Life Technologies, Grand Island, NY) and cultured for less than 6 months. GFP- and FOXP3-Tet-off MCF7 cells were established and maintained in 10 μg/ml doxycycline (Dox) as described previously (16, 17, 19). Specific primary antibodies were used to detect the following proteins: FOXP3 (ab450, Abcam, Cambridge, MA), Foxp3 (Poly2638b, BioLegend, San Diego, CA), NF-κB p65 (D14E12, Cell Signaling, Danvers, MA), IRAK1 (D51G7, Cell Signaling), TRAF6 (D21G3, Cell Signaling), EGFR (D38B1, Cell Signaling), Erk1/2 (H-72, Santa Cruz Biotechnology, Dallas, TX), p-Erk1/2 (E-4, Santa Cruz Biotechnology), Irak1 (H-273, Santa Cruz Biotechnology), Traf6 (H-274, Santa Cruz Biotechnology), p65 (D14E12, Cell Signaling), Irak1 (H-273, Santa Cruz Biotechnology), and Traf6 (H-274, Santa Cruz Biotechnology). The pEF1-FOXP3-V5 vector (20) or pEF1 empty vector was transfected into cells using FuGENE6 (Promega, Madison, WI). FOXP3 short hairpin RNAs (shRNAs) were described previously (20). Scramble control miR, miR-146a/b mimics, or specific anti-miR-146a/b inhibitors were obtained from Life Technologies. TNF-α (T6674, Sigma, St. Louis, MO) and Bay11-7082 (Sigma) were used for NF-κB activation and inhibition in cell culture, respectively. Lipopolysaccharide (LPS; O111B4, Sigma) was used for NF-κB activation in mice.
TaqMan miR assay
Expression levels of miR-146a/b were assessed using TaqMan MicroRNA Assay (Life Technologies). Human miR-146a/b and mouse miR-146a TaqMan primers and probes were purchased from Life Technologies. The average relative expression was determined using the comparative method (2-ΔCt) against the endogenous RNU6B (for human) or snoRNA202 (for mouse) controls.
Cell proliferation and apoptosis assays
Cell morphology, viability, and number of GFP- and FOXP3-Tet-off MCF7 cells were monitored at 0, 3, 5, 7, 10, and 14 days without Dox using a microscope and flow cytometry assays based on cell binding to Annexin V (561012, BD Biosciences, San Jose, CA) and 7-AAD (7-AAD; 555816, BD Biosciences). Since miR-146a/b inhibitors were effective for at least 4 days as tested (Fig. S1), transfection with miR-146a/b inhibitors was repeated every 4 days during cell proliferation.
Quantitative real-time PCR (qPCR)
Relative mRNA expression levels were determined using the comparative method (2-ΔCt) against endogenous GAPDH (for human) or Hprt (for mouse) controls. Primer sequences are listed in supplementary Table S2.
Western blot, quantitative ChIP, and co-IP
Western blotting and ChIP were performed as described previously (16-18). For co-IP, collected cells were washed with cold PBS and lysed in ice-cold buffer [20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, and 1% NP-40] supplemented with complete protease inhibitors (Sigma) on ice for 10 minutes. Lysates were aliquoted into two tubes and incubated with the designated antibody or an appropriate IgG control for 16 hours at 4°C. Protein A/G agarose (Invitrogen) was used to precipitate antibody-protein complexes.
NF-κB activation
NF-κB activation was determined by examining the expression profile of NF-κB target genes Bcl2l1 and Traf1/2 using qPCR and by western blot analysis of the accumulation of nuclear p65 (21). Analysis of NF-κB activation in mice was performed 12 hours after intraperitoneal injection of LPS (100 μL per mouse, 3 mg/mL solution), which enhances NF-κB signaling in cells, leading to increases in p65 loading (22).
Experimental animals
Transgenic Tg (MMTV-Cre) 4Mam/J mice with Cre under control of the mouse mammary tumor promoter (MMTV) and immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Male MMTV-Cre mice were crossed to female Foxp3flox/flox mice (20, 23) to generate breast Foxp3 conditional knockout (cKO, MMTV-Cre × Foxp3flox/flox) mice. All animal experiments were conducted in accordance with accepted standards of animal care and approved by the Institutional Animal Care and Use Committee of University of Alabama at Birmingham (UAB).
Immunohistochemistry (IHC)
The Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA) was used for immunostaining according to the manufacturer's protocol as described previously (16, 18).
Luciferase assay
The miR-146a promoter-luciferase gene vector was constructed by ligating the pGL2 vector (Promega) with the miR-146a promoter. Luciferase activity was measured as described previously (16-18, 20).
Electrophoretic mobility shift assay (EMSA)
Recombinant human FOXP3 (H00050943-P02, Abnova, Taipei, Taiwan) or nuclear extracts were prepared as described previously (24). Supershift analysis was performed using the Chemiluminescent EMSA Kit (Pierce Biotechnology, Rockford, IL) according to the manufacturer's protocol. Probe sequences are listed in supplementary Table S2. Mutant probes with mutation or deletion of a potential forkhead binding site were used for specificity control.
In vitro cell migration assay
Tet-off MCF7 cells were transfected with 100 nM scramble miR or inhibitors against miR-146a/b for 48 hours before scratch assays (25) and transwell migration assays (8-μM pore size; Millipore, Billerica, MA) (26).
In vivo tumor metastasis assay
A total of 1 × 104 control MDA-MB231 cells or MDA-MB231 cells stably expressing exogenous FOXP3 were implanted intravenously into 8-week-old female NSG mice. At 7 weeks after implantation, the mice were euthanized for histologic examination and miR analysis. The number of surface lesions over all lobes of the liver and lungs was scored before pathologic analysis. Tumor burden in the liver and lungs was quantified in two-step sections from each lobe (liver, left and right lobes; lung, left two lobes and right three lobes) in a blinded fashion by calculating the area of tumor tissue as a percentage of the total tissue area (27).
Human tissue specimens
Tumor specimens were recruited from the Tissue Procurement Shared Facility at the Comprehensive Cancer Center, UAB, with informed consent from all subjects in accordance with the requirements of the Institutional Review Board at UAB. The breast cancer specimens were collected from 20 patients who underwent primary surgery between January 2012 and June 2014. All patients had histologically confirmed breast cancer and had not received hormone or radiation therapy. Twenty breast cancer tissues were used for this study.
Statistical analysis
We compared the means of the variable using a two-tailed t test or a Mann-Whitney test between two groups and Fisher's protected least significant difference (PLSD) test among multiple groups. All data were entered into an access database using Excel 2010 and analyzed with SPSS (version 20; IBM, Armonk, NY) and StatView (version 5.0.1; SAS Institute Inc., Cary, NC).
