

Abstract
Purpose
Despite the prevalence and significant morbidity resulting from estrogen receptor positive (ER+) breast adenocarcinomas, there are only a few models of this cancer subtype available for drug development, and arguably none for studying etiology. Those models that do exist have questionable clinical relevance.
Methods
Given our goal of developing luminal models, we focused on six cell lines derived by minimal mutagenesis from normal human breast cells, and asked if any could generate clinically relevant xenografts, which we then extensively characterized.
Results
Xenografts of one cell line, 184AA3, consistently formed ER+ adenocarcinomas that had a high proliferative rate and other features consistent with “luminal B” intrinsic subtype. Squamous and spindle cell/mesenchymal differentiation was absent, in stark contrast to other cell lines that we examined or others have reported. We explored intratumoral heterogeneity produced by 184AA3 by immunophenotyping xenograft tumors and cultured cells, and characterized marker expression by immunofluorescence and flow cytometry. A CD44High subpopulation was discovered, yet their tumor forming ability was far less than CD44Low cells. Single cell cloning revealed the phenotypic plasticity of 184AA3, consistent with the intratumoral heterogeneity observed in xenografts. Characterization of ER expression in cultures revealed ER protein and signaling is intact, yet when estrogen was depleted in culture, and in vivo, it did not impact cell or tumor growth, analogous to therapeutically resistant ER+ cancers.
Conclusions
This model is appropriate for studies of the etiology of ovarian hormone independent adenocarcinomas, for identification of therapeutic targets, predictive testing and drug development.
Keywords: Luminal breast cancer models, xenograft, intratumoral heterogeneity, microenvironment
Introduction
A pressing goal for cancer researchers is recognizing different clinical forms and subtypes of tumors, and understanding how and why each type manifests. Defining the cellular origins and steps to malignant progression are critical to improved and personalized cancer-prevention and treatment strategies. Model systems with known origins, that faithfully recapitulate specific clinical cancer phenotypes and behaviors allow us to explore intricate tumor biology and dynamic relationships between tumor cells and their microenvironments; and most importantly, to investigate how these interactions influence clinical pathology and therapeutic response. No model is perfect, but some are more useful than others[1].
Our ability to explore biology of ‘luminal-type’ breast adenocarcinoma is limited unfortunately, due to the relatively few models available [2,3]. A notable deficiency, given the luminal subtype is by far the most prevalent form of breast cancer in the clinic. Over 70% of newly diagnosed breast malignancies are assigned to this class each year by positive staining for estrogen and progesterone receptors (ER/PR) or gene expression profiling[4,5]. Most women with these ER+ tumors respond well to hormone-targeted therapies, such as selective estrogen-receptor response modulators, aromatase inhibitors, luteinizing hormone-releasing hormone agents, ER down regulators, or prophylactic ovary removal. Nevertheless, roughly 30% of patients have luminal tumors that don't respond to treatment that leads to poorer outcomes[6]. Despite a better prognosis, the higher prevalence means that more women die each year from ER positive luminal breast cancers than all other subtypes combined[7]. This underscores how much we have to learn, and the lack of appropriate models is a fundamental obstacle.
Current models, such as established cell lines, are convenient tools for research and have taught us a great deal, but they do have drawbacks[8] –and none have generated the luminal adenocarcinomas of interest in vivo when xenografted into immunodeficient mice[9]. Grafting primary tumors directly into mice has been more successful in this regard, particularly in reproducing some features of the parent tumor. For reasons unknown however, the graft success-rate of ER+ luminal subtype tumors continues to be far lower than that for the basal subtypes[9]. As a result, there is even a paucity of patient-derived xenograft (PDX) models of luminal breast cancer[10,11].
To fill this model gap, we looked to several cell models of breast cancer progression. Isogenic progression-series of cell lines are potent tools—especially for etiological studies—as they include both the non-malignant precursor cell line and fully malignant derivatives to which comparisons can be drawn, with intermediate and parallel lines sometimes also available for study. These collections of cell lines allow exploration of early transformative events, adding insight into tumorigenic initiation, something PDX and other end-stage models, by their nature, cannot provide. For example, two human breast cancer progression series, HMT-3522-LBNL[12,13] and MCF10A[14], have been central to discovering the dominant role of microenvironmental factors in regulating cell phenotype and have revealed novel targets for breast cancer therapeutics[14,15]. Yet, neither is an ideal model of luminal breast cancer, as malignant lines from these series produce xenografts with squamous/basal-like histology, and not the ER+ adenocarcinomas seen most often in the clinic[13,16]. To solve this dilemma, we turned to another isogenic progression series, the 184 collection, to determine if any derivative lines in this series could form tumors with a luminal phenotype.
The 184 progression series[17,18] began with a culture of normal finite-lifespan cells and, following exposure to oncogenic agents, includes cell lines with either finite, extended, or immortal lifespans. Some immortal cell lines in the series display a transformed phenotype in culture, and recent genome sequencing in BaP-treated lines has revealed mutation patterns similar to clinical specimens[19]. Yet, we were uncertain of the tumor phenotypes that would emerge from most of the 184 derivatives, or whether they would form tumors at all.
To determine tumorigenicity of 184-derived cells, we orthotopically xenografted each cell line possessing anchorage independent growth into NOD scid gamma mice (NSG; NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) and monitored for tumor growth. Whereas most xenografts resulted in either squamous carcinomas or no tumors at all, one—184AA3—produced adenocarcinomas closely resembling clinical breast tumors. 184AA3 xenografts were invasive and expressed several key luminal markers—including estrogen receptor—yet were insensitive to hormone ablation via ovary removal. Here, we present and describe this novel model of ER+ luminal breast cancer.
Results
184AA3 xenografts produce ER+ adenocarcinomas
The primary founding culture of the 184 cell progression series was established from a reduction mammoplasty in 1980[17]. Many different sub-lines have since been produced, and several have acquired phenotypes associated with malignancy, such as anchorage-independent growth (AIG) when embedded in methylcellulose[18]. To determine in vivo tumorigenicity of these AIG+ lines; i.e., 184ZNMY3-N, 184B5ME, 184FMY2, 184AaGS1, 184AA2, and 184AA3; we xenografted each bilaterally into cleared #4 inguinal mammary fat pads of NSG immunodeficient mice. Injections included also primary-derived fibroblasts, 50% of which were treated with 0.3Gy X-ray radiation. Over the course of 15 months, four of six cell lines were found to produce tumors, these were: 184AaGS1(1/6 mice), 184ZNMY3-N (7/7 mice), 184AA2 (5/7 mice), and 184AA3 (5/5 mice). 184AaGS1, 184ZNMY3-N and 184AA2 produced similar histologies –all predominantly squamous, occasionally containing areas with calcifications and metaplasia (Fig. 1a). 184AA3-derived tumors however, were remarkable in that they formed adenocarcinomas phenotypically identical to human invasive ductal carcinomas. These would be scored as high combined histologic grade cancers with minimal tubule formation, large nuclei, and high proliferative rate (Fig. 1b). Xenografts exhibited large areas of variable ERα positivity—ranging from barely detectable to very intense—and a generally weak level of PR expression (Fig. S1a). Immunostaining highlighted clustered islands of tumor cells surrounded by streams of reactive stromal mesenchymal cells, similar to many human cancers; and in some cases, areas where tumor cells had invaded adipose and underlying muscle. In addition to ERα, cells exhibited strong expression of keratin19, keratin 5, and E-cadherin (indicative of ductal and not lobular differentiation); and to a lesser extent keratin 8/18 and keratin 6. Muc1 and Her2 were expressed also, as was p63 (Fig. S2a-c). Expression of ERα and a high proportion of Ki67+ cells indicates184AA3 xenografts likely reflect the Luminal B breast cancer subtype (fig. S1d).
Figure 1. 184AA3 ER+ adenocarcinomas resemble human breast tumors.

