

Abstract
NFAT5 plays a critical role in maintaining the renal functions. Its dis-regulation in the kidney leads to or is associated with certain renal diseases or disorders, most notably the urinary concentration defect. Hypertonicity, which the kidney medulla is normally exposed to, activates NFAT5 through phosphorylation of a signaling molecule or NFAT5 itself. Hypotonicity inhibits NFAT5 through a similar mechanism. More than a dozen of protein and lipid kinases have been identified to contribute to tonicity-dependent regulation of NFAT5. Hypertonicity activates NFAT5 by increasing its nuclear localization and transactivating activity in the early phase and protein abundance in the late phase. The known mechanism for inhibition of NFAT5 by hypotonicity is a decrease of nuclear NFAT5. The present article reviews the effect of each kinase on NFAT5 nuclear localization, transactivation and protein abundance, and the relationship among these kinases, if known. Cyclosporine A and tacrolimus suppress immune reactions by inhibiting the phosphatase calcineurin-dependent activation of NFAT1. It is hoped that this review would stimulate the interest to seek explanations from the NFAT5 regulatory pathways for certain clinical presentations and to explore novel therapeutic approaches based on the pathways. On the basic science front, this review raises two interesting questions. The first one is how these kinases can specifically signal to NFAT5 in the context of hypertonicity or hypotonicity, because they also regulate other cellular activities and even opposite activities in some cases. The second one is why these many kinases, some of which might have redundant functions, are needed to regulate NFAT5 activity. This review reiterates the concept of signaling through cooperation. Cells need these kinases working in a coordinated way to provide the signaling specificity that is lacking in the individual one. Redundancy in regulation of NFAT5 is a critical strategy for cells to maintain robustness against hypertonic or hypotonic stress.
Keywords: Tonicity enhancer binding protein, Osmotic response element binding protein, Phosphorylation, Kidney, Urinary concentration, Signal transduction, Nephropathy, Hypertonicity, Hypotonicity
Core tip: NFAT5 is critical for kidney functions. Its dis-regulation results in or is associated with the renal diseases and disorders. More than a dozen of kinases have been identified to contribute to tonicity-dependent regulation of NFAT5. The present review is focused on how these kinases regulate NFAT5 activity under the context of hypertonicity or hypotonicity. Understanding these regulatory mechanisms will have therapeutic implications. A precedent example is that recognition of the cyclosporine immunosuppressive effect resulted from inhibition of the phosphatase calcineurin-dependent activation of NFAT1 allows combination use of cyclosporine with other mechanistically different immunosuppressants to improve their therapeutic efficacy and reduce their side effects.
INTRODUCTION
Functions of NFAT5 in the kidney
The kidney medulla contributes to maintaining body fluid and electrolyte balance through concentration of urine. In order to achieve this goal, the medulla must establish two pre-requisites: Adequate water permeability alone the renal tubules and hypertonicity and hyperosmolality in the renal medullary interstitial fluid, which provide an osmolar gradient driving water absorption. NFAT5, nuclear factor of activated T cells 5[1] also named as TonEBP[2] and OREBP[3], is the primary transcription factor that is activated by hypertonicity in the mammalian system and plays a pivotal role in establishing these two conditions. NFAT5 activates expression of water channels aquaporin-2 (AQP-2), which dictates the apical water permeability of the collecting ducts[4-6] and aquaporin-1 (AQP-1), an important gene for water trafficking across the proximal tubules and descending limb of the loop of Henle[7], and urea transporter 1 (UTA1), a critical contributor for hyperosmolality in the renal medullary interstitium[5,6,8], and osmoprotective genes like betaine/glycine transporter 1 (BGT1), sodium-dependent myo-inositol transporter (SMIT) and aldose reductase (AR)[1-3,9], which are essential for the kidney medulla to survive in the hypertonic environment. Expression of a dominant negative mutant of NFAT5 in the kidney epithelial cells reduces expression of AQP-2 and UTA1 and impairs urinary concentration[5]. A majority of homozygous NFAT5 knockouts die embryonically[5], probably due to impaired development and function of cardiomycytes[10]. The survived knockouts have profound renal medullary hypotrophy with reduced expression of the osmoprotective gene[9]. Thus, NFAT5 is tightly regulated in the kidney medulla to ensure normal process of urinary concentration. Hypokalemia, cyclosporine A and lipopolysaccharides-induced urinary concentration defect is associated with reduced NFAT5 activity in the region[6,11,12]. Water restriction induces an increase of urinary excretion of sodium to prevent hypernatremia and rise in extracellular tonicity. In the primary rat renal medullary cells, NFAT5 is necessary for hypertonicity-induced increase of serum- and glucocorticoid-inducible kinase-dependent expression of the type A natriuretic peptide receptor[13]. This cascade might be a mechanism for dehydration-induced natriuresis[13].
Besides activation by hypertonicity, NFAT5 is also activated by hypoxia[14,15]. Renal ischemia for 30 min increases the mouse medullary mRNA abundance of NFAT5, which is protective against ischemia/reperfusion-induced acute kidney injury[14]. However, ischemia for 45 min in the rat kidney decreases NFAT5 mRNA and protein abundance in the medulla[16], but the functional consequence of the effect remains unknown[16]. NFAT5 mRNA is up-regulated in the kidney by unilateral ureteral obstruction[17]. NFAT5 involves in diabetic nephropathy. Haplotype association analysis of 718 type 1 diabetic patients reveals a significant association of NFAT5 with nephropathy[18]. High glucose increases NFAT5 transcriptional activity more in the peripheral blood mononuclear cells isolated from type 1 diabetes patients with nephropathy than in the cells isolated from the patients without nephropathy[19].
Phosphorylation of NFAT5
NFAT5 belongs to the family of the Rel transcription factors, including NFAT1-4 and NF-κB[1-3]. It is best known for its essential role in protecting cells from hypertonic stress. However, it has become clear that NFAT5 also has important functions outside hypertonicity[20]. Therefore, it is not surprising that NFAT5 is also expressed in the tissues that are not normally exposed to hypertonicity[21]. Hypertonicity activates NFAT5 by increasing its transactivation, nuclear localization and DNA binding and protein abundance[22]. Like many other biological processes, phosphorylation of NFAT5 regulates NFAT5 activation. High NaCl rapidly increases phosphorylation of NFAT5. NFAT5 has 216 serines, 15 tyrosines, and 111 threonines, all of which could be phosphorylated[22]. Through mass spectrometry, DNA mutation, immunocytochemistry and Western analyses, NFAT5 tyrosine 143 (Y143), threonine 135 (T135), serine 155 and 158 (S155 and S158) have been identified so far as the phosphorylation sites and play a critical role in regulation of NFAT5 activity. High NaCl increases phosphorylation of NFAT5-Y143, leading to increase of NFAT5 nuclear localization in cell culture[23-25], and phosphorylation of NFAT5-Y143 is increased in the normal rat renal inner medulla and the Brattleboro rat inner medulla treated with vasopressin, known to increase the renal medullary tonicity[25]. The similar phenomena are also observed with NFAT5-T135[26]. On the other hand, low NaCl increases phosphorylation of NFAT5-S155 and then S158, leading to reduced NFAT5 nuclear accumulation[26,27]. In contrast to the demonstration of regulation of NFAT5 nuclear distribution by direct phosphorylation, how phosphorylation regulates NFAT5 transactivating activity is elusive. Although a majority of kinases contributes to tonicity-dependent increase of NFAT5 transactivation (Figure 1), none of phosphorylation sites in the transactivation domain has been definitively identified. NFAT5-S1197, S1247 and S1367 lie in the NFAT5 transactivation domain. Over expression of the alanine mutants of these serine residues in HEK293 cells or AT cells, which have inactive ATM kinase, reduces NFAT5 transcriptional activity under isotonicity and/or hypertonicity[28]. However, whether high NaCl increases phosphorylation of these serine residues remains unknown.
Figure 1.
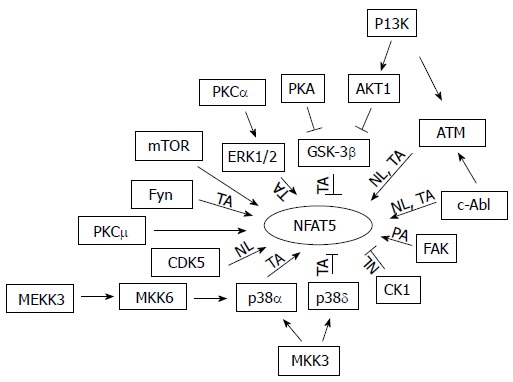
Summary of kinases known to regulate tonicity-dependent activation/inactivation of NFAT5 through an increase/decrease of its transactivating activity, nuclear localization and/or protein abundance. If none of these three steps appears with an arrow, this means that the mechanism is unknown. mTOR: Mammalian target of rapamycin; TA: Transactivating activity; NL: Nuclear localization; PA: Protein abundance; PKA: Protein kinase A; PKC: Protein kinase C; ERK: Extracellular signal-regulated kinase; CDK: Cyclin dependent kinase.
Signaling regulation through coordination
Consistent with the observations that phosphorylation regulates NFAT5 activation are that more than a dozen of kinases (Figure 1) and a few phosphatases have been identified to regulate NFAT5 transcriptional activity[22,23,29-32]. However, all of these kinases and phosphatases also regulate other cellular activities and even opposite activities. For example, p38 and ERK1/2 contribute to hypertonicity-induced activation of NFAT5, but hypotonicity, which is known to inhibit NFAT5 activity, also increases phosphorylation (activation) of these two kinases[30]. The pleiotropic effects of these kinases and phosphatases raise a question concerning how they can selectively signal to NFAT5 in the context of hypertonicity. Another question is why many signaling molecules are needed to regulate NFAT5 activity. Signaling through cooperation/committee might be a plausible explanation. This concept was originally put forward to describe how protein kinases and phosphatases in budding yeast capture and relay information in a coordinated way responding to a signal[33]. This concept can be viewed as that cells have a specific committee tasked for a specific perturbation. Each member in the committee is pre-decided when, where and how to act, so that cells can respond to the perturbation in a coordinated way[34]. The committee members are like different and redundant instrument players in an orchestra in which each one plays his/her instrument, maybe viewed as activation of a signaling molecule, in a coordinated way with other players for a specific music piece signaled by a conductor. This theory explains why each of the identified kinases is necessary for full activation of NFAT5, but none alone, is sufficient[22], why CDK5 is only required in the early phase of NFAT5 activation[26], and why over expression of catalytically active PKA only increases NFAT5 activity under isotonicity but not under hypertonicity[35], because neither a single player nor an over active player can play an orchestra piece. Numerous signaling molecules are required in order to form redundancy in signaling hypertonic stress. Redundancy is a critical strategy for cells to maintain robustness against internal and external perturbations[36], as multiple players are needed to produce desired volume from a particular instrument in orchestrating a music piece.
