

Abstract
Unresolved endoplasmic reticulum (ER) stress, with the subsequent persistent activation of the unfolded protein response (UPR) is a well-recognized mechanism of endothelial cell apoptosis with a major impact on the integrity of the endothelium during the course of cardiovascular diseases. As in other cell types, Ca2+ influx into endothelial cells can promote ER stress and/or contribute to mechanisms associated to it. In previous work we showed that in human coronary artery endothelial cells (HCAECs) the Ca2+-permeable non-selective cation channel Transient Receptor Potential Canonical 3 (TRPC3) mediates constitutive Ca2+ influx which is critical for operation of inflammatory signaling in these cells, through a mechanism that entails coupling of TRPC3 constitutive function to activation of Ca2+/calmodulin-dependent protein kinase II (CAMKII). TRPC3 has been linked to UPR signaling and apoptosis in cells other than endothelial, and CAMKII is a mediator of ER stress-induced apoptosis in various cell types, including endothelial cells. In the present work we used a pharmacological approach to examine whether in HCAECs TRPC3 and CAMKII also contribute to mechanisms of ER stress-induced apoptosis. The findings show for the first time that in HCAECs activation of the UPR and the subsequent ER stress-induced apoptosis exhibit a strong requirement for constitutive Ca2+ influx and that TRPC3 contributes to this process. In addition, we obtained evidence indicating that, similar to its roles in non-endothelial cells, CAMKII participates in ER stress-induced apoptosis in HCAECs.
Keywords: TRPC3 channels, endoplasmic reticulum stress, endothelial cell apoptosis
Graphical abstract
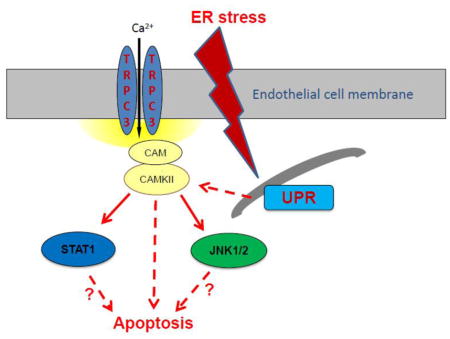
1. INTRODUCTION
The diversity of functions to which the endothelium is committed is in part due to the versatility and multi-functionality of the constituting endothelial cells. And it is by virtue of these properties that alterations in endothelial cell function are present in most, if not all, states of cardiovascular disease (reviewed by us in (Smedlund et al., 2012)). In the particular case of coronary artery disease, the endothelium lining the coronary arteries participates in an intricate interplay between mechanisms intrinsic to the arterial wall, genetic and environmental risk factors (Libby, 2012). The permanent exposure to hemodynamic changes and to the action of a diversity of circulating stressors imposes on the endothelial cells the necessity of maintaining a fine-tuned balance between apoptosis and survival in order to retain the integrity of the endothelium. Indeed, a number of systemic and local stressors acting on the endothelium can induce endoplasmic reticulum (ER) stress by causing perturbations in ER homeostasis, such as alteration of lipid biosynthesis, Ca2+ signaling or protein folding/trafficking, leading to activation of the unfolded protein response (UPR) ((Kratzer et al., 2014; Song and Zou, 2014) and references therein). The UPR is a highly conserved adaptive response aimed at alleviating ER stress and restore normal cell function. However, persistent activation of the UPR often leads to cell apoptosis. Indeed, sustained, unresolved ER stress is a well-recognized mechanism promoting apoptosis of endothelial and other cell types in the arterial wall during development and progression of cardiovascular disease (McAlpine and Werstuck, 2013; Zhou and Tabas, 2013). In endothelial cells, as in other eukaryotic cells, the UPR signals through three ER proteins, inositol-requiring enzyme 1α (IRE1α), PKR-like eukaryotic initiation factor 2α kinase (PERK) and activating transcription factor-6 (ATF6) (Hollien, 2013). The UPR then couples sustained ER stress to activation of pro-apoptotic signaling molecules, of which Ca2+/calmodulin-dependent protein kinase II (CAMKII) is a recognized mediator between ER stress and downstream execution steps in apoptosis signaling (Li et al., 2012; Timmins et al., 2009).
In a number of cell types, including endothelial cells, persistent and/or exacerbated Ca2+ influx can directly promote ER stress or contribute to mechanisms associated to it (Hecquet et al., 2014; Jambrina et al., 2003; Kim et al., 2011). In previous work from our laboratory we showed that in human coronary artery endothelial cells (HCAECs) basal or constitutive Ca2+ influx is an obligatory component of the inflammatory signaling that controls expression of the vascular cell adhesion molecule-1 (VCAM-1) and monocyte adhesion to these cells (Smedlund and Vazquez, 2008). In addition, we also demonstrated that the Ca2+-permeable non-selective cation channel Transient Receptor Potential Canonical 3 (TRPC3), a member of the TRPC family of cation channels (Liao et al., 2014; Vazquez et al., 2004a) is responsible for most of the constitutive Ca2+ influx into HCAECs, and that CAMKII couples TRPC3 constitutive function to activation of nuclear factor kappa B (NFkB)-dependent inflammatory signaling (Smedlund et al., 2010; Smedlund and Vazquez, 2008). Interestingly, in mouse epithelial cells from pancreatic and parotid acini TRPC3 activity is key to support UPR-dependent mechanisms associated to cell damage (Kim et al., 2011). Nevertheless, whether in HCAECs TRPC3 constitutive function also contributes to mechanisms of ER stress-induced apoptosis remains to be determined. To answer this question, in the present work we undertook a pharmacological approach to examine if constitutive Ca2+ influx in general and TRPC3 in particular participate in UPR activation and ER stress-induced apoptosis of HCAECs. Our findings show for the first time in an arterial-derived endothelial cell model that activation of the UPR and the subsequent ER stress-induced apoptosis exhibit a strong requirement for constitutive Ca2+ influx and that TRPC3 is an obligatory component of such process. In addition, we identify CAMKII as a potential mediator of ER stress-induced apoptosis in these cells.
2. MATERIALS AND METHODS
2.1. Cell culture
Culture conditions for human coronary artery endothelial cells (HCAECs, Lonza, CA) were as previously described by us (Smedlund et al., 2010; Smedlund and Vazquez, 2008). Briefly, HCAECs were grown in endothelial basal medium (EBM-2) supplemented with endothelial growth factors and 5% fetal bovine serum (FBS) at 37°C under humidified air (5% CO2). Human embryonic kidney (HEK293) cells stably expressing the human TRPC3 (kindly provided by Dr. J.W. Putney Jr., NIEHS, NC) were grown as we described in (Smedlund et al., 2010; Vazquez et al., 2004b).
2.2. In vitro TUNEL assay
apoptosis was evaluated by using the in situ cell death detection kit, TMR red (Roche, IN) as we described in (Smedlund et al., 2011). For TUNEL experiments treatment of HCAECs with the ER stressors was optimized to 16 hours. This allowed for sufficient number of TUNEL positive (apoptotic) cells to be counted in order to obtain reproducible and statistically testable data. Under these experimental conditions, necrotic cells, as assessed from both morphology and permeability to Trypan Blue, were not detected or present in a minimal number, and thus did not contribute significantly to the total number of TUNEL+ cells. When indicated, loading of the cells with the Ca2+ chelator BAPTA (1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis) was performed by incubating HCAECs during 30 min (37°C) in HEPES-buffered saline solution (HBSS, in mM: 140 NaCl, 4.7 KCl, 1 MgCl2, 10 glucose, 10 HEPES pH 7.4, 2 CaCl2) containing 0.1% bovine serum albumin and 10 μM of the cell permeable form of BAPTA, BAPTA/AM. The cells were then washed in BAPTA-free HBSS and then incubated in complete growth medium or endothelial basal medium under the experimental conditions indicated in the text and figure legends.
