

Abstract
Adjuvant therapy following breast cancer surgery generally consists of either a course of chemotherapy if the cancer lacks hormone receptors, or a course of hormonal therapy, otherwise. Here we report a correlation between adjuvant strategy, and mutated pathway patterns. In particular we find that for breast cancer patients, pathways enriched in non-synonymous mutations in the chemotherapy group, are distinct from those of the hormonal therapy group. We apply a recently developed method that identifies collaborative pathway groups for hormone and chemotherapy patients. A collaborative group of pathways is one in which each member is altered in the same–generally large–number of samples. In particular we find the following: (i) A chemotherapy group consisting of 3 pathways and a hormone therapy group consisting of 20, the members of the two groups being mutually exclusive. (ii) Each group is highly enriched in breast cancer drivers. (iii) The pathway groups are correlates of subtype-based therapeutic recommendations. These results suggest that patient profiling using these pathway groups can potentially enable the development of personalized treatment plans that may be more accurate and specific than those currently available.
Keywords: mutated pathway, chemotherapy, hormone therapy, breast cancer, MUDPAC
Introduction
Breast cancer is a highly heterogeneous disease consisting of distinct molecular subtypes having different prognostic and therapeutic responses (1). Of interest to this manuscript is that surgery is often followed by adjuvant therapy to diminish the chance of recurrence. Chemotherapy is used to stop the growth of cancer cells by either killing them or otherwise halting division (2); hormone therapy lowers estrogen levels or blocks its action (3). The choice between the two is usually made based on the levels of (4, 5) estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) (6). In general (4) Luminal-A is treated with hormone therapy; luminal B is often treated with chemotherapy, but occasionally with hormone therapy; and HER2 and basil-like subtypes are treated with chemotherapy.
Until now the choice of therapy has been guided by the levels of RNA transcripts of the three hormone receptor genes. Our understanding of cancer biology (7) and the state of computational science (8–15) has, however, now reached a point where a much fuller profile of the cancer cell can be used to guide the choice of therapy.
The development of cancer is a complex multi-step process that involves accumulation of multiple mutations that lead to dysfunction of cell signaling pathways responsible for cell growth and cell fate (16). In particular, mutations are not uniformly distributed across different cellular functions, but tend to cluster in a relatively well defined set of physiologically relevant pathways (17). Consequently it is now generally accepted that causal mechanisms underlying transformation generally reflect the behavior of functionally coupled sets of genes (18–20). An analysis of mutational heterogeneity in the context of cellular signaling and regulatory pathways can therefore add to our understanding of cancer progression, and its modulation by different therapeutic strategies (16).
We identified sets of mutated pathways that are found in a high percentage of TCGA (21) breast cancer samples derived from patients to whom adjuvant therapy had been, or was being, administered. We refer to these pathways as collaborative because they are mutated in a high percentage of the same samples, as opposed to pathways that are altered in different but overlapping sets of samples. This analysis was performed using a recently developed method, Mutational Driver Pathway Collaboration (MUDPAC) (20). Our goal was to determine whether different therapy groups–the chemotherapy group (CT) and the hormone therapy group (HT) have distinguishing sets of mutated pathways, and if they do, to determine the composition of the two groups. The central result of this paper is that we found strong correlation between altered processes (collaborative pathway groups) and therapy group. This suggests a useful addition to our knowledge of cancer subtypes, and provides a sound molecular basis for subtype recommendations, with perhaps some moderate shifts in recommendations. These observations could have important implications for cancer biology and therapy, which we discuss later. A flowchart of our method can be seen in Figure 1.
Figure 1. Flowchart of our method.