(a) H&E images of xenografts formed by the 4 tumor producing cell lines in the 184 progression series. Scale bars = 400μm (b) Comparison of an 184AA3 xenograft to a clinical breast cancer case. Both are ER+ adenocarcinomas that contain islands of tumor epithelium expressing luminal markers keratin 18(K18), E-Cadherin (E-Cad), Keratin 8 (K8), keratin 18 (K18), and Muc1. Smooth muscle actin (SMA) and vimentin (Vim) staining emphasizes the epithelial-stromal boundary in the clinical specimen. Keratin 14 (K14) is absent also in the clinical specimen. Scale bars = 200μm.
To test reproducibility of 184AA3 xenograft formation, we transplanted cells into a second set of mice under identical conditions and followed the mice over the course of 1 year. Tumors formed in 6 of 8 mice (8/16 glands), and exhibited the same histology and growth rates as in the prior experiment (Fig. 2). Most tumors were first noticed 80-100 days after transplantation, however one became palpable 2 months later, on day +158. To prevent central tumor necrosis, we sacrificed mice when a tumor on either side of the mouse reached a size of ∼ 5mm diameter. Histological examination of contralateral glands (that had no palpable tumor) in several instances revealed a small cluster of 184AA3 cells present at the injection site. Had mice not been sacrificed in these cases, its possible tumors may have eventually formed. For this reason, the actual success rate (per gland) of xenograft formation may be somewhat higher than that reported here. Histological examination again revealed adenocarcinomas (Fig. 2), and we observed ERα staining in each of the formed tumors, present at some level in all cases. Expression was again markedly heterogeneous and not always confined to the nucleus (Fig. 2, Fig. S2a). To show tumors were 184AA3-derived—and did not arise from host cells—we substituted GFP-tagged 184AA3 cells into the xenograft assay and subsequently observed GFP in the tumor epithelium of formed xenografts (Fig. S2b). Notably, stroma was GFP-negative, ruling out 184AA3 as its origin, such as through an epithelial to mesenchymal transition. These findings show 184AA3 cells reliably produce ER+ adenocarcinomas in this model system.
Figure 2. 184AA3 xenograft growth and ERα expression.

(a) 184AA3 growth in vivo. Tumor volumes of 184AA3 xenografts were calculated from bi-weekly caliper measurements. Each tumor is coded according to the mouse#, and side on which it arose; e.g., ‘1R’ indicates experiment mouse #1, right mammary gland. (b) H&E stained sections of 184AA3-derived tumors. Image borders are color-matched to the above growth curves; scale bar = 600μm. Inset: ERα staining with 100μm scale bar.
All xenografts to this point were made by co-injecting into mice both 184AA3 cells and isogenic 184 fibroblasts. The dependence on fibroblasts—or this particular strain of fibroblasts—for tumor formation was important for us to ascertain. We discovered this strain of fibroblasts was not a critical factor, as it could be replaced with a newly isolated line of FACS-sorted fibroblasts transduced with hTERT, WCH-N141-TERT, which produced tumors in 8 of 8 glands with essentially identical histology to those made with 184 fibroblasts. We discovered however, an absolute dependence on irradiated fibroblasts for xenograft formation. When fibroblasts were withheld, or were not irradiated, tumors did not form (0/8 glands in each case); but parallel grafts, with the 50% mixture of irradiated fibroblasts, formed tumors (13/14 glands) as they had in every prior experiment. These findings identify fibroblasts as an essential component of this model, and provide further evidence supporting a fundamental role of stroma in forming and shaping tumors.
Cellular Heterogeneity and CD44Low tumor initiating cells
Intratumoral heterogeneity in breast cancer is common and may be essential for virulent cancers to develop [20-22]. 184AA3 xenografts varied in their expression of ERα, and other markers, suggesting that they may be an advantageous model for exploring mechanisms of intratumoral heterogeneity (Fig. 1, Fig.S1). We explored origins of this heterogeneity by immunostaining both 2D and 3D cultures of 184-series cells using the lineage markers keratin 18 (K18) and keratin 14 (K14). Staining revealed a predominant basal phenotype for the normal early-passage (pre-stasis) 184 culture and post-stasis 184Aa strains, which became more heterogeneous in 184AA2, and more luminal in 184AA3 (Fig. 3a).
Figure 3. 184AA3 and 184 progression series characterization in culture.
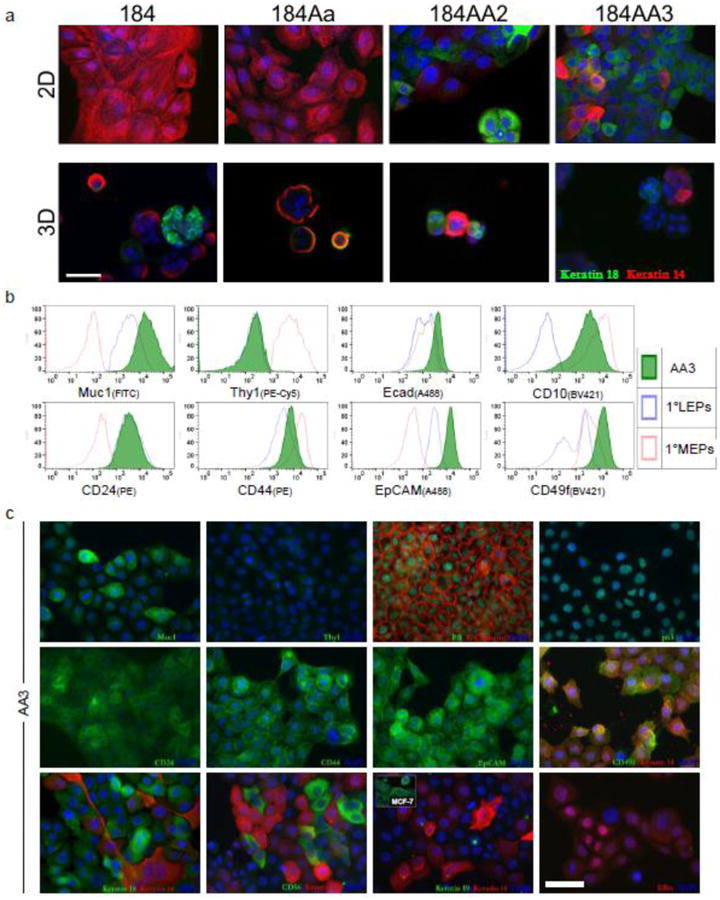
(a) Keratin 14 and 18 immunostaining of cells within the 184 progression series. Shown are the 184 primary culture (passage 4); 184Aa precursor line; and derivative sister lines 184AA2 and 184AA3, cultured on 2D substratum (a, top) and in 3D matrigel (a, bottom); scale bar = 50μm (b) Flow cytometry analysis of 184AA3 cells stained with the indicated conjugated antibodies (green-filled histogram plots). Primary luminal (LEPs, blue) and myoepithelial cells (MEPs, red) from reduction mammoplasty organoids serve as reference controls. (c) Immunostained 2D cultures of 184AA3 cells; scale bar = 50 μm.