Results
Regulation of miR-146a/b by FOXP3 contributes to tumor suppression through apoptosis in breast cancer cells
There are two subtypes of miRs. Intragenic miRs located within exons of host genes can be co-transcribed with their host genes by the same promoters (28-30), but transcriptional regulation of intergenic miRs is poorly understood because miR promoters are poorly characterized (28-30). However, the -2.0 kb region from 5′ pre-miR transcripts is considered the most likely promoter locus. Thus, we screened the FOXP3-binding peaks near the 5′ pre-miR transcripts (-2.0kb to 0kb) of intergenic miRs or near transcriptional start sites (-2.0 kb to 0 kb) of intragenic miR host genes. ChIP-seq analysis identified 43 candidate FOXP3-target miRs in the FOXP3-Tet-off MCF7 cells (Table S1). To test whether these miRs are regulated by FOXP3, we conducted TaqMan miR assays at 2 and 4 days after FOXP3 induction. FOXP3 expression increased dramatically 2 days after Dox removal (16, 17, 19) and was observed predominately in cell nuclei (Fig. S2). As shown in Table S1, a cutoff of at least a 1.5-fold change in miR expression after FOXP3 induction and no significant change (<1.5-fold) in the GFP-Tet-off MCF7 control cells was applied to identify potential FOXP3-target miRs.
FOXP3 induction by Dox removal significantly increased the expression levels of miR-146a (2.1-fold at 2 days, 2.8-fold at 4 days) in FOXP3-Tet-off MCF7 (ER+) cells but not in GFP-Tet-off MCF7 control cells (Fig. 1A and Table S1). MCF7 cells express low levels of endogenous miR-146a under basal conditions (4, 5, 31). FOXP3 induction led to even greater induction of miR-146b (4.4-fold at 2 days, 8.0-fold at 4 days) in FOXP3-Tet-off MCF7 cells (Fig. 1A). These observations were validated in FOXP3-transfected T47D (ER+), BT474 (ER+), and MDA-MB-468 (ER-) cells (2.6- to 6.5-fold miR-146a induction, 3.0- to 4.6-fold miR-146b induction) (Fig. 1B), which also express low levels of endogenous miR-146a/b (4, 5). We then observed the effects of inhibiting miR-146a/b on cell morphology, viability, and proliferation in FOXP3-Tet-off MCF7 cells during FOXP3 induction. Transfection with miR-146a/b inhibitors, individually or combined, partially mitigated the decrease in cell proliferation mediated by FOXP3 induction (Fig. 1C). Notably, FOXP3 induction promotes apoptosis in MCF7 cells (16), and miR-146a/b inhibitors also dramatically reduced the apoptosis observed upon FOXP3 induction for 7 days (Fig. 1D).
Figure 1.
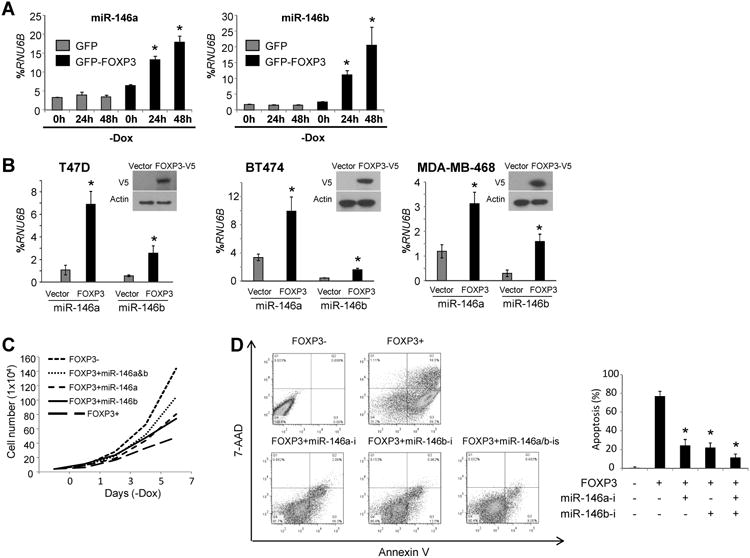
MiR-146a/b contribute to FOXP3-induced tumor suppression and apoptos is in breast cancer cells. A. Quantification of miR-146a/b expression in GFP- and FOXP3-Tet-off MCF7 cells. –Dox, Dox removal from the culture medium. B. Quantification of miR-146a/b expression in T47D (left panel), BT474 (middle panel), and MDA-MB-468 (right panel) cells. Representative western blot detecting FOXP3 expression in FOXP3-V5-transfected cells (insert in each panel). C. Graphic analysis of the effects of 100 nM scramble miR or miR-146a/b inhibitors on the growth of FOXP3-Tet-off MCF7 cells. D. Left panel: Representative flow cytometry plots generated by apoptosis analysis of FOXP3-Tet-off MCF7 cells with Annexin V/7-AAD staining. Right panel: Quantification of data. miR-146a-i, miR-146a inhibitor; miR-146b-i, miR-146b inhibitor; miR-146a/b-is, miR-146a/b inhibitors. All *, p < 0.05, two-tailed t test. Quantitative data are presented as the mean and SD of triplicates. All experiments were repeated three times.
FOXP3-miR-146-NF-κB axis in breast epithelial cells in vitro
Because miR-146a/b repress NF-κB through a negative feedback loop involving downregulation of IRAK1 and TRAF6 (2, 3), we tested whether FOXP3-induced miR-146a/b repress NF-κB activation in breast cancer cells. In FOXP3-Tet-off MCF7 cells, the expression levels of IRAK1 and TRAF6 decreased 3 days after FOXP3 induction, as did the level of nuclear p65 (Fig. 2A and B). Treatment with inhibitors of either miR-146a or miR-146b significantly blocked this repression (Fig. 2A and B). Furthermore, we analyzed FOXP3 and miR-146a/b transcriptional regulation of NF-κB target genes selected from the potential FOXP3-regulated genes identified in our previous study (19). From day 1 to day 4 after FOXP3 induction, CDKN1A expression increased, similar to our previous results (16, 19), whereas CXCR4 and MMP9 expression decreased. MiR-146a/b inhibitors did not interfere with the expression of these genes (Fig. 2C). The expression levels of BCL2L1 and TRAF1/2 were significantly downregulated from day 2 to day 5 after FOXP3 induction, but expression of these genes was rescued by miR-146a/b inhibitors (Fig. 2C). Because the cells undergo apoptosis after FOXP3 induction, all genes were downregulated from day 5 onward after Dox removal in FOXP3-Tet-off MCF7 cells, but not in GFP-Tet-off MCF7 cells (Fig. 1D and 2C). The expression levels of other anti-apoptotic genes, including BCL2, BCL10, and BIRC2/3, in FOXP3-Tet-off MCF7 cells did not change in response to miR-146a/b inhibition during FOXP3 induction (Fig. 2C and Fig. S3). EGFR is a potential target of miR-146a/b (9, 10), but the expression of this gene and its downstream target ERK1/2 did not change after FOXP3 induction or treatment with miR-146a/b inhibitors (Fig. 2A and B).
Figure 2.
