

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited monogenic kidney disease. Characterized by the development and growth of cysts that cause progressive kidney enlargement, it ultimately leads to end-stage renal disease. Approximately 85% of ADPKD cases are caused by mutations in the PKD1 gene, while mutations in the PKD2 gene account for the remaining 15% of cases. The PKD1 gene encodes for polycystin-1 (PC1), a large multi-functional membrane receptor protein able to regulate ion channel complexes, whereas polycystin-2 (PC2), encoded by the PKD2 gene, is an integral membrane protein that functions as a calcium-permeable cation channel, located mainly in the endoplasmic reticulum (ER). In the primary cilia of the epithelial cells, PC1 interacts with PC2 to form a polycystin complex that acts as a mechanosensor, regulating signaling pathways involved in the differentiation of kidney tubular epithelial cells. Despite progress in understanding the function of these proteins, the molecular mechanisms associated with the pathogenesis of ADPKD remain unclear. In this review we discuss how an imbalance between functional PC1 and PC2 proteins may disrupt calcium channel activities in the cilium, plasma membrane and ER, thereby altering intracellular calcium signaling and leading to the aberrant cell proliferation and apoptosis associated with the development and growth of renal cysts. Research in this field could lead to the discovery of new molecules able to rebalance intracellular calcium, thereby normalizing cell proliferation and reducing kidney cyst progression.
Keywords: Autosomal dominant polycystic kidney disease, Calcium signaling, cAMP, Cell growth, Non-capacitative calcium entry
Core tip: In the present article, we discuss: (1) the regulation of calcium signaling in the primary cilia of autosomal dominant polycystic kidney disease (ADPKD) cells and the downstream processes that lead to cystogenesis; (2) how calcium impairment promotes cell proliferation by activating different signaling pathways; (3) the activity of non-capacitative calcium entry channels, which in PKD1-silenced cells stimulates cell growth by Ca2+ oscillations and nuclear factor of activated T-cells activation, highlighting new findings showing the role of polycystin-2 in calcium oscillations; (4) the impairment of intracellular calcium signaling associated with apoptosis; and (5) the use of calcium channel blockers and calcium modulators in the treatment of ADPKD.
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited pathology of the kidneys, having an incidence of 1:500-1:1000 individuals. It accounts for roughly 10% of cases of end-stage renal disease[1,2], which results from the progressive bilateral development and expansion of fluid-filled cysts arising from the de-differentiation of renal tubule epithelial cells[1]. ADPKD is caused by the mutation of two genes: PKD1, which accounts for 85% of cases, and PKD2, associated with the remaining 15% of cases[1]. In ADPKD, the focal cyst development in the kidneys seems to be associated with a somatic second hit brought on by either loss of heterozygosity or other mutations in renal cyst lining epithelial cells[3,4]. PKD1 and PKD2 genes encode for polycystin-1 (PC1) and polycystin-2 (PC2) proteins, respectively[1,5]. PC1 is an integral membrane receptor with a large extracellular region consisting of a variety of domains involved in cell-cell and cell-matrix interactions. It also bears 11 transmembrane domains, and a short cytoplasmic segment containing motifs involved in signal transduction[1]. PC2, on the other hand, is an integral transmembrane protein mainly localized to the endoplasmic reticulum (ER); it is anchored to cell membranes by six transmembrane regions, and has two cytoplasmic N- and C-terminal tails. PC2 functions as a nonselective cation channel that transports calcium, and shows significant homology with transient receptor potential (TRP) channels[5-8]. Indeed, the polycystins and their homologous proteins are considered a new subfamily of TRP channels, and accordingly known as TRP polycystic proteins[1].
An interaction between PC1 and PC2 forms the so-called polycystin complex. This is mainly confined to the primary cilia of kidney epithelial cells, where it acts as a flow sensor, triggering intracellular calcium release via the activation of the PC2 channel in response to fluid-flow changes. Disruption of this complex impairs intracellular calcium influx, and leads to the development and expansion of kidney cysts[1,9,10].