To quantify marker expression, we stained 184AA3 cultures with a panel of antibodies and used flow cytometry to measure binding. For comparison, we stained also uncultured luminal and myoepithelial cells isolated directly from normal breast tissue. This juxtaposition revealed in 184AA3 an irregular staining pattern which is consistent with their malignant state, and reflected also the patterns observed in xenografts. 184AA3 cells expressed high levels of Muc1, CD24, EpCAM and E-cadherin; and lower levels of Thy 1 and CD44 –all reflecting a luminal-like phenotype (Fig. 3b). On the other hand, 184AA3 expressed CD10, which is observed normally in myoepithelial cells and, to a lesser extent, in fibroblasts. Microscopic examination of 184AA3 cultures immunostained with these antibodies and more, confirmed these results (Fig. 3c). 184AA3 cells expressed ERα, PR, Muc1, CD24, E-Cadherin, keratin 18, CD49f and CD44, all proteins found to some extent within normal luminal cells. 184AA3 cells lacked Thy1, which is consistent also with a luminal phenotype. However, keratin 19 (K19) was not expressed, whereas both p63 and K14 were, a pattern associated with normal myoepithelial cells, although the latter two were expressed heterogeneously. Expression of K19 and K14 was notably different in culture compared to that in xenografts (Fig. 3c vs. Fig. S1b,c). Absent in cultured cells, K19 was expressed abundantly in tumors; and conversely, K14 was present in culture but generally lost in xenografts. This indicates a shift to a more luminal-like phenotype in vivo. There were also other notable differences, such as with PR and ER. PR, expressed strongly in nuclei of cultured cells, was generally weak and diffuse in the tumors. In contrast, ER expression became much more pronounced and nuclear-localized in vivo. Whether altered expression observed in tumors is reflective of in vivo regulation or that of clonal selection is a mystery.
We explored the phenotypic plasticity of 184AA3 cells by testing whether single cells could reproduce the K14/K18 heterogeneity observed in cultures and xenografts. Indeed they could. When we FACS sorted single 184AA3 cells into individual wells with fibroblast feeder layers, 7/96 cells produced a colony after 14 days in culture. Each colony exhibited mixed staining for K14 and K18, indicating expression is conditional and that this heterogeneity could arise in vivo from clonal selection, as reported for tumor initiating cells (Fig. S3a).
Prior reports have demonstrated breast tumor initiating cells reside in tumor cell subpopulations expressing disproportionately high levels of CD44[23-25]. The CD44High/CD24Low phenotype has since become a widely reported marker of breast cancer stem cells (CSCs) or cells with more aggressive properties [26,24]. When we analyzed CD44 expression by flow cytometry, we discovered a small subpopulation of 184AA3 cells expressing CD44 at distinctly higher levels than the bulk of the culture. Inconspicuous in one dimensional FACS histogram plots, such as that provided in Fig 3b, this CD44High subpopulation was readily and repeatedly observed when we compared CD44 to other parameters, such as size (Forward scatter, FSC, Fig. 4a, FigS3b). When we sorted and cultured the CD44high and CD44low populations, they indeed exhibited distinct morphologies (Fig. 4a). 184AA3-CD44Low cells grew in large colonies with smooth borders, whereas the CD44high cells grew at roughly the same rate, but were more migratory and most often found as single cells or in small clusters (Fig.4a). This phenotype held as we expanded the culture in order to generate enough cells for xenografting into mice. Immediately prior to implantation, we re-examined CD44 expression levels in these two cultures and found their respective CD44 phenotypes had remained largely unchanged, even after 49 days in culture (Fig. 4a). These cells, along with the parental culture, were xenografted, and mice monitored for 5 months. Over the course of two independent experiments, we found both the 184AA3 parental line and CD44Low strain produced relatively the same proportion of tumors, respectively, 9/16 (56%) and 5/12(42%) tumors formed per gland injected (Fig. 4b). Surprisingly, the CD44High strain, which we had presumed to contain the CSC population, produced only a single tumor out of 10 injected glands. Its histology was similar to the others –all being ER+ adenocarcinomas (Fig. 4b). Tumor-initiating cells in 184AA3 appear thus, not in the CD44High subpopulation as anticipated, but in the remaining bulk of the population; cells that on average have less CD44 than normal myoepithelial cells (Figs. 4b, 3b).
Figure 4. Tumor initiation and culture phenotypes of 184AA3 CD44 subpopulations.
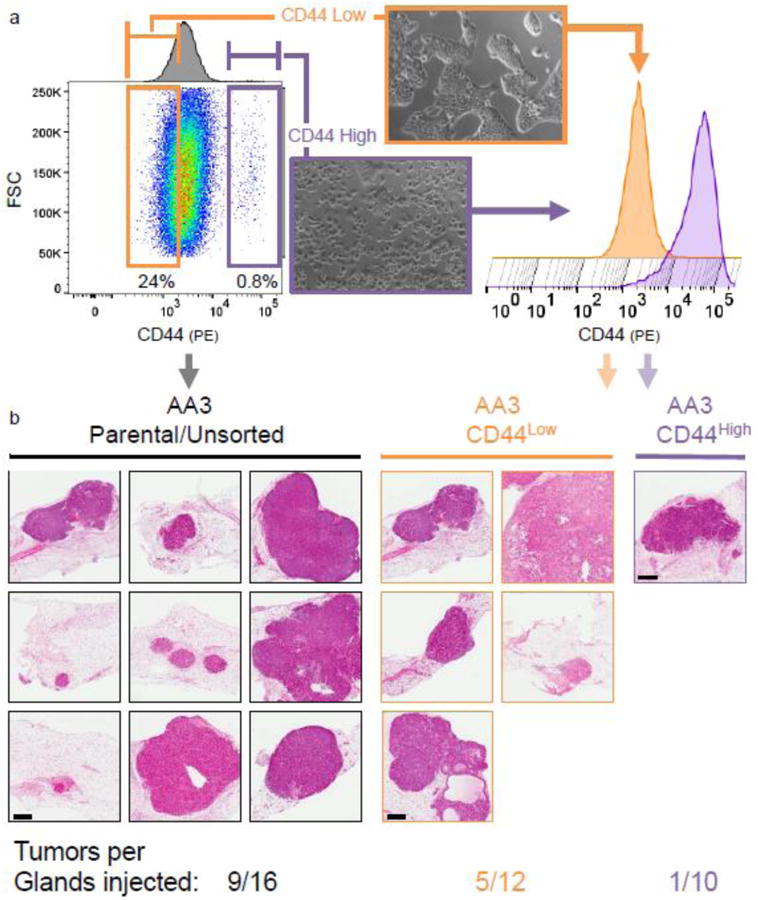
(a, left) 2D cultures of 184AA3 stained with phycoerythrin (PE) conjugated CD44 antibody and analyzed by flow cytometry. A discreet subpopulation, characterized by their elevated CD44 expression is present. Sorting gates were placed around this ‘CD44 High’ subpopulation (purple) and also around the lowest quartile of CD44 expressing cells, ‘CD44 Low’ (orange). (a, middle) morphologies of the FACS sorted cell populations in culture. (a, histograms on right) After expansion in culture for 49 days, and immediately prior to NSG xenograft implantation, CD44 expression in the CD44High and CD44low 184AA3 strains was evaluated by flow cytometry. (b) H&E sections of derived xenografts from injections of the unsorted parental 184AA3 culture and the CD44Low and CD44High 184AA3 strains, which respectively formed tumors in 9/16 (56%), 5/12(42%), and 1/10(10%) glands. scale bar = 600 μm
184AA3's estrogen receptor is functional, but not essential for growth
To ascertain the levels and functionality of ERα in 184AA3, we subjected the cells to a battery of tests, the first of which was to quantify levels of ERα message. Using qRT-PCR, we measured mRNA levels in cultures of 184AA3 and compared them to those expressed by MCF-7 and the basal cell line, MDA-MB-231, as well as freshly isolated FACS-sorted primary human breast luminal cells. ERα was expressed in all, although at vastly disparate levels (Fig. 5). Not surprisingly, mRNA levels were highest in MCF-7 and barely detectable—but present—in MDA-MB-231 cells. In 184AA3, ERα was 16-fold less than that in MCF-7, but notably matched levels found in normal luminal cells from tissue (Fig. 5a). Western blot analysis revealed protein and mRNA levels corresponded. ERα was by far expressed the most in MCF-7 cells, and after increasing the contrast of the western blot image, we detected ERα in 184AA3 cells as well (Fig. 5b). Less clear was a faint signal produced by MDA-MB-231. The band on the blot is just above detection threshold (measured digitally), but low expression is consistent with qRT-PCR mRNA levels (Figs. 5a,b). With knowledge of ERα mRNA and protein levels in 184AA3, we asked whether estrogen signaling was intact.
