

Abstract
PURPOSE
The mechanisms by which gut microbiota contribute to methylmercury metabolism remain unclear. Among a cohort of pregnant mothers, the objectives of our pilot study were to determine 1) associations between gut microbiota and mercury concentrations in biomarkers (stool, hair and cord blood) and 2) the contributions of gut microbial mercury methylation/demethylation to stool methylmercury.
METHODS
Pregnant women (36–39 weeks gestation, n=17) donated hair and stool specimens, and cord blood was collected for a subset (n=7). The diversity of gut microbiota was determined using 16S rRNA gene profiling (n=17). For 6 stool samples with highest/lowest methylmercury concentrations, metagenomic whole genome shotgun sequencing was employed to search for the mercury methylation gene (hgcA), and two mer operon genes involved in methylmercury detoxification (merA and merB).
RESULTS
Seventeen bacterial genera were significantly correlated (increasing or decreasing) with stool methylmercury, stool inorganic mercury, or hair total mercury; however, aside from one genus, there was no overlap between biomarkers. There were no definitive matches for hgcA or merB, while merA was detected at low concentrations in all six samples.
MAJOR CONCLUSIONS
Proportional differences in stool methylmercury were not likely attributed to gut microbiota through methylation/demethylation. Gut microbiota potentially altered methylmercury metabolism using indirect pathways.
Keywords: prenatal, mercury, gut, microbiota, microbiome, metabolism
Graphical Abstract
1. INTRODUCTION1
Microbes modulate the toxicity of mercury (Hg) through methylation of less toxic inorganic Hg(II) (IHg) and demethylation (i.e., detoxification) of methylmercury (MeHg) (Barkay et al 2003; Gilmour et al. 2013; Parks et al. 2013; Smith et al. 2015). The distal gastrointestinal tract is one of the most densely populated ecosystems, with greater than 1011–1012 organisms per mL of luminal content. Thus the gut is potentially an important reservoir for Hg cycling and MeHg metabolism.
Several animal and human studies confirmed gut microbiota modulate the enterohepatic cycling of MeHg. Co-consumption of high-fiber foods, including wheat and fruit, were associated with lower absorption of MeHg into tissues (Passos et al. 2007; Rowland et al. 1984, 1986), presumably due to higher elimination of MeHg. Treatment of animals with antibiotics reduced decomposition of MeHg in the large intestine compared to controls (Seko et al. 1981), and increased the half-time of MeHg elimination (Rowland et al. 1984), implicating gut microbes in MeHg metabolism. Microbial MeHg detoxification involves MerA, the mercuric reductase, and MerB, the organomercurial lyase (Barkay et al., 2003). Both genes encoding these proteins were recovered from human and non-human primate feces, verifying decomposition of Hg species by gut microbiota potentially occurred (Liebert et al. 1997). Tanzanian pregnant women who consumed probiotics daily (i.e., yogurt) had significantly lower blood Hg concentrations compared to controls, matched by age, nutritional status and fish intake (Bisanz et al. 2014). Methylation of IHg by gut microbes is also possible; to date, one commensal methanogen (Methanomassiliicoccus luminyensis) isolated from human feces (Dridi et al. 2012) contained the gene cluster (hgcA and hgcB) required for IHg methylation (Parks et al. 2013). However, the capacity of intestinal bacteria to methylate Hg was observed by some researchers (Rowland et al. 1975), but not others (Zhou et al. 2011). The mechanisms by which gut microbes contribute to MeHg cycling in the human body remain unclear.
This pilot study involves trimester 3 pregnant women because the fetus is the most vulnerable population to the deleterious effects due to MeHg exposure [National Research Council (NRC) 2000], and trimester 3 coincides with the period when brain growth is most rapid. Pregnancy is also associated with changes in gut microbiota composition (Koren et al. 2012; Santacruz et al. 2010), which may alter MeHg metabolism, and hence fetal MeHg exposure. The following hypotheses were investigated: 1) gut microbiota alter the speciation and toxicity of Hg through Hg(II) methylation and/or MeHg demethylation, and 2) gut microbiota affect MeHg bioavailability to the developing fetus by altering Hg speciation.
2. MATERIALS AND METHODS
Recruitment
Between November-December 2013, pregnant women from the Greenville Health System (Greenville, South Carolina, USA) were invited to participate, who were 36–39 weeks pregnant, at least 18 years of age, and in good general health. Nineteen mothers provided informed consent, including 17 mothers who donated both hair and stool samples (89%). Cord blood samples were obtained for a subset (n=7, 37%). Trimester 1 (8–12 weeks gestation) body mass index (BMI) (kg/m2), weight change during pregnancy, ethnicity, and antibiotic treatment were determined from the medical record (Table 1). Adherence to Institute of Medicine (IOM) of the National Academies Guidelines for healthy weight gain during pregnancy (IOM 2009) was determined from trimester 1 BMI and weight change at recruitment. Protocols were approved by the Institutional Review Boards at Greenville Health System and the University of South Carolina.
Table 1.
Demographic data for 17 participants.
| Average ± 1 SD (range) | |
|---|---|
| Gestation (weeks) | 37 ± 0.94 (36 – 39) |
| Trimester 1 BMI (kg/m2) | 29 ± 6.7 (16 – 40) |
| Weight gain (kg) | 14 ± 7.8 (1.4 – 33) |
| n (%) | |
| Ethnicity | |
| White | 7 (41) |
| Black | 7 (41) |
| Hispanic | 3 (18) |
| Weight class | |
| Underweight | 1 (5.9) |
| Normal | 3 (18) |
| Overweight | 5 (29) |
| Obese | 8 (47) |
| IOM Guidelines | |
| Below | 2 (12) |
| Within | 6 (35) |
| Above | 9 (53) |
| Antibiotic treatment previous 3 months | |
| No | 10 (59) |
| Yes | 6 (35) |
| Uncertain | 1 (5.9) |
Institute of Medicine (IOM), trimester 1 body mass index (BMI)
Biomarker collection and preservation
A hair sample was collected from the occipital region, which was tied with a string and stored in a plastic bag at room temperature. Mothers were given a stool collection kit including sterile collection containers (Thermo Fisher 02-544-208) and detailed instructions adapted from the Human Microbiome Project protocols [Human Microbiome Project (HMP) 2010]. Participants shipped stool samples overnight to the University of South Carolina. Upon receipt, stool samples were aliquoted using sterile microspatulas (Corning 3012) into 2 mL sterile cryovials (Thermo Fisher 50-476-502), and then a separate sample was aliquoted using acid-washed utensils into pre-weighed acid-washed 50 mL polypropylene vials. Vials were stored frozen (−80°C) until analysis. Whole blood was collected from the umbilical cord into vials (Becton Dickinson, K2EDTA, Royal Blue) and frozen at −20°C and then at −80°C.