Polycystins are able to regulate calcium channel activity not only in the cilia, but also in other cellular compartments, including the plasma membrane and ER. Indeed, PC1 and PC2 co-assembly has been seen to generate a cation-permeable current through the plasma membrane[11], and PC1 and PC2 are known to regulate intracellular calcium release in the ER through their interaction with the inositol 1,4,5-trisphosphate receptor (IP3R)[12-14]. In this context, PC2 enhances calcium release from the ER by stimulating the activity of the IP3 receptor, while PC1 inhibits this process by reducing PC2-IP3R interaction via a mechanism involving the stromal interaction molecule-1 (STIM1) and the PI3K/Akt pathway[12,15]. PC1 can also regulate other types of calcium channels, including non-capacitative calcium entry (NCCE) channels, which are able to generate intracellular calcium oscillations[16]. PC2, on the other hand, regulates intracellular calcium release by either interacting with the calcium channels TRPC1 and TRPV4 on the plasma membrane and in primary cilia, and/or through an association with ryanodine and IP3 receptors in the ER[8,17-19]. Moreover, PC2 appears to be able to generate a non-specific voltage-dependent cation current in native HEK293 kidney cells. This current is strongly associated with PC2 activity, and is completely abolished by the depletion of PC2 protein[20].
Taken together these findings suggest that PC1 and PC2 may affect calcium influx from different cellular compartments, including cilium boundaries (cilioplasm), plasma membrane and ER. Dysregulation of calcium signaling due to loss of polycystin function causes the aberrant activation of different pathways associated with abnormal cell proliferation and fluid secretion, thereby leading to the development and expansion of kidney cysts. However, the cascade of events that occur between polycystin dysfunction and kidney cyst formation in ADPKD is not yet fully understood.
In this review, we discuss the impact of polycystin loss of function on calcium signaling, which may alter different pathways associated with the cell growth and apoptosis that are a typical hallmark of ADPKD. Moreover, we report the potential effects of calcium dysregulation on kidney cyst formation and progression. Finally, we also discuss the state of the art in calcium channel modulators, able to restore normal calcium release and therefore appealing targets for ADPKD treatment.
ROLE OF THE POLYCYSTIN COMPLEX IN PRIMARY RENAL CILIA
It is well known that PC1 and PC2 co-localize in the primary cilia of kidney epithelial cells, performing a mechano-sensor function by transducing calcium signals in response to changes in tubular fluid flow. Loss or dysfunction of either PC1 or PC2 causes the inability of cells to sense mechanical stimuli due to bending of the cilia, which leads to abnormal cell morphology and polarity, and thereby contributes to renal cyst formation[10,21,22]. The calcium signaling triggered by fluid shear stress initiates in the primary cilia through PC2-dependent calcium release. This is initially confined to the cilioplasm, but, through the ryanodine receptor, the same calcium signal subsequently activates a cytosolic calcium response that induces calcium influx from intracellular stores[23].
PC2, as mentioned above, is also able to interact with the transient potential receptor channels TRPC1 and TRPV4. PC2 and TRPC1 assemble to form a heteromultimeric channel, not associated with PC1, which is activated in response to G-protein-coupled receptor stimulation, and shows a pattern of single-channel conductance distinct from that of the individual PC2 and TRPC1 channels[24]. Direct or indirect activation of the PC2/TRPC1 complex, either by cilium bending or through the activation of plasma membrane GPCRs, may affect the mechano-transduction of cilium-associated calcium signals[24]. PC2 can also form a heteromeric channel complex with TRPV4. This complex displays molecular mechano-sensor properties, being able to generate flow-induced calcium influx, which seems to be abolished by the depletion of TRPV4 channel in renal epithelial cells[18]. It is also plausible that polycystins cooperate with other proteins located in the cilium, such as cystin, polaris, inversin, and kinesin-II, as defects in these proteins may lead to the formation of kidney cysts[21,25].