Figure 5. ER expression and functionality in 184AA3 cells.
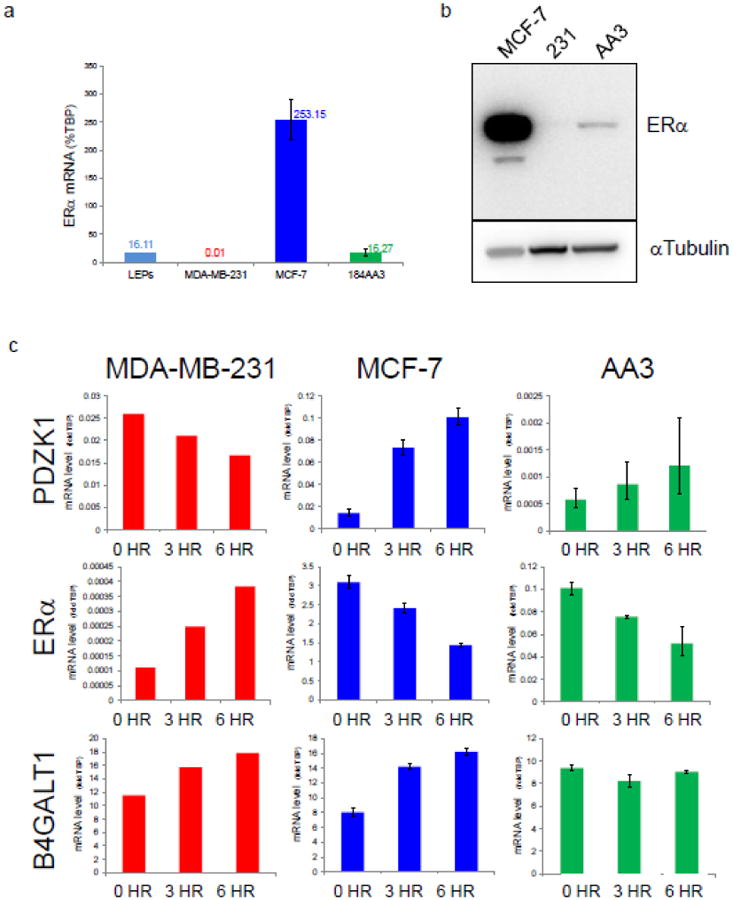
(a) transcript levels of ERα, as measured by qRT-PCR. Values are expressed relative to the internal control gene, TATA box-binding protein (TBP). 2D cultured MCF-7 and MDA-MB-231 breast cancer cell lines, and freshly sorted ‘luminal’ cells from normal tissue, serve as reference controls. (b, top) ERα protein levels in 184AA3, as measured by Western blot. The 66kDa canonical isoform is visible in all three cell lines; the 46kDa isoform is detected in MCF-7 only. (b, bottom) αTubulin loading control. (c) Gene expression changes resulting from 3-hour and 6-hour estradiol stimulation of 184AA3 and reference cell lines MDA-MB-231 and MCF-7. Transcript levels of PDZK1, ERα, and B4GALT were measured by qRT-PCR and are expressed as fold differences relative to TBP internal control gene.
As a steroid hormone receptor, ER is targeted—upon ligand binding—to specific DNA binding sites where it subsequently influences transcriptional activity. To determine if estrogen stimulation would induce transcriptional changes in 184AA3, we exposed cells to estradiol and monitored levels of several estrogen-regulated genes, again using MCF-7 and MDA-MB-231 as controls. Each cell line was placed into estrogen-free medium for 48 hours, then stimulated with 1μM 17-β estradiol. Three hours later, and then again at six hours post-stimulation, we collected RNA and measured expression of nine estrogen-regulated genes by qRT-PCR (Fig. 5c). As expected, the three cell lines expressed disparate levels of each gene, so results are displayed on separate graphs. We found each cell line responded to estrogen stimulation, although not always equally, nor with the same directional trend (Fig. S4). For example, when exposed to estrogen, both 184AA3 and MCF-7 increased expression of PDZK1 and repressed ERα mRNA, whereas MDA-MB-231 cells did exactly the opposite (Fig. 5c). However, MCF-7 and 184AA3 did not always respond in kind. For example, MCF-7 increased B4GALT1 when stimulated, whereas 184AA3 held levels constant. These and other differences in how each of the three cell lines respond to estrogen are interesting and likely reflect other cell-specific forms of gene regulation or ER variants with different functions (Fig. S4, Fig.5b). These results nevertheless show 184AA3 cells can sense and respond to estrogen stimulation, indicating ER is undoubtedly functional in these cells.
Indeed, when we immunostained 184AA3 cells for ER moments after estradiol exposure, we noticed the bulk of the receptor had translocated to the nucleus (Fig. 6b), providing further evidence that signaling is intact. This effect was even clearer in 184AA3XT, a ‘biofiltered’ 184AA3 derivative strain cultured from an 184AA3 xenograft that abundantly expressed ER (Him25.5R, Fig. S5). 184AA3XT were found to be less migratory and invasive in culture (Fig. S6 a,b), but surprisingly, and despite having similar growth rates to 184AA3 parental line in culture, formed tumors about three-times as fast (compare Figs. S6c & Fig. 2a). It is common for tumor or xenograft-derived strains such as 184AA3XT to be more efficient and quicker to form tumors than the parental strain, and these qualities may improve their practical utility in larger and more costly in vivo experiments.
Figure 6. 184AA3 estrogen-independent growth.

(a) Growth rates of MCF-7 (blue), MDA-MB-231(red), 184AA3 (dark green) and 184AA3XT (light green) cells cultured in standard medium (solid lines) or in estrogen-free medium (dashed lines). (b) ER translocation in 184AA3 and 184AA3XT. Cultures were exposed to estradiol for 15 minutes and then fixed and stained for ER. Scale bar = 10μm (b) Effect of ovary removal on 184AA3 tumor growth. 184AA3 cells were xenografted and allowed to grow to 3mm, at which time the tumor was biopsied and mice either ovariectomized or given a sham surgery. After recovery, the mice were monitored until tumors reached 8mm diameter. (graph on right) Time taken for tumors to reach the 8mm endpoint (from the time of biopsy, dichotomized by experimental group). There is not sufficient evidence to reject the claim that growth rates between the ovariectomized and sham mice are equivalent (p=0.17, Wilcoxon rank-sum test); it appears the ovaries, and thus ovarian hormones, do not affect 184AA3 tumor growth. (d) H&E staining, and ER & Ki-67 immunostained sections of a tumor-biopsy before (Pre-OVX) and 1 week after ovariectomy (Post-OVX tumor). Scale bar = 200μm.
Knowing ER is present and functional in 184AA3 cells, we sought to determine whether 184AA3 depended on estrogen for growth. MCF-7 cells, for example, are notoriously estrogen-dependent, and we were curious if 184AA3 and 184AA3XT cultures would display the same sensitivity. We cultured 184AA3 and 184AA3XT for six weeks, with or without estrogen, and compared their growth to both MCF-7 and MDA-MB-231 cell lines. The results were unexpected, but clear. Whereas MCF-7 cells stopped growing immediately in estrogen-free medium, 184AA3 and 184AA3XT were unaffected by estradiol depletion and unabashedly kept dividing (Fig. 6a), indicating estrogen signaling and growth are not linked within these cells.