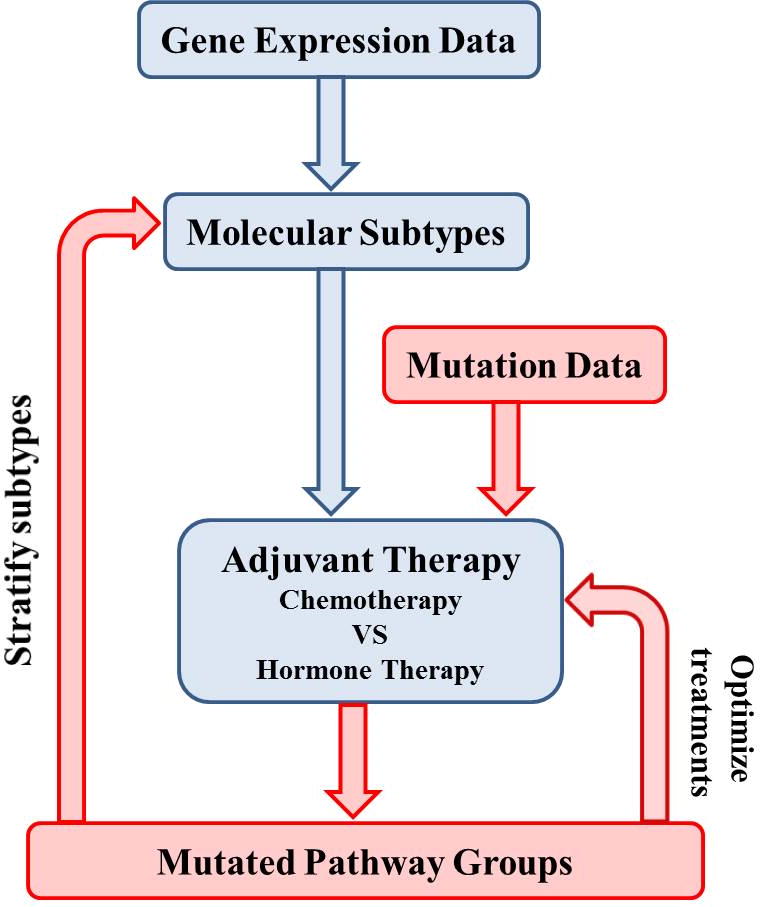
Current adjuvant therapy (chemotherapy and hormone therapy) generally follows molecular subtypes which are induced from gene expression data (shown in blue flow path). In this paper, we apply mutation data into chemotherapy and hormone therapy patients respectively (shown in red flow path) and identify mutated pathways for each therapy group. The two mutated pathway groups are mutual exclusive to each other. We hope mutated pathway groups could be considered as a signature in the future to stratify subtypes and optimize therapeutic treatments.
Materials and Methods
Somatic mutation data
Breast cancer tissue somatic mutations (.maf file) were downloaded from TCGA on March, 2013. All mutations in .maf file are sequenced and annotated before any systemic treatment and therapy (22).
Tumor sample selection
Tissue sampling is carried out by the TCGA project; no further tissue sampling is performed in this study. Sample subtype information was obtained from the supplementary table of (21) where PAM50 is used to stratify subtypes based on mRNA data. The therapy treatment and drug information of each patient was downloaded from breast cancer TCGA clinical data released in July, 2013. Only chemotherapy and hormone therapy patients are included is this study, which are the only adjuvant treatments with a sufficient number of samples in TCGA (>50). Cooperative pathways are identified using samples from patients who have been treated by chemotherapy or hormone therapy, but not both. After filtering, 56 chemotherapy samples remained (34 basal-like, 10 HER2+, 9 Luminal-A, 3 Luminal-B) with 3971 mutations in 3186 genes, and 51 hormone therapy samples remained (1 basal-like, 0 HER2+, 40 Luminal-A, 10 Luminal-B), with 2049 mutations in 2049 genes.
Pathway data
269 pathways (.xml and .gene files) were downloaded from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [77] released on June 2013; 200 of them are used in this study after excluding 3 global metabolic pathways and 66 human disease pathways.
MUDPAC
MUDPAC is a method that identifies mutated collaborative pathway-groups, i.e. pathways that are altered in a high percentage of the same samples of a certain cancer or cancer subtype (20). It consists of two steps. The first step is an identification of pathways showing statistically significant differences between non-synonymous mutation group and synonymous mutation group, based on some characteristics of mutations, such as mutation frequency, functional mutation score, mutual exclusivity, and network topology. The second step uses a greedy search for collaborative pathways. Two more criteria are considered when selecting a new pathway into collaborative set. First, the newly selected pathway along with all pathways already in collaborative set, should have a Maximal Coverage Rate (MCR) higher than the highest mutation rate of genes in this new pathway by a given threshold (3% in this study). Second, permutation is used to test whether MCR of this new pathway is significantly higher than a background MCR (p<0.05 in this study).