2.3. Cell lysis and immunoblotting
essentially as we described in (Tano et al., 2011; Tano and Vazquez, 2011). Briefly, following cell lysis, solubilized proteins were separated in 10% acrylamide gels, electrotransferred to PVDF membranes and immunoblotted with the indicated primary antibody. After incubation with HRP-conjugated secondary antibodies, immunoreactive bands were visualized by ECL (Amersham, PA). Primary antibodies against IRE1α, BiP, CHOP, phospho-JNK1/2 (Thr183/Tyr185), total JNK1/2, phospho-STAT1 (Ser727) and total STAT1, were all from Cell Signaling (MA); the antibody against phospho-CAMKII (Thr286) was from Abcam (MA); the antibody against GAPDH was from Santa Cruz Biotechnology (Santa Cruz, CA) and the anti-TRPC3 antibody was from Alomone Labs (Jerusalem, Israel). In our hands, two different commercially available pan-CAMKII antibodies gave inconsistent, low quality results; thus, phospho-CAMKII levels were normalized by GAPDH.
2.4. Real-time PCR (RT-PCR)
as we described in ref. (Tano et al., 2014). Briefly, cDNA was evaluated by semi-quantitative real-time PCR (qRT-PCR) using TrueAmp SYBR green qPCR supermix (Applied Biosystems, CA). The relative amount of mRNA was calculated by comparison to the corresponding standards and normalized relative to 18S rRNA. Results are expressed as mean ± SEM (relative quantitation, RQ). Sequences of primers used are as follows: IRE1α (F: AGAGAAGCAGCAGACTTTGTC; R: GTTTTGGTGTCGTACATGGTGA), PERK (F: GGAAACGAGAGCCGGATTTATT; R: ACTATGTCCATTATGGCAGCTTC), BiP (F: CATCACGCCGTCCTATGTCG; R: CGTCAAAGACCGTGTTCTCG), CHOP (F: GGAAACAGAGTGGTCATTCCC; R: CTGCTTGAGCCGTTCATTCTC), ERO1α (F: GCCAGGTTAGTGGTTACTTGG; R: GGCCTCTTCAGGTTTACCTTGT), sXBP1 (F: TGCTGAGTCCGCAGCAGGTG; R: GCTGGCAGGCTCTGGGGAAG, as described in (van Schadewijk et al., 2012)), TRPC3 (F: AGCAGCTCTTGACGATCTGG; R: GCACAACGAGACACTTGATAGC), 18S (F: GGCGTCCCCCAACTTCTTA; R: GGGCATCACAGACCTGTTATTG). Primerbank (Spandidos et al., 2010; Wang et al., 2012) identification numbers: IRE1α: 153946420c3; PERK: 284005480c3; BiP: 305855105c1; CHOP: 304282228c1; ERO1α: 7657068c2; TRPC3: 194733734c3. Primers for 18S were from Eurofins MWG Operon Inc. (#7060139, Huntsville, AL).
2.5. Real-time Fluorescence measurements of cytosolic Ca2+ and Ba2+
coverslip-plated HCAECs loaded with Fura-2 were used to monitor changes of intracellular Ca2+ or Ba2+ by real-time fluorescence with a CCD camera-based imaging system (Intracellular Imaging Inc., Cincinnati OH) as we described in (Smedlund et al., 2010; Smedlund and Vazquez, 2008). Loading of cells with Fura-2/AM was performed at room temperature to minimize dye compartmentalization (see for instance (Baldi C, 2003)) and thus we proceeded with the recordings at room temperature to avoid unnecessary temperature changes immediately before measurements. All treatments were in HEPES-buffered saline solution (HBSS, in mM: 140 NaCl, 4.7 KCl, 1 MgCl2, 10 glucose, 10 HEPES pH 7.4, 2 CaCl2). “Nominally Ca2+-free medium” means HBSS with no Ca2+ added (free Ca2+ ≪5 μM). Calculated Ca2+ values are only estimative (for a thorough explanation see (Smedlund et al., 2010)) and thus for simplicity, the ratio of Fura-2 fluorescence (510 nm emission) when excited at 340 and 380 nm (F340/F380) was used to calculate rates of changes in fluorescence (discussed in (Smedlund et al., 2010; Vazquez et al., 2004b)).
2.6. Statistical analysis
values shown are mean ± SEM; the corresponding number of independent experiments (biological replicates, “n”) is indicated in the figure legends. Comparison of mean values between groups was performed with a two-tail Student’s t test using Prism Graph Pad version 6 for Windows 2007 (Graph Pad Software, San Diego CA). The statistical analysis of rates of Ba2+ entry was performed as described in detail in (Vazquez et al., 2004b) P values of less than 0.05 were considered significant.
3. RESULTS
3.1. Suppression of constitutive Ca2+ influx impairs UPR activation and reduces ER stress-induced apoptosis in HCAECs
As a first step to examine whether constitutive Ca2+ influx contributes to ER stress-dependent activation of the UPR and/or of ER stress-induced apoptosis in HCAECs, we investigated the effect of abolishing Ca2+ influx on activation of the UPR and subsequent apoptosis of HCAECs when the cells were subjected to conditions that lead to sustained and irreversible ER stress. HCAECs were exposed to the direct ER stressors thapsigargin (1 μM), which causes depletion of ER Ca2+ content by irreversible inhibition of the sarco-endoplasmic reticulum Ca2+ ATPase (SERCA), or tunicamycin (10 μg/ml), which causes accumulation of proteins in the ER by inhibiting N-glycosylation. These treatments were conducted in the presence or absence of the non-specific cation channel blocker SKF96,365 (“SKF”, 30 μM); we have previously shown that in HCAECs this concentration of SKF completely suppresses both constitutive and regulated Ca2+ influx (Smedlund and Vazquez, 2008). Following treatments, the cells were processed for evaluation of apoptosis using a terminal deoxynucleotidyl-transferase-dUTP nick end labeling (TUNEL) assay, as we described in detail in ((Tano et al., 2012); see also Methods). As shown in Figure 1, a significant number of TUNEL positive cells were observed after exposure of HCAECs to thapsigargin or tunicamycin, indicative of ER stress-induced apoptosis. Whereas treatment with SKF alone did not alter the number of apoptotic cells compared to control conditions, the channel blocker drastically reduced thapsigargin-induced apoptosis (>60%), and almost completely prevented that induced by tunicamycin. To determine if the effect of Ca2+ influx blockade on ER stress-induced apoptosis was correlated with changes in the extent of activation of the UPR signaling, we examined the expression levels of typical ER stress markers when constitutive Ca2+ entry was prevented by SKF treatment. HCAECs were treated with thapsigargin in the presence or absence of SKF and processed for evaluation of mRNA and/or protein levels of IRE1α, PERK, the chaperone glucose-regulated protein-78/Immunoglobulin-binding Protein (BiP) and the UPR-induced transcriptional factor CCAAT/enhancer binding protein homologous protein (CHOP). Notably, SKF caused a significant reduction in the mRNA for all of these UPR components, as well as in the mRNA for endoplasmic reticulum oxidoreductase-1α (ERO1α), a transcriptional target of CHOP (Figure 2A). Only IRE1α and CHOP mRNA levels were somewhat augmented by treatment of the cells with SKF alone, but this effect was minor compared to that of thapsigargin. In addition, western blot analysis of lysates from HCAECs treated under the conditions above, showed complete abrogation of thapsigargin-induced expression of IRE1α, BiP and CHOP proteins when the channel blocker SKF was present (Figure 2B). IRE1α activation can be assessed by its endoribonuclease activity which leads to selective splicing of the mRNA for the transcription factor X-box binding protein 1 (XBP1) into an active spliced form (sXBP1) (Sovolyova et al., 2014). When HCAECs were treated with SKF, the thapsigargin-induced mRNA levels for sXBP1 were markedly reduced, supporting the notion of a correlation between reduced total IRE1α expression and activity (0.79 ± 0.11, 11.81 ± 0.96 and 7.90 ± 0.73, for normalized sXBP1 mRNA levels under control, thapsigargin, and thapsigargin+SKF conditions, respectively; n=3, P=0.033 for the difference between thapsigargin and thapsigargin + SKF). Altogether, these findings suggested that constitutive Ca2+ influx was required for activation of the UPR and ER stress-induced apoptosis in HCAECs.