In the primary cilia, PC1/PC2, PC2/TRPC1 and PC2/TRPV4 complexes regulate the calcium signaling activated by cilium deflection due to changes in fluid flow. Surprisingly, however, depletion of TRPC1 and TRPV4 is not associated with cyst formation, despite it altering ciliary calcium signaling. Hence, the impairment of ciliary calcium signaling alone is not sufficient to trigger kidney cyst development, a process which, instead, seems to be closely linked to the activity/function of PC1 and PC2 proteins. In fact, a recent study has shown that ablation of the cilia in both PC1- and PC2-deficient cells reduces cyst growth, suggesting that the loss of cilia may cause milder cyst progression than in the cilia-equipped ADPKD cells[26]. Moreover, cilia-dependent cyst growth is not associated with the activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase, mammalian target of rapamycin (mTOR) or cyclic adenosinemonophosphate (cAMP) pathways[26]. As a whole, these findings suggest that the polycystins govern ciliary signaling by an unknown mechanism when the normal kidney epithelial cell phenotype is maintained. In ADPKD, the inactivation of polycystins alters cilia-dependent signaling, thereby promoting the formation of the characteristic kidney cysts.
CALCIUM SIGNALING AND CELL PROLIFERATION IN ADPKD CELLS
ADPKD is strongly associated with the altered cell proliferation of cystic kidney epithelial cells that represents a typical hallmark of the disease. The disruption of calcium signaling associated with PC1 and PC2 deficiency could be the primary event behind the increased cell growth seen in ADPKD. Indeed, we do know that calcium restriction in ADPKD cells causes cAMP-dependent activation of the B-Raf/mitogen-activated protein kinase kinase (MEK)/extracellular-signal-regulated kinases (ERK) pathway, which results in increased cell growth[27]. Moreover, this reduction in intracellular calcium levels also inhibits the activity of AKT kinase, a negative regulator of B-Raf[27]. In cystic cells, normal growth can be restored by increasing their cytosolic calcium concentration, which increases AKT activity and inhibits cAMP-dependent B-Raf/ERK activation[28].
Low intracellular calcium levels may also stimulate the activity of the ciliary calcium-sensitive adenylyl cyclases AC5 and AC6, as well as the plasma-membrane-anchored AC6, leading to the elevation of cAMP[29,30]. Therefore, loss of polycystin function may promote the activity of AC5/6 by reducing intracellular Ca2+ release from the cilia, ER and plasma membrane[29,30] (Figure 1). Indeed, it has recently been reported that the double knockout of PKD1 and AC6 genes decreases cystogenesis, improves renal function and increases survival in a mouse model of ADPKD. These improvements in renal function occur through a reduction in cAMP levels and inhibition of the B-Raf/MEK/ERK pathway, suggesting that AC6 could be a key mediator of cyst formation in ADPKD[31]. In addition, cAMP elevation may activate the cAMP-response element-binding protein, which promotes cell proliferation in an epidermal growth factor receptor (EGFR)-activation-dependent manner by stimulating expression of the EGF-like peptide amphiregulin[32]. EGFR signaling is dependent upon a mechanism involving the sequential activation of Ras, Raf-1, MEK and ERK. It can converge on the same pathway activated by cAMP, thereby leading to activation of the ERK kinases that promote cell proliferation in ADPKD cells[33] (Figure 1). Furthermore, the abnormal activity of mTOR kinase has been observed to contribute to increased cell proliferation and cyst formation in ADPKD cyst-lining epithelial cells. In normal kidney epithelial cells mTOR activity is inhibited by PC1, which interacts with TSC1/TSC2, an inhibitory complex of mTOR, preventing its inactivation. Conversely, in ADPKD cells polycystin dysfunction promotes mTOR activation by inhibition of the TSC1/TSC2 complex, through a mechanism involving the cAMP-dependent B-Raf/ERK pathway[34,35] (Figure 1).
Figure 1.