Potential clinical significance of this review
The present review is focused on how kinases contribute to tonicity-dependent activation/inactivation of NFAT5, since this area is the most studied one. Understanding these regulatory pathways will have therapeutic implications. A good example is the mechanism by which cyclosporine A and tacrolimus suppress immune reactions. These two medications inhibit the phosphatase calcineurin, which leads to inhibition of nuclear translocation of NFAT1, NFAT2 and NFAT4, resulting in suppression of expression of the proinflamatory cytokines[37]. This mechanism has helped understanding both the therapeutic and side effects including renal toxic effects of the medications[37] and is critical for the combined use of cyclosporine A and tacrolimus with other mechanistically different immunosuppressants to improve their therapeutic efficacy and reduce their side effects[38]. Interestingly, cyclosporine A induces urinary concentration defect, which is ascribed to the decrease in NFAT5 activity in the rat kidney[12,39]. The effect is mediated through inhibition of calcineurin remains unknown. Another example is that the anti-diabetes medication metformin was shown to induce apoptosis in the kidney medulla of both normally hydrated and dehydrated type 2 diabetic mice, probably by inhibition of NFAT5 through activation of 5’-AMP-activated protein kinase[40]. This observation raises safety concern for metformin in the dehydrated diabetic patients[41].
Diabetic nephropathy is one of the most severe complications of diabetes with resultant increases of morbidity and mortality. Its treatment has posed a formidable challenge to the medical and scientific communities. Numerous novel therapeutic approaches promisingly found in animal studies have not been successfully translated into clinical practices[42]. For example, pyridoxamine, which showed a potent effect in blocking formation of advanced glycosylated end product in animal models has failed in clinical trials[42,43]. AR, a transcriptional target of NFAT5, is a rate-limiting enzyme of the polyol pathway, which plays a crucial role in the pathogenesis of diabetic complications including diabetic nephropathy[44,45]. Thus, targeting the regulatory network of NFAT5 may be an alternative approach to treat diabetic nephropathy. In this regard, the extract from plant Aralia elata has been recently shown to prevent neuronal death by downregulating NFAT5 and AR in mice with diabetic retinopathy, although which regulatory pathway it affects remains unknown[46].
Mitogen-activated protein kinases
Mitogen-activated protein kinases (MAPKs) have three major families: p38, extracellular signal-regulated kinases (ERKs) and c-Jun NH2-terminal protein kinases (JNKs). Each family has multiple isoforms. They are the most studied kinases in tonicity-dependent activation of NFAT5, which was recently reviewed[30]. The present review only summarizes the salient points of that review. p38 has four isoforms: p38α[47], p38β[48], p38γ[49] and p38δ[50]. p38α contributes to tonicity-dependent activation of NFAT5, whereas p38δ does the opposite[51]. The imidazole derivatives such as SB203580 inhibit p38α and p38β, but not p38δ[50,52]. SB203580, its analogs, p38α dominant negative mutant or siRNAs uniformly inhibits hypertonicity-induced NFAT5 transcriptional activity[53-63]. Because p38α is also called p38, it has been often concluded that p38 signals hypertonicity-induced activation of NFAT5. However, this conclusion causes confusion for interpretation of the effect of the p38 upstream kinase MKK3 and phosphatase MKP-1 on NFAT5 activity. Over expression of MKK3 dominant negative mutant[64] or MKP-1[51] inhibits p38 without significantly affecting NFAT5 transcriptional activity. This paradox can be explained by the interpretation that inhibition of the positive effect of p38α by SB203580, a dominant negative mutant[54] or its siRNAs[51] unmasks an inhibitory effect of p38δ, whereas the dominant negative mutant of MKK3[50,65] and MKP-1[51] reduce both p38α and p38δ activities, therefore, causing no significant change in NFAT5 activity[51,64]. Based on this theory, it is not surprising that another p38 upstream kinase MKK6[13,66] and the MKK3 and MKK6 upstream kinase MEKK3[67] have been demonstrated to contribute to tonicity-dependent activation of NFAT5, because although both MKK3 and MKK6 activate p38α under hypertonicity[68,69], MKK3 strongly activates p38δ, whereas MKK6 does not[70].
Although p38 is the most studied MAPKs in the context of tonicity-dependent regulation of NFAT5, the exact mechanism underlying this effect is far from clear. Whether p38 is critical for tonicity-dependent activation of the transcription factor is even questionable. Knockdown of Rac1 or OSM by its siRNAs reduces high NaCl-induced NFAT5 transcriptional activity, but increases phosphorylation of p38 at both basal and hypertonic levels in HEK293 cells[66]. It should be noted that an opposite effect of knockdown of Rac1 or OSM on phosphorylation of p38 in the same type of cells was reported[68]. Although whether activation of p38 is regulated by cell volume or intracellular ionic strength remains unclear, hypotonicity, which reduces nuclear NFAT5, presumably NFAT5 activity[27,71], also activates p38 in various types of cells[72-75]. These observations call for more attention to which isoform of p38 when the effect of the kinase on NFAT5 is examined.
The chemical inhibitors of MEK-ERK1/2 PD98059 and U-0126 inhibit high NaCl-induced activation of NFAT5 in nucleus pulposus cells[62], renal carcinoma cells[55] and possibly in mIMCD3 cells[76]. ERK2 siRNA reduces high NaCl-dependent NFAT5 transcriptional activity in nucleus pulposus[62] and in HEK293 cells[29]. It is reasonably concluded that ERK1/2, or at least ERK2, contributes to tonicity-dependent activation of NFAT5[30], although it is not clear why PD98059 fails to inhibit NFAT5 transcriptional activity in the primary splenocytes[61]. The effect of JNK on tonicity-dependent activation of NFAT5 is elusive and also least studied. Both lack of effect[64] and a positive effect of JNK1/2[55] on NFAT5 have been reported.
Like the effect on p38, hypotonicity also increases phosphorylation of ERK1/2 in human keratinocytes[73], mIMCD3 cells[77], renal epithelial A6 cells[72], although inhibition of ERK by hypotonic stress in A6 cells was also reported[78]. Therefore, the mechanism for how ERK1/2 contributes to tonicity-dependent activation of NFAT5 remains to be elucidated. In an overly simplified term, p38 and ERK1/2 can signal both hypertonic and hypotonic responses, depending on which committee they are in.
AGC protein kinases
Based on sequence alignments of the catalytic domains, the term AGC kinase was first used in 1995 to define a subgroup of serine/threonine protein kinases that were most related to cAMP-dependent protein kinase 1 (PKA; also known as PKAC), cGMP-dependent protein kinase (PKG; also known as CGK1α) and protein kinase C (PKC)[79]. It was later realized that the group of AGC protein kinases includes more than 60 protein kinases in the human genome, classified into 14 families: PDK1, AKT/PKB, SGK, PKA, PKG, PKC, PKN/PRK, RSK, NDR, MAST, YANK, DMPK, GRK and SGK494[80]. AGC kinases regulate a wide array of important cellular functions. Therefore, their mutation and dysregulation contribute to the pathogenesis of various human diseases, including kidney diseases[80].
(1) PKA exists as a heterotetramer composed of two regulatory subunits and two catalytic subunits. A pseudosubstrate motif in the regular subunits binds to the substrate-binding site of the catalytic domain. Upon activation, two molecules of cAMP bind to each regulatory subunit, allowing the release of active catalytic subunits[80]. PKA is the first AGC kinase demonstrated contributing to tonicity-dependent activation of NFAT5[35]. Hypertonicity induced by high NaCl increases PKA activity. An inhibitor of PKA (H89, 10 μmol/L) and dominant-negative PKA catalytic subunit reduce NFAT5 transcriptional activity associated with a decrease of NFAT5 transactivating activity in HepG2 cells[35]. Further, overexpression of the catalytic subunit of PKA (PKAc) alone increases NFAT5 transactivating and transcriptional activities under the isotonic condition[35]. Subsequent studies indicate that PKA contributes to tonicity-dependent activation of NFAT5 by suppressing the negative effect of GSK-3β on the transcription factor through increasing the inhibitory phosphorylation of GSK-3β at serine 9[81]. PKA has also been suggested to contribute to tonicity-dependent activation of NFAT5 in the primary splenocytes, based on the inhibitory effect of H89[61]. H89 at 2 μmol/L failed to inhibit high NaCl-induced increase of protein abundance of HSP70, a transcriptional target of NFAT5[82,83], in NIH3T3 cells[84]. This is probably due to that the concentration of H89 is too low. Whether hypertonicity-induced activation of PKA requires cAMP is not clear. High NaCl does not significantly alter cAMP level in HepG2 cells where the effect of PKA on NFAT5 is observed[35] or in LLC-PK1 cells[85], but increases cAMP level in mIMCD3 cells[86] and neutrophils[87]. Yet, the lack of the effect of forskolin (increasing intracellular cAMP) or dibutyryl-cAMP (a mimic of cAMP) on NFAT5 transcriptional activity in HepG2 cells let investigators conclude that the effect of PKA on NFAT5 is cAMP-independent[35]. A precedent example is that activation of NF-κB by PKA is independent of cAMP[35]. It is not clear why hypertonicity does not increase PKA activity in mpkCCD14 cells[88].