Figure 1.

Human coronary artery endothelial cells (HCAECs) were maintained in endothelial basal medium (EBM-2) alone (Control) or EBM-2 containing thapsigargin (“Thaps”, 1 μM) or tunicamycin (“Tun”, 10 μg/ml) for 24 hours in the presence or absence of the non-selective channel blocker SKF96,365 (“SKF”, 30 μM), and then processed for in vitro TUNEL assay. Treatment with SKF alone was included as a control. P values for the differences between treatments with and without SKF are shown. Differences between treatments with thapsigargin or tunicamycin and the control had P <0.0001. All values are mean ± SEM (n=3–5).
Figure 2.


HCAECs were maintained in EBM-2 alone (Control) or EBM-2 containing the ER stressor thapsigargin (“Thaps”, 1 μM) for 24 hours in the presence or absence of SKF96,365 (“SKF”, 30 μM); treatment with SKF alone was included as a control. In A), following treatments total RNA and cDNA were prepared as described in Methods. Expression levels of the UPR components IRE1α, PERK, BiP, CHOP and ERO1α were examined by qRT-PCR. Graphs represent mean ± SEM of at least three independent experiments performed in triplicates. B) After the treatments described above, cells were processed for immunodetection of IRE1α (~130 kDa), BiP (~75 kDa) or CHOP (~27 kDa) in whole cell lysates. Membranes were reprobed for GAPDH (~37 kDa) to control for protein loading. Blots are representative of three independent experiments.
3.2. Buffering of intracellular Ca2+ impairs UPR activation and reduces ER stress-induced apoptosis in HCAECs
Influx of Ca2+ through plasma membrane Ca2+ permeable channels can lead to transient or sustained increases in cytosolic Ca2+, which can be either localized to sub-membrane domains or spread for quite few micrometers throughout the cytosol ((Vazquez et al., 2010) and references therein). As an alternative to evaluate the contribution of constitutive Ca2+ influx to UPR activation and ER stress-induced apoptosis, but without global blockade of Ca2+-permeable channels, we examined the effect of strong cytosolic Ca2+ buffering on those events. To achieve this, we loaded HCAECs with the membrane permeable form of the Ca2+ chelator BAPTA, BAPTA-AM (10 μM) for 30 min before exposing the cells to the ER stressors. Under these conditions, both the release and influx phases of the ATP-dependent biphasic Ca2+ response in HCAECs were abolished, indicating that any changes in cytosolic Ca2+ were efficiently buffered by BAPTA (Figure 3). Notably, in BAPTA-loaded HCAECs treated with thapsigargin (1μM) or tunicamycin (10 μg/mL) for 16 h to induce irreversible ER stress, apoptosis was markedly reduced when compared to cells treated with the ER stressors alone (Figure 4A). This was positively correlated with a decrease in the extent of UPR activation, as evidenced by decreased levels of both mRNA and protein of IRE1α, BiP and CHOP in BAPTA-loaded cells subjected to ER stress (Figures 4B and 4C).
Figure 3.

In HCAECs stimulation of P2Y2 receptors with ATP induces a typical biphasic Ca2+ response consisting of a transient phospholipase C-dependent IP3-mediated release of Ca2+ from internal Ca2+ stores (first phase) followed by Ca2+ influx (second phase) across the plasma membrane (Smedlund and Vazquez, 2008). This response is illustrated in this figure. Fura-2 loaded HCAECs were challenged with ATP (100 μM) in nominally Ca2+-free medium to induce the first phase of the Ca2+ response; once the Ca2+ transient was over, Ca2+ (2 mM) was added to the bath to make evident the second, Ca2+ influx phase. Alternatively, the cells were loaded with 10 μM BAPTA/AM (as described in Methods) before being loaded with Fura-2 and subjected to the protocol described above (discontinuous trace). Traces are averages of 18–21 cells and are representative of 3 independent experiments.
Figure 4.


HCAECs were maintained during 16 h in EBM-2 alone (Control) or EBM-2 containing thapsigargin (“Thaps”, 1 μM) or tunicamycin (“Tun”, 10 μg/ml), as indicated; alternatively, the cells were loaded with 10 μM BAPTA/AM (as described in Methods), washed and then kept for 16 h in EBM-2 alone (“BAPTA” control) or EBM-2 containing thapsigargin (“Thaps+BAPTA”) or tunicamycin (“Tun+BAPTA) at the concentrations indicated above. In A), the cells were processed for in vitro TUNEL assay. Differences between “Thaps” and “Tun” treatments and their corresponding control (“Control”) had P <0.0001. All values are mean ± SEM (n=3–5). In B) and C), following treatments expression levels of the UPR components IRE1α and BiP (B), or of the UPR-induced transcriptional factor CHOP (C), were examined by qRT-PCR and by immunoblotting. Bar graphs represent data (mean ± SEM) of at least three independent experiments, each performed in triplicates. Apparent molecular weights are as follows: IRE1α (~130 kDa), BiP (~75 kDa) and CHOP (~27 kDa). Membranes were reprobed for GAPDH (~37 kDa) to control for protein loading. Blots are representative of three independent experiments.