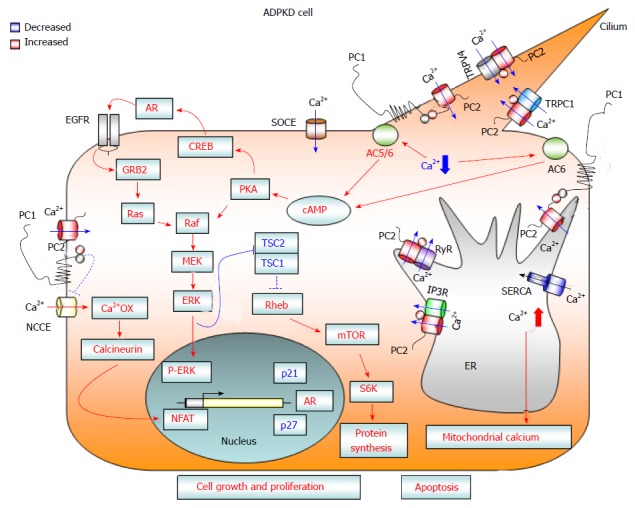
Diagram showing calcium-dependent dysregulated signaling pathways that promote cell proliferation and apoptosis in autosomal dominant polycystic kidney disease cells. Loss of PC1 and/or PC2 function causes a reduction in cytosolic calcium influx from three different cellular compartments: (1) the primary cilium after mechanical stimuli; (2) the endoplasmic reticulum, in an IP3R- and RyR-dependent manner; and (3) the plasma membrane, through a reduction in SOCE channel activity. The reduced concentration of cytosolic calcium may activate Ca2+ sensitive adenylyl cyclases 5 and 6, leading to a rise in cAMP. Increased levels of cAMP cause the activation of B-Raf/MEK/ERK and CREB/AREG/EGFR pathways, as well as stimulating mTOR signaling, through the active form of ERK kinases that inactivate the TSC1/TSC2 complex. Moreover, deficiency of PC1 and/or PC2 enhances the activity of NCCE channels, which, by increasing calcium oscillation frequency, results in the activation of the transcription factor NFAT. The abnormal activation of these signaling pathways promotes cell proliferation and kidney cyst formation. In addition, the reduction in Ca2+ influx from the ER to the cytosol caused by a deficiency in PC2 channel activity brings about an imbalance in ER calcium concentration, resulting in ER Ca2+ overload. The increased ER calcium concentration sensitizes kidney cystic cells to apoptotic stimuli by abnormal ER calcium release, which may induce mitochondrial damage and thereby lead to cytochrome C release and activation of apoptosis. AC 5/6: Adenylyl cyclase 5/6; AR: Amphiregulin; Ca2+ OX: Calcium oscillations; cAMP: Cyclic adenosine monophosphate; CREB: cAMP response element binding transcription factor; EGFR: Epidermal growth factor receptor; ER: Endoplasmic reticulum; ERK: Extracellular-signal-regulated kinases; GRB2: Growth factor receptor-bound protein 2; IP3R: Inositol 1,4,5-trisphosphate receptor; MEK: Mitogen-activated protein kinase kinase; mTOR: Mammalian target of rapamycin; NCCE: Non-capacitative calcium channel entry; NFAT: Nuclear factor of activated T-cells; PKA: Protein kinase A; PC1: Polycystin-1; PC2: Polycystin-2; S6K: Ribosomal S6 kinase; Raf: Rapidly accelerated fibrosarcoma kinase; Ras: Rat sarcoma viral oncogene homolog family; Rheb: Ras homolog enriched in brain; RyR: Ryanodine receptor; SERCA: Sarcoplasmic endoplasmic reticulum calcium ATPase; SOCE: Store-operated calcium channel entry; TRPC1: Transient receptor potential channel 1; TRPV4: Transient receptor potential cation channel subfamily V member 4; TSC: Tuberous sclerosis complex.