Hg analyses for hair
Hair samples corresponding to trimester 3 were analyzed, based on the monthly growth rate of hair (Loussouarn et al. 2005), assuming a 10-day lag between MeHg intake and absorption into the hair shaft (Cernichiari et al. 1995). Hair samples were washed in 1% (v/v) 2-mercaptoethanol, shaken for 1 hour, then triple rinsed with double-distilled (DDI)-H20, and air-dried overnight in a Class II Biosafety Hood (Baker Company, Sanford, ME, USA). Hair total Hg (THg) (~10 mg) was analyzed with a portable Hg vapor analyzer (Lumex, Model RA-915+/PYRO-915+, St. Petersburg, Russia) using thermal decomposition and atomic absorption spectrophotometry [U.S. Environmental Protection Agency (USEPA) Method 7473] (USEPA 2007). There was insufficient hair for MeHg analysis.
Hg analyses for stools
For THg, stool specimens (~0.6 g) were digested in 10 mL of freshly prepared 7:3 nitric:sulfuric acid, samples were gently refluxed for 3 hours in a water bath (85°C), then oxidized with 0.2 N bromine monochloride. Excess oxidant was neutralized with hydroxylamine hydrochloride, and Hg was further reduced with stannous chloride, converting IHg to Hg(0). Quantification was by gold amalgamation followed by cold vapor atomic fluorescence spectrometry (CVAFS) (Brooks Rand Model III, Seattle, WA, USA) (EPA Method 1631) (USEPA 2002). For MeHg, stool samples (~0.6 g) were leached in 1.5 ml 1 M copper sulfate, 7.5 mL 25% nitric acid, and 10 mL dichloromethane (CH2Cl2) and MeHg was back-extracted into DDI-H2O (Liang et al. 2004). MeHg extracts were analyzed following EPA Method 1630 (USEPA 2001) using gas chromatography (GC)-CVAFS (Brooks Rand Model III). Stool THg and MeHg concentrations were reported in dry weight, which was determined after drying a subsample at 105°C overnight.
Hg analyses for cord blood
For THg, ~0.7 g of blood were analyzed using EPA Method 7473 (22) (as described above). For blood MeHg, the same steps for stool MeHg were followed, except leaching procedures differed: ~0.6 g of blood were dried in a 70°C oven overnight, then leached in 2 mL 25% potassium hydroxide:methanol (w/v) for 3 hours in a 75°C oven, then 10 mL CH2Cl2 and 2 mL hydrochloric acid were added (Liang et al. 2000). MeHg was quantified by EPA Method 1630 (EPA 2001) using GC-CVAFS (as described above).
Hg quality assurance/quality control (QA/QC)
QA/QC parameters are summarized in Table S1. Matrix-specific detection levels were based on the average mass of sample analyzed (hair: 0.01 g, stool: 0.7 g, and blood: 0.6 g) and the region of the calibration curve where there was a significant change in sensitivity, including hair (THg: 9.5 ng/g), stool (THg: 0.033 ng/g, MeHg: 0.0017 ng/g), and blood (THg: 0.14 μg/l, MeHg: 0.0017 μg/l). Two values were below the detection level, and half the detection level was imputed.
Gut microbiota and 16S rRNA gene profiling
Frozen stool samples (−80°C) were shipped overnight to the Alkek Center for Metagenomics and Microbiome Research (CMMR) at Baylor College of Medicine. Microbial genomic DNA was extracted using the PowerSoil DNA Isolation Kit according to the manufacturer’s instructions (MoBio Laboratories, CA, USA), and the concentration and purity of the extracted DNA were evaluated through gel electrophoresis and PicoGreen assays (Invitrogen, NY, USA).
For all stool samples (n=17), the 16S rDNA V4 region was amplified by PCR using bacteria/archaeal primers 515F and 806R (see Table S2). Sequencing was performed on the MiSeq platform (Illumina, CA, USA) using the 2 × 250 bp paired-end protocol, which yielded pair-end reads that almost completely overlapped. The primers contained adapters for MiSeq sequencing and dual-index barcodes so that the PCR products were pooled and sequenced directly (Caporaso et al. 2012), targeting at least 15,000 reads per sample. The 16S rRNA gene pipeline data incorporates phylogenetic and alignment-based approaches to maximize data resolution. The read pairs were demultiplexed based on unique molecular barcodes, and merged using USEARCH v7.0.1001 (Edgar 2010). The CMMR pipeline for 16S analysis leverages the QIIME (Quantitative Insights Into Microbial Ecology) software package (Caporaso et al. 2010), as well as custom analytic packages. 16S rRNA gene sequences were classified into Operational Taxonomic Units (OTUs) at a similarity cutoff value of 97% using the UPARSE pipeline in QIIME and the SILVA Database (Quast et al. 2013), and abundances were recovered by mapping the demultiplexed reads to the UPARSE OTUs database. An OTU table was constructed for taxonomic summaries and calculate alpha- and beta-diversity (Lozupone and Knight 2005).