To mimic anti-hormonal therapy and study effects of estrogen depletion in vivo, we tested the consequences ovary removal had on 184AA3 tumor growth. Mice were xenografted with 184AA3, and in order to study effects on growth rather than on tumor initiation, we allowed tumors to develop until they reached 3mm in diameter prior to treatment. At that point, a fine needle biopsy of the tumor was taken and mice were alternated into one of two experimental arms; i.e., they either had their ovaries removed or received a control sham surgery. The mice were monitored afterward, and when tumors ultimately reached 8mm diameter, the mice were sacrificed and tumors removed. The uterus was removed also, and each uterine lining was examined to determine the ovarian hormone status of each animal. Similar to what we saw in culture, estrogen depletion had no effect on tumor growth. 184AA3 tumors grew at a rate independent of ovarian hormones, with no statistical difference between ovariectomized mice and controls (p=0.17, Fig. 6b). In fact, the median time it took to reach the endpoint trended less in ovariectomized mice than controls (22 vs. 33 days, Fig. 6b). There were no noticeable differences in histology either. However, to rule out possible transient effects, we performed another set of xenografts and ovariectomies using the same experimental design, except animals were euthanized and tumors collected at fixed times after ovariectomy; i.e, 0.5, 1, 2, 3, 4, 5, 6, 7, and 8 weeks later. A total of 43 biopsy-tumor pairs were collected and each was analyzed for prevalence, intensity and cellular localization of ER and Ki67. We observed no ovariectomy- or sham-dependent differences at any of the time points (Fig. 6c). These results indicate 184AA3 cells have indeed somehow uncoupled ER signaling from growth, as evidenced in culture and analogous to clinical tumors that are refractory to anti-hormonal therapies.
Discussion
Tractable models for researching the causes and cures of the most prevalent form of breast cancer are wanting. The histologies of current xenograft models seldom reflect what we see in the clinic, throwing into question whether such models can be predictive. Here, we describe a cell line, 184AA3, which reliably and reproducibly forms adenocarcinomas when xenografted as described here. Tumors produced by 184AA3 likely reflect the luminal B subtype as assessed by their aggressiveness and expression of luminal markers, which includes ER. Yet, much like the 30% of clinical luminal tumors that prove fatal, another distinguishing characteristic of AA3-derived tumors is that they also do not respond to hormonal ablation.
Estrogen receptor expression is rarely observed in cultured human breast cells [27], notably lacking in primary cultures derived from normal reduction mammoplasty tissues. Only a few established cancer-derived cell lines express ER, the most well-known being MCF-7, which is quite aberrant as it has amplified the gene 20+ times over and produces over-abundant levels of the protein. Our finding of ER+ in cultures of 184AA3 (Fig. 3c) and in xenografts (Figs. 1&2; Figs. S1&2), establishes this model as a potentially valuable tool for study of ER function and regulation using a non-amplified ER gene. The tissue architecture, including invasive aspects, stromal involvement, and hormone-deprivation resistance of 184AA3 tumors will likely prove useful for identification, testing and development of therapeutics targeting aggressive luminal breast cancers.
Adding to the utility of this system, 184AA3 is part of a progression series. 184AA3 was derived from the 184Aa strain, itself a derivative of cells cultured from reduction mammoplasty tissue. Precursor cultures, and even sister lines, that are part of the 184 progression series, are available for study, giving the 184AA3 model added potential as a system for research into the etiology of refractory luminal adenocarcinomas. For instance, recent publications call into question whether mammary tumors are derived from luminal epithelial cells. The 184Aa cells from which 184AA3 was derived are K14(+)/K18/K19 (-)– a surprisingly basal phenotype for the progenitors of a line that generates ER+ luminal adenocarcinomas. Indeed we and others have observed generation of ER+ tumors (though not adenocarcinomas) from transformed primary human myoepithelial cells, raising the possibility that tumorigenicity may involve lineage de-differentiation, then re-differentiation in humans as has been reported for PIK3CAmutant tumors in mice [28,29].
Moving beyond the cytokeratins, the intratumoral heterogeneity of this model is notable. Judah Folkman once noted that heterogeneity is almost a universal feature for tumors to succeed[30], and a growing body of literature attests to its role in tumor survival, growth, and therapeutic resistance[21,31]. It is likely that the phenotypic plasticity of 184AA3 cells contributes to the mechanism that generates the intratumoral heterogeneity observed in 184AA3 xenografts, as outgrowths of single cell clones of 184AA3 are also phenotypically heterogeneous (note that we cannot distinguish intrinsic potential of the cell line from potential that is dependent on fibroblastic factors as 7 of the 8 single cell clones of 184AA3 were grown on fibroblast feeder layers). Dissecting the origins of heterogeneity and the factors regulating it, be it clonal diversification or microenvironmental factors, are important avenues for future research that can be addressed using this system.
Further, it is clear that etiology and tumor phenotype do not rely on the epithelial cell alone. That tumorigenicity is a group activity is evidenced here by the xenografting requirement for irradiated fibroblasts. This observation reinforces prior work in dissecting the role of stroma in promoting tumorigenicity[32], which has been extensively elaborated upon, including a recent report identifying factors required for tumorigenicity that are elicited from the stroma by transformed epithelial cells, clarifying a mechanism of malignant cooperation [33].
Returning to the question of which cells are tumor-initiating, we note that the CD44+ subpopulation of 184AA3 was essentially non-tumorigenic, while the bulk of the cells efficiently generated tumors. This contradicts the early work of Al Hajj and colleagues [23] (2003), and reinforces that tumor initiation can arise from populations not previously identified as stem-like [34].
Our discovery of the conditions under which 184AA3 cells generate clinically-relevant luminal tumors is an important step towards defining and overcoming the remaining obstacles that have until now prevented development of models of luminal breast cancer. The fact that this progression model culminates in xenografts that are ovarian-hormone and estradiol independent, proliferative, and invasive suggests its use to learn which factors of the model promote these aggressive phenotypes, and thus which aspects may be changed to instead generate more benign phenotypes. Such observations can form the bases of novel clinical therapeutics.
Methods
Mice
NOD scid gamma mice (‘NSG’, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were obtained from Jackson Labs (stock#005557). Animal use protocols were obtained and procedures followed in strict accordance with guidelines established by the Lawrence Berkeley National Laboratory Animal Welfare and Research Committee (AWRC).
Breast tissues
Breast tissues from reduction mammoplasties and tumors were acquired from the Cooperative Human Tissue Network (CHTN), a program funded by the National Cancer Institute. All specimens were collected with patient consent; reduction mammoplasties were reported negative for proliferative breast disease by board-certified pathologists. Use of anonymous samples was granted exemption status by the University of California at Berkeley Institutional Review Board, in accordance with the Code of Federal Regulations 45 CFR 46.101.
Cell lines
The primary culture and derivation of strains and cell lines in the 184 progression series have been described previously [17,35,18,36]. For this study, we have cultured normal finite pre-stasis 184 HMEC, the post-stasis 184Aa strain resulting from benzo(a)pyrene exposure of primary 184, and immortally transformed cell lines derived from184 that had been shown capable of growing in anchorage independent conditions, namely: 184ZNMY3-N, 184B5ME, 184FMY2, 184AaGS1, 184AA2, and 184AA3. 184 primary-derived fibroblasts were established as described[37]. WCH-N141-TERT fibroblasts were FACS sorted from reduction mammoplasty tissue and transduced with pLXSN-hTERT[G418], a kind gift from Judith Campisi. Fibroblasts and all HMEC were routinely cultured in M87A medium [38], unless otherwise noted in figure legends. MDA-MB-231 and MCF-7 were obtained directly from the American Type Culture Collection (ATCC). 3D cultures were established by seeding cells into 100% Growth factor depleted Matrigel (Invitrogen).
Antibodies
Supplemental Table 1 provides all necessary information regarding antibodies used in this study, including clone information, marker conjugation, supplier, and references to figures where they were used.