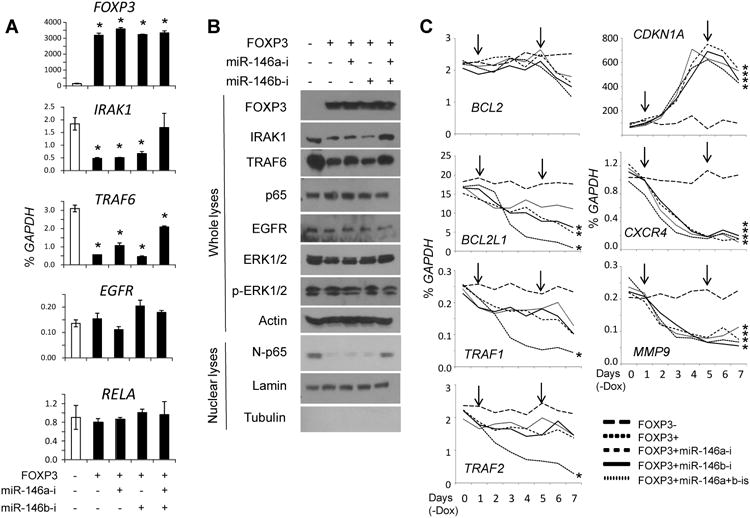
FOXP3-miR-146-NF-κB axis in breast cancer cells. A. Quantification of FOXP3, IRAK1, TRAF6, EGFR, and RELA mRNA levels at 72 hours after FOXP3 induction. Data are presented as the mean and SD of triplicates. All *, p < 0.05, two-tailed t test. B. Representative western blots detecting the expression of FOXP3, IRAK1, TRAF6, EGFR, ERK1/2, p-ERK1/2, p65, and nuclear p65 (N-p65) 72 hours after FOXP3 induction. C. Quantification of NF-κB target gene mRNA expression after FOXP3 induction. Down arrows indicate transfection with scramble miR or miR-146a/b inhibitors. *, p < 0.05, Fisher's PLSD test. All experiments were repeated three times.
To validate the endogenous FOXP3-miR-146a/b-NF-κB axis in breast epithelial cells, we tested the effect of the FOXP3 shRNAs on miR-146a/b expression in the normal breast epithelial cell line MCF10A. FOXP3 shRNAs caused a substantial reduction in the expression of miR-146a/b (Fig. 3A). Correspondingly, IRAK1 and TRAF6 transcripts were significantly elevated by FOXP3 shRNAs, and this increase was suppressed by co-transfection of miR-146a/b mimics (Fig. 3B). Similarly, IRAK1 and TRAF6 protein expression increased after FOXP3 inhibition, before or after stimulation with TNF-α to activate NF-κB (Fig. 3C). Furthermore, expression of miR-146a/b was dramatically induced by TNF-α at 8 hours, especially in FOXP3-expressing cells (Fig. 3C and D). Although nuclear p65 levels did not increase after FOXP3 silencing, p65 was present in nuclei for up to 8 hours after treatment with TNF-α (Fig. 3C). Additionally, the mRNA and protein expression levels of NF-κB-target genes BCL2L1 and TRAF1/2 were significantly elevated after FOXP3 silencing when cells were stimulated with TNF-α (Fig. 3E and F).
Figure 3.
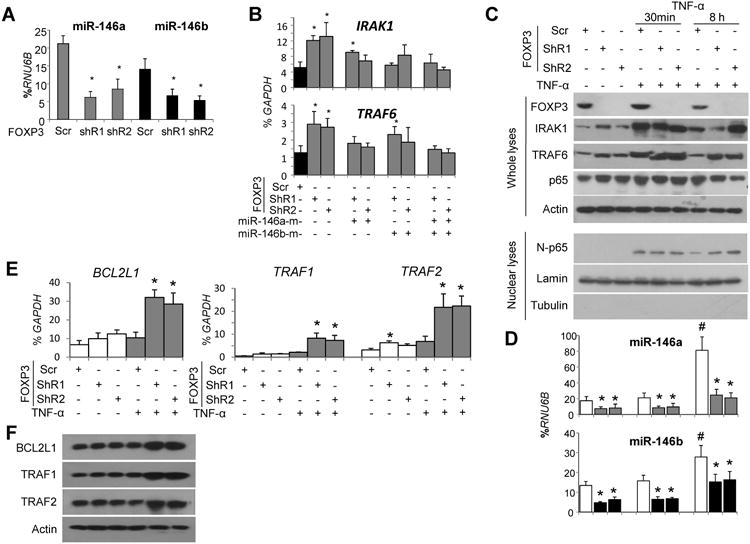
FOXP3-miR-146-NF-κB axis in MCF10A cells. Quantification of miR-146a/b (A) and IRAK1 and TRAF6 (B) expressionin cells transfected with vector control, FOXP3 shRNA, or miR-146a/b mimics (final concentration, 100 nM). C and D. Representative western blots detecting the expression of FOXP3, IRAK1, TRAF6, p65, and nuclear p65 (N-p65) and quantification of miR-146a/b expression in cells transfected with FOXP3 shRNA, before and after treatment with TNF-α (20 ng/mL). E and F. Quantification of NF-κB-target gene mRNA and protein expression, before and after treatment of cells with TNF-α for 8 hours. Scr, scramble miR; ShR, short hairpin RNA; miR-146a-m, miR-146a mimic; miR-146b-m, miR-146b mimic. All *, p < 0.05 for Scr vs. ShR; #, p < 0.05 for 8 hours vs. 0 or 30 minutes of TNF-α treatment. All p values were calculated by two-tailed t test. Quantitative data are presented as the mean and SD of triplicates. All experiments were repeated three times.
Foxp3-miR-146-NF-κB axis in breast epithelial cells in vivo
To validate the Foxp3-miR-146-NF-κB axis in breast epithelial cells in vivo, we created breast-specific Foxp3cKO mice (Fig. 4A). We observed approximately 90% deletion of the Foxp3 locus (Fig. 4B) and>90% reduction of Foxp3 mRNA in microdissected breast epithelial cells of 12-week-old non-gestational Foxp3cKO mice compared with MMTV-Cre control mice (Fig. 4C). Reduced expression of Foxp3 protein was confirmed by IHC (Fig. 4E). This deletion also caused a >2-fold decrease in miR-146a expression in the microdissected breast epithelial cells of both 12-week-old non-gestational and 20-week-old gestationa lmice (Fig. 4D).
Figure 4.