Whole genome shotgun (WGS) sequencing
For 6 (of 17) samples, metagenomic libraries were subjected to WGS (Illumina), including 3 samples with high (≥ median) stool MeHg (# 134, 146, and 157) and 3 with low (< median) stool MeHg (# 101, 118, and 163). To search Hg methylation genes, nucleotide sequence reads were converted into amino acid sequences by six-frame translation. Although both HgcA and HgcB are considered essential for microbial Hg methylation (Parks et al. 2013; Smith et al. 2015), most organisms possessing HgcA also contain HgcB. Sequences of 77 HgcA homologs from bacteria and archaea with complete genome sequences (http://www.esd.ornl.gov/programs/rsfa/data.shtml) were obtained from GenBank. A 30-amino-acid-stretch encompassing the highly conserved ‘cap helix’ region of HgcA, containing the conserved motif TxG[IV]N[VI]WCA[AGS][GA][KE] (Parks et al 2013; Smith et al. 2015) was used to search for the presence of HgcA in the metagenomic libraries. For MerA and MerB, a reference database was created using the found sequences in the KEGG (Kyoto Encyclopedia of Genes and Genomes) database (Kanehisa et al. 2014). Metagenomic libraries were searched using USearch’s usearch_local method with target coverage set to 0.8 (80% coverage) and ID threshold of 0.5 (50%).
Statistics
Alpha- and beta-diversity were used to examine associations between gut microbiota and dichotomized (high: ≥median, low: < median) Hg concentrations in biomarkers (stool MeHg, stool IHg, and hair THg). Alpha-diversity (within sample diversity) was measured with the Chao1 index (richness) (Chao 1984), the Shannon diversity index, and the observed number of sequences per sample. Beta-diversity (between sample diversity), defined as a measure of the evolutionary distance between gut microbiotas, was evaluated with the Unifrac distance metric (Lozupone and Knight, 2005).
Bivariate associations between Hg biomarkers (continuous, not dichotomized) and phylum- and genus-level gut microbiota abundances were assessed using Spearman’s and Pearson’s correlation. Spearman’s correlation was calculated with the untransformed variables. For Pearson’s correlation, a log10-transformation was applied to right-skewed variables for gut microbiota abundances after increasing all values by 0.001% due to the number of 0’s; 0.001% was considered a minimum detection level. For one genus (unclassified 00r39 Peptostreptococcaceae), 0.001% was disconnected from the remaining observations, and therefore this value was not imputed for 0. Bivariate associations were also determined between categorical data from the medical record and Hg biomarkers (hair, stool and cord blood), and phylum- and genus-level gut microbiota abundances using Wilcoxon rank-sum test or Student’s 2-tailed t-test. Multiple test comparisons were applied using the Benjamini-Hochberg False Discovery Rate (FDR) procedure (Yekutieli and Benjamini, 1999), using a q-value of 0.20. Statistical analyses were completed using the R-platform or Stata (Version 9.2, College Station, TX, USA).
Accession numbers
This study has been deposited to the U.S. National Institutes of Health Database for Genotypes and Phenotypes (dbGaP) and the data are available through accession number phs000970.v1.p1.
3. RESULTS
Demographics
Demographic data and body measurements are included in Table 1. Trimester 1 BMI averaged 29 kg/m2 (range 16–40 kg/m2), including 76% of pregnant mothers classified as overweight or obese (i.e., BMI ≥ 25 kg/m2). More than half the mothers exceeded IOM guidelines for healthy weight gain during pregnancy (IOM 2009). Gestational weight gain was similar for obese and non-obese mothers (t-test, p=0.80).
Biomarker Hg concentrations
Hair and cord blood THg and/or MeHg indicated low dietary intake of MeHg through fish consumption for most mothers. Hair THg concentrations averaged 57 ng/g (Table 2), which was 5–10 times lower compared to cohorts of U.S. pregnant women (average: 290–550 ng/g) (Oken et al. 2005; Xue et al. 2007). Cord blood THg averaged 2.9 times lower compared to blood Hg for U.S. adults (0.99 μg/L, n=10,673) (Nielsen et al. 2014), while the geometric mean for blood Hg was 1.9 times lower than Rhode Island mothers (this study: 0.28 μg/L; Rhode Island: 0.52 μg/L, n=538, from King et al. 2012).
Table 2.
Summary statistics for total mercury, methylmercury and percent methylmercury (of total mercury) in biomarkers, including maternal hair, maternal stool, and cord blood.
| Parameter | Hair (ng/g) (n=17) | Stool (ng/g) (n=17) | Cord Blood (μg/L) (n=7) | |||
|---|---|---|---|---|---|---|
| Average (range) | Median | Average (range) | Median | Average (range) | Median | |
| THg | 57 (BDL-230) | 27 | 150 (2.1–810) | 30 | 0.34 (BDL-0.67) | 0.35 |
| MeHg | NA | NA | 0.097 (0.0025–0.39) | 0.060 | 0.23 (0.061–0.73) | 0.18 |
| %MeHg (of THg) | NA | NA | 0.78 (0.0058–5.8) | 0.12 | 69 (25–124) | 50 |
Below detection level (BDL), methylmercury (MeHg), not applicable (NA), total mercury (THg)
Biomarker Hg correlations
Maternal hair THg and cord blood MeHg (or THg) are established biomarkers for fetal MeHg exposure (Cernichiari et al 1995; NRC 2000). Hair THg and cord blood MeHg concentrations were highly correlated when log10-transformed (Pearson’s rho=0.91, p<0.01, n=7) (Figure 1). Stool MeHg was more positively correlated with cord blood MeHg (Pearson’s rho=0.58, p=0.18, n=7) than hair THg (Pearson’s rho=0.16, p=0.54, n=17) (when log10-transformed); however, both were non-significant (Figure 1). Results did not differ when the sample size was limited to mothers with cord blood data (Pearson’s rho=0.24, p=0.61, n=7). These data suggested stool MeHg did not contribute significantly to fetal MeHg exposure.
Figure 1.