Xenograft transplantation
Xenograft protocol was generally as reported by Lim and colleagues[39], with the exception that the epithelial cells were pre-clustered on low attachment plates and other minor exceptions; details as follows. NSG mice (described above) were housed for stabilization and observation for 3 days prior to transplantation surgeries. The day before transplantation surgery, subconfluent passage 50 cultures of epithelial cells were dissociated with 0.05% trypsin and aggregated on low attachment polyhema-coated plates in M87A medium containing 5% growth factor reduced Matrigel™ (Invitrogen). Fibroblasts, either 184-Fb or N141-hTERT primary tissue derived fibroblasts, were plated onto two sets of plates. On the day of surgery, half of the fibroblast cultures were exposed to 0.3Gy X-ray radiation, then all the fibroblast cultures were harvested and combined to yield a tube of 50% 0.3 Gy irradiated fibroblasts. One sterile Eppendorf tube was then prepared as follows for each of the mammary glands to be transplanted; each tube contained a mixture of 2.5e5 irradiated and 2.5e5 non-irradiated fibroblasts. 2.5e5 pre-clustered 184AA3 cells were then added to each tube, and the mixture adjusted to 50% Matrigel™ and 20μl final volume containing 1μl surgical tracking dye. Both #4 inguinal fat pads of 3-week old NSG mice were cleared of their epithelial rudiments, and the cell mixture injected into the cleared pad. All animal procedures were performed in compliance with animal safety and pain prevention guidelines.
Ovariectomy protocol
For the ovariectomy experiments, xenografts were performed as described immediately above. When the first tumor in a mouse reached 3 mm diameter, a biopsy was taken using a 16 gauge biopsy needle, and the mouse was either ovariectomized or given a sham surgery. The animals were euthanized at the time points post biopsy as stated in the text, and the tumors and reproductive tracts were resected. Reproductive tracts were microscopically evaluated to determine whether the animal had effectively been ovariectomized. Portions of each tumor were flash-frozen in cell freezing medium, while both the remainder of the tumor and each of the biopsy samples were fixed, paraffin embedded and further processed at the UC Davis Mouse Pathology Laboratory for histological and immunohistochemical (IHC) analyses. Sections of each tumor and each biopsy were immunostained with antibodies to ERa and Ki67. A modified version of the Imagescope “IHC Nuclear” algorithm[36] was used to quantify intensity of immunostain signal in each epithelial cell nucleus in representative sections of each sample. Results of these analyses were then compared for each tumor and associated biopsy pair to determine whether ovariectomy-dependent changes were detectable in the 184AA3 tumors.
Flow Cytometry and Fluorescence-Activated Cell Sorting (FACS)
Analysis and cell sorting was performed using a BD FACS Vantage cytometer (FACSDIVA software, version 5.0.3). Prior to staining, we dissociated the cells, rinsed and re-suspended them in PBS/1% bovine serum albumin (Sigma # A8412). Following their filtration through 40μm nylon mesh strainers (BD Biosciences), the cells were placed into several tubes and combinations of the following antibodies were added: Muc1-FITC, Thy1-PE-Cy5, E-cadherin-Alexafluor 488, CD10-Brilliant Violet 421, CD24-PE, Cd44-PE, EpCAM-Alexafluor488, and CD49f- Brilliant Violet 421; amounts of each determined by empirical titration. Viable cells were selected by using either DAPI or To-Pro-3 dye exclusion, and we used forward scatter (height) vs. side scatter (width) gating to exclude cell doublets. Compensation was calculated using beads custom-labeled with each fluorophor (APC anti-mouse bead kit (Molecular Probes/Invitrogen). Negative controls consisted of unlabeled beads and cells incubated with conjugated isotype control antibodies. Primary cells from tissue-derived organoids served as positive controls and as a comparator sample, prepared as previously described[40]. Supplementary Figure 7 illustrates gates and full backgating used to sort the 184AA3-CD44High and 184AA3-CD44Low subpopulations. Cells were sorted twice—using conservative sorting masks—to ensure purity. For single cell cloning experiments, single viable 184AA3 cells were FACS sorted directly into a 96-well dish containing a subconfluent monolayer of 30Gy X-ray irradiated primary-derived fibroblasts, WCH-N141 TERT ZsGreenhigh. Visual inspection of wells from a parallel sort, where a feeder layer was not used, allowed us to confirm both the targeting of the sort stream and seeding of single cells.
Migration and Invasion
To measure transwell migration, we suspended 105 cells into 200μl of their respective media: M87A for 184AA3 and 184AA3xt, H14 for S1 cells, DMEM (4.5g/L glucose) for MDA-MB-231 and MCF7. Suspensions were placed into the top chamber of tissue-culture treated transwells containing a polyethylene terephthalate (PET) membrane with 8.0μm pore size (Corning, #353097). In the bottom chamber, we placed 300 μl of medium supplemented with 10% fetal bovine serum (FBS). The non-malignant human breast cell line, HMT3522-LBNL-S1 and malignant MDA-MB-231 cell lines served respectfully as non-invasive and invasive reference controls. Cells were incubated at 37°C for 24, 48, and 72 hours. At the end of each timepoint, we fixed the cells and stained them with 0.5% Toluidine Blue in 2% w/v Na2CO3. The top chamber was wiped with a cotton swab and we examined the underside of the transwell and counted cells that had migrated through the pores. To quantify, we counted the cells by hand (if only a few hundred cells had migrated) or extrapolated from images taken in each of four quadrants. Invasion was similarly measured, except a matrigel coating was applied and allowed to set prior to seeding cells(20 μl of 6% matrigel per chamber, incubated 1hr at 37°C).
qRT-PCR
RNAs from cell lines and sorted cells were isolated using silica-based spin-column extraction kits according to the manufacturer's protocol (RNeasy mini kit, Qiagen). All RNA samples were DNase I treated (DNA free, Ambion) to remove traces of genomic DNA. Complimentary DNA (cDNA) was synthesized by random-hexamer primed reverse transcription using Thermoscript™ reverse transcription kit (Invitrogen) and the manufacturer's standard protocol. Water was substituted for enzyme to provide negative controls. Transcript levels were measured by quantitative real-time PCR (qRT-PCR) using the Lightcycler® 480 system (Roche), Sybr Green chemistry (Roche #04707516001) and primers provided in Supplemental Table 2. Primer sets were designed to span intron/exon borders to prevent amplification from genomic DNA (except E2IG4, which has only 2 exons). ERα primers amplify mRNA sequence encoding amino acids 387-418 of the protein, an area common to all known functional splice variants. Substitution of PCR-grade water for cDNA template served as an additional negative control. Transcripts were amplified in parallel, along with a stably expressed reference gene, TBP, in triplicate reactions using equal amounts of 5x diluted cDNA (1ul per reaction). Relative levels of transcripts were calculated using the delta Ct method and normalized to those of the TBP reference transcript using the formula: %TBP = 2 - (Ct GENE - CtTBP) × 100%.
Estrogen response assays
To explore estrogen signaling in 184AA3 cells and determine if the cells could sense and respond to estrogen, we stimulated the cells with estradiol and monitored transcript levels of known estrogen-sensitive genes. For 48 hours, 184AA3 cells and the reference cell lines, MCF-7 and MDA-MB-231, were cultured in estrogen-free medium. After this time, the medium was refreshed with that containing 1uM 17-β estradiol. RNA was collected at 0-hour (untreated) and at 3 and 6 hours post-estradiol exposure. Transcripts were measured by qRT-PCR, as described above. Primers of the estrogen-sensitive genes are provided in Supplementary Table 2. We validated the assay by comparing results of MCF-7 to those published previously [41]. Nuclear translocation of ER was measured by observing the localization of ER in estradiol stimulated and unstimulated cells. We seeded fifty thousand cells (AA3 and 184AA3XT, in parallel) onto fibronectin coated (2μg/cm2) glass coverslips and cultured them overnight to allow for attachment. After this time, cells were thoroughly washed in PBS and placed into estrogen free medium for 24 hours (phenol red-free DMEM/F12 plus 1% charcoal-stripped FBS). The cells were then stimulated for 15 minutes in medium containing 10nM 17-β estradiol. Cells were fixed in 4% PFA, immunostained for ERα, and compared to unstimulated controls. Images were captured on an Olympus FV-1000 confocal microscope.