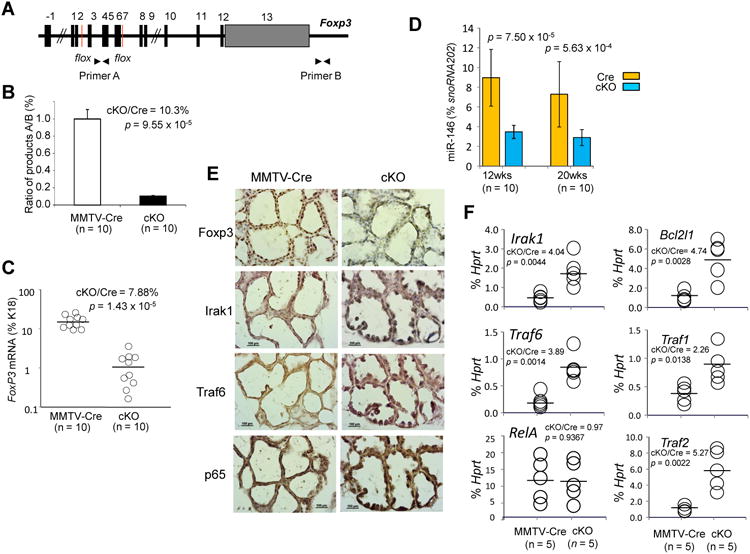
Foxp3-miR-146-NF-κB axis in mouse breast in vivo. A. Diagram of the floxed Foxp3 locus with the two primers, A and B, used to measure the ratio of deleted to undeleted alleles by PCR. B. Ratio of product A to product B DNA isolated from breast epithelial cells in mice to evaluate Foxp3 deletion (p value by a two-tailed t test). C. Quantification of relative Foxp3 mRNA levels in breast epithelial cells from mice (p value by a Mann-Whitney test). D. Quantification of miR-146a expression in breast epithelial cells from mice (p value by a Mann-Whitney test). E and F. Protein and mRNA expression of Irak1, Traf6, and p65 in breast tissue of mice 12 hours after LPS stimulation (p value by a two-tailed t test). MMTV-Cre or Cre, MMTV-Cre+FoxP3wt/wt; cKO, Foxp3cKO; cKO/Cre, ratio of levels in Foxp3cKO mice to levels in MMTV-Cre mice. Horizontal lines represent the median value. Error bar, SD. All experiments were repeated two times.
To observe the effect of breast-specific Foxp3 deletion on NF-κB activation, we injected the mice with LPS 12 hours before sacrifice. Expression of Irak1, Traf6, and p65 substantially increased in the breast epithelial cells of Foxp3cKO mice compared with those of MMTV-Cre control mice (Fig. 4E). Microdissected breast epithelial cells from Foxp3cKO mice also had a >3-fold increase in the mRNA levels of Irak1 and Traf6 and NF-κB target genes Bcl2l1 and Traf1/2 but no change in RelA mRNA levels (Fig. 4F).
The mechanism of miR-146a/b induction by FOXP3 in breast cancer cells
Using our previous ChIP-seq data (19), we found that FOXP3 directly binds to the promoter region of the human miR-146a host gene DQ658414 (Fig. S4A). MiR-146a, located within exon 2 of DQ658414 (Fig. 5A), shares the promoter of this gene and is thus transcriptionally expressed with it (2). Interestingly, the FOXP3-binding sites are adjacent to NF-κB-binding elements 1 and 2 (2) (Fig. 5C). To confirm the FOXP3-binding sites, we performed a ChIP assay with qPCR analysis (16-18). FOXP3 bound the proximal promoter region of themiR-146a host gene in MCF7 cells (Fig. 5B). However, no FOXP3-binding signal was identified in the intergenic miR-146b locus (-20 kb/+10kb) (Fig. S4B). Although an miR promoter locus can occasionally be far more than 10 kb from the 5′ pre-miR transcript (28, 29), no significant FOXP3-binding sites were identified between -17kb and +1kb from the 5′ pre-miR-146b transcript (Fig. S5).
Figure 5.
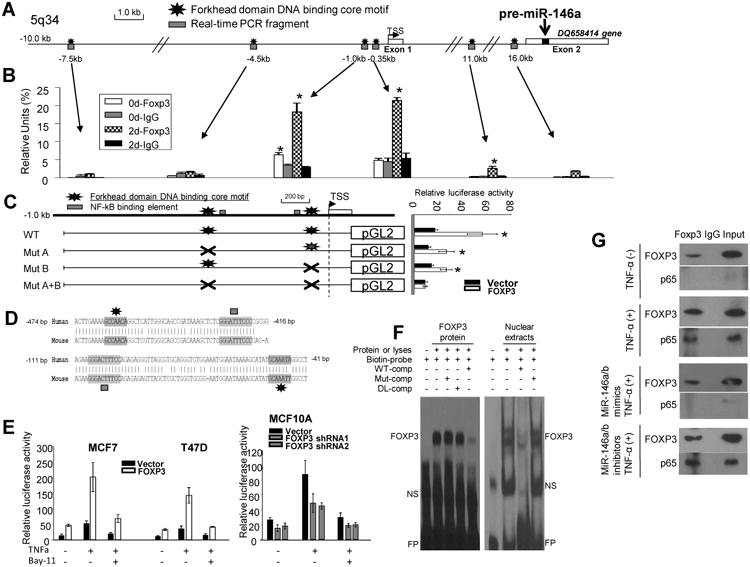
Molecular mechanism for the transcriptional regulation of human miR-146a by FOXP3 in breast cancer cells. A. Diagram showing the location of miR-146a (green box) within exon 2 of DQ658414 (yellow boxes) on chromosome 5. MiR-146a and DQ658414 share the same promoter region with few forkhead consensus motifs (black stars). PCR-amplified DNA fragments are depicted as red boxes. B. Quantification of the amounts of DNA precipitated, expressed as a percentage of the total input DNA, in ChIP analysis of FOXP3-binding sites in the promoter region of miR-146a. C. Diagram depicting the miR-146a promoter with two identified FOXP3-binding motifs (black stars). The WT-miR-146a-pGL2 reporter vector or vectors with either deletion (saltire) (-455 to -449 bp: GCCAACA or -52 to -46 bp: GCAAATA) of the pGL2 reporter were transfected into MCF7 cells in conjunction with either the pEF1 control vector or the pEF1-FOXP3 vector. Right panel: Quantification of luciferase activity in cells transfected with the respective vectors showed containing mutated miR-146a promoters, with or without FOXP3. WT, wild-type; Mut, deletion of FOXP3-binding motif. D. The alignment of human and mouse FOXP3 proximal promoters indicating identified FOXP3- and NF-κB-binding motifslocated within the conserved regions. E. Quantification of luciferase activity before and after treatment with TNF-α (20 ng/mL) for 30 minutes or plus Bay 11-7082 (10 μM) 30 minutes before treatment with TNF-α in cells. F. Specific binding of FOXP3 to the first forkhead-binding motif (GCCAACA) in the miR-146a promoter. Human FOXP3 recombinant protein (left panel) or nuclear extracts from FOXP3-Tet-off MCF7 cells cultured without Dox (FOXP3+; right panel) were preincubated with biotin-labeled WT or mutant probes in the presence of an unlabeled WT or mutant competitors. WT, biotin-labeled WT probe; Mut, probe with mutant forkhead-binding motif; DL, probe with deletion of forkhead-binding motif; Comp, unlabeled competitor; NS, nonspecific band; FP, free probe. G. Protein-protein interaction between FOXP3 and p65 in FOXP3-Tet-off MCF7 cells. Nuclear lysates were prepared after FOXP3 induction (48 hours) with or without TNF-α (30 minutes), miR-146a/b mimics (24 hours), or miR-146a/b inhibitors (24 hours) and then precipitated with antibodies specific for either FOXP3, p65, or IgG control. All *, p < 0.05, two-tailed t test. Quantitative data are presented as the mean and SD of triplicates. All experiments were repeated three times.