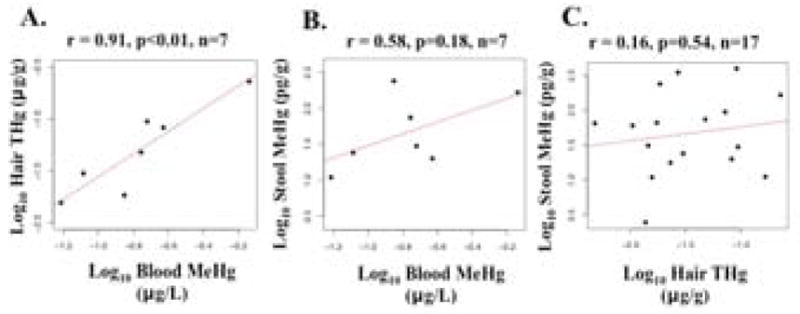
Bivariate scatterplots relating total mercury (THg) or methylmercury (MeHg) concentrations between a) hair-cord blood, b) stool-cord blood, and c) stool-hair, including Pearson’s correlation (rho).
Medical record data and Hg biomarkers
There were no significant associations between Hg biomarkers [hair THg, stool MeHg, and stool IHg (IHg=THg-MeHg)], and categorical data from the medical record, including 1) trimester 1 weight status (obese/non-obese), 2) weight gain during pregnancy (excessive weight gain versus below or within IOM guidelines), and 3) antibiotic treatment (yes versus no or uncertain) (Wilcoxon rank-sum test, p = 0.15–0.92, n=17; t-test, p = 0.17–0.90, n=17). The latter results were surprising because antibiotic treatment altered the enterohepatic cycling of Hg in mice (Rowland et al. 1984; Seko et al. 1981). For both animal studies, MeHg-chloride was administered near the same time as antibiotics (seven days after antibiotics started, Rowland et al., 1984; two days before antibiotics started, Seko et al., 1981). In the present study, antibiotic treatment occurred within the previous three months and possibly did not coincide with dietary MeHg intake.
16S rRNA gene profiling of gut microbes
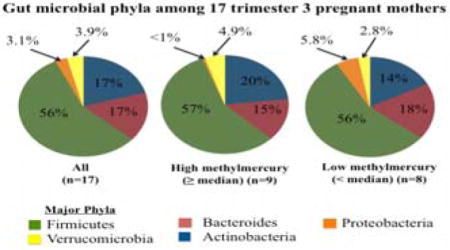
The total number of mapped reads for 17 stool samples was 356,456, averaging 20,968 reads per sample; analyses were performed on an equal number of reads per sample (15,928). 16S rRNA reads were assigned to 7 phyla, including the following 5 phyla representing 98 ± 0.94 % of gut microbes. On average (± 1 SD), Firmicutes comprised 56% ± 17%, followed by Bacteroidetes (17% ± 17%), Actinobacteria (17% ± 19%), Verrucomicrobia (3.9% ± 7.2%), and Proteobacteria (3.1% ± 6.9%). The 7 phyla were represented by 81 genera (averaging 52 ± 7.2 genera per mother), including 21 genera with abundances >1% (Figure S1). The most abundant genera were Bifidobacterium (15% ± 18%), Bacteroides (11% ± 13%), Alistipes (4.1 ± 5.3), Subdoligranulum (5.7% ± 4.6%), Blautia (5.5% ± 4.3) unclassified Lachnospiraceae spp. (4.8% ± 3.9%), and Akkermansia spp. (3.9% ± 7.2%). The latter was the sole representative of the Verrucomicrobia phylum, and therefore results/discussion for Verrucomicrobia were omitted.
Indices for microbial richness (i.e., alpha-diversity) did not differ significantly for gut microbiotas with high (≥ median) and low (< median) concentrations of hair THg and stool IHg (p=0.44–0.85) (Figure S2). Microbial richness estimated by the Chao1 index was significantly higher for mothers with high stool MeHg concentrations (p<0.05), while there were no significant differences using other indices (p=0.29–0.50). Chao1 is more sensitive to rare species, whereas the Shannon index estimates the uniformity of sequences (Chao 1984), suggesting differences between high/low stool MeHg are due to the presence/absence of rare gut microbial species. Beta-diversity patterns were visualized using Principal Coordinates Analysis (PCoA) (Figure S3). For all three outcomes (stool MeHg, stool IHg, and hair THg) beta-diversity did not differ between high/low groups using unweighted and weighted Unifrac distance metrics (p=0.11–0.64).
Medical record data and gut microbiota
Like Hg biomarkers, there were no significant associations between gut microbiota phyla and categorical data from the medical record, including 1) trimester 1 weight status (obese/non-obese), 2) weight gain during pregnancy (excessive weight gain versus below or within IOM guidelines), and 3) antibiotic treatment (yes versus no or uncertain) (Wilcoxon rank-sum test, p=0.07–0.92). Among gut microbiota genera, abundance of Blautia (phylum: Firmicutes) was significantly higher for mothers who gained excessed weight (Wilcoxon rank-sum, p<0.05), and unclassified members of the genus Ruminococcaceae (phylum: Firmicutes) were significantly higher for mothers prescribed antibiotics (Wilcoxon rank-sum, p<0.05); however all correlations were not significant at a FDR of 20%. The abundance of Akkermansia spp. was higher (but not significantly) for mothers that gained excessive weight compared to normal weight (Wilcoxon rank-sum, p=0.09). This trend differed from Santacruz et al. (2010), who reported significantly lower abundance of A. muciniphilia among Spanish pregnant women that gained excessive weight (n=16) compared to normal weight (n=34).