The expression of full-length PC1 has been shown to inhibit intracellular calcium release in response to ATP in Madin-Darby canine kidney (MDCK) cells, in a mechanism that involves the interaction of STIM1 with IP3R, and reduces the association between PC2 and the IP3 receptor[15]. Moreover, PC1 seems able to regulate intracellular calcium release and PC2-IP3R-STIM1 interaction through the PI3K/Akt signaling pathway[15]. The exogenous expression of the C-terminal fragment of PC1 (PC1-Cter) could function as a dominant negative effector, causing an increased intracellular calcium release in response to ATP treatment, as seen in HEK-293 cells[36]. Furthermore, PC1-Cter-expressing cells not only exhibit increased levels of basal calcium, but also show enhanced cell proliferation, which is associated with the activation of ERK kinases[37]. Consistently, the transfection of HEK-293 cells with the C-terminal tail of PC1 has been observed to cause an increase in both basal and intracellular calcium release, leading to the activation of the nuclear factor of activated T-cells (NFAT)[38]. Moreover, NFAT activation, associated with increased cell proliferation in HEK-293 cells, is also observed after the downregulation of PC1 by RNA interference[16]. NFAT activation occurs through a rise in the frequency of intracellular calcium oscillations, caused by the increased activity of NCCE channels[16] (Figure 1). In HEK293 cells, these calcium oscillations can be increased by either reduced or undetectable levels of PC1, but are only induced by the absence of PC2 (Figure 2A). Normal calcium oscillations can be restored in PKD1-deficient cells via the reintroduction of mouse wild-type PC1, and in PKD2 knockout cells via the transfection of full-length PC2 (reference[16] and Figure 2B). These findings suggest that PC1 could negatively regulate NCCE channels in a mechanism involving PC2 expression.
Figure 2.
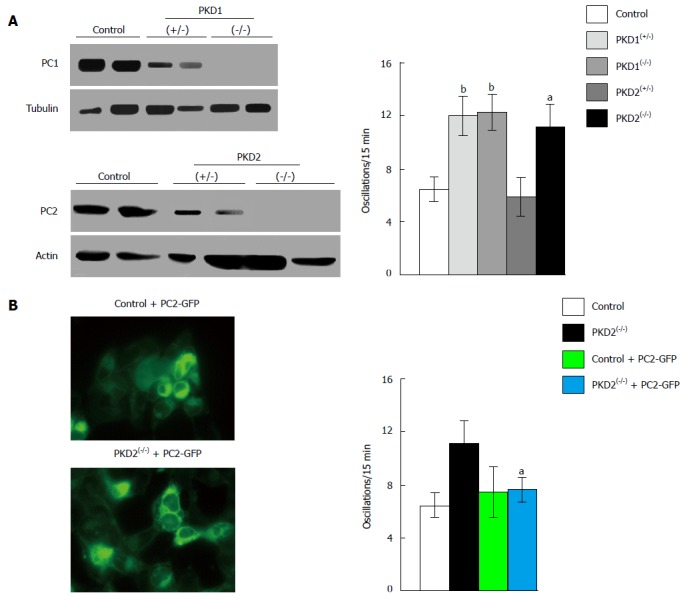
Downregulation of PKD1 and PKD2 genes increases fetal bovine serum-induced calcium oscillations in HEK293 cells. A: The stable transfection of HEK293 cells with plasmids containing specific anti-PKD1 and anti-PKD2 sequences causes a partial (+/-) or complete (-/-) downregulation of PC1 and PC2 expression compared with HEK293 cells stably transfected with scramble sequences (control). PKD1 and PKD2 gene silencing was evaluated by Western blotting using anti-PC1 and anti-PC2 antibodies. Calcium oscillations were increased in both partially (+/-) and fully (-/-) cells silenced for the PKD1 gene, as well as in fully (-/-) PKD2-silenced cells, as compared with scramble-treated cells (control). The number of oscillations/15 min were: 12 ± 1.5 in PKD1(+/-) cells, 12.2 ± 1.42 in PKD1(-/-) cells and 11.13 ± 1.79 in PKD2(-/-) cells, vs 6.39 ± 1.09 in control cells (bP < 0.01; aP < 0.05); B: The expression of full-length exogenous PC2 fused with GFP in PKD2(-/-) cells restores normal calcium oscillations (11.13 ± 1.79 oscillations/15 min in PKD2(-/-) cells vs 7.72 ± 1.07 in PKD2(-/-) cells transiently transfected with PKD2-GFP cDNA; aP < 0.05). Western blotting, oscillation recording and cell imaging were performed as previously reported[16]. Data, obtained from three different experiments analyzing at least 45 cells for every HEK293 clone, are represented as mean ± standard deviation. Analysis of data was performed using Student’s t test, and differences were considered significant at a value of P < 0.05. PKD: Polycystic kidney disease; HEK293: Human embryonic kidney cells; GFP: Green fluorescent protein; PC: Polycystin.