We continued to verify the regulation of miR-146a by direct binding of FOXP3 to its promoter using a dual-luciferase reporter assay in MCF7 cells (17, 18) and found that transfection of FOXP3 significantly induced the transcriptional activity of themiR-146a promoter (Fig. 5C). We have theoretically predicted the potential forkhead-binding motifs (RYMAAYA) in the miR-146a promoter region (19) (Fig. 5C and D). Sequence alignment analysis revealed two conserved regions in themiR-146a proximal promoter that contain two forkhead-binding motifs surrounding the highest ChIP signal (Fig. 5B) and two NF-κB-binding sites (Fig. 5C and D). Deletion of either forkhead-binding motif abrogated miR-146a promoter activity in the luciferase assay (Fig. 5C). To test whether FOXP3 interacts with NF-κB to regulate the transcriptional activity of the miR-146a promoter, we analyzed luciferase expression after stimulation with TNF-α in MCF7 and T47D cells. TNF-α significantly increased the promoter activity of miR-146a in the absence of exogenous FOXP3 and dramatically enhanced the FOXP3-mediated induction of promoter activity. This induction was significantly blocked by treatment with the NF-κB inhibitor Bay11-7082 (Fig. 5E). Additionally, FOXP3 silencing significantly reduced the TNF-α-induced promoter activity of miR-146a in MCF10A cells (Fig. 5E). The specific binding of FOXP3 to two forkhead-binding motifs in the miR-146a promoter was also validated by a gel-shift assay using either recombinant FOXP3 or nuclear extract from FOXP3-induced MCF7 cells (Fig. 5F and Fig. S6). Furthermore, a direct interaction of FOXP3 with NF-κB p65 was observed after stimulation of FOXP3-Tet-off MCF7 cells with TNF-α, and this interaction was attenuated by exogenous miR-146a/b mimics (Fig. 5G).
FOXP3-induced miR-146a/b inhibit tumor growth but not tumor metastasis
We next investigated the effects of FOXP3-induced miR-146a/b on tumor cell migration, invasion, and metastasis. In in vitro scratch assays with FOXP3-Tet-off MCF7 cells, cell migration was inhibited by FOXP3 induction, but this inhibition was not reversed by miR-146a/b inhibitors (Fig. 6A). Similarly, cell migration was significantly reduced after FOXP3 induction and was not rescued by miR-146a/b inhibitors (Fig. 6B).
Figure 6.
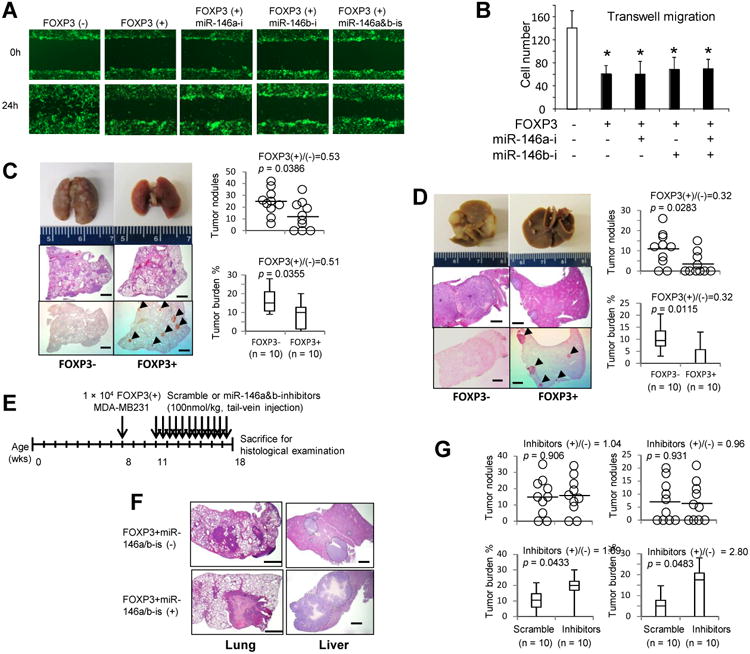
Effects of FOXP3 on tumor growth, migration, and metastasis. A. Representative images of scratch assays with FOXP3-Tet-off MCF7 cells cultured with (FOXP3-) or without Dox (FOXP3+) for 24 hours and then transfected with 100 nM scramble miR or miR-146a/b inhibitors. B. Quantification of migratory cells in transwell assays performed with FOXP3-Tet-off MCF7 cells cultured with (FOXP3-) or without Dox (FOXP3+) for 24 hours and then transfected with miR-146a/b inhibitors (p value by a two-tailed t test). Data are presented as the mean and SD of triplicate samples. All in vitro experiments were repeated three times. C and D. MDA-MB231 cells stably expressing FOXP3 or a control vector were injected intravenously into immunodeficient NSG mice at 8 weeks of age. At 7 weeks after injection, the lungs (C) and livers (D) were removed (left top panels) and stained with H&E (left middle panels) and for the detection of FOXP3 (left bottom panels). Arrowheads indicate the FOXP3+ tumors. Dotted lines circumscribe metastatic lesions in livers (left middle panels in D). Scale bar, 1 mm. Right top graphs: Quantification of gross metastatic nodules. Right bottom graphs: Quantification of tumor burden (tumor area/tissue area × 100%). E. Timeline of therapeutic delivery of miR-146a/b inhibitors. F. Representative images of low-power magnification of H&E-stained sections used for analyses of size and distribution of metastatic tumors in the lung (left panels) and liver (right panels) from miR-146a/b-treated and control mice. Scale bar, 1 mm. Dotted lines circumscribe metastatic lesions in liver sections (right panels). G. Quantification of gross tumor nodules and burden of the lungs (left panels) and livers (right panels) from miR-146a/b-treated and control mice. Data are shown graphically with black circles representing the number of tumor nodules from each mouse and columns representing the mean and SD of tumor burden. Horizontal lines represent the average or median value. p values were determined by two-tailed t test or Mann-Whitney test. Wks, weeks; miR-146a/b-is, miR-146a/b inhibitors. All in vivo experiments were repeated two times.
MDA-MD321 metastatic breast cancer cells express endogenous miR-146a/b (4, 5, 31) but only a low level of endogenous FOXP3 (18). Expression of exogenous FOXP3, which localized in nuclei, significantly increased the expression of miR-146a/b in these cells (Fig. S7). To test the role of FOXP3-induced miR-146a/bin tumor metastasis in vivo, we intravenously injected MDA-MB231 wild-type (WT) cells or MDA-MB231 cells stably expressing FOXP3 into NSG mice. At 7 weeks after injection, significant reductions in the number and size of lung and liver metastases were observed in the mice injected with FOXP3-overexpressing cells (Fig. 6C and D). Intravenous injection of miR-146a/b inhibitors beginning 3 weeks after implantation of FOXP3-overexpressing MDA-MB231 cells (Fig. 6E) increased the size of lung and liver metastases substantially, but the number of metastases did not change (Fig. 6F and G). Significant increases of IRAK1 and TRAF6 transcripts in the tumors were also detected after treatment with miR-146a/b inhibitors (Fig. S8).