(2) The PKC family has 10 isoforms and can be divided into three categories based on their structure and biochemical properties: classical or conventional PKC (cPKC), including PKCα, PKCβI, PKCβII, and PKCγ; novel PKC (nPKC), including PKCδ, PKCε, PKCη, and PKCθ; and atypical PKC (aPKC), including PKCζ and PKCλ. PKC is a primary target of diacylglycerol, which is produced by phospholipase C (PLC)-catalyzed hydrolysis of lipid phosphatidylinositol-(4,5)-bisphosphate [PtdIns(4,5)P2]. Diacylglycerol binds a conserved C1 domain in PKC, resulting in the plasma membrane translocation and activation of the kinase[89]. High NaCl increases PLCγ1 activity[24], diacylglycerol and total PKC activity[90]. PKC inhibitors reduce NFAT5 transcriptional activity in mIMCD3[91] and NIH3T3 cells[84]. We recently identified PKCα involved in regulation of NFAT5 activity[29]. Acute hypertonic stress with high NaCl increases PKCα activity in HEK293 cells. Knockdown of PKCα by its siRNAs decreases NFAT5 transcriptional activity mediated by reduction of NFAT5 transactivating activity, but not by NFAT5 nuclear localization or protein abundance[29]. More interestingly, PKCα activity is elevated in the kidney inner medulla due to increase of its protein abundance. Knockout of PKCα reduces expression of NFAT5-targeted genes AR and betain/glycine transporter 1, associated with reduced expression of NFAT5 protein abundance[29]. This is the first demonstration showing that a signaling molecule regulates NFAT5 in the kidney inner medulla. The effect of PKCα on NFAT5 is relayed by ERK1/2 in HEK293 cells and possible in the kidney inner medulla, since knockdown of PKCα attenuates high NaCl-induced phosphorylation of ERK1/2 and has no additional inhibition on NFAT5 in the presence of ERK2 siRNAs in HEK293 cells, and knockout of the kinase reduces phosphorylation of ERK1/2 in the kidney inner medulla[29]. PKCα was previously demonstrated to contribute to regulation of urinary concentration[92,93], possibly by increasing high NaCl-dependent phosphorylation of urea transporters[93] and urea permeability[94] in the inner medullary collecting ducts. Our recent observations provide a possible additional mechanism for the effect of PKC on urinary concentration[29].
PKD1, also called PKCmu, is one of three members of PKD kinase family that is closely related to PKC. PKC activates PKD through direct phosphorylation of S744 and S748 in the activation loop of PKD[95]. PKD is highly mobile and functions as a “communicator”between different subcellular compartments[95]. General PKC inhibitors, Go6976 and GF109203X, and siRNA-mediated knockdown of PKD1 reduce high NaCl-induced increase of protein abundance of HSP70[84]. The general inhibitors reduce high NaCl-induced NFAT5 mobility shift and have no significant effect on NFAT5 nuclear localization[84]. The latter effect is consistent with the lack of effect of PKCα and ERK1/2 on NFAT5 nuclear accumulation. PKD acts upstream of ERK1/2 under certain contexts[95]. However, it remains unclear whether PKD1 involves in PKCα-ERK1/2 signaling activation of NFAT5, since elimination of high NaCl-induced phosphorylation of ERK1/2 by PD98059 (20 micromol/L) does not reduce tonicity-dependent increase of HSP70 protein abundance[84].
(3) The AKT protein kinase family comprises three highly related isoforms encoded by different genes. Despite the shared common, multi-step mechanism of activation downstream of class IA PI3 kinases, these isoforms play different roles in signaling, as revealed by distinct phenotypes displayed by genetically modified animals, identification of isoform-specific substrates and association with discrete subcellular locations[96]. Inhibition of phosphorylation of AKT1-S473 by a general AKT inhibitor, triciribine, or by a PI3K inhibitor wortmannin reduces high NaCl-induced expression of AR, BGT1 and SMIT[97]. Co-expression of the catalytically active AKT1 with GSK3 in the GSK3-/- mouse embryonic fibroblasts reverses the inhibitory effect of GSK3β on NFAT5. These data indicate that AKT1 contributes to tonicity-dependent activation of NFAT5 by attenuating the inhibitory effect of GSK3[81]. Whether hypertonicity/hyperosmolality activates AKT remains controversial. Hypertonicity activates AKT, including direct measurements of increased AKT activity in NIH 3T3 and CHO cells[98]. High NaCl increases phosphorylation (activation) of AKT1-S473 in mCCDcl1 and HepG2 cells[97]. Also, high NaCl increases phosphorylation of AKT-S174 in Madin-Darby canine kidney cells, so does dehydration in the rat inner medulla[99]. In contrast, high sorbitol decreases the kinase activity in HEK293 and COS cells[100], and high sucrose decreases the kinase activity in Swiss 3T3 cells, despite increases of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) abundance and PI3K activity[101]. In the latter study, failure of the increased PIP3 to activate AKT was ascribed to concomitant activation of an inhibitory pathway. It is worthwhile to note that these studies were done with different hypertonicity/hyperosmolality-inducers in different types of cells.
Ataxia telangiectasia-mutated, c-Abl and phosphatidylinositide 3-kinase-IA
Ataxia telangiectasia-mutated (ATM) is a DNA damage-inducible serine/threonine kinase belonging to the PI3K-like kinase family[102]. PI3K is a family of lipid kinases that phosphorylate the 3’-position hydroxyl of the D-myo-inositol head group to generate specific phosphoinositide forms[103]. Based on their in vitro lipid substrate specificity, structure, and mode of regulation, PI3Ks can be divided into three main classes. Class I, which has class IA and B, synthesizes phosphatidylinositol (3,4)-bisphosphate [PtdIns(3,4)P2] and phosphatidylinositol (3,4,5)-trisphosphate [PtdIns (3,4,5) P3][103]. It is a heterodimer composed of a p110 catalytic subunit and a p85 regulatory subunit[103]. c-Abl belongs to a family of non-receptor tyrosine kinases, which has two members, c-Abl and Arg (Abl-related gene)[104]. These three different types of kinases are reviewed together, because evidence already exists that they act in coordination to regulate high NaCl-induced activation of NFAT5. It has been proposed for a while that hypertonicity/hyperosmolality-induced damages interplay with hypertonicity/hyperosmolality-induced responses[22,105,106]. The role of ATM in regulation of NFAT5 activity is an example of this theory. High NaCl damages DNA[107]. High NaCl activates ATM, most likely through high NaCl-induced DNA damage, although it is difficult to directly approve it[28]. ATM contributes to high NaCl-induced activation of NFAT5 through increasing NFAT5 transactivating activity[28] and nuclear localization[108]. Phosphatidylinositide 3-kinase-IA (PI3K-IA) contributes to tonicity-dependent activation of NFAT5 by increasing its transactivation, since over expression of a dominant negative mutant of p85 or by siRNA-mediated knockdown of p110α reduces NFAT5 transcriptional and transactivating activities[109]. PI3K-IA acts as an upstream kinase to mediate high NaCl- and ionizing radiation-induced activation of ATM as measured by the stimulatory phosphorylation of ATM[109]. Since NaCl-induced increase of NFAT5 activity is reduced equally by inhibition of ATM and PI3K-IA, and the effects are not additive, it is concluded that the effect of PI3K-IA on tonicity-dependent activation of NFAT5 is mediated by ATM[109]. However, it is not clear why PI3K-IA is not involved in high NaCl-induced increase of nuclear NFAT5[109]. High NaCl increases c-Abl kinase activity. Like ATM, c-Abl regulates tonicity-dependent activation of NFAT5 through increasing NFAT5 transactivating activity and nuclear localization[25]. The effect of c-Abl on NFAT5 nuclear distribution is also mediated by direct phosphorylation of NFAT5-Y143[25]. Over expression of a c-Abl kinase dead mutant abolishes high NaCl-induced phosphorylation (activation) of S1981 of ATM, and high NaCl-induced NFAT5 nuclear accumulation is greatly enhanced in AT cells, which lack active ATM, when wild-type ATM is transfected[25]. These data indicate that c-Abl regulates NFAT5 activity through ATM. However, it is unlikely that the protein tyrosine kinase c-Abl directly phosphorylates ATM-S1981. Further, the relationship between PI3K-IA and c-Abl in signaling activation of NFAT5 remains unknown.
Mammalian target of rapamycin
Mammalian target of rapamycin (mTOR) is a serine-threonine kinase belonging to the phosphatidylinositol kinase-related kinase family[110]. It has two multi-protein complex isoforms, mTORC1 and mTORC2. The mTORC1 is composed of regulatory-associated protein of mTOR (Raptor), PRAS40 (also known as Akt substrate 1) and mLST8. mTORC1 is rapamycin-sensitive. mTORC2 combines rapamycin-insensitive companion of mTOR, mSIN1, Protor and mLST8[110]. mTOR controls cell growth and division in part through regulating ribosomal p70 S6 kinase and the eukaryotic translation initiation factor 4E binding proteins[110]. It is well-known that mTORC1 is activated by PI3K-AKT axis[110]. Whether this mechanism is also present in the hypertonic setting is not clear. High NaCl increases PI3K-IA kinase activity in HEK293 cells[109] and phosphorylation (activation) of AKT1-S473 in mCCDcl1 or HepG2 cells[97], but analyses of diagnostic substrates downstream mTORC1 by phosphorylated-S235/236 in the ribosomal subunit S6, and phosphorylation-dependent electrophoretic mobility shift of 4E-BP1 and mTORC2 by phosphorylation of S473 of AKT show that hypertonicity partially inhibits both complexes in the immortalized wild-type adenosine monophosphate-activated protein kinase (AMPK) mouse embryonic fibroblasts[111]. The discrepancy could be due to different types of cells used. Nevertheless, based on the inhibitory effects of the mTOR inhibitors, torin1 and rapamycin, on high NaCl-induced expression of NFAT5-targeted genes and NFAT5 transcriptional reporter activity, it is concluded that mTOR contributes to tonicity-dependent activation of NFAT5. The effect of mTOR is probably due to facilitating a transcription-permissive condition for NFAT5 by enhancing histone H4 acetylation and the recruitment of RNA polymerase II[111]. It should be pointed out that in human colon cancer cell lines under an isotonic condition, NFAT5 activates expression of a DNA damage-response kinase, REDD1, which in turn inhibits mTOR signaling[112].