Bacterial diversity and Hg biomarkers
For continuous Hg biomarkers (stool MeHg, stool IHg, and hair THg), significant associations were observed between stool MeHg and Proteobacteria using Pearson’s correlation (rho = −0.61, p<0.01), but not Spearman’s correlation (rho = −0.45, respectively, p=0.07). However, this inverse association was driven by a single observation; when this observation was removed Pearson’s correlation was no longer significant (p=0.13). Seventeen genera (of 81) were significantly correlated (Spearman’s and/or Pearson’s) with stool MeHg, stool IHg or hair THg (p<0.05), including three genera that were significantly correlated at a FDR of 20% (Figure 2, Table S3). Two genera were positively associated with stool MeHg (both were unclassified members of the Ruminococcaceae genus), and one genus was inversely associated with stool IHg (Moryella). There was no overlap between taxonomic groups significantly correlated with stool MeHg, stool IHg or hair THg, aside from one genus (Subdoligranulum) (Figure 2, Table S3).
Figure 2.
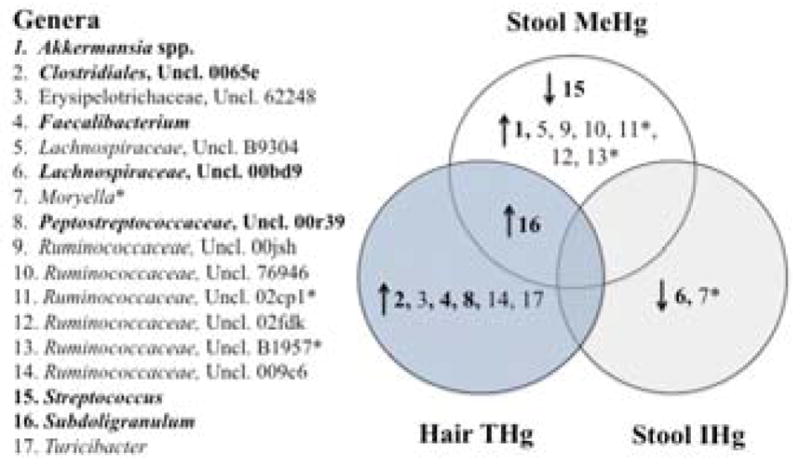
Venn diagram including gut microbial genera that were significantly correlated with stool methylmercury (MeHg), hair total mercury (THg), or stool inorganic mercury(II) (IHg) using Spearman’s and/or Pearson’s rho (n=17) (p<0.05). (*) indicates significance at a False Discovery Rate of 20%, arrows represent the direction of correlation, and bolded numbers indicate relative abundance was >1% (see Table S3).
Metagenomic WGS results
Among 6 stool samples, including 3 with low MeHg and 3 with high MeHg, there were no definitive matches for HgcA. Although some of the matches overall showed a high level of sequence identity to known HgcA sequences, significant sequence variability was observed near the strictly conserved cysteine in the ‘cap-helix’ region, which was expected to be the least variable region (Parks et al. 2013; Smith et al. 2015). For MerA, low abundant hits (<0.001%) were observed across all six samples with good identity scores, while MerB produced no hits.
4. DISCUSSION
Stool MeHg and dietary MeHg intake
Results suggested stool MeHg concentrations did not likely reflect biotransformation by gut microbiota through Hg(II) methylation or demethylation, thus rejecting hypothesis 1. To date, one commensal methanogen (M. luminyensis) isolated from human feces contained the gene cluster required for Hg(II) methylation (Dridi et al. 2012; Parks et al. 2013). In this study, the proportional abundance of this genus was less than 0.001%, suggesting: 1) the abundance of M. luminyensis was too low to be detected, or 2) dietary MeHg intake contributed to stool MeHg. For 6 samples, including 3 with the highest/lowest stool MeHg content, both hgcA and merB genes were not present. Microbial methylation/demethylation did not likely contribute to the net stool MeHg concentrations, supporting the second assumption. Alternatively, MeHg was demethylated abiotically, for example, via phagocytosis (Suda et al. 1992). Gut microbiota possibly demethylated MeHg using a different metabolic pathway (i.e., oxidative demethylation), which was reported in anaerobic freshwater sediment (Barkay et al. 2003; Oremland et al. 1991); however no specific genes have been identified for this process.
Ishihara (2000) published the only other study to date (to the best of our knowledge) including IHg and MeHg concentrations for stool specimens from 4 Japanese men, who ingested fish regularly (unlike most mothers in this study). Stool MeHg averaged 7.0 ng MeHg/g wet weight and stool %MeHg (of THg) averaged 17%, while hair THg averaged 4800 ng/g (Ishihara 2000), which were 72, 22, and 84 times higher than corresponding values for this study (Table 2). The magnitude difference for stool MeHg was likely higher (i.e., 200–300, not 72), because Ishihara (2000) recorded stool Hg concentrations in wet weight (median wet:dry ratio for this study = 3.50).
The distal gut is considered the primary site where MeHg is demethylated to IHg, which is less likely to be absorbed through the intestinal wall and thus excreted, reducing human MeHg body burden (Clarkson and Magos 2006). Results from this study and Ishihara (2000) indicated not all MeHg was demethylated before excretion. Combined with the analysis of hair/blood/stool (Figure 1), stool MeHg did not correlate significantly with fetal MeHg exposure, i.e., MeHg was not bioavailable.
Hg biomarkers and gut microbial taxa
Seventeen genera were significantly associated with Hg biomarkers (stool MeHg, stool IHg, and hair THg), and aside from one genus (Subdoligranulum), there was no overlap between biomarkers (Figure 2). A majority of genera were not significantly associated with Hg biomarkers at a FDR of 20%; however these modest associations suggested other pathways were possibly more important for MeHg metabolism and exposure. For some genera their functions are well known, which are discussed below.
Akkermansia spp. were positively correlated with stool MeHg (when log10-transformed) (Pearson’s rho=0.50, p<0.05). To date, no species in this phylum (Verrucomicrobia) have been identified, which contained the gene cluster required to methylate IHg (i.e., hgcA and hgcB) (Parks et al. 2013). The intestinal tract is covered by a mucus layer, which insulates gut microbes from host tissues and protects the epithelium from pathogenic microorganisms, as well as toxins and acids (Derrien et al. 2004). A. muciniphila is the dominant human bacterium that resides within the mucus layer and degrades mucin, providing usable energy to non-mucolytic bacteria (Derrien et al. 2004). Enrichment of A. muciniphila is associated with stronger gut barrier function (Png et al. 2010), and is considered a biomarker for a healthy intestine (Belzer and de Vos 2012).