Validation of a FOXP3-miR-146-NF-κB axis in human breast cancer
We have demonstrated that nuclear FOXP3 is expressed in normal breast epithelial cells but is lost in 70-80% of breast cancer cells in human breast cancer samples (18). In cancer cells that do express FOXP3, it localizes predominately to the cytoplasm. In contrast, FOXP3 localizes predominately to the nuclei of normal cells (16-18, 20). The cytoplasmic localization is associated with the loss of tumor inhibition (32). Thus, we used IHC analysis with a specific human FOXP3 antibody to obtain two breast cancer sample groups, one group of 8 nuclear FOXP3+ samples and one group of 12 FOXP3- samples (Fig. 7A). Expression levels of miR-146a/b are significantly higher in microdissected cells from nuclear FOXP3+ samples (approximately 2.3- and 2.2-fold, respectively) than in those from FOXP3- samples (Fig. 7B). Conversely, mRNA expression of IRAK1 and TRAF6 was significantly lower in microdissected nuclear FOXP3+ cancer cells (approximately 2.0- and 1.7-fold, respectively) than in FOXP3- cancer cells (Fig. 7C). Therefore, FOXP3 defects are likely a major determinant of miR-146a/b levels and their target signaling in human breast cancer.
Figure 7.
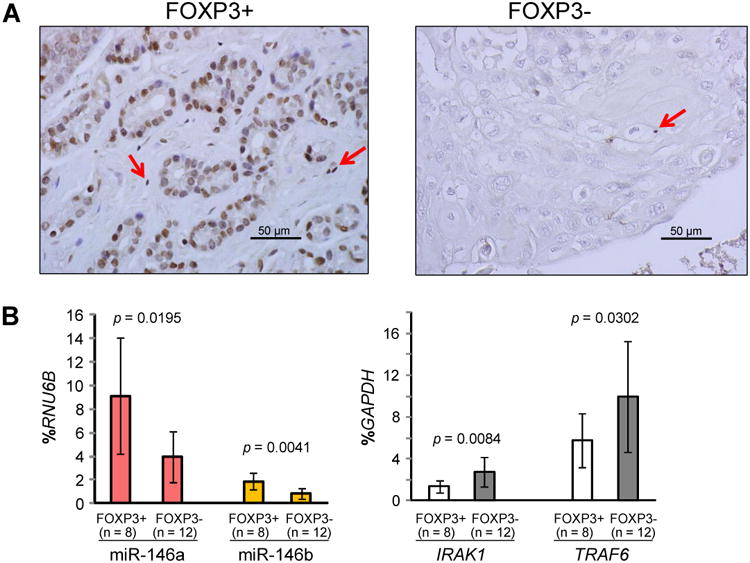
Effects of FOXP3 defects on miR-146a/b, IRAK1 and TRAF6 mRNA levels in human breast cancer samples. A. Representative IHC analysis with a specific antibody against human FOXP3 used to identify nuclear FOXP3+ and FOXP3- primary breast cancer samples. Red arrows indicate Tregs with FOXP3 staining. B. Quantification of miR-146a/b expression in microdissected nuclear FOXP3+ cancer cells and FOXP3- cancer cells. C. Quantification of IRAK1 and TRAF6 mRNA expression in microdissected nuclear FOXP3+ cancer cells and FOXP3- cancer cells. All p values were calculated by two-tailed t test. All experiments were repeated two times.
Discussion
FOXP3-regulated miRs and their regulatory effects remain largely unexamined in Tregs and cancer cells. Here, our preliminary examination of potential FOXP3-targeted miRs in breast cancer cells identified a functional FOXP3-miR-146-NF-κB axis in breast epithelial cells. Specifically, miR-146a/b contribute at least in part to FOXP3-mediated suppression of tumor growth in breast cancer cells, although FOXP3 has an miR-146a/b-independent tumor suppressive role during tumor metastasis. MiR146a/b are both substantially upregulated after FOXP3 induction in breast epithelial cells. However, our results indicate that only miR-146a is targeted directly by FOXP3 in MCF7 cells. Although miR-155 was reported to be induced by FOXP3 in Tregs and cancer cells (33, 34) and miR-7 and miR-183 were identified as potential FOXP3 targets in human breast cancer BT549 cells (33) and leukemia U937 cells (35), respectively, these miRs were not identified as potential FOXP3 targets in our assays.
Previous studies reported data consistent with the existence of an miR-146-NF-κB negative feedback regulation loop (2, 3). Our in vitro and in vivo analyses suggest that FOXP3-mediated induction of miR-146a/b results in downregulation of IRAK1 and TRAF6 and subsequently inhibits NF-κB activation, leading to tumor suppression in breast cancer cells. Furthermore, we identified two forkhead-binding motifs in the proximal promoter region of miR-146a that are required for transcriptional regulation of miR-146a by FOXP3. Interestingly, the forkhead-binding motifs are adjacent to NF-κB binding sites (2), and our data show that FOXP3 interacts synergistically with NF-κB to induce transcriptional activity of miR-146a, even though FOXP3 and NF-κB have opposing functions in tumorigenesis. Recent studies found that FOXP3 and NF-κB co-localize in the nuclei of gastric cancer cells after TNF-α stimulation, and FOXP3 interacts with NF-κB and represses its activation, suggesting FOXP3 as a negative regulator of NF-κB activation (36, 37). Our data also validated a direct interaction between FOXP3 and NF-κB in MCF7 cells after stimulation with TNF-α. The attenuation of this interaction by miR146a/b mimics suggests a mechanism by which FOXP3 regulates the miR-146-NF-κB negative feedback loop in breast epithelial and cancer cells. Although miR-146b was also induced by FOXP3, we did not identify any significant forkhead-binding motifs in the potential promoter region of this miR. However, it remains possible that miR-146b is indirectly regulated by FOXP3 through an as yet undiscovered mechanism. Interestingly, direct targeting of miR-146b by STAT3 can induce miR-146b to inhibit NF-κB activation in breast cancer cells (38). Although STAT3 is not a FOXP3-target gene (19), FOXP3 acts as a co-transcriptional factor with STAT3 in tumor-induced Tregs (39), perhaps suggesting a FOXP3/STAT3-miR-146b-NF-ĸB axis in breast cancer cells.