Each of the missense mutations in .maf file is assigned a functional mutation score by MutationAssessor (23) based on hg19 reference genome. Since MutationAssessor can only evaluate missense polymorphisms, the functional scores for the remaining mutations are assessed following the same criteria in MUDPAC: the highest score that can be calculated using MutationAssessor is assigned to all indels, nonsense mutations and splice site mutations; the lowest score from MutationAssessor is allocated to synonymous mutations; the average score of all missense mutations is assigned to other missense mutations that can’t be calculated using MutationAssessor.
MuSiC (24) is used to calculate the total number of bases for each gene having available alignment data, as part of the input required by MUDPAC. MuSiC uses Broad Institute’s analysis infrastructure Firehose to count bases with sufficient coverage of each gene from the given wiggle tract format file. Wiggle files contain dense, continuous data such as GC percent, probability scores, and transcriptome data, and were downloaded from Broad Institute Firehose webpage on Aug 2012. The thresholds for sufficient coverage are at least 8 fold read depth in normal tissue, and at least 14 fold read depth in cancer tissue.
Statistical analysis
The Fisher exact test is used to identify KEGG pathways that are statistically enriched in mutated genes of identified pathways compared to the human genome background, and to assess correlations between pathway groups and therapy.
Results
Collaborative pathway groups from hormone therapy and chemotherapy patients are mutually exclusive
Altered physiological functions in CT and HT groups were examined separately using MUDPAC. We identified 3 collaborative pathways (Figure 2A) in CT, having a Maximal Coverage Rate (MCR) of approximately 79%, which means these three pathways are altered in the same 44 (of 56) chemotherapy patients. Of the 51 patients who received only hormone therapy, 20 collaborative pathways (Figure 2B) were identified with a MCR of 57%, which results in a much longer and more stable plateau. None of these 20 are among the 3 pathways that form the CT group (Figure 2C).
Figure 2. Mutated pathways in chemotherapy and hormone therapy.
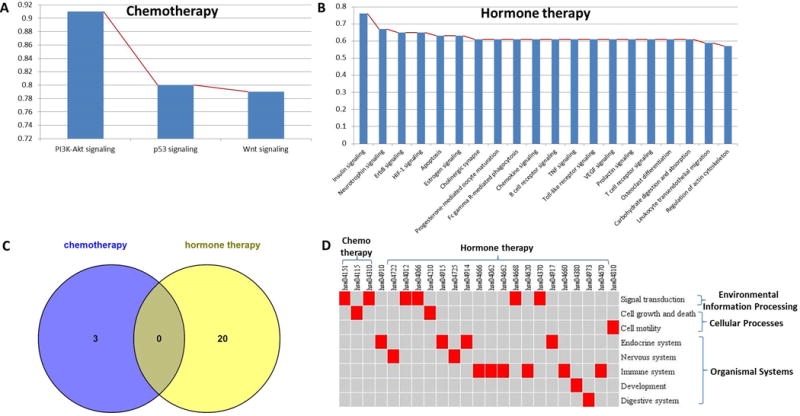
(A) Mutated pathway collaboration in chemotherapy: 79% chemo patients can simultaneously have 3 pathways mutated. (B) Mutated pathway collaboration in hormone therapy: 57% hormone patients can simultaneously have 20 pathways mutated. (C) Venn diagram of two pathway sets showing there is no common pathways between different therapy treatments. (D) Relationships between identified pathways and their KEGG pathway category, with each column is the identified pathway ID, each row represents a KEGG pathway category with higher level category bracketed on right. A red cell means this pathway belongs to the certain category, otherwise not.
All three mutated pathways (PI3K-Akt signaling, p53 signaling, Wnt signaling) in the chemo-therapy pathway group (CTPG) are TP53 related. This is consistent with the sample distribution within this therapy group, which has 34 (61%) basal-like samples dominated by the mutated TP53 suppressor gene (21), which is known to be associated with the basal-like subtype.