3.3. The selective TRPC3 blocker pyrazole-10 (Pyr10) reduces UPR activation and ER stress-induced apoptosis in HCAECs
Previous studies from our laboratory showed that in HCAECs native TRPC3 accounts for most of the constitutive Ca2+ influx and only partially for receptor (P2Y2)-stimulated cation entry, and that both processes are completely prevented by SKF (Smedlund and Vazquez, 2008). However, SKF is not specific for TRPC3 (Smyth et al., 2006; Trebak et al., 2003b). In recent years the 3,5-bis (trifluoromethyl)-pyrazole derivative Pyr10 has been identified as a highly selective and potent inhibitor of TRPC3 (Schleifer et al., 2012). We confirmed the ability of Pyr10 (2 μM) to effectively block TRPC3 by examining the extent of inhibition of the barium (Ba2+) influx induced by 1-oleyl-2-acetyl-sn-glycerol (OAG, 100 μM) in HEK293 cells overexpressing the human form of TRPC3 (Vazquez et al., 2004b), which is a hallmark of TRPC3 function (rates of Ba2+ influx were: 0.030 ± 0.002, 0.82 ± 0.005 and 0.017 ± 0.004 ratio units/min, for control, OAG-treated and OAG-treated+Pyr10 cells, respectively, n=22–29 cells; Figure 5A). Of importance, at this concentration Pyr10 did not affect store-operated Ca2+ entry (SOCE) in HCAECs (Figure 5B), in line with our previous observations that TRPC3 does not contribute to SOCE in these cells (Vazquez and Putney, 2006). Remarkably, Pyr10 reduced P2Y2 (ATP)- and OAG-induced Ba2+ influx in HCAECs (Figures 6A and 6B) to the same extent as knockdown of TRPC3 in these cells does (Smedlund and Vazquez, 2008; Vazquez and Putney, 2006). Together, these observations provide strong evidence that Pyr10 acts as a selective TRPC3 channel blocker in HCAECs and that it can be used as a pharmacological tool to evaluate the roles of this channel in ER stress-dependent apoptosis in these cells. We next took advantage of this selectivity of Pyr10 to examine whether inhibition of TRPC3 had an impact on the UPR and on ER stress-induced apoptosis in HCAECs. The cells were treated with thapsigargin (1μM) or tunicamycin (10 μg/mL) for 16 hours in the presence or absence of Pyr10 (2 μM), and the expression levels of IRE1α, BiP and CHOP were evaluated by Western blot. Notably, treatment of HCAECs with Pyr10 resulted in a marked reduction of ER stress-induced expression of all three UPR markers (Figure 7A). The basal expression levels of IRE1α and BiP were somewhat reduced by treatment with Pyr10, probably evidencing a requirement for TRPC3-mediated constitutive Ca2+ influx in the proper maintenance of tonic expression of these proteins. When HCAECs were treated with Pyr10, the thapsigargin-induced mRNA levels for sXBP1 were significantly reduced (0.84 ± 0.20, 48.83 ± 2.13 and 27.15 ± 3.61, for normalized sXBP1 mRNA levels under control, thapsigargin, and thapsigargin+Pyr10 conditions, respectively; n=3, P=0.03 for the difference between thapsigargin and thapsigargin + Pyr10). Remarkably, as shown in Figure 7B, treatment with Pyr10 resulted in more than fifty percent reduction in apoptosis of HCAECs, as compared to cells subjected to ER stress in the absence of the TRPC3 blocker. Treatment with Pyr10 alone did not alter apoptotic rates compared to controls. In addition, Pyr10 did not have any effect on staurosporine-induced apoptosis, which proceeds for the most part in a UPR-independent manner (Figure 7B). Overall, these findings indicate that in HCAECs TRPC3, presumably through its constitutive function, contributes to mechanisms associated to ER stress-induced apoptosis. The expression levels of TRPC3 mRNA or protein were not altered under the above ER stress conditions (normalized mRNA levels: 1.06 ± 0.01, 0.96 ± 0.03, 1.3 ± 0.10, for control, thapsigargin and tunicamycin treatments, respectively; n=3, P=0.198 for thapsigargin vs. control, and P=0.075 for tunicamycin vs. control; see also western blot for TRPC3 in figure 8).
Figure 5.


A) Fura-2 loaded HEK293 cells stably overexpressing the human TRPC3 (Vazquez et al., 2004b) were maintained in nominally Ca2+-free medium and, when indicated, OAG (100 μM) and Ba2+ (2 mM) were added to the bath to evidence OAG-induced TRPC3-mediated Ba2+ influx. Alternatively, cells were subjected to the same protocol but in the presence of the TRPC3 selective blocker Pyr10 (2 μM) as indicated. Traces are averages of 10–20 cells and are representative of 3 independent experiments. Values for rates of Ba2+ influx are provided in the text. B) Fura-2 loaded HCAECs were exposed to the SERCA inhibitor thapsigargin (1 μM) in nominally Ca2+-free medium to deplete intracellular Ca2+ stores. Once the Ca2+ transient was over, Ca2+ (2 mM) was added to the bath to evidence the store-operated Ca2+ influx. Alternatively, cells were subjected to the same protocol but in the presence of the TRPC3 selective blocker Pyr10 (2 μM) as indicated. Traces are averages of 18–22 cells and are representative of 3 independent experiments.
Figure 6.


A) Fura-2 loaded HCAECs were challenged with ATP (100 μM) in nominally Ca2+-free medium in the absence or presence of the TRPC3 selective blocker Pyr10 (2 μM) as indicated. Once the Ca2+ transient (IP3-mediated Ca2+ release) was over, Ba2+ (2 mM) was added to the bath to evaluate unidirectional cation (Ba2+) influx. “Control” cells represent Fura-2 loaded HCAECs that were maintained in nominally Ca2+-free medium –no ATP stimulation- and at the indicated time, exposed to Ba2+ (2 mM) to evaluate constitutive Ba2+ influx. Traces are averages of 19–21 cells and are representative of 3 independent experiments. B) HCAECs were maintained in nominally Ca2+-free medium and in the presence of the protein kinase C inhibitor Go6976 (5 μM) to minimize the protein kinase Cα-dependent inhibition of native TRPC3 induced by exogenous synthetic diacylglycerols (Lievremont et al., 2005; Trebak et al., 2004; Trebak et al., 2003a; Vazquez and Putney, 2006). After 5 min, OAG (100 μM) and Ba2+ (2 mM) were added to the bath to trigger OAG-induced TRPC3-mediated Ba2+ influx. Alternatively, cells were subjected to the same protocol but in the presence of the TRPC3 selective blocker Pyr10 (2 μM). Shown are values for rates of Ba2+ influx (ratio units/min) expressed as mean ± SEM. n=20–22 cells.
Figure 7.

HCAECs were maintained in EBM-2 alone (Control) or EBM-2 containing the ER stressors thapsigargin (“Thaps”, 1 μM) or tunicamycin (“Tun”, 10 μg/ml) for 16 hours, in the presence or absence of the selective TRPC3 blocker pyrazole-10 (Pyr10, 2 μM) as indicated; treatment with Pyr10 alone was included as a control. In A), following treatments the cells were processed for immunodetection of IRE1α (~130 kDa), BiP (~75 kDa) or CHOP (~27 kDa) in whole cell lysates. Membranes were reprobed for GAPDH (~37 kDa) to control for protein loading. Blots are representative of three independent experiments. In B) the cells were processed for in vitro TUNEL assay. Where indicated HCAECs were incubated in EBM-2 containing staurosporine (“St”, 1 μM) for 16 hours, in the presence or absence of Pyr10 (2 μM). Differences between “Thaps” and “Tun” treatments and their corresponding control (“Control”) had P<0.001. Differences between staurosporine treatments and the control had P<0.001. All values are mean ± SEM (n=3–5). “ns”: not statistically significant.