We hypothesize that gut microbiota contributed indirectly to MeHg metabolism through improved gut barrier function, which prevented re-absorption of MeHg through the intestinal epithelial layer and concentrated MeHg in the stool. Using an in vitro model, Vázquez et al. (2013) reported the mucus layer represented a barrier to transport of MeHg and inorganic Hg(II), trapping 70% and 40%, respectively, supporting an association between intestinal permeability and Hg absorption into systemic circulation. Higher absorption of other metals (cadmium and lead) was reported for germfree mice compared to controls, which was attributed to differences in the gut barrier function and expression of host-genes such as metallothioneins (Breton et al. 2013). Therefore, gut microbiota may reduce MeHg bioavailability and fetal exposure through indirect pathways (i.e., intestinal permeability), as well as direct pathways (i.e., methylation/demethylation).
Other genera were significantly associated with stool MeHg (positive: Subdoligranulum, negative: Streptococcus spp.). Streptococcus spp. is one of the most dominant microbes in breast milk, and some Streptococcus spp. are associated with greater pathogen resistance among preterm neonates (Martín et al. 2004). Both genera were previously reported in maternal stool and breast milk, and the authors suggested the maternal gut was a source of microbiota to breast milk (Jost et al. 2014). From this study, both positive and inverse associations indicate dietary MeHg intake may impact maternal stool gut microbiota, and possibly breast milk microbiota composition, which should be further investigated.
5. CONCLUSIONS
Methylation of IHg and reductive detoxification of MeHg mediated by the mer operon are two known pathways that microbes use to alter Hg cycling (Barkay et al. 2003; Gilmour et al. 2013; Liebert et al. 1997; Parks et al. 2013; Smith et al. 2015). It is thought that gut microbes demethylate MeHg and reduce bioavailability to cross the intestine into systemic circulation (Clarkson and Magos 2006). Although this was a small data set, results from this pilot study suggested gut microbes possibly reduced MeHg bioavailability and exposure by indirect pathways. This conclusion is supported in part by an absence of hgcA and merB in 6 stool samples with highest/lowest stool MeHg concentrations, and in part by associations between Hg biomarkers and gut microbial taxa with known functions (e.g., Akkermansia spp.). Future research should consider both direct and indirect pathways by which gut microbiota affect MeHg metabolism and exposure. Direct pathways include microbial methylation/demethylation and indirect pathways include changes in the gut barrier function.
Findings from this analysis are limited. A food frequency questionnaire was not administered, and dietary MeHg intake was inferred from Hg biomarkers. In addition, diet profoundly affects the microbiome (Wu et al. 2011), and observed associations between Hg biomarkers and gut microbiota taxa may be confounded by diet. Dietary MeHg intake was low for most mothers, and therefore results were not necessarily applicable to populations ingesting more fish. Most mothers were overweight or obese, which possibly affected MeHg metabolism (Rothenberg et al. 2015). However, aside from one genus (Blautia), there were no significant associations between obesity and gut microbiota, and there were no genera significant at the FDR of 20%. Stool specimens do not accurately reflect microbial activity in proximal regions of the gastrointestinal tract (Eckburg et al. 2005), where IHg methylation and/or demethylation possibly occurred, and may not represent the mucosa-associated bacteria (Png et al. 2010). This is a cross-sectional study, which precludes us from making any inferences about direction or causality. Lastly, this study was limited to a few weeks in late-stage pregnancy and may only be suggestive of windows of susceptibility to toxic insult from MeHg for certain endpoints, while missing susceptibility windows for other endpoints.
In spite of these limitations, profiles of gut microbes differed dramatically between Hg biomarkers, and their roles in the transformation of MeHg and/or reducing MeHg exposure should be elucidated.
Supplementary Material
Highlights.
Mercury methylation/demethylation did not likely contribute to stool methylmercury.
Maternal stool mercury was not correlated with maternal hair or cord blood mercury.
Seventeen bacterial genera were correlated with mercury in maternal stool or hair.
For these correlations, gut microbiota functions (utilized or affected) are unknown.
Acknowledgments
We wish to acknowledge all the anonymous reviewers, whose comments greatly improved this manuscript. The authors thank Chuan Hong and Si Chen for laboratory assistance, and Susan Korrick for insightful discussions. The authors are grateful to Lee Higdon and Allison Moore at Greenville Health System, as well as Tulin Ayvaz, Ginger A. Metcalf, Donna M. Muzny, and Richard A. Gibbs at the Human Genome Sequencing Center at Baylor College of Medicine, for their contributions.
This research was supported in part by grants to S.E. Rothenberg from the U.S. National Institute Of Environmental Health Sciences (Award: R15 ES022409), the U.S. National Institute of Health Loan Repayment Program (Awards: L30 ES023165), and the University of South Carolina Department of Environmental Health Sciences. This work was also supported in part by the U.S. Department of Energy Office of Science, Biological and Environmental Research, Subsurface Biogeochemical Research Program through grant DE SC0006809 to the Oak Ridge National Laboratory (ORNL) Mercury Scientific Focus Area. ORNL is managed by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. By accepting the article for publication, the publisher acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The U.S. Department of Energy will provide public access to these results of federally sponsored research in accordance with the Public Access Plan (http://energy.gov/downloads/doe-public-access-plan). The content is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. National Institutes of Health or the U.S. Department of Energy.