In contrast to these three pathways, which play key roles in signal transduction and cell growth (Figure 2D), the twenty pathways in the hormone therapy pathway group (HTPG) tend to have an organismal systems classification (Figure 2D), including the endocrine, immune, central nervous, development and digestive systems. More than half are either immune system or endocrine system related: 30% for the former (6 pathways: Fc gamma R-mediated phagocytosis, Chemokine signaling, B cell receptor signaling, Toll-like receptor signaling, T cell receptor signaling, Leukocyte transendothelial migration), and 20% for the latter (4 pathways: Insulin signaling, Estrogen signaling, Progesterone-mediated oocyte maturation, Prolactin signaling).
The gene sets that trigger pathway dysfunction in the two therapy groups are largely distinct from one other. The top 10 genes with highest mutation frequency in CT and HT are shown in Table 1.
Table 1.
Top 10 highly mutated genes in collaborative pathways of chemotherapy and hormone therapy groups. Genes are listed with mutation frequency descending order, with mutation frequency shown in brackets.
| Gene rank | Chemotherapy | Hormone therapy |
|---|---|---|
| 1 | TP53 (75%) | PIK3CA (53%) |
| 2 | TTN (21%) | MAP3K1 (20%) |
| 3 | PIK3CA (18%) | GATA3 (10%) |
| 4 | OBSCN (13%) | MLL3 (10%) |
| 5 | MUC4 (11%) | OBSCN (10%) |
| 6 | USH2A (11%) | LAMA5 (8%) |
| 7 | AHCTF1 (9%) | PHKA2 (8%) |
| 8 | CACNA1B (9%) | RYR2 (8%) |
| 9 | CSMD2 (9%) | SPEN (8%) |
| 10 | DNAH5 (9%) | TP53 (8%) |
Of the 10 mutated genes in each group that occur with the highest frequency, 3–TP53, PIK3CA, OBSCN–are common to the two groups. TP53 ranks first with a frequency of 75% in the CT but 10th (frequency of 8%) in HT. PIK3CA ranks first in HT with a frequency of 53%, but has a frequency of only 18% in CT. OBSCN has comparable mutation rates in the two groups. However, the mutation type and functional mutational score for OBSCN are different in the two groups. OBSCN has 10 mutations (1 silent, 6 missense mutations, 1 frame shift mutation, 2 nonstop mutations) in CT with an average mutation score of 1.84, but it only has 5 mutations (2 silent mutations, 2 missense mutations, 1 nonsense mutation) in HT with an average functional score of 0.44. This suggests that comparable frequencies do not necessarily imply comparable physiological impact.
We also compared all the genes in the identified pathways of the two therapy groups with 87 plausible breast cancer genes (25): 37 confirmed drivers, and another 50 driver candidates identified with high likelihood computationally. Of the 532 genes in the CTPG, 27 are among the 87 plausible drivers (Fisher exact test p<10−15). Of the 1059 genes in the HTPG, 40 are among the 87 plausible drivers (Fisher exact test p<10−15). These results suggest that the mutated genes in the identified pathways are highly enriched in drivers. For the two sets of drivers–27 genes for CTPG, and 40 for HTPG – 21 are common. The remaining 6 (19) are unique to the chemo (hormone) therapy patients.
Pathway groups are correlates of therapy recommendations
The choice of adjuvant therapy, as noted above, generally follows subtype (4): the Luminal-A subtype is treated with hormone therapy; the Luminal-B subtype with chemotherapy, but occasionally with hormone therapy, and the HER2 and basil-like subtypes with chemotherapy. The patient population in this analysis deviates somewhat from these guidelines, showing 9 of the 49 luminal-A patients are in chemotherapy. The deviation for luminal-B is more severe, with only 3 of the 13 in chemotherapy. We will comment on this below.