Our data show FOXP3-mediated inhibition of cell proliferation and tumor growth is partly blocked by miR-146a/b inhibitors, suggesting a contribution of miR-146a/b to FOXP3-triggered tumor suppression. Although a previous study suggested that ectopic miR-146a/b downregulate expression of EGFR in MDA-MB231 cells, inhibiting invasion and migration in vitro and suppressing experimental lung metastasis in vivo (4), our results do not support a similar role for FOXP3-induced miR-146a/b in MCF7 cells. Although FOXP3 also inhibited cell migration and tumor metastasis, this inhibition was not disturbed by miR-146a/b inhibitors in breast cancer cells. Interestingly, when compared with the expression levels of miR-146a/b in MCF10A normal breast epithelial cells, the expression levels are low in less invasive breast cancer cell lines such as MCF7 and T47D and high in more invasive breast cancer cell lines such as MDA-MB-231 (4, 5, 31), suggesting miR-146a/b functions may be lost in tumor development but not in tumor metastasis. In contrast, the expression levels of CXCR4 and MMP9, which promote tumor metastasis, were dramatically reduced by FOXP3 in MCF7 cells (40, 41). CXCR4 has been reported as a potential target of miR-146a (11), but miR-146a did not contribute to the FOXP3-induced regulation of CXCR4 in MCF7 cells. Thus, our future studies will examine whether CXCR4 and MMP9 bypass miR-146a to contribute to FOXP3-mediated suppression of tumor metastasis.
This study also reveals the involvement of the FOXP3-miR-146-NF-κB axis in apoptotic signaling in breast cancer cells. MiR-146a/b, through repression of IRAK1 and TRAF6, reduce NF-κB activation (2, 3), which can inhibit apoptosis in cancer cells by inducing anti-apoptotic factors such as BCL2 and BCL2L1 (42-44). Interestingly, BCL2L1 expression was reduced by FOXP3 through miR-146a/b, but BCL2 expression was not. BCL2L1 is a well-characterized apoptotic inhibitor that can promote evasion of autophagic cell death (45, 46). Notably, BCL2L1 is likely to be regulated by p65, whereas BCL2 is not (42, 47). In fact, our data show that nuclear p65 but not total p65 is reduced after FOXP3-miR-146 induction in breast cancer cells, suggesting the FOXP3-miR-146 axis inhibits NF-κB activation through specific repression of p65 nuclear translocation, which may also contribute to FOXP3-miR-146-induced apoptosis. Likewise, NF-κB-targeting genes TRAF1/2, which can inhibit apoptosis (43), were also inhibited by FOXP3 through miR-146a/b in breast epithelial cells. Thus, the identification of FOXP3-miR-146-NF-κB axis-induced apoptosis advances our understanding of FOXP3-mediated tumor suppression.
In summary, miR-146a/b function in FOXP3-mediated tumor suppression, especially in regulating tumor growth and apoptosis (Fig. S9). Identification of the FOXP3-miR-146-NF-κB axis in tumorigenes is reveals new therapeutic targets for cancers with FOXP3 defects.
Supplementary Material
Acknowledgments
We thank Dr. Alexander Rudensky for the Foxp3flox/flox mice and Dr. Erin Thacker and Lisa L.H. Nguyen for editorial assistance in preparing this manuscript.
Grant support: Research reported in this publication was supported by the National Institutes of Health/National Cancer Institute (CA164688, CA179282 and CA118948 for L.W.), the Department of Defense (PC130594 for L.W. and W.H.Y.), the UAB Faculty Development Grant (for R.L.), the Larsen Endowment Fellowship Program Grant (for W.H.Y.) and the Mercer University Seed Grant (for W.H.Y.).
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Authors' Contributions: Conception and design: L. Wang, R. Liu, C. Liu
Development of methodology: R. Liu, C. Liu, W.H. Yang, F. Tang, D. Chen
Acquisition of data (provided animals, provided facilities, etc.): R. Liu, L. Wang, C.M. Dugas
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): D. Chen, L. Wang, R. Liu, C. Liu, C.M. Dugas
Writing, review, and/or revision of the manuscript: L. Wang, R. Liu
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): F. Tang, W.H. Yang, D. Chen, P. Zheng, Y. Liu, X. Liu, C.G. Liu
Study supervision: L. Wang, R. Liu
References cited
- 1.Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142:914–29. doi: 10.1016/j.cell.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–6. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene. 2008;27:5643–7. doi: 10.1038/onc.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hurst DR, Edmonds MD, Scott GK, Benz CC, Vaidya KS, Welch DR. Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis. Cancer Res. 2009;69:1279–83. doi: 10.1158/0008-5472.CAN-08-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo D, Wilson JM, Harvel N, Liu J, Pei L, Huang S, et al. A systematic evaluation of miRNA:mRNA interactions involved in the migration and invasion of breast cancer cells. J Transl Med. 2013;11:57. doi: 10.1186/1479-5876-11-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011;208:1189–201. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao JL, Rao DS, Boldin MP, Taganov KD, O'Connell RM, Baltimore D. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci U S A. 2011;108:9184–9. doi: 10.1073/pnas.1105398108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Starczynowski DT, Morin R, McPherson A, Lam J, Chari R, Wegrzyn J, et al. Genome-wide identification of human microRNAs located in leukemia-associated genomic alterations. Blood. 2011;117:595–607. doi: 10.1182/blood-2010-03-277012. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Vandenboom TG, 2nd, Wang Z, Kong D, Ali S, Philip PA, et al. miR-146a suppresses invasion of pancreatic cancer cells. Cancer Res. 2010;70:1486–95. doi: 10.1158/0008-5472.CAN-09-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu B, Wang N, Wang X, Tong N, Shao N, Tao J, et al. MiR-146a suppresses tumor growth and progression by targeting EGFR pathway and in a p-ERK-dependent manner in castration-resistant prostate cancer. Prostate. 2012;72:1171–8. doi: 10.1002/pros.22466. [DOI] [PubMed] [Google Scholar]
- 11.Wang D, Liu D, Gao J, Liu M, Liu S, Jiang M, et al. TRAIL-induced miR-146a expression suppresses CXCR4-mediated human breast cancer migration. FEBS J. 2013;280:3340–53. doi: 10.1111/febs.12323. [DOI] [PubMed] [Google Scholar]
- 12.Mei J, Bachoo R, Zhang CL. MicroRNA-146a inhibits glioma development by targeting Notch1. Molecular and cellular biology. 2011;31:3584–92. doi: 10.1128/MCB.05821-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin SL, Chiang A, Chang D, Ying SY. Loss of mir-146a function in hormone-refractory prostate cancer. Rna. 2008;14:417–24. doi: 10.1261/rna.874808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Li D, Li M, Ye M, Ding L, Cai H, et al. MicroRNA-146a targets PRKCE to modulate papillary thyroid tumor development. International journal of cancer Journal international du cancer. 2014;134:257–67. doi: 10.1002/ijc.28141. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Xu B, Moran MS, Zhao Y, Su P, Haffty BG, et al. 53BP1 functions as a tumor suppressor in breast cancer via the inhibition of NF-kappaB through miR-146a. Carcinogenesis. 2012;33:2593–600. doi: 10.1093/carcin/bgs298. [DOI] [PubMed] [Google Scholar]