Figure 8.
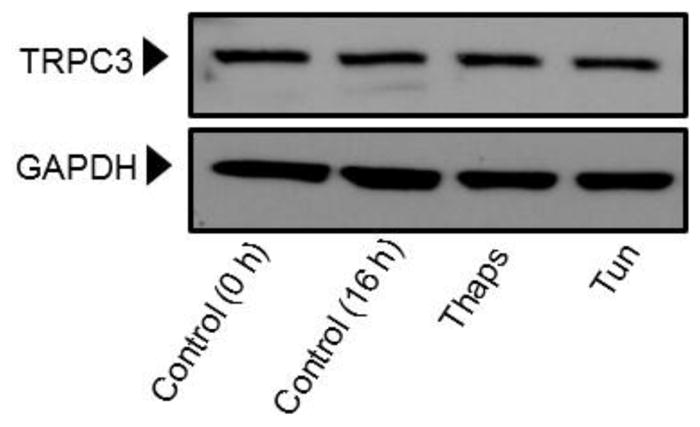
HCAECs were maintained in EBM-2 alone (Control) or EBM-2 containing the ER stressors thapsigargin (“Thaps”, 1 μM) or tunicamycin (“Tun”, 10 μg/ml) for 16 hours. Following treatments the cells were processed for immunodetection of TRPC3 (~100 kDa) in whole cell lysates. Membranes were reprobed for GAPDH (~37 kDa) to control for protein loading. Blots are representative of three independent experiments.
3.4. Calcium/calmodulin-dependent protein kinase II (CAMKII) participates in ER stress-induced apoptosis in HCAECs
In a previous study we showed that in HCAECs the Ca2+ influx that supports activation of CAMKII is contributed to a great extent by the constitutive function of TRPC3 (Smedlund et al., 2010). Furthermore, CAMKII is a well-recognized mediator of ER stress-induced apoptosis in a number of cell types, including endothelial cells (Li et al., 2012; Timmins et al., 2009). To examine if and to what extent CAMKII contributes to ER stress-induced apoptosis in HCAECs, the cells were treated with thapsigargin or tunicamycin for 16 hours and in the presence or absence of the selective CAMKII inhibitor KN93 (10 μM;(Sumi et al., 1991)) before being processed for evaluation of apoptosis by the TUNEL assay. As shown in Figure 9, whereas inhibition of CAMKII did not affect the extent of apoptosis under control conditions, the number of apoptotic cells was significantly lower (~50% reduction) when ER stress was induced in the presence of KN93; treatment with KN92 (10 μM), an inactive analogue of KN93, did not affect apoptosis under control conditions nor that induced by ER stress (Figure 9A and 9B; representative images of TUNEL+ cells are shown in supplemental figure I). Of importance, treatment of HCAECs with KN93 resulted in a marked reduction of CAMKII activation, as evidenced by the extent of phosphorylation on Thr-286 of CAMKII (Figures 10A and 10B), strengthening the notion that the decrease in apoptosis observed with KN93 treatment was indeed subsequent to reduced CAMKII activity. Notably, Pyr10 completely prevented ER stress-dependent activation of CAMKII (Figure 11), suggesting that TRPC3, similar to its role in inflammatory signaling in these cells, is required to support activation of the kinase under conditions of ER stress.
Figure 9.


HCAECs were maintained in EBM-2 alone (Control) or EBM-2 containing thapsigargin (“Thaps”, 1 μM) or tunicamycin (“Tun”, 10 μg/ml) in the presence or absence of the selective CAMKII inhibitor KN93 (“KN93”, 10 μM, panel A) or its inactive analogue KN92 (“KN92”, 10 μM, panel B) and then processed for in vitro TUNEL assay. P values for the differences between treatments with and without KN93 are shown. Differences between treatments with thapsigargin or tunicamycin and the control had P <0.001. All values are mean ± SEM (n=3). ns: not statistically significant. *P=0.011, **P=0.03, for the differences between thapsigargin vs. control and tunicamycin vs. control, respectively.
Figure 10.


A) HCAECs were maintained in EBM-2 alone (Control) or EBM-2 containing thapsigargin (“Thaps”, 1 μM, 10 min) or tunicamycin (“Tun”, 10 μg/ml, 30 min) in the presence or absence of the selective CAMKII inhibitor KN93 (“KN93”, 10 μM), and then processed for immunodetection of phospho-CAMKII (Thr286; ~50 kDa), phospho-STAT1 (Ser727; ~91 kDa) or phospho-JNK1/2 (Thr-183/Tyr-185; ~46/54 kDa) in whole cell lysates, as indicated. Membranes were reprobed for GAPDH (~37 kDa) or total STAT1 (~91 kDa) or total JNK1/2 (~46/54 kDa), as indicated, to control for protein loading. Immunodetection of CAMKII and STAT1 was always performed on the same blot, and thus the same GAPDH blot applies as a loading control for both proteins. Blots are representative of three independent experiments. B) Bar graphs show normalized densitometry values for three independent experiments. P values for the difference respect to the control are shown on top of the bars. ns: not statistically significant.
Figure 11.

HCAECs were maintained in EBM-2 alone (Control) or EBM-2 containing tunicamycin (“Tun”, 10 μg/ml, 30 min) or thapsigargin (“Thaps”, 1 μM, 10 min) and in the presence or absence of the TRPC3 selective blocker Pyr10 (2 μM), as indicated. Following treatments, cells were processed for immunodetection of phospho-CAMKII (Thr286; ~50 kDa) in whole cell lysates. Membranes were reprobed for GAPDH (~37 kDa) to control for protein loading.
3.5. CAMKII is required for activation of the Signal transducer and activator of transcription 1 (STAT1) and c-Jun N-terminal kinases 1/2 (JNK1/2) under conditions of ER stress
In diverse cell types the pro-apoptotic actions of CAMKII are, to some extent, driven through CAMKII-dependent activation of STAT1 and JNK1/2, which in turn control expression of pro-apoptotic and anti-apoptotic genes (Lim et al., 2008; Nair et al., 2002; Timmins et al., 2009). To explore the possibility that also in HCAECs CAMKII drives activation of STAT1 and JNK1/2 under conditions of ER stress, we treated the cells with thapsigargin (1 μM) or tunicamycin (10 μg/mL) for 10- and 30-min respectively and in the presence or absence of the CAMKII inhibitor KN93 (10μM). These early time points allow for a better appreciation of the contribution of ER stress and constitutive Ca2+ influx to activation of these signaling proteins in HCAECs, without interference from non-ER stress and/or non-Ca2+ dependent processes that might also contribute later on in the process. After treatment, whole-cell lysates were analyzed by western blot to determine the activation status of STAT1 and JNK1/2 as indicated, respectively, by the extent of phosphorylation of Ser-727 (STAT1) and Thr-183/Tyr-185 (JNK1/2). Whereas both ER stressors induced activation of STAT1 and JNK1/2 (Figures 10A and 10B), phosphorylation of these signaling molecules was more pronounced in cells exposed to tunicamycin compared to that in cells treated with thapsigargin. Remarkably, phosphorylation of STAT1 on Ser-727 and that of JNK1/2 on Thr-183/Tyr-185 were almost completely suppressed when HCAECs were treated with the CAMKII inhibitor KN93 (Figures 10A and 10B).