The subtypes and their therapy recommendations are approximately correlated. Thus, for basal-like subtype patients having chemotherapy, 95 out of 102 pathways (34 basal-like patients × 3 identified pathways in chemotherapy) are mutated in CT while 7 are not; 264 out of 680 (34 basal-like patients × 20 identified pathways in hormone therapy) pathways are mutated in HT while 416 are not. Consequently basal-like patients having chemotherapy are significantly more likely to be mutated in CTPG than in HTPG (p<10−15). Similar results are observed for HER2+ (p=7.35e-4) and Luminal-A (p=2.10e-8), which are recommended for chemo and hormone therapy respectively. But there are no significant correlations found in Luminal-B patients, no matter in CT (p=0.6119) or in HT (p=0.7489).
There are, as expected, strong correlations between the mutated pathway groups seen in patients, and the kind of therapy they receive. The distribution of mutations between HTPG and CTPG for CT patients is highly significant. In particular, the probability that the observed distribution of mutations between the two pathway groups is due to chance, given the patients are in chemotherapy (Figure 3A) is p<10−15. This follows from a Fisher exact test, based on the observation that for CT, (i) 141 out of 168 (56×3) CTPG pathways are mutated, and 27 are not; and (ii) 503 of 1120 (56×20) HTPG mutated, and 617 are not (Figure 3A; CT columns).
Figure 3. Mutated pathways and genes distributions for each patient.
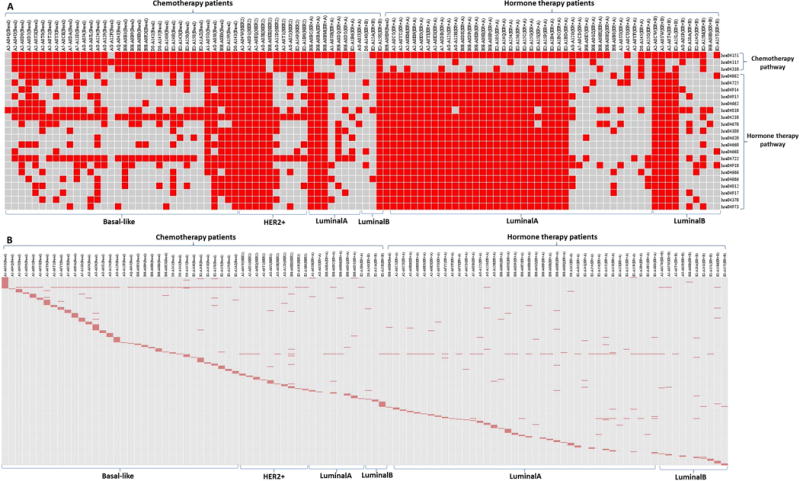
(A) Mutated pathway distribution for each patient. Each column is an individual patient, with chemotherapy and hormone therapy being grouped separately, molecular subtypes are displayed at the bottom. All analyses are done based on 49 Luminal-A patients, 9 in CT and 40 in HT, but this figure only shows 8 in CT and 38 in HT since the 1 in CT and 2 in HT do not have any identified pathways mutated. For other subtypes, the number of patients analyzed and the number of patients showing in figure are identical. Each row is an identified pathway, with pathways of CT and HT grouped separately. Pathway ID and name are: hsa04151-PI3K-Akt signaling; hsa04115-p53 signaling; hsa04310-Wnt signaling; hsa04910-Insulin signaling; hsa04722-Neurotrophin signaling; hsa04012-ErbB signaling; hsa04066-HIF-1 signaling; hsa04210-Apoptosis; hsa04915-Estrogen signaling; hsa04725-Cholinergic synapse; hsa04914-Progesterone-mediated oocyte maturation; hsa04666-Fc gamma R-mediated phagocytosis; hsa04062-Chemokine signaling; hsa04662-B cell receptor signaling; hsa04668-TNF signaling; hsa04620-Toll-like receptor signaling; hsa04370-VEGF signaling; hsa04917-Prolactin signaling; hsa04660-T cell receptor signaling; hsa04380-Osteoclast differentiation; hsa04973-Carbohydrate digestion and absorption; hsa04670-Leukocyte transendothelial migration; hsa04810-Regulation of actin cytoskeleton. A red cell means the patient has the pathway mutated (with at least one mutated gene in this pathway), otherwise not. (B) Mutated gene distribution for each patient. Each column is an individual patient, with patients of CT and HT being grouped separately, molecular subtypes are displayed at the bottom. Each row is a mutated gene in identified pathways. A red cell means the patient has the gene mutated, otherwise not.