Footnotes
Abbreviations: BMI, body mass index; CH2Cl2, dichloromethane; CMMR, Alkek Center for Metagenomics and Microbiome Research; CVAFS, cold vapor atomic fluorescence spectrometry; DDI-H2O, double-distilled water; FDR, False Discovery Rate; GC, gas chromatography; Hg, mercury; HMP, Human Microbiome Project; IHg, inorganic mercury(II); IOM, Institute of Medicine of the National Academies; KEGG, Kyoto Encyclopedia of Genes and Genomes; MeHg, methylmercury; NRC, National Research Council; ORNL, Oak Ridge National Laboratory; OTU, Operational Taxonomic Units; PCoA, Principal Coordinates Analysis; QA/QC, Quality Assurance/Quality Control; QIIME, Quantitative Insights Into Microbial Ecology; THg, total mercury; USEPA, U.S. Environmental Protection Agency; WGS, whole genome shotgun
The authors have no actual or potential conflicts of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barkay T, Miller SM, Summers AO. Bacterial resistance from atoms to ecosystems. FEMS Microbiol Ecol. 2003;27:355–384. doi: 10.1016/S0168-6445(03)00046-9. [DOI] [PubMed] [Google Scholar]
- Belzer C, de Vos WM. Microbes inside- from diversity to function: the case of Akkermansia. ISME J. 2012;6:1449–1458. doi: 10.1038/ismej.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisanz JE, Enos MK, Mwanga JR, Changalucha J, Burton JP, Gloor GB, Reid G. Randomized open-label pilot study of the influence of probiotics and the gut microbiome on toxic metal levels in Tanzanian pregnant women and school children. mBio. 2014;5 doi: 10.1128/mBio.01580-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton J, Daniel C, Dewulf J, Pothion S, Froux N, Sauty M, Thomas P, Pot B, Foligné B. Gut microbiota limits heavy metals burden caused by chronic oral exposure. Toxicol Lett. 2013;222:132–138. doi: 10.1016/j.toxlet.2013.07.021. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knight D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernichiari E, Brewer R, Myers GJ, Marsh DO, Lapham LW, Cox C, Shamlaye CF, Berlin M, Davidson PW, Clarkson TW. Monitoring methylmercury during pregnancy: maternal hair predicts fetal brain exposure. Neurotoxicology. 1995;16:705–710. [PubMed] [Google Scholar]
- Chao A. Nonparametric-estimation of the number of classes in a population. Scand J Stat. 1984;11:265–270. [Google Scholar]
- Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Crit Rev Toxicol. 2006;36:609–662. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermania muciniphila gen. nov., sp. Nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54:1469–1476. doi: 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- Dridi B, Fardeau M-L, Ollivier B, Raoult D, Drancourt M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int J Syst Evol Microbiol. 2012;62:1902–1907. doi: 10.1099/ijs.0.033712-0. [DOI] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Gilmour CC, Podar M, Bullock AL, Graham AM, Brown SD, Somenahally AC, Johs A, Hurt RA, Jr, Bailey KL, Elias DA. Mercury methylation by novel microorganisms from new environments. Environ Sci Technol. 2013;47:11810–11820. doi: 10.1021/es403075t. [DOI] [PubMed] [Google Scholar]
- Human Microbiome Project (HMP) [accessed 5 September 2015];Manual of Procedures, Core Microbiome Sampling, Protocol A, HMP Protocol #07-001, Version 11.0. 2010 Mar 29; 2010. Available: http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/GetPdf.cgi?id=phd002854.2.
- Institute of Medicine of the National Academies (IOM) [accessed 5 September 2015];Weight gain during pregnancy: reexamining the guidelines. 2009 Available: http://www.iom.edu/en/Reports/2009/Weight-Gain-During-Pregnancy-Reexamining-the-Guidelines.aspx. [PubMed]
- Ishihara N. Excretion of methyl mercury in human feces. Arch Environ Health. 2000;55:44–47. doi: 10.1080/00039890009603384. [DOI] [PubMed] [Google Scholar]
- Jost T, Lacroix C, Braegger CP, Rochat F, Chassard C. Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding. Environ Microbiol. 2014;16:2891–2904. doi: 10.1111/1462-2920.12238. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King E, Shih G, Ratnapradipa D, Quilliam DN, Morton J, Magee SR. Mercury, lead, and cadmium in umbilical cord blood. J Environ Health. 2012;75:38–43. [PubMed] [Google Scholar]
- Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Backhed HK, Gonzalez A, Werner JJ, Angenent LT, Knight R, Backhed F, Isolauri E, Salminen S, Ley RE. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150:470–480. doi: 10.1016/j.cell.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Evens C, Lazoff S, Woods JS, Cernichiari E, Horvat M, Martin MD, DeRouen T. Determination of methyl mercury in whole blood by ethylation-GC-CVAFS after alkaline digestion-solvent extraction. J Anal Toxicol. 2000;24:328–332. doi: 10.1093/jat/24.5.328. [DOI] [PubMed] [Google Scholar]
- Liang L, Horvat M, Feng X, Shang L, Li H, Pang P. Re-evaluation of distillation and comparison with HNO3 leaching/solvent extraction for isolation of methylmercury compounds from sediment/soil samples. Appl Organomet Chem. 2004;18:264–270. [Google Scholar]
- Liebert CA, Wireman J, Smith T, Summers AO. Phylogeny of mercury resistance (mer) operons of gram-negative bacteria isolated from the fecal flora of primates. Appl Environ Microbiol. 1997;63:1066–1076. doi: 10.1128/aem.63.3.1066-1076.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loussouarn G, El Rawadi C, Genain G. Diversity of hair growth profiles. Intl J Dermatol. 2005;44(Suppl):6–9. doi: 10.1111/j.1365-4632.2005.02800.x. [DOI] [PubMed] [Google Scholar]
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín R, Langa S, Reviriego C, Jiménez E, Marín ML, Olivares M, Boza J, Jimenez J, Fernández L, Xaus J, Rodríguez JM. The commensal microflora of human milk: new perspectives for food bacteriotherapy and probiotics. Trends Food Sci Tech. 2004;15:121–127. [Google Scholar]