4. DISCUSSION
Members of the TRPC group of non-selective cation channels (TRPC1-7) are among the most important Ca2+-permeable channels expressed in endothelium where they participate in numerous events associated to cardiovascular physiology and disease (Liao et al., 2014; Smedlund et al., 2012). In all expression systems where they have been studied, TRPCs form non-voltage gated, Ca2+-permeable channels that are activated downstream of receptor-stimulated phospholipases (Liao et al., 2014; Vazquez et al., 2004a). Besides this receptor-regulated function, TRPC3 is the only TRPC member that also forms channels with significant constitutive function that is manifest both in overexpression and native expression conditions (Trebak et al., 2003b; Vazquez et al., 2010; Vazquez et al., 2004a). Our previous studies demonstrating that in HCAECs TRPC3-mediated constitutive Ca2+ influx is obligatory within the inflammatory signaling that controls expression of VCAM-1 and monocyte adhesion to these cells (Smedlund et al., 2010; Smedlund and Vazquez, 2008) revealed potential pathophysiological relevance of the constitutive function of this TRPC. The in vivo relevance of these findings was indeed validated by recent studies in Apoe knockout mice with endothelial-specific overexpression of human TRPC3 (Smedlund et al., 2015). In these mice, advanced atherosclerotic lesions in the aortic sinus were of bigger size, increased cellularity and exhibited augmented endothelial inflammation compared to lesions in non-transgenic mice (Smedlund et al., 2015).
The studies in the present work show that in HCAECs full activation of the UPR that follows irreversible ER stress and the subsequent cell apoptosis, exhibit a strong dependence upon constitutive Ca2+ influx and TRPC3 function. In HCAECs native TRPC3 accounts for most of the constitutive Ca2+ influx and this is completely abrogated by the non-selective channel blocker SKF (Smedlund and Vazquez, 2008). However, despite ample evidence showing that SKF is a potent TRPC3 blocker (reviewed in (Smyth et al., 2006; Trebak et al., 2003b)) this compound is not specific for TRPC3. Hence, that UPR activation and ER stress-induced apoptosis are sensitive to SKF is not conclusive pharmacological evidence of TRPC3 being involved in these processes. Due to the lack of selective blockers for TRPC channels, for more than a decade our ability to assign a specific cellular event to a particular TRPC was limited to the use of genetic tools. In recent years the so called “pyrazole derivatives” emerged as promising, potent small molecule inhibitors of TRPC channels (Kiyonaka et al., 2009; Schleifer et al., 2012). Among these, Pyr3 and Pyr10 were found to be selective for TRPC3 with Pyr10 possessing the highest selectivity for this channel. The activating mechanism for TRPC3 has been –and still is- a matter of debate (reviewed by us in (Smedlund et al., 2012; Vazquez et al., 2010)). Depending on a number of factors such as the cell type, the expression levels of the TRPC3 protein and the signaling repertoire of a particular cell type under specific experimental conditions, receptor dependent activation of TRPC3 channels can occur through store-operated –i.e., requiring depletion of ER Ca2+ stores- and/or non-store-operated mechanisms (Liao et al., 2014; Trebak et al., 2007). Whichever the activating mechanism, TRPC3 consistently exhibits constitutive function (Vazquez et al., 2010). Pyr10 has been shown to efficiently and selectively block the store-operated function of TRPC3 in rat dorsal root ganglion neurons (Alkhani et al., 2014), the non-store operated activity of TRPC3 in HEK293 cells (Schleifer et al., 2012) and, as recently shown by us, the TRPC3 constitutive function in mouse bone marrow-derived macrophages (Solanki et al., 2014). Thus, Pyr10 suppresses TRPC3 channel function in a variety of cell types and, most importantly, it does so regardless of the channel’s operating mode. In HCAECs native TRPC3 forms channels that also contribute, although partially, to receptor-activated Ca2+ influx (Smedlund and Vazquez, 2008) but does not form store-operated Ca2+ channels (Vazquez and Putney, 2006) and does not respond to direct application of exogenous synthetic diacylglycerols, unless pharmacological inhibition of endogenous protein kinase C is pre-set (Vazquez and Putney, 2006; Vazquez et al., 2010). In line with all these functional features of endogenous TRPC3 channels in HCAECs, we found that Pyr10 does not inhibit store-operated Ca2+ entry, but partially reduces ATP-induced Ba2+ influx and almost completely abrogates the ability of OAG to promote Ba2+ influx in cells pre-treated with a protein kinase C inhibitor, a hallmark of endogenous TRPC3 function in these cells (Vazquez and Putney, 2006). Remarkably, in HCAECs Pyr10 affects these processes to the same extent as knockdown of TRPC3 in these cells does (Smedlund and Vazquez, 2008; Vazquez and Putney, 2006).
This evidence provides confidence about the use of Pyr10 as a selective TRPC3 channel blocker in HCAECs. Therefore, the findings in the present studies showing that in these cells Pyr10 effectively suppresses UPR activation and ER stress-induced apoptosis, strongly suggest that it is TRPC3, presumably through its constitutive function, that supports the Ca2+ influx required for full activation of these processes. We have previously shown that in HCAECs activation of CAMKII is supported to a great extent by the constitutive function of native TRPC3 (Smedlund et al., 2010). Based on this and on the well-recognized role of CAMKII as a mediator of ER stress-induced apoptosis in both endothelial and non-endothelial cell types (Li et al., 2012; Timmins et al., 2009), we examined if CAMKII was involved in the mechanism associated to ER stress-induced apoptosis in HCAECs. Our findings show that selective suppression of CAMKII activation by the KN93 compound results in a marked reduction in ER stress-induced apoptosis, both under thapsigargin and tunicamycin treatments. This is important, because thapsigargin and tunicamycin cause ER stress by, respectively, Ca2+-dependent –i.e., ER Ca2+ store depletion- and Ca2+-independent mechanisms, supporting the notion that the participation of CAMKII is related, for the most part, to ER stress. Of importance, under our experimental conditions tunicamycin, which does not alter intracellular Ca2+, was able to induce CAMKII activation in as early as thirty minutes –sufficient to cause ER stress- supporting a scenario where constitutive Ca2+ entry provides, at least in part, for the early activation of this kinase. Remarkably, treatment with Pyr10 completely abolished both tunicamycin- and thapsigargin-induced CAMKII activation, indicating that TRPC3, through its constitutive function, is required for activation of this kinase under conditions of ER stress, and suggests operation of a TRPC3/CAM/CAMKII axis, similar to the coupling of TRPC3 to NFkB-dependent inflammatory signaling in these cells (Smedlund et al., 2010). Remarkably, inhibition of CAMKII resulted in almost complete abrogation of ER stress-induced activation of STAT1 and JNK1/2, two well-established mediators of ER stress-induced apoptosis in other cell types. Although if and to what extent STAT1 and JNK1/2 contribute to apoptosis of HCAECs was not examined here, these findings suggest that, should these molecules be pro-apoptotic in these cells, they are likely to operate downstream CAMKII, as is the case for example in peritoneal macrophages (Lim et al., 2008; Timmins et al., 2009).