Similar results apply to patients receiving hormone therapy (Figure 3A; HT columns): 69 out of 153 (51×3) CTPG pathways are mutated while 84 are not, and 677 out of 1020 (51×20) HTPG pathways are mutated while 343 of them not. The probability that mutations are equally likely in the two pathway groups, given that patients are in hormone therapy is p=4×10−7.
These results provide a previously unnoticed molecular rationale for the choice of therapy based on cancer subtype. In addition, as discussed below, the structure of Figure 3A suggests well defined stratifications within the standard subtypes. The implications of these observations for therapeutic choices are discussed below.
Pathway groups discriminate between subtypes
One of the 3 CTPG pathways– phosphatidylinositol 3′ –kinase (PI3K)-Akt – is altered in almost all patients, while the other 2 are mutated in a high percentage of basal-like (32/35) and HER-2+ (9/10) patients, and only occasionally in luminal-A (5/49) and luminal-B (4/13) patients, irrespective of whether they received hormone or chemotherapy therapy. Consequently, these two pathways discriminate extremely well between HER2+ and basal like subtypes on the one hand, and Luminal-A and Luminal-B subtypes on the other (Fisher exact test p<10−15). Since the first two subtypes received chemotherapy, and rarely hormone therapy, these pathway groups also discriminate between the two treatment groups.
The situation for the HTPG is more complicated. Basal-like and HER2+ are typically treated with chemotherapy, and the basal-like samples do show a relatively low frequency of mutated HTPG pathways. For basal like patients, therefore, the frequency of HTPG pathways mutated is a good correlate of treatment of choice. We also find that 29 of the 49 Luminal-A patients have virtually all HTPG pathways mutated. Three out of the 9 Luminal-A patients in chemotherapy are among them. This suggests potential utility of the pathway groups, since the standard recommendation for Luminal-A patients is hormone therapy. Of the 40 Luminal-A patients who received hormone therapy, 26 have mutated HTGP pathways, so again there’s reasonable correlation between the frequency of mutated pathway groups and recommended treatment. This is seen strikingly in Figure 3A.
For HER2+, however, there appears to be little if any correlation. Half the HER2+ patients (all of whom are in chemotherapy) have mutations in almost all pathways in HTPG, and half have mutations in only a small percentage of pathways. There also appears to be little or no relation between HTPG pathways and therapy group for Luminal-B patients, nor does the assignment of Luminal-B to therapy group conform to subtype classification (10 in HT and 3 in CT; whereas guidelines would have it roughly reversed). Consequently the classification of Luminal-B sheds no light on HTPG.
Pathway groups stratify subtypes
The Luminal-A pattern provides strong statistical evidence that the population of Luminal-A patients in this study can be split into two distinct groups: 29 patients have almost all 20 pathways from HTPG mutated (577 mutated, 3 not), while 17 patients show relatively infrequent mutations (65 mutated, 275 not). The probability that this pattern is the result of chance is p<10−15. This again suggests that traditional subtypes can be stratified further, and that higher resolution can be identified by pathway groups. Similarly, although the HER2+ pattern does not support correlation with traditional clinical guidelines for therapy, it does suggest two distinct subpopulations.
Discussion
We demonstrated that HT and CT samples are characterized by mutually exclusive sets of mutated pathways, that are strong correlates of subtype-based therapy recommendation. Furthermore, genes in these altered pathways are highly enriched in breast cancer drivers and may provide further guidelines for the personalized therapy. Since all the mutations reported in this paper are from DNA sequenced and annotated prior to surgery or therapy, using tissue from the primary tumor at the initial site of the cancer, the results are not a consequence of mutations that might be induced by therapy.