- 16.Liu R, Wang L, Chen G, Katoh H, Chen C, Liu Y, et al. FOXP3 up-regulates p21 expression by site-specific inhibition of histone deacetylase 2/histone deacetylase 4 association to the locus. Cancer Res. 2009;69:2252–9. doi: 10.1158/0008-5472.CAN-08-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuo T, Liu R, Zhang H, Chang X, Liu Y, Wang L, et al. FOXP3 is a novel transcriptional repressor for the breast cancer oncogene SKP2. J Clin Invest. 2007;117:3765–73. doi: 10.1172/JCI32538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuo T, Wang L, Morrison C, Chang X, Zhang H, Li W, et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell. 2007;129:1275–86. doi: 10.1016/j.cell.2007.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katoh H, Qin ZS, Liu R, Wang L, Li W, Li X, et al. FOXP3 orchestrates H4K16 acetylation and H3K4 trimethylation for activation of multiple genes by recruiting MOF and causing displacement of PLU-1. Mol Cell. 2011;44:770–84. doi: 10.1016/j.molcel.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Liu R, Li W, Chen C, Katoh H, Chen GY, et al. Somatic single hits inactivate the X-linked tumor suppressor FOXP3 in the prostate. Cancer Cell. 2009;16:336–46. doi: 10.1016/j.ccr.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nature reviews. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 22.Magness ST, Jijon H, Van Houten Fisher N, Sharpless NE, Brenner DA, Jobin C. In vivo pattern of lipopolysaccharide and anti-CD3-induced NF-kappa B activation using a novel gene-targeted enhanced GFP reporter gene mouse. Journal of immunology. 2004;173:1561–70. doi: 10.4049/jimmunol.173.3.1561. [DOI] [PubMed] [Google Scholar]
- 23.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–41. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Wang CY, Cusack JC, Jr, Liu R, Baldwin AS., Jr Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med. 1999;5:412–7. doi: 10.1038/7410. [DOI] [PubMed] [Google Scholar]
- 25.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nature protocols. 2007;2:329–33. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 26.McSherry EA, Brennan K, Hudson L, Hill AD, Hopkins AM. Breast cancer cell migration is regulated through junctional adhesion molecule-A-mediated activation of Rap1 GTPase. Breast cancer research : BCR. 2011;13:R31. doi: 10.1186/bcr2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schramek D, Kotsinas A, Meixner A, Wada T, Elling U, Pospisilik JA, et al. The stress kinase MKK7 couples oncogenic stress to p53 stability and tumor suppression. Nature genetics. 2011;43:212–9. doi: 10.1038/ng.767. [DOI] [PubMed] [Google Scholar]
- 28.Corcoran DL, Pandit KV, Gordon B, Bhattacharjee A, Kaminski N, Benos PV. Features of mammalian microRNA promoters emerge from polymerase II chromatin immunoprecipitation data. PLoS One. 2009;4:e5279. doi: 10.1371/journal.pone.0005279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baer C, Claus R, Plass C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res. 2013;73:473–7. doi: 10.1158/0008-5472.CAN-12-3731. [DOI] [PubMed] [Google Scholar]
- 30.Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG, et al. Chromatin structure analyses identify miRNA promoters. Genes & development. 2008;22:3172–83. doi: 10.1101/gad.1706508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Lu H, Li T, Yu L, Liu G, Peng X, et al. Kruppel-like factor 8 promotes tumorigenic mammary stem cell induction by targeting miR-146a. Am J Cancer Res. 2013;3:356–73. [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Liu R, Ribick M, Zheng P, Liu Y. FOXP3 as an X-linked tumor suppressor. Discov Med. 2010;10:322–8. [PMC free article] [PubMed] [Google Scholar]
- 33.McInnes N, Sadlon TJ, Brown CY, Pederson S, Beyer M, Schultze JL, et al. FOXP3 and FOXP3-regulated microRNAs suppress SATB1 in breast cancer cells. Oncogene. 2012;31:1045–54. doi: 10.1038/onc.2011.293. [DOI] [PubMed] [Google Scholar]
- 34.Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–40. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- 35.Liu WH, Chang LS. Suppression of Akt/Foxp3-mediated miR-183 expression blocks Sp1-mediated ADAM17 expression and TNFalpha-mediated NFkappaB activation in piceatannol-treated human leukemia U937 cells. Biochem Pharmacol. 2012;84:670–80. doi: 10.1016/j.bcp.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 36.Hao Q, Li W, Zhang C, Qin X, Xue X, Li M, et al. TNFalpha induced FOXP3-NFkappaB interaction dampens the tumor suppressor role of FOXP3 in gastric cancer cells. Biochem Biophys Res Commun. 2013;430:436–41. doi: 10.1016/j.bbrc.2012.11.039. [DOI] [PubMed] [Google Scholar]
- 37.Hao Q, Zhang C, Gao Y, Wang S, Li J, Li M, et al. FOXP3 inhibits NF-kappaB activity and hence COX2 expression in gastric cancer cells. Cellular signalling. 2014;26:564–9. doi: 10.1016/j.cellsig.2013.11.030. [DOI] [PubMed] [Google Scholar]
- 38.Xiang M, Birkbak NJ, Vafaizadeh V, Walker SR, Yeh JE, Liu S, et al. STAT3 induction of miR-146b forms a feedback loop to inhibit the NF-kappaB to IL-6 signaling axis and STAT3-driven cancer phenotypes. Science signaling. 2014;7:ra11. doi: 10.1126/scisignal.2004497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hossain DM, Panda AK, Manna A, Mohanty S, Bhattacharjee P, Bhattacharyya S, et al. FoxP3 acts as a cotranscription factor with STAT3 in tumor-induced regulatory T cells. Immunity. 2013;39:1057–69. doi: 10.1016/j.immuni.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 40.Douglass S, Meeson AP, Overbeck-Zubrzycka D, Brain JG, Bennett MR, Lamb CA, et al. Breast cancer metastasis: demonstration that FOXP3 regulates CXCR4 expression and the response to CXCL12. J Pathol. 2014;234:74–85. doi: 10.1002/path.4381. [DOI] [PubMed] [Google Scholar]
- 41.Nakahira K, Morita A, Kim NS, Yanagihara I. Phosphorylation of FOXP3 by LCK downregulates MMP9 expression and represses cell invasion. PLoS One. 2013;8:e77099. doi: 10.1371/journal.pone.0077099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dong QG, Sclabas GM, Fujioka S, Schmidt C, Peng B, Wu T, et al. The function of multiple IkappaB : NF-kappaB complexes in the resistance of cancer cells to Taxol-induced apoptosis. Oncogene. 2002;21:6510–9. doi: 10.1038/sj.onc.1205848. [DOI] [PubMed] [Google Scholar]
- 43.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–3. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 44.Weintraub SJ, Manson SR, Deverman BE. Resistance to antineoplastic therapy. The oncogenic tyrosine kinase-Bcl-x(L) axis. Cancer cell. 2004;5:3–4. doi: 10.1016/s1535-6108(03)00338-6. [DOI] [PubMed] [Google Scholar]
- 45.Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
- 46.Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. The FEBS journal. 2011;278:403–13. doi: 10.1111/j.1742-4658.2010.07965.x. [DOI] [PubMed] [Google Scholar]
- 47.Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol Cell Biol. 2000;20:2687–95. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.