Under the ER stress conditions used in the present studies the expression levels of TRPC3 were not affected. In HCAECs TRPC3 expression is augmented by pro-inflammatory factors, and this is followed by an increased number of plasma membrane TRPC3 channels operating in constitutive mode (Smedlund et al., 2010; Smedlund and Vazquez, 2008). The initial purpose of the UPR is to trigger an adaptive response to the ER stressing environment and re-establish normal ER function. However, both thapsigargin and tunicamycin treatments result in sustained and irreversible ER stress that derives in cell apoptosis, similar to the scenario that endothelial cells encounter in, for instance, advanced atherosclerosis (Zeng et al., 2009; Zhou and Tabas, 2013). Hence, it is possible that under ER stress conditions other than those utilized here changes in TRPC3 expression may occur, the direction of the change likely dictating the final impact on the UPR response.
In summary, the present studies provide pharmacological evidence on the impact of TRPC3 constitutive function in mechanisms of ER stress-induced apoptosis of coronary endothelial cells of human origin. Interestingly, in mouse epithelial cells from pancreatic and parotid acini TRPC3 function was found to be necessary for activation of the PERK branch of the UPR that promotes cell damage (Kim et al., 2011). In podocytes, TRPC6-mediated Ca2+ entry mediates albumin overload-induced apoptosis (Chen et al., 2011) and in mouse dopaminergic neurons, neurotoxin-induced activation of the UPR and subsequent cell apoptosis require downregulation of the store-operated channel TRPC1 (Selvaraj et al., 2012). We recently showed that in polarized macrophages the constitutive function of TRPC3 is part of the signaling mediating ER stress-dependent apoptosis (Solanki et al., 2014), a major mechanism regulating macrophage death and necrosis in atherosclerosis. Altogether, these observations reveal novel roles of TRPC channels in mechanisms of ER stress-induced apoptosis, and are suggestive of a previously unforeseen therapeutic potential of these channel forming proteins. Studies using mouse models of disease in which endothelial expression of TRPC3 is manipulated, as we recently described in (Smedlund et al., 2015), will provide the appropriate context to appreciate the contribution of endothelial TRPC3 to mechanisms of ER stress in an inflammatory setting.
Supplementary Material
Representative images of TUNEL experiments performed under the conditions described in Figure 9. Nuclei are stained blue (DAPI) and TUNEL+ cells manifest with red, rather fragmented and/or condensed nuclei. Magnification: 20x.
{kind=link}
Acknowledgments
This work was supported by NIH grant R01HL111877-03 (to G.V.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alkhani H, Ase AR, Grant R, O’Donnell D, Groschner K, Seguela P. Contribution of TRPC3 to store-operated calcium entry and inflammatory transductions in primary nociceptors. Mol Pain. 2014;10:43. doi: 10.1186/1744-8069-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi CVG, Boland R. Capacitative calcium influx in human epithelial breast cancer and non-tumorigenic cells occurs through Ca2+ entry pathways with different permeabilities to divalent cations. Journal of cellular biochemistry. 2003;88:1265–1272. doi: 10.1002/jcb.10471. [DOI] [PubMed] [Google Scholar]
- Chen S, He FF, Wang H, Fang Z, Shao N, Tian XJ, Liu JS, Zhu ZH, Wang YM, Wang S, Huang K, Zhang C. Calcium entry via TRPC6 mediates albumin overload-induced endoplasmic reticulum stress and apoptosis in podocytes. Cell calcium. 2011;50:523–529. doi: 10.1016/j.ceca.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Hecquet CM, Zhang M, Mittal M, Vogel SM, Di A, Gao X, Bonini MG, Malik AB. Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis. Circ Res. 2014;114:469–479. doi: 10.1161/CIRCRESAHA.114.302414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J. Evolution of the unfolded protein response. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2013;1833:2458–2463. doi: 10.1016/j.bbamcr.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Jambrina E, Alonso R, Alcalde M, del Carmen Rodriguez M, Serrano A, Martinez AC, Garcia-Sancho J, Izquierdo M. Calcium influx through receptor-operated channel induces mitochondria-triggered paraptotic cell death. The Journal of biological chemistry. 2003;278:14134–14145. doi: 10.1074/jbc.M211388200. [DOI] [PubMed] [Google Scholar]
- Kim MS, Lee KP, Yang D, Shin DM, Abramowitz J, Kiyonaka S, Birnbaumer L, Mori Y, Muallem S. Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity. Gastroenterology. 2011;140:2107–2115. 2115 e2101–2104. doi: 10.1053/j.gastro.2011.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, Hamachi I, Mori Y. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5400–5405. doi: 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzer A, Giral H, Landmesser U. High-density lipoproteins as modulators of endothelial cell functions: alterations in patients with coronary artery disease. Cardiovascular research. 2014;103:350–361. doi: 10.1093/cvr/cvu139. [DOI] [PubMed] [Google Scholar]
- Li J, Wang P, Yu S, Zheng Z, Xu X. Calcium entry mediates hyperglycemia-induced apoptosis through Ca(2+)/calmodulin-dependent kinase II in retinal capillary endothelial cells. Mol Vis. 2012;18:2371–2379. [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Abramowitz J, Birnbaumer L. The TRPC family of TRP channels: roles inferred (mostly) from knockout mice and relationship to ORAI proteins. Handb Exp Pharmacol. 2014;223:1055–1075. doi: 10.1007/978-3-319-05161-1_14. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lievremont JP, Numaga T, Vazquez G, Lemonnier L, Hara Y, Mori E, Trebak M, Moss SE, Bird GS, Mori Y, Putney JWJ. The role of canonical transient receptor potential 7 in B-cell receptor-activated channels. J Biol Chem. 2005;280:35346–35351. doi: 10.1074/jbc.M507606200. [DOI] [PubMed] [Google Scholar]
- Lim WS, Timmins JM, Seimon TA, Sadler A, Kolodgie FD, Virmani R, Tabas I. Signal transducer and activator of transcription-1 is critical for apoptosis in macrophages subjected to endoplasmic reticulum stress in vitro and in advanced atherosclerotic lesions in vivo. Circulation. 2008;117:940–951. doi: 10.1161/CIRCULATIONAHA.107.711275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlpine CS, Werstuck GH. The development and progression of atherosclerosis: evidence supporting a role for endoplasmic reticulum (ER) stress signaling. Cardiovasc Hematol Disord Drug Targets. 2013;13:158–164. doi: 10.2174/1871529x11313020009. [DOI] [PubMed] [Google Scholar]