The discovery that the altered cellular processes that drive transformation in patients having hormone therapy, have a substantially different biology than those that do not, may open an opportunity for the identification of new therapies. Particularly, we found that 100% of the pathways in chemotherapy are TP53 related, while 70% of pathways in hormone therapy are involved in organismal system, especially in immune system (30%) and endocrine system (20%). Both immune (26, 27) and endocrine systems (28–30) can work primarily through estrogen receptors, regulating cells in pathways from these two systems could therefore lead to the successful development of therapeutic concepts in breast cancer. In brief, we hope our identified pathways could be considered as a signature to classify patients for their treatments in the future, as an auxiliary of current protocol, after we could get enough samples to test. Moreover, speaking of each individual therapy option, like hormone therapy for example, beyond targeting estrogen related genes to lower or stop estrogen level, our mutated pathways could also open up many other treatment options by targeting other mutations that up or down-regulate estrogen related genes or initially trigger the cancer.
The mutated genes in our identified pathways tend to be sample specific (Figure 3B). Except for TP53 and PIK3CA, which have the highest mutated rates in chemotherapy (75% in CT and 8% in HT) and hormone therapy (53% in HT and 18% in CT) groups respectively, almost all the other genes are dispersed among samples, even for subtypes that have similar pathway patterns (i.e. have a significantly large number of mutated pathways in common). For example 4 of the HER2+ patients (TCGA-A2-A04W, TCGA-A2-A0D1, TCGA-A2-A0EQ, TCGA-AO-A0JE) and 3 of the Luminal-B patients (TCGA-A2-A0CW, TCGA-A2-A0SW, TCGA-E2-A15K) have all collaborative pathways mutated (3 in CT, 20 in HT); i.e. they have identical mutation patterns. But the mutated genes in these pathways are specific to each patient: there are totally 12 genes mutated in HER2+ (PIK3CA, TP53, ABL2, ATR, NKD2, TXK, XCR1, ADCY8, FLT4, PPP3CA, STAT5B, IRF9), total 15 genes mutated in Luminal-B (PIK3CA, TP53, RPS6KA6, TCF7, FIGF, PAK7, PIK3R1, BCL2L11, CCNB1, FGFR2, FN1, MYH10, NFATC4, PIK3R3, TRIP10). Besides PIK3CA which is mutated in all 7 patients, and TP53 which is mutated in 6 out of 7 patients, all the remaining genes are mutated in only one patient, and no two genes are mutated in the same patient.
The conventional way to determine whether to apply chemo or hormone therapy is according to the breast cancer molecular subtypes, which are induced by gene regulation level of several molecular factors like estrogen receptor, progesterone receptor, and human epidermal growth factor. This strategy has been developed for decades and greatly helped physicians to draw up clinical schemes, which are also evidenced by our identified mutated pathways: different molecular subtypes tend to have distinct mutation patterns, and are approximately concordant with pathways of their recommended therapy groups.
The subtype information including biological process and molecular functions in epigenetic alone, however, may not fully uncover the whole picture. Mutated drivers, including both genes and pathways that initiate cancer development, may supplement this picture. We present in Results that even for the same subtype like Luminal-A, the mutational landscape is diverse between patients in CT and HT. This may advise that current treatment protocol need to be personalized by integrating genetic factors like driver genes and driver pathways.
One limitation of this paper is what we reported are exploratory results only. We discussed summarized clinical information from TCGA with observation and detailed interpretation, but without very strong statistical support due to small portion of available samples in TCGA (around 50 samples for each therapy), and without further verification in new samples due to limited existing clinical sources.
Taken together, our findings suggest that systematic mutation analysis of breast cancer can reveal pathway dysfunction status and mutated gene portraits that may not easily be discovered in transcriptome level. This brings new insights about personalized treatment and targeted agents.
Acknowledgments
This work is supported by National Institutes of Health (R01GM103502-05).
Footnotes
Disclosure of Potential Conflicts of Interest
All authors disclosed no potential conflicts of interest.
Contributor Information
Yang Liu, Email: yliubu@bu.edu.
Zhenjun Hu, Email: zjhu@bu.edu.
Charles DeLisi, Email: charlesdelisi@gmail.com.