Src family kinases
Src kinase is a family of non-receptor tyrosine kinases that regulate a wide variety of cellular activities such as cell adhesion and motility, carcinogenesis, immune cell function, and even learning and memory. This family has 12 members: c-Src, Fyn, Yes, Yrk, Lyn, Hck, Fgr, Blk, Lck, Brk, Srm, and Frk (with Frk/Rak and Iyk/Bsk subfamilies), 11 of which are found in humans[113]. Src family kinases exhibit a common modular architecture dominated by so-called “SRC homology,” or SH domain. SH1 is the catalytic domain. In the inactive state, a key tyrosine in this domain (Y416) blocks the substrate binding site. When autophosphorylated, this residue is displaced and substrate access is unimpeded. SH2 and SH3 are protein-protein interaction domains shared not only among the members but also with many other signaling proteins[114]. The effects of hypertonicity on activities of Src kinases are heterogeneous. Hypertonicity increases Fyn activity and phosphorylation of its targets[115,116], whereas it inhibits c-Src activity[116]. The involvement of Src family kinases in regulation of NFAT5 was suggested by the observation that a Src family kinase inhibitor PP2 reduces NFAT5 transactivating activity and protein abundance in the colon cancer cells[117]. More convincing evidence comes from studies of Fyn. Using PP2, Fyn dominant negative mutant and Fyn null cells, Ko et al[54] demonstrated that Fyn contributes to hypertonicity-induced activation of NFAT5 by increasing its transactivating activity.
Focal adhesion kinase
Focal adhesion kinase (FAK) is a mechanosensitive non-receptor protein tyrosine kinase that is widely expressed. In response to integrin engagement as occurs in hypertonicity-induced cell shrinkage, FAK is autophosphorylated and activated at Y397, which entails diverse intracellular events. This function makes FAK a central signaling component downstream of integrin[118]. FAK is abundant in the renal papilla, and furosemide, known to reduce the renal medullary interstitial tonicity, decreases phosphorylation of FAK-Y397 in the region[32]. Hypertonicity increases time-dependent phosphorylation of FAK-Y397 in HEK293 cells[32]. FAK contributes to hypertonicity-induced increase of NFAT5 transcriptional activity[32]. The mechanism underlying this effect is unique, because FAK affects neither hypertonicity-induced increase of nuclear NFAT5 nor NFAT5 transactivating activity. Instead, the effect is mediated by contribution of FAK to hypertonicity-induced increase of NFAT5 protein abundance through stabilizing its mRNA, which depends on NFAT5 3′-UTR[32]. Integrin alpha1beta1 is necessary for hypertonicity-induced full activation of NFAT5 in the inner medullary collecting duct cells[119]. Integrin alpha1-null mice have impaired ability to accumulate organic osmolytes in the inner medulla due to decreased expression of NFAT5-targeted osmoprotective genes and develop early tubular necrosis and increased apoptosis of renal medullary cells following dehydration[119]. Although integrin regulates NFAT5 activity in renal cells and possible in the renal medulla[119] and carcinoma cells[32,117,120,121], whether the effect is through FAK remains to be determined. Besides autophosphorylation at Y397, FAK can be also phosphorylated at multiple tyrosine residues by Src family kinases[118]. FAK is constitutively active in a renal cell carcinoma cell line Caki-1 under an isotonic condition. This is probably due to a high Src kinase activity in the cells[55]. The high activities of Src and FAK in Caki-1 cells are in part responsible for the high basal activity of NFAT5 in the cells as compared with that in the non-cancerous proximal tubule cell line HK-2[55].
Cyclin dependent kinases
The human kinome reveals that the serine/threonine kinase Cyclin dependent kinase (CDK) family has 26 members, of which 21 are classified as CDKs and five form a more distant group of CDK-like kinases[122,123]. CDKs regulate the cell division cycle, apoptosis, transcription and differentiation. Each CDK serves its function by recognizing its specific substrate or other protein effector through the divergent spots located in an overall conserved architecture[122,123]. In HEK293 cells, high NaCl activates CDK5, which directly phosphorylates NFAT5-T135. Phosphorylation of NFAT5-T135 is also increased in the rat renal inner medulla[26]. Inhibition of CDK5 by its siRNA or an inhibitor reduces the increase in NFAT5 transcriptional activity that has occurred by 4 h after NaCl is raised, associated with inhibition of NFAT5 nuclear accumulation at that time, but does not reduce either NFAT5 activity or nuclear NFAT5 after 16 h. This is because high NaCl increases the overall abundance of NFAT5 protein at the later time, which eventually raises its effective level in the nucleus, but the early effect of high NaCl on NFAT5 nuclear localization requires CDK5[26]. CDK5 has no significant effect on NFAT5 transactivating activity. This is special, because a majority of signaling molecules identified so far affects NFAT5 transactivation activity without altering NFAT5 nuclear localization (reviewed above). Besides CDK5, CDK9 also regulates NFAT5. The targeted proteomics shows that CDK9 is physically associated with DDX5/17, a RNA helicase important in alternative RNA splicing of NFAT5. CDK9 is necessary for DDX5 recruitment to NFAT5 as measured by chromatin immunoprecipitation[124].
Glycogen synthase kinase 3β, Casein kinase 1 and 5’-AMPK
In contrast to kinases reviewed above that contribute to high NaCl-induced activation of NFAT5, Glycogen synthase kinase 3β (GSK3β), Casein kinase 1 (CK1) and AMPK actually inhibit tonicity-dependent activation of NFAT5. Therefore, these three kinases are reviewed together. GSK3β is a ubiquitously expressed serine/threonine kinase originally characterized as phosphorylating and inactivating glycogen synthase, the rate-limiting enzyme of glycogen synthesis[125]. Since then, GSK3β has been found to regulate a wide variety of biological processes such as function of neurons[126], immunological responses[127], cardiac hypertrophy[128] and cancer[129]. The pleiotropic effects of GSK3β involve regulation of many transcription factors, such as cAMP response element-binding protein, neurogenin 2, SMAD1, c-Jun, β-catenin[126] and NFAT1-4[127]. GSK3β is unique because unlike most other protein kinases it is most active in cells’ resting state, contributing to inhibition of its target transcription factors. When the cells are stimulated, GSK3β is inhibited, resulting in activation of its substrates. The activity of GSK3β is inhibited by phosphorylation of serine residues, of which, serine 9 is most studied[126]. This mechanism is not exceptional in tonicity-dependent activation of NFAT5. GSK3β inhibits NFAT5 transcriptional activity by reducing NFAT5 transactivating activity and protein abundance under the normal tonicity. High NaCl increases phosphorylation of GSK3β-S9 and decreases GSK3β activity, which results in an increase of NFAT5 transcriptional activity mediated by the increment of NFAT5 transactivating activity, but not by NFAT5 nuclear localization or protein abundance[81]. The lack of the effect of GSK3β on NFAT5 nucleo-cytoplasmic trafficking is in contrast to its effect on NFAT1-4. GSK3β phosphorylates the serines in serine-proline repeats, conserved in the amino terminus of NFAT1-4, resulting in promotion of nuclear exit of NFAT1-4 and inhibition of NFAT1-4 transcriptional activity[130]. Unlike NFAT1-4, NFAT5 does not contain serine-proline repeats in its amino terminus[2,131]. Instead, its nucleo-cytoplasmic distribution is regulated by phosphorylation of other amino acids in the terminus such as tyrosine 143[23-25], threonine 135[26] and serines 155 and 158[27]. The difference in amino acid composition explains why GSK-3β affects nuclear localization of NFAT5 differently from that of NFAT1-4.
The stimulatory effect of PKA, PI3K and AKT1 on NFAT5 is dependent on their attenuation of the GSK3β inhibitory effect on the transcription factor[81]. Therefore, GSK3β integrates, at least in part, the effects of PKA, PI3K and AKT1 on NFAT5. However, GSK3β is not involved in the effect of p38α on NFAT5, because co-expression of p38α and its constitutively active upstream kinase MKK6 does not increase phosphorylation of GSK3β-S9 or reverse the inhibitory effect of GSK3β-S9 on NFAT5[81], despite the observations in other settings that p38α inhibits GSK3β activity[132]. On the other hand, low NaCl reduces the inhibitory phosphorylation of GSK3β-S9, which leads to reduction of NFAT5 mRNA and protein abundance in the mouse inner medullary collecting duct cells[133]. It is worth noting that the inhibitory effect of high NaCl on GSK3β may be cell-dependent, because high NaCl reduces the phosphorylation of GSK3β-S9 and increases the kinase activity in several tumor cell lines[134] and decreases the phosphorylation of GSK3β-S9 in the renal medullary interstitial cells[135]. It would be interesting to know the effect of high NaCl on NFAT5 activity in these cells.
CK is a group of serine/threonine kinases that can be divided into CK1 and CK2 families based on their high homology in their catalytic domains[136]. In vertebrates, seven CK1 isoforms (α, β, γ1, γ2, γ3, δ and ε) and several splice variants for CK1α, δ, ε and γ3 have been identified[136]. This family of kinases has been shown to phosphorylate key regulatory molecules involved in a wide array of cellular activities such as cell cycle, cytokinesis, chromosome and microtubule dynamics and transcription and translation[136]. NFAT5 nucleocytoplasmic trafficking is regulated by the dual phosphorylation of serine 155 and 158[27]. Hypotonicity increases phosphorylation of NFAT5-S155, which primes the phosphorylation of serine 158, leading to reduction of nuclear NFAT5[27]. Unlike GSK3β, which has no significant effect on NFAT5 cellular trafficking[81], CK1α1L increases phosphorylation of NFAT5-S158, contributing to hypotonicity-induced decrease of nuclear NFAT5[27].
The serine/threonine kinase AMPK is a major cellular energy sensor that exists as a heterotrimer composed of a catalytic α subunit and each of regulatory β and γ subunits[137]. A high level of AMP or a low level of ATP activates AMPK through phosphorylation of the kinase, resulting in inhibition of energy consumption and stimulation of energy production, which leads to restoration of energy homeostasis[137]. Hypertonicity inhibits the kinase as measured by phosphorylation of the enzyme in the renal medullary interstitial cells (RMIC)[138]. Pharmacological activators of AMPK reduce high NaCl-induced NFAT5 nuclear localization and expression of NFAT5-targeted genes in the cultured RMIC and increases dehydration-induced apoptosis in the mice medulla, suggesting that AMPK inhibits tonicity-dependent activation of NFAT5[138]. Further, the anti-diabetes medication metformin activates AMPK and inhibits NFAT5 transcriptional activity in RMIC and increases RMIC apoptosis in both normally hydrated and dehydrated type 2 diabetes mice[40]. However, since metformin and the pharmacological activators have other effects besides activation of AMPK, whether AMPK inhibits tonicity-dependent activation of NFAT5 needs to be confirmed with a specific way of manipulating the kinase.