Taken as a whole, the evidence above suggests that the increased cell proliferation, fluid secretion and kidney cyst development seen in ADPKD may arise due to either the loss of polycystin complex function or an imbalance in the PC1/PC2 ratio causing intracellular calcium changes that trigger the B-Raf/MEK/ERK signaling cascade, as well as mTOR and NFAT pathways (Figure 1). However, the different effects on intracellular calcium concentration and downstream events observed with PC1 fragments and full-length PC1 expression suggest that further investigations are needed to clarify the function of polycystins in the regulation of calcium signaling.
CALCIUM SIGNALING AND APOPTOSIS IN ADPKD CELLS
In ADPKD, cyst formation and expansion rely on multiple mechanisms, including apoptosis, whose levels are higher in kidney cells from patients with ADPKD with respect to healthy individuals[39]. As apoptosis is one of the multiple cellular processes regulated by calcium signaling, this increase in apoptosis may be associated with abnormal intracellular calcium influx. Indeed, it has been demonstrated that cell sensitivity to apoptotic stimuli can be enhanced by calcium accumulation in the ER of renal epithelial cells deprived of functional PC2[40]. Conversely, expression of the PC2 protein, which functions as a calcium channel, inhibits apoptosis by lowering ER calcium levels[40]. Therefore, polycystin dysfunction appears to bring about an imbalance in ER calcium concentration through a reduction in the activity of the PC2 channel, causing calcium overload in the ER. This increase in ER calcium concentration, and its subsequent release, sensitizes cystic kidney cells to apoptotic stimuli. The excess calcium released from the ER is absorbed by the mitochondria, potentially causing damage that may lead to the release of cytochrome C, which activates the programmed cell death (Figure 1). In light of these findings, it seems that PC2 may function as an anti-apoptotic calcium channel in kidney epithelial cells[40]. Likewise, programmed cell death in kidney cells may be also regulated by PC1. In fact, apoptosis is prevented in MDCK cells, through the activation of the phosphatidylinositol 3-kinase/Akt signaling pathway, by the expression of full-length PC1[41,42].
CALCIUM CHANNELS AS A TARGET FOR ADPKD THERAPY
Drugs able to inhibit mTOR and cAMP-related pathways have already completed clinical trials. In particular, the use of the vasopressin V2 receptor Tolvaptan has led to significant improvements in renal function[43], although treatment with mTOR signaling pathway inhibitors did not yield satisfactory results[44]. Investigation into the calcium modulator molecules as an alternative treatment for ADPKD has also begun. To this end, significant results have already been achieved in preclinical trials of triptolide, an active diterpene that induces intracellular calcium release through a PC2-dependent mechanism. Treatment with this molecule improved renal function in a mouse model for ADPKD, inhibiting cyst expansion by restoring normal calcium signaling and cell proliferation[45,46]. Conversely, retrospective studies have shown that treating ADPKD patients with calcium channel blockers provokes a worsening of renal function, as compared to untreated patients, by reducing the glomerular filtration rate[47]. However, treatment of PKD2(-/WS25) ADPKD mice with R-568, a type-2 calcimimetic molecule that triggers the activation of calcium-sensing receptors, showed no detectable effect on cystogenesis[48]. Nonetheless, despite the unsatisfactory results yielded by current therapeutic interventions relying on calcium channel modulators, it is worthwhile continuing this line of research, as further studies into other calcium regulators may lead to the discovery of more efficient drugs.