References
- 1.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 2.Abe O, Abe R, Enomoto K, Kikuchi K, Koyama H, Masuda H, et al. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687–717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 3.van’t Veer LJ, Dai HY, van de Vijver MJ, He YDD, Hart AAM, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 4.Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn HJ, et al. Strategies for subtypes-dealing with the diversity of breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol. 2011;22(8):1736–47. doi: 10.1093/annonc/mdr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu ZQ, Zhang XS, Zhang SH. Breast tumor subgroups reveal diverse clinical prognostic power. Sci Rep-Uk. 2014:4. doi: 10.1038/srep04002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Abreu FB, Schwartz GN, Wells WA, Tsongalis GJ. Personalized therapy for breast cancer. Clin Genet. 2014;86(1):62–7. doi: 10.1111/cge.12381. [DOI] [PubMed] [Google Scholar]
- 7.Loeb KR, Loeb LA. Significance of multiple mutations in cancer. Carcinogenesis. 2000;21(3):379–85. doi: 10.1093/carcin/21.3.379. [DOI] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 9.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Li M, Cheung YM, Sham PC, Ng MK. SKM-SNP: SNP markers detection method. Journal of biomedical informatics. 2010;43(2):233–9. doi: 10.1016/j.jbi.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Ng M. Shrunken methodology to genome-wide SNPs selection and construction of SNPs networks. BMC systems biology. 2010;4(Suppl 2):S5. doi: 10.1186/1752-0509-4-S2-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Lee YF, Ng MK. SNP and gene networks construction and analysis from classification of copy number variations data. BMC bioinformatics. 2011;12(Suppl 5):S4. doi: 10.1186/1471-2105-12-S5-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu QY, Ye YM, Liu Y, Ng MK. SNP Selection and Classification of Genome-Wide SNP Data Using Stratified Sampling Random Forests. Ieee T Nanobiosci. 2012;11(3):216–27. doi: 10.1109/TNB.2012.2214232. [DOI] [PubMed] [Google Scholar]
- 14.Jia PL, Liu Y, Zhao ZM. Integrative pathway analysis of genome-wide association studies and gene expression data in prostate cancer. BMC systems biology. 2012:6. doi: 10.1186/1752-0509-6-S3-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu ZJ, Chang YC, Wang Y, Huang CL, Liu Y, Tian F, et al. VisANT 4.0: Integrative network platform to connect genes, drugs, diseases and therapies. Nucleic Acids Res. 2013;41(W1):W225–W31. doi: 10.1093/nar/gkt401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bild AH, Yao G, Chang JT, Wang QL, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439(7074):353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 17.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 18.Chin L, Meyerson M, Aldape K, Bigner D, Mikkelsen T, VandenBerg S, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Hu ZJ. Identification of collaborative driver pathways in breast cancer. Bmc Genomics. 2014;15 doi: 10.1186/1471-2164-15-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The Somatic Genomic Landscape of Glioblastoma. Cell. 2013;155(2):462–77. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):E118–U85. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dees ND, Zhang QY, Kandoth C, Wendl MC, Schierding W, Koboldt DC, et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012;22(8):1589–98. doi: 10.1101/gr.134635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Tian F, Hu Z, DeLisi C. Evaluation and integration of cancer gene classifiers: identification and ranking of plausible drivers. Sci Rep. 2015;5:10204. doi: 10.1038/srep10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooke PS, Selvaraj V, Yellayi S. Genistein, estrogen receptors, and the acquired immune response. J Nutr. 2006;136(3):704–8. doi: 10.1093/jn/136.3.704. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt M, Bohm D, von Torne C, Steiner E, Puhl A, Pilch H, et al. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68(13):5405–13. doi: 10.1158/0008-5472.CAN-07-5206. [DOI] [PubMed] [Google Scholar]
- 28.Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, et al. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocr Rev. 2009;30(4):293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith IE, Dowsett M. Drug therapy: Aromatase inhibitors in breast cancer. New Engl J Med. 2003;348(24):2431–42. doi: 10.1056/NEJMra023246. [DOI] [PubMed] [Google Scholar]
- 30.Weigel MT, Ghazoui Z, Dunbier A, Pancholi S, Dowsett M, Martin LA. Preclinical and clinical studies of estrogen deprivation support the PDGF/Abl pathway as a novel therapeutic target for overcoming endocrine resistance in breast cancer. Breast cancer research: BCR. 2012;14(3):R78. doi: 10.1186/bcr3191. [DOI] [PMC free article] [PubMed] [Google Scholar]