Summary and perspective
NFAT5 is clearly critical for kidney functions. Emerging evidence has shown that its dis-regulation results in or is associated with the renal diseases and disorders. Figure 1 summarizes currently known protein and lipid kinases that involve in regulation of tonicity-dependent activation of NFAT5. More are expected to come. Cells need these kinases working together to orchestrate a specific signal to NFAT5 in response to hypertonic or hypotonic perturbation. These kinases could fulfill their assignments by their different activation duration and strength, and their network with each other as well as with other signaling molecules and scaffolds in a specific subcellular location and time. Further work is needed to provide direct pieces of evidence to support this hypothesis. A vast majority of these kinases were identified in cultured cells. They need to be tested directly in the kidney to determine whether they have the same functions in vivo. Inhibition of NFAT5 results in the urinary concentration defect, indicative of a decrease in the renal medullary interstitial tonicity[5]. The decrease of the renal medullary tonicity inhibits NFAT5 activity[139]. It is difficult to dissect whether the effect of knockout of a kinase, even when it is done in the kidney epithelium-specific manner, on NFAT5 is from the direct effect on the transcription factor or from an indirect effect secondary to alteration of tonicity in the renal medullary interstitium. This challenge calls for a new technology to address how NFAT5 is regulated in the kidney medulla.
ACKNOWLEDGMENTS
The author thanks Ms. Yao Xiao for her secretarial assistance in preparation of this review.
Footnotes
Conflict-of-interest statement: The author declares no conflict-of-interest for this manuscript.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: September 8, 2015
First decision: September 29, 2015
Article in press: December 11, 2015
P- Reviewer: de Oliveira JMF, Friedman EA, Kelesidis T S- Editor: Qiu S L- Editor: A E- Editor: Jiao XK
References
- 1.Lopez-Rodríguez C, Aramburu J, Rakeman AS, Rao A. NFAT5, a constitutively nuclear NFAT protein that does not cooperate with Fos and Jun. Proc Natl Acad Sci USA. 1999;96:7214–7219. doi: 10.1073/pnas.96.13.7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyakawa H, Woo SK, Dahl SC, Handler JS, Kwon HM. Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc Natl Acad Sci USA. 1999;96:2538–2542. doi: 10.1073/pnas.96.5.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ko BC, Turck CW, Lee KW, Yang Y, Chung SS. Purification, identification, and characterization of an osmotic response element binding protein. Biochem Biophys Res Commun. 2000;270:52–61. doi: 10.1006/bbrc.2000.2376. [DOI] [PubMed] [Google Scholar]
- 4.Li SZ, McDill BW, Kovach PA, Ding L, Go WY, Ho SN, Chen F. Calcineurin-NFATc signaling pathway regulates AQP2 expression in response to calcium signals and osmotic stress. Am J Physiol Cell Physiol. 2007;292:C1606–C1616. doi: 10.1152/ajpcell.00588.2005. [DOI] [PubMed] [Google Scholar]
- 5.Lam AK, Ko BC, Tam S, Morris R, Yang JY, Chung SK, Chung SS. Osmotic response element-binding protein (OREBP) is an essential regulator of the urine concentrating mechanism. J Biol Chem. 2004;279:48048–48054. doi: 10.1074/jbc.M407224200. [DOI] [PubMed] [Google Scholar]
- 6.Küper C, Fraek ML, Müller HH, Beck FX, Neuhofer W. Sepsis-induced urinary concentration defect is related to nitric oxide-dependent inactivation of TonEBP/NFAT5, which downregulates renal medullary solute transport proteins and aquaporin-2. Crit Care Med. 2012;40:1887–1895. doi: 10.1097/CCM.0b013e31824e1186. [DOI] [PubMed] [Google Scholar]
- 7.Lanaspa MA, Andres-Hernando A, Li N, Rivard CJ, Cicerchi C, Roncal-Jimenez C, Schrier RW, Berl T. The expression of aquaporin-1 in the medulla of the kidney is dependent on the transcription factor associated with hypertonicity, TonEBP. J Biol Chem. 2010;285:31694–31703. doi: 10.1074/jbc.M109.093690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakayama Y, Peng T, Sands JM, Bagnasco SM. The TonE/TonEBP pathway mediates tonicity-responsive regulation of UT-A urea transporter expression. J Biol Chem. 2000;275:38275–38280. doi: 10.1074/jbc.M004678200. [DOI] [PubMed] [Google Scholar]
- 9.López-Rodríguez C, Antos CL, Shelton JM, Richardson JA, Lin F, Novobrantseva TI, Bronson RT, Igarashi P, Rao A, Olson EN. Loss of NFAT5 results in renal atrophy and lack of tonicity-responsive gene expression. Proc Natl Acad Sci USA. 2004;101:2392–2397. doi: 10.1073/pnas.0308703100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mak MC, Lam KM, Chan PK, Lau YB, Tang WH, Yeung PK, Ko BC, Chung SM, Chung SK. Embryonic lethality in mice lacking the nuclear factor of activated T cells 5 protein due to impaired cardiac development and function. PLoS One. 2011;6:e19186. doi: 10.1371/journal.pone.0019186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeon US, Han KH, Park SH, Lee SD, Sheen MR, Jung JY, Kim WY, Sands JM, Kim J, Kwon HM. Downregulation of renal TonEBP in hypokalemic rats. Am J Physiol Renal Physiol. 2007;293:F408–F415. doi: 10.1152/ajprenal.00502.2006. [DOI] [PubMed] [Google Scholar]
- 12.Lim SW, Ahn KO, Sheen MR, Jeon US, Kim J, Yang CW, Kwon HM. Downregulation of renal sodium transporters and tonicity-responsive enhancer binding protein by long-term treatment with cyclosporin A. J Am Soc Nephrol. 2007;18:421–429. doi: 10.1681/ASN.2006060664. [DOI] [PubMed] [Google Scholar]
- 13.Chen S, Grigsby CL, Law CS, Ni X, Nekrep N, Olsen K, Humphreys MH, Gardner DG. Tonicity-dependent induction of Sgk1 expression has a potential role in dehydration-induced natriuresis in rodents. J Clin Invest. 2009;119:1647–1658. doi: 10.1172/JCI35314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao S, Bellner L, Zhao H, Ratliff BB, Darzynkiewicz Z, Vio CP, Ferreri NR. NFAT5 is protective against ischemic acute kidney injury. Hypertension. 2014;63:e46–e52. doi: 10.1161/HYPERTENSIONAHA.113.02476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villanueva S, Suazo C, Santapau D, Pérez F, Quiroz M, Carreño JE, Illanes S, Lavandero S, Michea L, Irarrazabal CE. NFAT5 is activated by hypoxia: role in ischemia and reperfusion in the rat kidney. PLoS One. 2012;7:e39665. doi: 10.1371/journal.pone.0039665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cha SA, Park BM, Jung YJ, Kim SM, Kang KP, Kim W, Kim SH. Regional heterogeneity of expression of renal NPRs, TonEBP, and AQP-2 mRNAs in rats with acute kidney injury. Peptides. 2015;69:33–39. doi: 10.1016/j.peptides.2015.03.026. [DOI] [PubMed] [Google Scholar]
- 17.Feger M, Alesutan I, Castor T, Mia S, Musculus K, Voelkl J, Lang F. Inhibitory effect of nh4cl treatment on renal Tgfβ1 signaling following unilateral ureteral obstruction. Cell Physiol Biochem. 2015;37:955–964. doi: 10.1159/000430222. [DOI] [PubMed] [Google Scholar]
- 18.Kavanagh DH, Savage DA, Patterson CC, McKnight AJ, Crean JK, Maxwell AP, McKay GJ. Haplotype association analysis of genes within the WNT signalling pathways in diabetic nephropathy. BMC Nephrol. 2013;14:126. doi: 10.1186/1471-2369-14-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang B, Hodgkinson AD, Oates PJ, Kwon HM, Millward BA, Demaine AG. Elevated activity of transcription factor nuclear factor of activated T-cells 5 (NFAT5) and diabetic nephropathy. Diabetes. 2006;55:1450–1455. doi: 10.2337/db05-1260. [DOI] [PubMed] [Google Scholar]
- 20.Halterman JA, Kwon HM, Wamhoff BR. Tonicity-independent regulation of the osmosensitive transcription factor TonEBP (NFAT5) Am J Physiol Cell Physiol. 2012;302:C1–C8. doi: 10.1152/ajpcell.00327.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, Ferraris JD, Brooks HL, Brisc I, Burg MB. Expression of osmotic stress-related genes in tissues of normal and hyposmotic rats. Am J Physiol Renal Physiol. 2003;285:F688–F693. doi: 10.1152/ajprenal.00028.2003. [DOI] [PubMed] [Google Scholar]
- 22.Burg MB, Ferraris JD, Dmitrieva NI. Cellular response to hyperosmotic stresses. Physiol Rev. 2007;87:1441–1474. doi: 10.1152/physrev.00056.2006. [DOI] [PubMed] [Google Scholar]