CONCLUSION
Polycystin complex, formed by the interaction between PC1 and PC2, may function as a calcium-permeable receptor-channel complex able to regulate intracellular calcium signaling. As both PC1 and PC2 are mutated in ADPKD, and in light of their effects on cell proliferation and apoptosis, considered typical hallmarks of ADPKD, it is highly plausible that such mutations play a central role in the disease. In complex or alone, PC1 and PC2 can both act in different cellular compartments, including the plasma membrane, endoplasmic reticulum and primary cilium, but the downstream effects of their dysfunction in ADPKD have still not been clarified. Nevertheless, it is known that functional loss of either PC1 or PC2 causes calcium signaling disruption, which is considered a primary event for kidney cyst formation in ADPKD. Although intracellular Ca2+ alteration abnormally activates several pathways that stimulate cell proliferation in ADPKD cystic cells, including cAMP-dependent B-Raf/MEK/ERK signaling and mTOR, EGFR and NFAT pathways, these are not activated in the cyst formation process associated with ciliary signaling impairment. Further investigation is therefore required to clarify the function of polycystins, and in turn identify new targets for ADPKD treatment.
ACKNOWLEDGMENTS
We greatly thank Professor Harris PC (Mayo Clinic, Rochester, MN, United States) for performing Western blot analyses, and Professor Witzgall R (Institute for Molecular and Cellular Anatomy, University of Regensburg, Germany) for providing full-length PKD2-GFP plasmid.
Footnotes
Supported by University of Ferrara local funds: FAR 2012, 2013, 2014 and Regione Emilia Romagna grant (Ricerca Regione-Università) 2007-2009.
Conflict-of-interest statement: The authors declare that they have no commercial, personal, political, intellectual or religious conflict of interests regarding the data or scientific reports included in the present article. There is no conflict of interest associated with the senior author or other coauthors contributed their efforts in this paper.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: September 10, 2015
First decision: October 8, 2015
Article in press: December 11, 2015
P- Reviewer: Saleem M, Sands JM, Yorioka N S- Editor: Gong XM L- Editor: A E- Editor: Jiao XK
References
- 1.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321–337. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cornec-Le Gall E, Audrézet MP, Le Meur Y, Chen JM, Férec C. Genetics and pathogenesis of autosomal dominant polycystic kidney disease: 20 years on. Hum Mutat. 2014;35:1393–1406. doi: 10.1002/humu.22708. [DOI] [PubMed] [Google Scholar]
- 3.Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–987. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- 4.Wu G, D’Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, Maeda Y, Le TC, Hou H, Kucherlapati R, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93:177–188. doi: 10.1016/s0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 5.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 6.Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich BE, Somlo S. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol. 2002;4:191–197. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 7.González-Perrett S, Kim K, Ibarra C, Damiano AE, Zotta E, Batelli M, Harris PC, Reisin IL, Arnaout MA, Cantiello HF. Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc Natl Acad Sci USA. 2001;98:1182–1187. doi: 10.1073/pnas.98.3.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsiokas L, Arnould T, Zhu C, Kim E, Walz G, Sukhatme VP. Specific association of the gene product of PKD2 with the TRPC1 channel. Proc Natl Acad Sci USA. 1999;96:3934–3939. doi: 10.1073/pnas.96.7.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet. 1997;16:179–183. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]
- 10.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 11.Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408:990–994. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Wright JM, Qian F, Germino GG, Guggino WB. Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J Biol Chem. 2005;280:41298–41306. doi: 10.1074/jbc.M510082200. [DOI] [PubMed] [Google Scholar]