Immunoblots
ERα protein expression was measured in 184AA3, MDA-MB-231 and MCF-7 cells (Fig. 5b). Cells were lysed in in 4% SDS/PBS containing proteinase inhibitor cocktail set I and phosphatase inhibitor cocktail set I [Calbiochem]. After 10 second sonication and centrifugation, the protein concentration in the supernatant was measured using the BioRad DC™ Protein assay. Proteins were separated by SDSPAGE (40μg/lane) on 4-20% Tris-Glycine Gels and transferred to nitrocellulose membranes. A protein standard was included (Precision Plus Protein™ Kaleidoscope standard, BioRad #161-0375). Blots were probed with antibodies specific to ER and alpha tubulin (Supplemental Table 1). After washing and probing with HRP-conjugated secondary antibodies, blots were developed using SuperSignal West Femto chemiluminescence reagent (Pierce Biotechnology) and imaged with a FluorChem HD2 imager (Cell Biosciences/ProteinSimple). To permit accurate comparison to the protein standards, we used Photoshop (Adobe Systems) to merge the chemiluminescent image to the color image of the standards. Contrast adjustment of the captured 16-bit image was also needed, and was applied uniformly.
Immunofluorescence
Immunofluorescence was performed on monolayer cell cultures, smears of 3-dimensional cultures, and frozen tumor sections. Specimens were fixed in 4% paraformaldehyde (5 minutes, 23°C), followed by a 10 minute treatment with 4% formaldehyde+0.5% saponin, then washed in staining buffer (0.5% w/v saponin, 10% v/v goat serum in PBS). After overnight incubation with primary antibodies (diluted in staining buffer), the specimens were rinsed thoroughly and treated with anti-mouse and anti-rabbit secondary antibodies, respectively conjugated with Alexafluor 488 and Alexfluor 594 (Invitrogen), diluted 1:400 (1 hour, 37°C). Nuclei were stained using 0.3 μM DAPI (Molecular Bioprobes) and mounted with Fluormount G mounting medium (Southern Biotech). We captured and processed images using an AxioImager fluorescent microscope and Axiovision (Zeiss) and/or Photoshop (Adobe) software. If contrast adjustments were needed, they were applied uniformly. Primary antibodies are provided in Supplementary Table 1.
Immunohistochemistry
Processing of xenograft specimens, including paraffin embedding, sectioning, and all staining, was performed by the Mutant Mouse Pathology Laboratory at the Center for Comparative Medicine at the University of California, Davis. Four micrometer thick paraffin sections were stained with Mayer's hemotoxylin and eosin or immunostained as described previously,[42] with some antibody-dependent and empirical based modifications; e.g., lot-based differences in antibody dilutions. Antigen retrieval was performed at 125°C in pH 6.0 citrate buffer, using a Decloaking Chamber (Biocare Medical, Concord, CA) pressurized to 15psi. The total incubation time was 45 min. Antibodies used for immunohistochemistry are provided in Supplementary Supplementary Table 1. Specimens were incubated with primary antibodies overnight at room temperature in a humidified chamber. Slides were scanned at 20× magnification using an Aperio® XT slide scanner (Leica Biosystems), imported into the Aperio Spectrum database, and visualized with Aperio® Imagescope software.
Statistical Analysis
JMP 7 (SAS Institute) and Prism5 (GraphPad) statistical software packages were used for all statistical analyses. In all cases, error bars indicate the standard deviation of at least three multiple biological replicates. For qRT-PCR, standard deviations of Ct values from triplicate reactions were used to determine percent relative error and then propagated using the square root of the sum of squares method.
Supplementary Material
Acknowledgments
For their invaluable technical assistance during this project, we thank Maria Rojec, Dinah Groesser, Alvin Lo, Sun-Young Lee, Xuefei Tian, and Eva Lee (Lawrence Berkeley National Laboratory). We appreciate also the expertise and help given by Judith Walls and Ed Hubbard (University of California, Davis Center for Comparative Medicine). We express special gratitude also to Michelle Scott of the LBNL flow cytometry and Advanced microscopy facility for her expert technical advice and assistance. Grant support: Innovator award to M.J.B. from the U.S. Department of Defense (W81XWH0810736 and W81XWH12M9532) and in part by National Cancer Institute awards (R37CA064786, R01CA140663, U54CA112970) and by grants from the U.S. Department of Energy, Office of Biological and Environmental Research and Low Dose Scientific Focus Area (contract no. DE-AC02-05CH1123) and the Breast Cancer Research Foundation. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Conflicts of interest: The authors declare that they have no conflict of interest.
Animal Welfare: All procedures performed in studies involving animals were in accordance with the ethical standards of the Lawrence Berkeley National Laboratory, at which all such studies were conducted.
References
- 1.Box GE, Draper NR. Empirical model-building and response surfaces. Vol. 424. Wiley; New York: 1987. [Google Scholar]
- 2.Hines WC, Yaswen P, Bissell MJ. Modelling breast cancer requires identification and correction of a critical cell lineage-dependent transduction bias. Nature communications. 2015;6:6927. doi: 10.1038/ncomms7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagner KU. Models of breast cancer: quo vadis, animal modeling? Breast Cancer Res. 2004;6(1):31–38. doi: 10.1186/bcr723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Brien KM, Cole SR, Tse CK, Perou CM, Carey LA, Foulkes WD, Dressler LG, Geradts J, Millikan RC. Intrinsic breast tumor subtypes, race, and long-term survival in the Carolina Breast Cancer Study. Clin Cancer Res. 2010;16(24):6100–6110. doi: 10.1158/1078-0432.CCR-10-1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tran B, Bedard PL. Luminal-B breast cancer and novel therapeutic targets. Breast Cancer Res. 2011;13(6):221. doi: 10.1186/bcr2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, Snider J, Ballman K, Weber J, Chen K, Koboldt DC, Kandoth C, Schierding WS, McMichael JF, Miller CA, Lu C, Harris CC, McLellan MD, Wendl MC, DeSchryver K, Allred DC, Esserman L, Unzeitig G, Margenthaler J, Babiera GV, Marcom PK, Guenther JM, Leitch M, Hunt K, Olson J, Tao Y, Maher CA, Fulton LL, Fulton RS, Harrison M, Oberkfell B, Du F, Demeter R, Vickery TL, Elhammali A, Piwnica-Worms H, McDonald S, Watson M, Dooling DJ, Ota D, Chang LW, Bose R, Ley TJ, Piwnica-Worms D, Stuart JM, Wilson RK, Mardis ER. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486(7403):353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burdall SE, Hanby AM, Lansdown MR, Speirs V. Breast cancer cell lines: friend or foe? Breast Cancer Res. 2003;5(2):89–95. doi: 10.1186/bcr577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kabos P, Finlay-Schultz J, Li C, Kline E, Finlayson C, Wisell J, Manuel CA, Edgerton SM, Harrell JC, Elias A, Sartorius CA. Patient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen-dependent gene signatures. Breast Cancer Res Treat. 2012;135(2):415–432. doi: 10.1007/s10549-012-2164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, Factor R, Matsen C, Milash BA, Nelson E, Neumayer L, Randall RL, Stijleman IJ, Welm BE, Welm AL. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17(11):1514–1520. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cottu P, Bieche I, Assayag F, El Botty R, Chateau-Joubert S, Thuleau A, Bagarre T, Albaud B, Rapinat A, Gentien D, de la Grange P, Sibut V, Vacher S, Hatem R, Servely JL, Fontaine JJ, Decaudin D, Pierga JY, Roman-Roman S, Marangoni E. Acquired resistance to endocrine treatments is associated with tumor-specific molecular changes in patient-derived luminal breast cancer xenografts. Clin Cancer Res. 2014;20(16):4314–4325. doi: 10.1158/1078-0432.CCR-13-3230. [DOI] [PubMed] [Google Scholar]