- 23.Zhou X, Gallazzini M, Burg MB, Ferraris JD. Contribution of SHP-1 protein tyrosine phosphatase to osmotic regulation of the transcription factor TonEBP/OREBP. Proc Natl Acad Sci USA. 2010;107:7072–7077. doi: 10.1073/pnas.1002795107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irarrazabal CE, Gallazzini M, Schnetz MP, Kunin M, Simons BL, Williams CK, Burg MB, Ferraris JD. Phospholipase C-gamma1 is involved in signaling the activation by high NaCl of the osmoprotective transcription factor TonEBP/OREBP. Proc Natl Acad Sci USA. 2010;107:906–911. doi: 10.1073/pnas.0913415107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gallazzini M, Yu MJ, Gunaratne R, Burg MB, Ferraris JD. c-Abl mediates high NaCl-induced phosphorylation and activation of the transcription factor TonEBP/OREBP. FASEB J. 2010;24:4325–4335. doi: 10.1096/fj.10-157362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallazzini M, Heussler GE, Kunin M, Izumi Y, Burg MB, Ferraris JD. High NaCl-induced activation of CDK5 increases phosphorylation of the osmoprotective transcription factor TonEBP/OREBP at threonine 135, which contributes to its rapid nuclear localization. Mol Biol Cell. 2011;22:703–714. doi: 10.1091/mbc.E10-08-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu S, Wong CC, Tong EH, Chung SS, Yates JR, Yin Y, Ko BC. Phosphorylation by casein kinase 1 regulates tonicity-induced osmotic response element-binding protein/tonicity enhancer-binding protein nucleocytoplasmic trafficking. J Biol Chem. 2008;283:17624–17634. doi: 10.1074/jbc.M800281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irarrazabal CE, Liu JC, Burg MB, Ferraris JD. ATM, a DNA damage-inducible kinase, contributes to activation by high NaCl of the transcription factor TonEBP/OREBP. Proc Natl Acad Sci USA. 2004;101:8809–8814. doi: 10.1073/pnas.0403062101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Ferraris JD, Klein JD, Sands JM, Burg MB, Zhou X. PKC-α contributes to high NaCl-induced activation of NFAT5 (TonEBP/OREBP) through MAPK ERK1/2. Am J Physiol Renal Physiol. 2015;308:F140–F148. doi: 10.1152/ajprenal.00471.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou X. Regulation of tonicity-dependent activation of NFAT5 by mitogen-activated protein kinases. Abdomen. 2015;2:e767. [Google Scholar]
- 31.Zhou X, Wang H, Burg MB, Ferraris JD. High NaCl-induced inhibition of PTG contributes to activation of NFAT5 through attenuation of the negative effect of SHP-1. Am J Physiol Renal Physiol. 2013;305:F362–F369. doi: 10.1152/ajprenal.00218.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neuhofer W, Küper C, Lichtnekert J, Holzapfel K, Rupanagudi KV, Fraek ML, Bartels H, Beck FX. Focal adhesion kinase regulates the activity of the osmosensitive transcription factor TonEBP/NFAT5 under hypertonic conditions. Front Physiol. 2014;5:123. doi: 10.3389/fphys.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, Lin ZY, Breitkreutz BJ, Stark C, Liu G, et al. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levy ED, Landry CR, Michnick SW. Cell signaling. Signaling through cooperation. Science. 2010;328:983–984. doi: 10.1126/science.1190993. [DOI] [PubMed] [Google Scholar]
- 35.Ferraris JD, Persaud P, Williams CK, Chen Y, Burg MB. cAMP-independent role of PKA in tonicity-induced transactivation of tonicity-responsive enhancer/ osmotic response element-binding protein. Proc Natl Acad Sci USA. 2002;99:16800–16805. doi: 10.1073/pnas.222659799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitano H. Biological robustness. Nat Rev Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- 37.Azzi JR, Sayegh MH, Mallat SG. Calcineurin inhibitors: 40 years later, can’t live without. J Immunol. 2013;191:5785–5791. doi: 10.4049/jimmunol.1390055. [DOI] [PubMed] [Google Scholar]
- 38.Aliabadi A, Cochrane AB, Zuckermann AO. Current strategies and future trends in immunosuppression after heart transplantation. Curr Opin Organ Transplant. 2012;17:540–545. doi: 10.1097/MOT.0b013e328358000c. [DOI] [PubMed] [Google Scholar]
- 39.Lim SW, Li C, Sun BK, Han KH, Kim WY, Oh YW, Lee JU, Kador PF, Knepper MA, Sands JM, et al. Long-term treatment with cyclosporine decreases aquaporins and urea transporters in the rat kidney. Am J Physiol Renal Physiol. 2004;287:F139–F151. doi: 10.1152/ajprenal.00240.2003. [DOI] [PubMed] [Google Scholar]
- 40.Zheng S, Liu J, Han Q, Huang S, Su W, Fu J, Jia X, Du S, Zhou Y, Zhang X, et al. Metformin induces renal medullary interstitial cell apoptosis in type 2 diabetic mice. J Diabetes. 2014;6:132–146. doi: 10.1111/1753-0407.12105. [DOI] [PubMed] [Google Scholar]
- 41.von Mach MA, Gauer M, Meyer S, Omogbehin B, Schinzel H, Kann PH, Weilemann LS. Antidiabetic medications in overdose: a comparison of the inquiries made to a regional poisons unit regarding original sulfonylureas, biguanides and insulin. Int J Clin Pharmacol Ther. 2006;44:51–56. doi: 10.5414/cpp44051. [DOI] [PubMed] [Google Scholar]
- 42.Burney BO, Kalaitzidis RG, Bakris GL. Novel therapies of diabetic nephropathy. Curr Opin Nephrol Hypertens. 2009;18:107–111. doi: 10.1097/MNH.0b013e3283249c51. [DOI] [PubMed] [Google Scholar]
- 43.Fernández Fernández B, Elewa U, Sánchez-Niño MD, Rojas-Rivera JE, Martin-Cleary C, Egido J, Ortiz A. 2012 update on diabetic kidney disease: the expanding spectrum, novel pathogenic insights and recent clinical trials. Minerva Med. 2012;103:219–234. [PubMed] [Google Scholar]
- 44.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 45.Iso K, Tada H, Kuboki K, Inokuchi T. Long-term effect of epalrestat, an aldose reductase inhibitor, on the development of incipient diabetic nephropathy in Type 2 diabetic patients. J Diabetes Complications. 2001;15:241–244. doi: 10.1016/s1056-8727(01)00160-x. [DOI] [PubMed] [Google Scholar]
- 46.Kim SJ, Yoo WS, Kim H, Kwon JE, Hong EK, Choi M, Han Y, Chung I, Seo S, Park J, et al. Aralia elata prevents neuronal death by downregulating tonicity response element binding protein in diabetic retinopathy. Ophthalmic Res. 2015;54:85–95. doi: 10.1159/000437356. [DOI] [PubMed] [Google Scholar]
- 47.Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 48.Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, Han J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta) J Biol Chem. 1996;271:17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- 49.Li Z, Jiang Y, Ulevitch RJ, Han J. The primary structure of p38 gamma: a new member of p38 group of MAP kinases. Biochem Biophys Res Commun. 1996;228:334–340. doi: 10.1006/bbrc.1996.1662. [DOI] [PubMed] [Google Scholar]
- 50.Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, Di Padova F, Ulevitch RJ, Han J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J Biol Chem. 1997;272:30122–30128. doi: 10.1074/jbc.272.48.30122. [DOI] [PubMed] [Google Scholar]
- 51.Zhou X, Ferraris JD, Dmitrieva NI, Liu Y, Burg MB. MKP-1 inhibits high NaCl-induced activation of p38 but does not inhibit the activation of TonEBP/OREBP: opposite roles of p38alpha and p38delta. Proc Natl Acad Sci USA. 2008;105:5620–5625. doi: 10.1073/pnas.0801453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Avitzour M, Diskin R, Raboy B, Askari N, Engelberg D, Livnah O. Intrinsically active variants of all human p38 isoforms. FEBS J. 2007;274:963–975. doi: 10.1111/j.1742-4658.2007.05644.x. [DOI] [PubMed] [Google Scholar]
- 53.Nadkarni V, Gabbay KH, Bohren KM, Sheikh-Hamad D. Osmotic response element enhancer activity. Regulation through p38 kinase and mitogen-activated extracellular signal-regulated kinase kinase. J Biol Chem. 1999;274:20185–20190. doi: 10.1074/jbc.274.29.20185. [DOI] [PubMed] [Google Scholar]
- 54.Ko BC, Lam AK, Kapus A, Fan L, Chung SK, Chung SS. Fyn and p38 signaling are both required for maximal hypertonic activation of the osmotic response element-binding protein/tonicity-responsive enhancer-binding protein (OREBP/TonEBP) J Biol Chem. 2002;277:46085–46092. doi: 10.1074/jbc.M208138200. [DOI] [PubMed] [Google Scholar]
- 55.Küper C, Beck FX, Neuhofer W. NFAT5-mediated expression of S100A4 contributes to proliferation and migration of renal carcinoma cells. Front Physiol. 2014;5:293. doi: 10.3389/fphys.2014.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–522. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsin YH, Tang CH, Lai HT, Lee TH. The role of TonEBP in regulation of AAD expression and dopamine production in renal proximal tubule cells upon hypertonic challenge. Biochem Biophys Res Commun. 2011;414:598–603. doi: 10.1016/j.bbrc.2011.09.128. [DOI] [PubMed] [Google Scholar]
- 58.Kojima R, Taniguchi H, Tsuzuki A, Nakamura K, Sakakura Y, Ito M. Hypertonicity-induced expression of monocyte chemoattractant protein-1 through a novel cis-acting element and MAPK signaling pathways. J Immunol. 2010;184:5253–5262. doi: 10.4049/jimmunol.0901298. [DOI] [PubMed] [Google Scholar]
- 59.Kino T, Takatori H, Manoli I, Wang Y, Tiulpakov A, Blackman MR, Su YA, Chrousos GP, DeCherney AH, Segars JH. Brx mediates the response of lymphocytes to osmotic stress through the activation of NFAT5. Sci Signal. 2009;2:ra5. doi: 10.1126/scisignal.2000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee JH, Kim M, Im YS, Choi W, Byeon SH, Lee HK. NFAT5 induction and its role in hyperosmolar stressed human limbal epithelial cells. Invest Ophthalmol Vis Sci. 2008;49:1827–1835. doi: 10.1167/iovs.07-1142. [DOI] [PubMed] [Google Scholar]
- 61.Morancho B, Minguillón J, Molkentin JD, López-Rodríguez C, Aramburu J. Analysis of the transcriptional activity of endogenous NFAT5 in primary cells using transgenic NFAT-luciferase reporter mice. BMC Mol Biol. 2008;9:13. doi: 10.1186/1471-2199-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsai TT, Guttapalli A, Agrawal A, Albert TJ, Shapiro IM, Risbud MV. MEK/ERK signaling controls osmoregulation of nucleus pulposus cells of the intervertebral disc by transactivation of TonEBP/OREBP. J Bone Miner Res. 2007;22:965–974. doi: 10.1359/jbmr.070322. [DOI] [PubMed] [Google Scholar]
- 63.Gallazzini M, Karim Z, Bichara M. Regulation of ROMK (Kir 1.1) channel expression in kidney thick ascending limb by hypertonicity: role of TonEBP and MAPK pathways. Nephron Physiol. 2006;104:126–135. doi: 10.1159/000095855. [DOI] [PubMed] [Google Scholar]