- National Research Council (NRC) Toxicological Effects of Methylmercury. National Academy Press; Washington. D.C: 2000. [last accessed 5 September 2015]. Available: http://www.nap.edu/openbook.php?isbn=0309071402. [Google Scholar]
- Nielsen AJ, Kit BK, Aoki Y, Ogden CL. Seafood consumption and blood mercury concentrations in adults aged ≥ 20 y2007–2010. Am J Clin Nutr. 2014;99:1066–1070. doi: 10.3945/ajcn.113.077081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oken E, Wright RO, Kleinman KP, Bellinger D, Amarasiriwardena CJ, Hu H, Rich-Edwards JW, Gillman MW. Maternal fish consumption, hair mercury, and infant cognition in a U.S. cohort. Environ Health Perspect. 2005;113:1376–1380. doi: 10.1289/ehp.8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oremland RS, Culbertson CW, Winfrey MR. Methylmercury decomposition in sediments and bacterial cultures: involvement of methanogens and sulfate reducers in oxidative demethylation. Appl Environ Microbiol. 1991;57:130–137. doi: 10.1128/aem.57.1.130-137.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks JM, Johs A, Podar M, Bridou R, Hurt RA, Smith SD, Tomanicek SJ, Qian Y, Brown SD, Brandt CC, Palumbo AV, Smith JC, Wall JD, Elias DA, Liang L. The genetic basis for bacterial mercury methylation. Science. 2013;339:1332–1335. doi: 10.1126/science.1230667. [DOI] [PubMed] [Google Scholar]
- Passos CJ, Mergler D, Fillion M, Lemire M, Mertens F, Guimaraes JRD, Philibert A. Epidemiologic confirmation that fruit consumption influences mercury exposure in riparian communities in the Brazilian Amazon. Environ Res. 2007;105:183–193. doi: 10.1016/j.envres.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Png WC, Linden SK, Gilshenan KS, Zoentendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin TH. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Amer J Gastroenterol. 2010;105:2420–2428. doi: 10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(Database issue):D590–596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg SE, Korrick SA, Fayad R. The influence of obesity on blood mercury levels for U.S. non-pregnant adults and children: NHANES 2007–2010. Environ Res. 2015;138:173–180. doi: 10.1016/j.envres.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland IR, Grasso P, Davies MJ. The methylation of mercuric chloride by human intestinal bacteria. Experientia. 1975;31:1064–1065. doi: 10.1007/BF02326961. [DOI] [PubMed] [Google Scholar]
- Rowland IR, Robinson RD, Doherty RA. Effects of diet on mercury metabolism and excretion in mice given methylmercury: role of gut flora. Arch Environ Health. 1984;39:401–408. doi: 10.1080/00039896.1984.10545872. [DOI] [PubMed] [Google Scholar]
- Rowland IR, Mallett AK, Flynn J, Hargreaves RJ. The effect of various dietary fibres on tissue concentration and chemical form of mercury after methylmercury exposure in mice. Arch Toxicol. 1986;59:94–98. doi: 10.1007/BF00286730. [DOI] [PubMed] [Google Scholar]
- Santacruz A, Collado MC, Garcia-Valdes L, Segura MT, Martin-Lagos JA, Anjos T, Marti-Romero M, Lopez RM, Florido J, Campoy C, Sanz Y. Gut mcirobiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Brit J Nutr. 2010;104:83–92. doi: 10.1017/S0007114510000176. [DOI] [PubMed] [Google Scholar]
- Seko Y, Miura T, Takahashi M, Koyama T. Methyl mercury decomposition in mice treated with antibiotics. Acta Pharmacol Toxicol. 1981;49:259–265. doi: 10.1111/j.1600-0773.1981.tb00903.x. [DOI] [PubMed] [Google Scholar]
- Smith SD, Bridou R, Johs A, Parks JM, Elias DA, Hurt RA, Jr, Brown SD, Podar M, Wall JD. Site-directed mutagenesis of HgcA and HgcB reveals amino acid residues important for mercury methylation. Appl Environ Microbiol. 2015;81:3205–3217. doi: 10.1128/AEM.00217-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda I, Totoki S, Uchida T, Takahashi H. Degradation of methyl and ethyl mercury into inorganic mercury by phagocytic-cells. Arch Toxicol. 1992;66:40–44. doi: 10.1007/BF02307268. [DOI] [PubMed] [Google Scholar]
- U.S. Environmental Protection Agency (USEPA) Method 1630, Methyl Mercury in Water by Distillation, Aqueous Ethylation, Purge and Trap, and CVAFS. 2001 EPA 821-R-01-020. [Google Scholar]
- U.S. Environmental Protection Agency (USEPA) Method 1631, Revision E: Mercury in water by oxidation, purge and trap, and cold vapor atomic fluorescence spectrometry. 2002 EPA-821-R-02-019. [Google Scholar]
- U.S. Environmental Protection Agency (USEPA) Method 7473, Mercury in Solids and Solutions by Thermal Decomposition Amalgamation, and Atomic Absorption Spectrophotometry 2007 [Google Scholar]
- Vázquez M, Calatayud M, Vélez D, Devesa V. Intestinal transport of methylmercury and inorganic mercury in various models of Caco-2 and HT29-MTX cells. Toxicology. 2013;311:147–153. doi: 10.1016/j.tox.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Wu GD, Chen J, Hoffman C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, Sinha R, Gilroy E, Gupta K, Baldassano R, Nessel L, Li H, Bushman FD, Lewis JD. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue F, Holzman C, Rahbar MH, Trosko K, Fischer L. Maternal fish consumption, mercury levels, and risk of preterm delivery. Envrion Health Perspect. 2007;115:42–47. doi: 10.1289/ehp.9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekutieli D, Benjamini Y. Resampling-based false discovery rate controlling multiple test procedures for correlated test statistics. J Stat Plan Inference. 1999;82:171–196. [Google Scholar]
- Zhou X, Wang L, Sun X, Yang X, Chen C, Wang Q, Yang X. Cinnabar is not converted into methylmercury by human intestinal bacteria. J Ethnopharmacol. 2011;135:110–115. doi: 10.1016/j.jep.2011.02.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.