- 12.Briand P, Petersen OW, Van Deurs B. A new diploid nontumorigenic human breast epithelial cell line isolated and propagated in chemically defined medium. In Vitro Cell Dev Biol. 1987;23(3):181–188. doi: 10.1007/BF02623578. [DOI] [PubMed] [Google Scholar]
- 13.Rizki A, Weaver VM, Lee SY, Rozenberg GI, Chin K, Myers CA, Bascom JL, Mott JD, Semeiks JR, Grate LR, Mian IS, Borowsky AD, Jensen RA, Idowu MO, Chen F, Chen DJ, Petersen OW, Gray JW, Bissell MJ. A human breast cell model of preinvasive to invasive transition. Cancer Res. 2008;68(5):1378–1387. doi: 10.1158/0008-5472.CAN-07-2225. doi:68/5/1378 [pii] 10.1158/0008-5472.CAN-07-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50(18):6075–6086. [PubMed] [Google Scholar]
- 15.Park CC, Zhang H, Pallavicini M, Gray JW, Baehner F, Park CJ, Bissell MJ. Beta1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res. 2006;66(3):1526–1535. doi: 10.1158/0008-5472.CAN-05-3071. doi:66/3/1526 [pii] 10.1158/0008-5472.CAN-05-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. The American journal of pathology. 1996;148(1):313–319. [PMC free article] [PubMed] [Google Scholar]
- 17.Stampfer MR, Bartley JC. Induction of transformation and continuous cell lines from normal human mammary epithelial cells after exposure to benzo[a]pyrene. Proc Natl Acad Sci U S A. 1985;82(8):2394–2398. doi: 10.1073/pnas.82.8.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stampfer MR, LaBarge MA, Garbe JC. Cell and Molecular Biology of Breast Cancer. Springer; 2013. An integrated human mammary epithelial cell culture system for studying carcinogenesis and aging; pp. 323–361. [Google Scholar]
- 19.Severson PL, Vrba L, Stampfer MR, Futscher BW. Exome-wide mutation profile in benzo[a]pyrene-derived post-stasis and immortal human mammary epithelial cells. Mutat Res Genet Toxicol Environ Mutagen. 2014:775–776. 48–54. doi: 10.1016/j.mrgentox.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heppner GH, Miller BE. Tumor heterogeneity: biological implications and therapeutic consequences. Cancer Metastasis Rev. 1983;2(1):5–23. doi: 10.1007/BF00046903. [DOI] [PubMed] [Google Scholar]
- 21.Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514(7520):54–58. doi: 10.1038/nature13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6(12):924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 23.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R, Jr, Badve S, Nakshatri H. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006;8(5):R59. doi: 10.1186/bcr1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65(13):5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 26.Biddle A, Gammon L, Fazil B, Mackenzie IC. CD44 Staining of Cancer Stem-Like Cells Is Influenced by Down-Regulation of CD44 Variant Isoforms and Up-Regulation of the Standard CD44 Isoform in the Population of Cells That Have Undergone Epithelial-to-Mesenchymal Transition. PLoS One. 2013;8(2):e57314. doi: 10.1371/journal.pone.0057314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graham JD, Mote PA, Salagame U, Balleine RL, Huschtscha LI, Clarke CL. Hormone-responsive model of primary human breast epithelium. Journal of mammary gland biology and neoplasia. 2009;14(4):367–379. doi: 10.1007/s10911-009-9160-6. [DOI] [PubMed] [Google Scholar]
- 28.Koren S, Reavie L, Couto JP, De Silva D, Stadler MB, Roloff T, Britschgi A, Eichlisberger T, Kohler H, Aina O, Cardiff RD, Bentires-Alj M. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature. 2015;525(7567):114–118. doi: 10.1038/nature14669. [DOI] [PubMed] [Google Scholar]
- 29.Van Keymeulen A, Lee MY, Ousset M, Brohee S, Rorive S, Giraddi RR, Wuidart A, Bouvencourt G, Dubois C, Salmon I, Sotiriou C, Phillips WA, Blanpain C. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature. 2015;525(7567):119–123. doi: 10.1038/nature14665. [DOI] [PubMed] [Google Scholar]
- 30.Bissell MJ, Bartholomew JC, Folkman J, Smith H, Stampfer M. Culture systems for studying malignancy. Cancer Res;(United States) 1979;39 [Google Scholar]
- 31.Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques RA, Rabinovitch PS, Reid BJ. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet. 2006;38(4):468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- 32.Thomasset N, Lochter A, Sympson CJ, Lund LR, Williams DR, Behrendtsen O, Werb Z, Bissell MJ. Expression of autoactivated stromelysin-1 in mammary glands of transgenic mice leads to a reactive stroma during early development. The American journal of pathology. 1998;153(2):457–467. doi: 10.1016/S0002-9440(10)65589-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang P, Lo A, Huang Y, Huang G, Liang G, Mott J, Karpen GH, Blakely EA, Bissell MJ, Barcellos-Hoff MH, Snijders AM, Mao JH. Identification of genetic loci that control mammary tumor susceptibility through the host microenvironment. Scientific reports. 2015;5:8919. doi: 10.1038/srep08919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J, Villadsen R, Sorlie T, Fogh L, Gronlund SZ, Fridriksdottir AJ, Kuhn I, Rank F, Wielenga VT, Solvang H, Edwards PA, Borresen-Dale AL, Ronnov-Jessen L, Bissell MJ, Petersen OW. Tumor initiating but differentiated luminal-like breast cancer cells are highly invasive in the absence of basal-like activity. Proc Natl Acad Sci U S A. 2012;109(16):6124–6129. doi: 10.1073/pnas.1203203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stampfer MR, Garbe J, Nijjar T, Wigington D, Swisshelm K, Yaswen P. Loss of p53 function accelerates acquisition of telomerase activity in indefinite lifespan human mammary epithelial cell lines. Oncogene. 2003;22(34):5238–5251. doi: 10.1038/sj.onc.1206667. [DOI] [PubMed] [Google Scholar]
- 36.Cardiff RD, Hubbard NE, Engelberg JA, Munn RJ, Miller CH, Walls JE, Chen JQ, Velasquez-Garcia HA, Galvez JJ, Bell KJ, Beckett LA, Li YJ, Borowsky AD. Quantitation of fixative-induced morphologic and antigenic variation in mouse and human breast cancers. Lab Invest. 2013;93(4):480–497. doi: 10.1038/labinvest.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labarge MA, Garbe JC, Stampfer MR. Processing of human reduction mammoplasty and mastectomy tissues for cell culture. J Vis Exp. 2013;(71) doi: 10.3791/50011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garbe JC, Bhattacharya S, Merchant B, Bassett E, Swisshelm K, Feiler HS, Wyrobek AJ, Stampfer MR. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res. 2009;69(19):7557–7568. doi: 10.1158/0008-5472.CAN-09-0270. doi:0008-5472.CAN-09-0270 [pii] 10.1158/0008-5472.CAN-09-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, Feleppa F, Huschtscha LI, Thorne HJ, Fox SB, Yan M, French JD, Brown MA, Smyth GK, Visvader JE, Lindeman GJ. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15(8):907–913. doi: 10.1038/nm.2000. doi:nm.2000 [pii] 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 40.Hines WC, Su Y, Kuhn I, Polyak K, Bissell MJ. Sorting Out the FACS: A Devil in the Details. Cell reports. 2014;6(5):779–781. doi: 10.1016/j.celrep.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 41.Wang DY, Fulthorpe R, Liss SN, Edwards EA. Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1. Mol Endocrinol. 2004;18(2):402–411. doi: 10.1210/me.2003-0202. [DOI] [PubMed] [Google Scholar]
- 42.Toth M, Gervasi DC, Fridman R. Phorbol ester-induced cell surface association of matrix metalloproteinase-9 in human MCF10A breast epithelial cells. Cancer Res. 1997;57(15):3159–3167. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.