- 64.Kültz D, Garcia-Perez A, Ferraris JD, Burg MB. Distinct regulation of osmoprotective genes in yeast and mammals. Aldose reductase osmotic response element is induced independent of p38 and stress-activated protein kinase/Jun N-terminal kinase in rabbit kidney cells. J Biol Chem. 1997;272:13165–13170. doi: 10.1074/jbc.272.20.13165. [DOI] [PubMed] [Google Scholar]
- 65.Enslen H, Raingeaud J, Davis RJ. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J Biol Chem. 1998;273:1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- 66.Zhou X, Izumi Y, Burg MB, Ferraris JD. Rac1/osmosensing scaffold for MEKK3 contributes via phospholipase C-gamma1 to activation of the osmoprotective transcription factor NFAT5. Proc Natl Acad Sci USA. 2011;108:12155–12160. doi: 10.1073/pnas.1108107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Padda R, Wamsley-Davis A, Gustin MC, Ross R, Yu C, Sheikh-Hamad D. MEKK3-mediated signaling to p38 kinase and TonE in hypertonically stressed kidney cells. Am J Physiol Renal Physiol. 2006;291:F874–F881. doi: 10.1152/ajprenal.00377.2005. [DOI] [PubMed] [Google Scholar]
- 68.Uhlik MT, Abell AN, Johnson NL, Sun W, Cuevas BD, Lobel-Rice KE, Horne EA, Dell’Acqua ML, Johnson GL. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol. 2003;5:1104–1110. doi: 10.1038/ncb1071. [DOI] [PubMed] [Google Scholar]
- 69.Cuevas BD, Abell AN, Johnson GL. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene. 2007;26:3159–3171. doi: 10.1038/sj.onc.1210409. [DOI] [PubMed] [Google Scholar]
- 70.Remy G, Risco AM, Iñesta-Vaquera FA, González-Terán B, Sabio G, Davis RJ, Cuenda A. Differential activation of p38MAPK isoforms by MKK6 and MKK3. Cell Signal. 2010;22:660–667. doi: 10.1016/j.cellsig.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 71.Woo SK, Dahl SC, Handler JS, Kwon HM. Bidirectional regulation of tonicity-responsive enhancer binding protein in response to changes in tonicity. Am J Physiol Renal Physiol. 2000;278:F1006–F1012. doi: 10.1152/ajprenal.2000.278.6.F1006. [DOI] [PubMed] [Google Scholar]
- 72.Taruno A, Niisato N, Marunaka Y. Hypotonicity stimulates renal epithelial sodium transport by activating JNK via receptor tyrosine kinases. Am J Physiol Renal Physiol. 2007;293:F128–F138. doi: 10.1152/ajprenal.00011.2007. [DOI] [PubMed] [Google Scholar]
- 73.Kippenberger S, Loitsch S, Guschel M, Müller J, Kaufmann R, Bernd A. Hypotonic stress induces E-cadherin expression in cultured human keratinocytes. FEBS Lett. 2005;579:207–214. doi: 10.1016/j.febslet.2004.11.077. [DOI] [PubMed] [Google Scholar]
- 74.Tilly BC, Gaestel M, Engel K, Edixhoven MJ, de Jonge HR. Hypo-osmotic cell swelling activates the p38 MAP kinase signalling cascade. FEBS Lett. 1996;395:133–136. doi: 10.1016/0014-5793(96)01028-9. [DOI] [PubMed] [Google Scholar]
- 75.Niisato N, Taruno A, Marunaka Y. Involvement of p38 MAPK in hypotonic stress-induced stimulation of beta- and gamma-ENaC expression in renal epithelium. Biochem Biophys Res Commun. 2007;358:819–824. doi: 10.1016/j.bbrc.2007.04.192. [DOI] [PubMed] [Google Scholar]
- 76.Umenishi F, Schrier RW. Hypertonicity-induced aquaporin-1 (AQP1) expression is mediated by the activation of MAPK pathways and hypertonicity-responsive element in the AQP1 gene. J Biol Chem. 2003;278:15765–15770. doi: 10.1074/jbc.M209980200. [DOI] [PubMed] [Google Scholar]
- 77.Zhang Z, Yang XY, Cohen DM. Hypotonicity activates transcription through ERK-dependent and -independent pathways in renal cells. Am J Physiol. 1998;275:C1104–C1112. doi: 10.1152/ajpcell.1998.275.4.C1104. [DOI] [PubMed] [Google Scholar]
- 78.Niisato N, Ohta M, Eaton DC, Marunaka Y. Hypotonic stress upregulates β- and γ-ENaC expression through suppression of ERK by inducing MKP-1. Am J Physiol Renal Physiol. 2012;303:F240–F252. doi: 10.1152/ajprenal.00198.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- 80.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 81.Zhou X, Wang H, Burg MB, Ferraris JD. Inhibitory phosphorylation of GSK-3β by AKT, PKA, and PI3K contributes to high NaCl-induced activation of the transcription factor NFAT5 (TonEBP/OREBP) Am J Physiol Renal Physiol. 2013;304:F908–F917. doi: 10.1152/ajprenal.00591.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Woo SK, Lee SD, Na KY, Park WK, Kwon HM. TonEBP/NFAT5 stimulates transcription of HSP70 in response to hypertonicity. Mol Cell Biol. 2002;22:5753–5760. doi: 10.1128/MCB.22.16.5753-5760.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heo JI, Lee MS, Kim JH, Lee JS, Kim J, Park JB, Lee JY, Han JA, Kim JI. The role of tonicity responsive enhancer sites in the transcriptional regulation of human hsp70-2 in response to hypertonic stress. Exp Mol Med. 2006;38:295–301. doi: 10.1038/emm.2006.35. [DOI] [PubMed] [Google Scholar]
- 84.Lim YS, Lee JS, Huang TQ, Seo JS. Protein kinase Cmu plays an essential role in hypertonicity-induced heat shock protein 70 expression. Exp Mol Med. 2008;40:596–606. doi: 10.3858/emm.2008.40.6.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hasler U, Nunes P, Bouley R, Lu HA, Matsuzaki T, Brown D. Acute hypertonicity alters aquaporin-2 trafficking and induces a MAPK-dependent accumulation at the plasma membrane of renal epithelial cells. J Biol Chem. 2008;283:26643–26661. doi: 10.1074/jbc.M801071200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee SM, Lee YJ, Yoon JJ, Kang DG, Lee HS. Effect of Poria cocos on hypertonic stress-induced water channel expression and apoptosis in renal collecting duct cells. J Ethnopharmacol. 2012;141:368–376. doi: 10.1016/j.jep.2012.02.048. [DOI] [PubMed] [Google Scholar]
- 87.Orlic T, Loomis WH, Shreve A, Namiki S, Junger WG. Hypertonicity increases cAMP in PMN and blocks oxidative burst by PKA-dependent and -independent mechanisms. Am J Physiol Cell Physiol. 2002;282:C1261–C1269. doi: 10.1152/ajpcell.00479.2001. [DOI] [PubMed] [Google Scholar]
- 88.Hasler U, Vinciguerra M, Vandewalle A, Martin PY, Féraille E. Dual effects of hypertonicity on aquaporin-2 expression in cultured renal collecting duct principal cells. J Am Soc Nephrol. 2005;16:1571–1582. doi: 10.1681/ASN.2004110930. [DOI] [PubMed] [Google Scholar]
- 89.Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab. 2010;298:E395–E402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhuang S, Hirai SI, Ohno S. Hyperosmolality induces activation of cPKC and nPKC, a requirement for ERK1/2 activation in NIH/3T3 cells. Am J Physiol Cell Physiol. 2000;278:C102–C109. doi: 10.1152/ajpcell.2000.278.1.C102. [DOI] [PubMed] [Google Scholar]
- 91.Zhao H, Tian W, Cohen DM. Rottlerin inhibits tonicity-dependent expression and action of TonEBP in a PKCdelta-independent fashion. Am J Physiol Renal Physiol. 2002;282:F710–F717. doi: 10.1152/ajprenal.00303.2001. [DOI] [PubMed] [Google Scholar]
- 92.Yao L, Huang DY, Pfaff IL, Nie X, Leitges M, Vallon V. Evidence for a role of protein kinase C-alpha in urine concentration. Am J Physiol Renal Physiol. 2004;287:F299–F304. doi: 10.1152/ajprenal.00274.2003. [DOI] [PubMed] [Google Scholar]
- 93.Klein JD, Martin CF, Kent KJ, Sands JM. Protein kinase C-α mediates hypertonicity-stimulated increase in urea transporter phosphorylation in the inner medullary collecting duct. Am J Physiol Renal Physiol. 2012;302:F1098–F1103. doi: 10.1152/ajprenal.00664.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Y, Klein JD, Froehlich O, Sands JM. Role of protein kinase C-α in hypertonicity-stimulated urea permeability in mouse inner medullary collecting ducts. Am J Physiol Renal Physiol. 2013;304:F233–F238. doi: 10.1152/ajprenal.00484.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang QJ. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol Sci. 2006;27:317–323. doi: 10.1016/j.tips.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 96.Toker A, Marmiroli S. Signaling specificity in the Akt pathway in biology and disease. Adv Biol Regul. 2014;55:28–38. doi: 10.1016/j.jbior.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roth I, Leroy V, Kwon HM, Martin PY, Féraille E, Hasler U. Osmoprotective transcription factor NFAT5/TonEBP modulates nuclear factor-kappaB activity. Mol Biol Cell. 2010;21:3459–3474. doi: 10.1091/mbc.E10-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Konishi H, Matsuzaki H, Tanaka M, Ono Y, Tokunaga C, Kuroda S, Kikkawa U. Activation of RAC-protein kinase by heat shock and hyperosmolarity stress through a pathway independent of phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1996;93:7639–7643. doi: 10.1073/pnas.93.15.7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Terada Y, Inoshita S, Hanada S, Shimamura H, Kuwahara M, Ogawa W, Kasuga M, Sasaki S, Marumo F. Hyperosmolality activates Akt and regulates apoptosis in renal tubular cells. Kidney Int. 2001;60:553–567. doi: 10.1046/j.1523-1755.2001.060002553.x. [DOI] [PubMed] [Google Scholar]
- 100.Meier R, Thelen M, Hemmings BA. Inactivation and dephosphorylation of protein kinase Balpha (PKBalpha) promoted by hyperosmotic stress. EMBO J. 1998;17:7294–7303. doi: 10.1093/emboj/17.24.7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Van der Kaay J, Beck M, Gray A, Downes CP. Distinct phosphatidylinositol 3-kinase lipid products accumulate upon oxidative and osmotic stress and lead to different cellular responses. J Biol Chem. 1999;274:35963–35968. doi: 10.1074/jbc.274.50.35963. [DOI] [PubMed] [Google Scholar]