- Nair JS, DaFonseca CJ, Tjernberg A, Sun W, Darnell JE, Chait BT, Zhang JJ. Requirement of Ca2+ and CaMKII for Stat1 Ser-727 phosphorylation in response to IFN-γ. Proceedings of the National Academy of Sciences. 2002;99:5971–5976. doi: 10.1073/pnas.052159099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleifer H, Doleschal B, Lichtenegger M, Oppenrieder R, Derler I, Frischauf I, Glasnov TN, Kappe CO, Romanin C, Groschner K. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca(2+) entry pathways. Br J Pharmacol. 2012;167:1712–1722. doi: 10.1111/j.1476-5381.2012.02126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj S, Sun Y, Watt JA, Wang S, Lei S, Birnbaumer L, Singh BB. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. The Journal of clinical investigation. 2012;122:1354–1367. doi: 10.1172/JCI61332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedlund K, Bah M, Vazquez G. On the role of endothelial TRPC3 channels in endothelial dysfunction and cardiovascular disease. Cardiovasc Hematol Agents Med Chem. 2012;10:265–274. doi: 10.2174/187152512802651051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedlund K, Tano JY, Margiotta J, Vazquez G. Evidence for operation of nicotinic and muscarinic acetylcholine receptor-dependent survival pathways in human coronary artery endothelial cells. Journal of cellular biochemistry. 2011;112:1978–1984. doi: 10.1002/jcb.23169. [DOI] [PubMed] [Google Scholar]
- Smedlund K, Tano JY, Vazquez G. The constitutive function of native TRPC3 channels modulates vascular cell adhesion molecule-1 expression in coronary endothelial cells through nuclear factor kappaB signaling. Circ Res. 2010;106:1479–1488. doi: 10.1161/CIRCRESAHA.109.213314. [DOI] [PubMed] [Google Scholar]
- Smedlund K, Vazquez G. Involvement of native TRPC3 proteins in ATP-dependent expression of VCAM-1 and monocyte adherence in coronary artery endothelial cells. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:2049–2055. doi: 10.1161/ATVBAHA.108.175356. [DOI] [PubMed] [Google Scholar]
- Smedlund KB, Birnbaumer L, Vazquez G. Increased size and cellularity of advanced atherosclerotic lesions in mice with endothelial overexpression of the human TRPC3 channel. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E2201–2206. doi: 10.1073/pnas.1505410112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth JT, DeHaven WI, Jones BF, Mercer JC, Trebak M, Vazquez G, Putney JJW. Emerging perspectives in store-operated Ca2+ entry: Roles of Orai, Stim and TRP. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2006;1763:1147–1160. doi: 10.1016/j.bbamcr.2006.08.050. [DOI] [PubMed] [Google Scholar]
- Solanki S, Dube PR, Tano JY, Birnbaumer L, Vazquez G. Reduced endoplasmic reticulum stress-induced apoptosis and impaired unfolded protein response in TRPC3-deficient M1 macrophages. American journal of physiology Cell physiology. 2014;307:C521–531. doi: 10.1152/ajpcell.00369.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Zou MH. Redox regulation of endothelial cell fate. Cell Mol Life Sci. 2014;71:3219–3239. doi: 10.1007/s00018-014-1598-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sovolyova N, Healy S, Samali A, Logue Susan E. Stressed to death –mechanisms of ER stress-induced cell death. Biological Chemistry. 2014:1. doi: 10.1515/hsz-2013-0174. [DOI] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic acids research. 2010;38:D792–D799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochemical and biophysical research communications. 1991;181:968–975. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- Tano JY, Lee RH, Vazquez G. Involvement of calmodulin and calmodulin kinase II in tumor necrosis factor alpha-induced survival of bone marrow derived macrophages. Biochemical and biophysical research communications. 2012;427:178–184. doi: 10.1016/j.bbrc.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tano JY, Smedlund K, Lee R, Abramowitz J, Birnbaumer L, Vazquez G. Impairment of survival signaling and efferocytosis in TRPC3-deficient macrophages. Biochemical and biophysical research communications. 2011;410:643–647. doi: 10.1016/j.bbrc.2011.06.045. [DOI] [PubMed] [Google Scholar]
- Tano JY, Vazquez G. Requirement for non-regulated, constitutive calcium influx in macrophage survival signaling. Biochemical and biophysical research communications. 2011;407:432–437. doi: 10.1016/j.bbrc.2011.03.048. [DOI] [PubMed] [Google Scholar]
- Tano JY, Solanki S, Lee RH, Smedlund K, Birnbaumer L, Vazquez G. Bone marrow deficiency of TRPC3 channel reduces early lesion burden and necrotic core of advanced plaques in a mouse model of atherosclerosis. Cardiovascular research. 2014;101:138–144. doi: 10.1093/cvr/cvt231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, Tabas I. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. The Journal of clinical investigation. 2009;119:2925–2941. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M, Hempel N, Wedel BJ, Smyth JT, Bird GS, Putney JW. Negative Regulation of TRPC3 Channels by Protein Kinase C-mediated Phosphorylation of Serine 712. Mol Pharmacol. 2004 doi: 10.1124/mol.104.007252. [DOI] [PubMed] [Google Scholar]
- Trebak M, Lemonnier L, Smyth JT, Vazquez G, Putney JW., Jr Phospholipase C-coupled receptors and activation of TRPC channels. Handb Exp Pharmacol. 2007:593–614. doi: 10.1007/978-3-540-34891-7_35. [DOI] [PubMed] [Google Scholar]
- Trebak M, St JBG, McKay RR, Birnbaumer L, Putney JW., Jr Signaling mechanism for receptor-activated canonical transient receptor potential 3 (TRPC3) channels. The Journal of biological chemistry. 2003a;278:16244–16252. doi: 10.1074/jbc.M300544200. [DOI] [PubMed] [Google Scholar]
- Trebak M, Vazquez G, Bird GS, Putney JW., Jr The TRPC3/6/7 subfamily of cation channels. Cell calcium. 2003b;33:451–461. doi: 10.1016/s0143-4160(03)00056-3. [DOI] [PubMed] [Google Scholar]
- van Schadewijk A, van’t Wout EF, Stolk J, Hiemstra PS. A quantitative method for detection of spliced X-box binding protein-1 (XBP1) mRNA as a measure of endoplasmic reticulum (ER) stress. Cell stress & chaperones. 2012;17:275–279. doi: 10.1007/s12192-011-0306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez G, Putney JW. Role of Canonical Transient Receptor Potential Channels (TRPC) in Receptor-Dependent Regulation of Vascular Cell Adhesion Molecule-1 In Human Coronary Artery Endothelium. Art Thr Vasc Biol. 2006;26:93–94. [Google Scholar]
- Vazquez G, Tano JY, Smedlund K. On the potential role of source and species of diacylglycerol in phospholipase-dependent regulation of TRPC3 channels. Channels (Austin, Tex) 2010;4:232–240. doi: 10.4161/chan.4.3.12058. [DOI] [PubMed] [Google Scholar]
- Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney J, James W. The mammalian TRPC cation channels. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2004a;1742:21–36. doi: 10.1016/j.bbamcr.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Vazquez G, Wedel BJ, Kawasaki BT, Bird GS, Putney JW., Jr Obligatory role of Src kinase in the signaling mechanism for TRPC3 cation channels. The Journal of biological chemistry. 2004b;279:40521–40528. doi: 10.1074/jbc.M405280200. [DOI] [PubMed] [Google Scholar]
- Wang X, Spandidos A, Wang H, Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic acids research. 2012;40:D1144–D1149. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Zampetaki A, Margariti A, Pepe AE, Alam S, Martin D, Xiao Q, Wang W, Jin ZG, Cockerill G, Mori K, Li YS, Hu Y, Chien S, Xu Q. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:8326–8331. doi: 10.1073/pnas.0903197106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou AX, Tabas I. The UPR in atherosclerosis. Semin Immunopathol. 2013;35:321–332. doi: 10.1007/s00281-013-0372-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative images of TUNEL experiments performed under the conditions described in Figure 9. Nuclei are stained blue (DAPI) and TUNEL+ cells manifest with red, rather fragmented and/or condensed nuclei. Magnification: 20x.