- 102.Sirbu BM, Cortez D. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol. 2013;5:a012724. doi: 10.1101/cshperspect.a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jean S, Kiger AA. Classes of phosphoinositide 3-kinases at a glance. J Cell Sci. 2014;127:923–928. doi: 10.1242/jcs.093773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sirvent A, Benistant C, Roche S. Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biol Cell. 2008;100:617–631. doi: 10.1042/BC20080020. [DOI] [PubMed] [Google Scholar]
- 105.Lamitina T, Huang CG, Strange K. Genome-wide RNAi screening identifies protein damage as a regulator of osmoprotective gene expression. Proc Natl Acad Sci USA. 2006;103:12173–12178. doi: 10.1073/pnas.0602987103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Choe KP, Strange K. Genome-wide RNAi screen and in vivo protein aggregation reporters identify degradation of damaged proteins as an essential hypertonic stress response. Am J Physiol Cell Physiol. 2008;295:C1488–C1498. doi: 10.1152/ajpcell.00450.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dmitrieva NI, Cai Q, Burg MB. Cells adapted to high NaCl have many DNA breaks and impaired DNA repair both in cell culture and in vivo. Proc Natl Acad Sci USA. 2004;101:2317–2322. doi: 10.1073/pnas.0308463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang Z, Ferraris JD, Irarrazabal CE, Dmitrieva NI, Park JH, Burg MB. Ataxia telangiectasia-mutated, a DNA damage-inducible kinase, contributes to high NaCl-induced nuclear localization of transcription factor TonEBP/OREBP. Am J Physiol Renal Physiol. 2005;289:F506–F511. doi: 10.1152/ajprenal.00417.2004. [DOI] [PubMed] [Google Scholar]
- 109.Irarrazabal CE, Burg MB, Ward SG, Ferraris JD. Phosphatidylinositol 3-kinase mediates activation of ATM by high NaCl and by ionizing radiation: Role in osmoprotective transcriptional regulation. Proc Natl Acad Sci USA. 2006;103:8882–8887. doi: 10.1073/pnas.0602911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yip PY. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Transl Lung Cancer Res. 2015;4:165–176. doi: 10.3978/j.issn.2218-6751.2015.01.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ortells MC, Morancho B, Drews-Elger K, Viollet B, Laderoute KR, López-Rodríguez C, Aramburu J. Transcriptional regulation of gene expression during osmotic stress responses by the mammalian target of rapamycin. Nucleic Acids Res. 2012;40:4368–4384. doi: 10.1093/nar/gks038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhou Y, Wang Q, Weiss HL, Evers BM. Nuclear factor of activated T-cells 5 increases intestinal goblet cell differentiation through an mTOR/Notch signaling pathway. Mol Biol Cell. 2014;25:2882–2890. doi: 10.1091/mbc.E14-05-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Puls LN, Eadens M, Messersmith W. Current status of SRC inhibitors in solid tumor malignancies. Oncologist. 2011;16:566–578. doi: 10.1634/theoncologist.2010-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cohen DM. SRC family kinases in cell volume regulation. Am J Physiol Cell Physiol. 2005;288:C483–C493. doi: 10.1152/ajpcell.00452.2004. [DOI] [PubMed] [Google Scholar]
- 115.Kapus A, Di Ciano C, Sun J, Zhan X, Kim L, Wong TW, Rotstein OD. Cell volume-dependent phosphorylation of proteins of the cortical cytoskeleton and cell-cell contact sites. The role of Fyn and FER kinases. J Biol Chem. 2000;275:32289–32298. doi: 10.1074/jbc.M003172200. [DOI] [PubMed] [Google Scholar]
- 116.Kapus A, Szászi K, Sun J, Rizoli S, Rotstein OD. Cell shrinkage regulates Src kinases and induces tyrosine phosphorylation of cortactin, independent of the osmotic regulation of Na+/H+ exchangers. J Biol Chem. 1999;274:8093–8102. doi: 10.1074/jbc.274.12.8093. [DOI] [PubMed] [Google Scholar]
- 117.Chen M, Sastry SK, O’Connor KL. Src kinase pathway is involved in NFAT5-mediated S100A4 induction by hyperosmotic stress in colon cancer cells. Am J Physiol Cell Physiol. 2011;300:C1155–C1163. doi: 10.1152/ajpcell.00407.2010. [DOI] [PubMed] [Google Scholar]
- 118.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 119.Moeckel GW, Zhang L, Chen X, Rossini M, Zent R, Pozzi A. Role of integrin alpha1beta1 in the regulation of renal medullary osmolyte concentration. Am J Physiol Renal Physiol. 2006;290:F223–F231. doi: 10.1152/ajprenal.00371.2004. [DOI] [PubMed] [Google Scholar]
- 120.Jauliac S, López-Rodriguez C, Shaw LM, Brown LF, Rao A, Toker A. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat Cell Biol. 2002;4:540–544. doi: 10.1038/ncb816. [DOI] [PubMed] [Google Scholar]
- 121.Chen M, Sinha M, Luxon BA, Bresnick AR, O’Connor KL. Integrin alpha6beta4 controls the expression of genes associated with cell motility, invasion, and metastasis, including S100A4/metastasin. J Biol Chem. 2009;284:1484–1494. doi: 10.1074/jbc.M803997200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Endicott JA, Noble ME. Structural characterization of the cyclin-dependent protein kinase family. Biochem Soc Trans. 2013;41:1008–1016. doi: 10.1042/BST20130097. [DOI] [PubMed] [Google Scholar]
- 123.Lolli G. Structural dissection of cyclin dependent kinases regulation and protein recognition properties. Cell Cycle. 2010;9:1551–1561. doi: 10.4161/cc.9.8.11195. [DOI] [PubMed] [Google Scholar]
- 124.Yang J, Zhao Y, Kalita M, Li X, Jamaluddin M, Tian B, Edeh CB, Wiktorowicz JE, Kudlicki A, Brasier AR. Systematic Determination of Human Cyclin Dependent Kinase (CDK)-9 Interactome Identifies Novel Functions in RNA Splicing Mediated by the DEAD Box (DDX)-5/17 RNA Helicases. Mol Cell Proteomics. 2015;14:2701–2721. doi: 10.1074/mcp.M115.049221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hughes K, Pulverer BJ, Theocharous P, Woodgett JR. Baculovirus-mediated expression and characterisation of rat glycogen synthase kinase-3 beta, the mammalian homologue of the Drosophila melanogaster zeste-white 3sgg homeotic gene product. Eur J Biochem. 1992;203:305–311. doi: 10.1111/j.1432-1033.1992.tb19860.x. [DOI] [PubMed] [Google Scholar]
- 126.Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539–551. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang H, Brown J, Martin M. Glycogen synthase kinase 3: a point of convergence for the host inflammatory response. Cytokine. 2011;53:130–140. doi: 10.1016/j.cyto.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cheng H, Woodgett J, Maamari M, Force T. Targeting GSK-3 family members in the heart: a very sharp double-edged sword. J Mol Cell Cardiol. 2011;51:607–613. doi: 10.1016/j.yjmcc.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mills CN, Nowsheen S, Bonner JA, Yang ES. Emerging roles of glycogen synthase kinase 3 in the treatment of brain tumors. Front Mol Neurosci. 2011;4:47. doi: 10.3389/fnmol.2011.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275:1930–1934. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 131.Graef IA, Gastier JM, Francke U, Crabtree GR. Evolutionary relationships among Rel domains indicate functional diversification by recombination. Proc Natl Acad Sci USA. 2001;98:5740–5745. doi: 10.1073/pnas.101602398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kaidanovich-Beilin O, Woodgett JR. GSK-3: Functional Insights from Cell Biology and Animal Models. Front Mol Neurosci. 2011;4:40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Quadri S, Siragy HM. Regulation of (pro)renin receptor expression in mIMCD via the GSK-3β-NFAT5-SIRT-1 signaling pathway. Am J Physiol Renal Physiol. 2014;307:F593–F600. doi: 10.1152/ajprenal.00245.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Perez-Pinera P, Menendez-Gonzalez M, del Valle M, Vega JA. Hypertonicity activates GSK3beta in tumor cells. Mol Cell Biochem. 2006;291:93–100. doi: 10.1007/s11010-006-9201-z. [DOI] [PubMed] [Google Scholar]
- 135.Rao R, Hao CM, Breyer MD. Hypertonic stress activates glycogen synthase kinase 3beta-mediated apoptosis of renal medullary interstitial cells, suppressing an NFkappaB-driven cyclooxygenase-2-dependent survival pathway. J Biol Chem. 2004;279:3949–3955. doi: 10.1074/jbc.M309325200. [DOI] [PubMed] [Google Scholar]
- 136.Schittek B, Sinnberg T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol Cancer. 2014;13:231. doi: 10.1186/1476-4598-13-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hardie DG. AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem Soc Trans. 2011;39:1–13. doi: 10.1042/BST0390001. [DOI] [PubMed] [Google Scholar]
- 138.Han Q, Zhang X, Xue R, Yang H, Zhou Y, Kong X, Zhao P, Li J, Yang J, Zhu Y, et al. AMPK potentiates hypertonicity-induced apoptosis by suppressing NFκB/COX-2 in medullary interstitial cells. J Am Soc Nephrol. 2011;22:1897–1911. doi: 10.1681/ASN.2010080822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sheen MR, Kim JA, Lim SW, Jung JY, Han KH, Jeon US, Park SH, Kim J, Kwon HM. Interstitial tonicity controls TonEBP expression in the renal medulla. Kidney Int. 2009;75:518–525. doi: 10.1038/ki.2008.601. [DOI] [PubMed] [Google Scholar]