- 13.Sammels E, Devogelaere B, Mekahli D, Bultynck G, Missiaen L, Parys JB, Cai Y, Somlo S, De Smedt H. Polycystin-2 activation by inositol 1,4,5-trisphosphate-induced Ca2+ release requires its direct association with the inositol 1,4,5-trisphosphate receptor in a signaling microdomain. J Biol Chem. 2010;285:18794–18805. doi: 10.1074/jbc.M109.090662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Santoso NG, Yu S, Woodward OM, Qian F, Guggino WB. Polycystin-1 interacts with inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling with implications for polycystic kidney disease. J Biol Chem. 2009;284:36431–36441. doi: 10.1074/jbc.M109.068916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santoso NG, Cebotaru L, Guggino WB. Polycystin-1, 2, and STIM1 interact with IP(3)R to modulate ER Ca release through the PI3K/Akt pathway. Cell Physiol Biochem. 2011;27:715–726. doi: 10.1159/000330080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aguiari G, Trimi V, Bogo M, Mangolini A, Szabadkai G, Pinton P, Witzgall R, Harris PC, Borea PA, Rizzuto R, et al. Novel role for polycystin-1 in modulating cell proliferation through calcium oscillations in kidney cells. Cell Prolif. 2008;41:554–573. doi: 10.1111/j.1365-2184.2008.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang P, Luo Y, Chasan B, González-Perrett S, Montalbetti N, Timpanaro GA, Cantero Mdel R, Ramos AJ, Goldmann WH, Zhou J, et al. The multimeric structure of polycystin-2 (TRPP2): structural-functional correlates of homo- and hetero-multimers with TRPC1. Hum Mol Genet. 2009;18:1238–1251. doi: 10.1093/hmg/ddp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Köttgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol. 2008;182:437–447. doi: 10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc Natl Acad Sci USA. 2007;104:6454–6459. doi: 10.1073/pnas.0610324104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pelucchi B, Aguiari G, Pignatelli A, Manzati E, Witzgall R, Del Senno L, Belluzzi O. Nonspecific cation current associated with native polycystin-2 in HEK-293 cells. J Am Soc Nephrol. 2006;17:388–397. doi: 10.1681/ASN.2004121146. [DOI] [PubMed] [Google Scholar]
- 21.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13:2508–2516. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 22.Nauli SM, Rossetti S, Kolb RJ, Alenghat FJ, Consugar MB, Harris PC, Ingber DE, Loghman-Adham M, Zhou J. Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol. 2006;17:1015–1025. doi: 10.1681/ASN.2005080830. [DOI] [PubMed] [Google Scholar]
- 23.Jin X, Mohieldin AM, Muntean BS, Green JA, Shah JV, Mykytyn K, Nauli SM. Cilioplasm is a cellular compartment for calcium signaling in response to mechanical and chemical stimuli. Cell Mol Life Sci. 2014;71:2165–2178. doi: 10.1007/s00018-013-1483-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bai CX, Giamarchi A, Rodat-Despoix L, Padilla F, Downs T, Tsiokas L, Delmas P. Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO Rep. 2008;9:472–479. doi: 10.1038/embor.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ong AC, Wheatley DN. Polycystic kidney disease--the ciliary connection. Lancet. 2003;361:774–776. doi: 10.1016/S0140-6736(03)12662-1. [DOI] [PubMed] [Google Scholar]
- 26.Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet. 2013;45:1004–1012. doi: 10.1038/ng.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279:40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 28.Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol. 2006;17:178–187. doi: 10.1681/ASN.2005060645. [DOI] [PubMed] [Google Scholar]
- 29.Choi YH, Suzuki A, Hajarnis S, Ma Z, Chapin HC, Caplan MJ, Pontoglio M, Somlo S, Igarashi P. Polycystin-2 and phosphodiesterase 4C are components of a ciliary A-kinase anchoring protein complex that is disrupted in cystic kidney diseases. Proc Natl Acad Sci USA. 2011;108:10679–10684. doi: 10.1073/pnas.1016214108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25:18–32. doi: 10.1681/ASN.2013040398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE. Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. J Am Soc Nephrol. 2014;25:232–237. doi: 10.1681/ASN.2013010077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aguiari G, Bizzarri F, Bonon A, Mangolini A, Magri E, Pedriali M, Querzoli P, Somlo S, Harris PC, Catizone L, et al. Polycystin-1 regulates amphiregulin expression through CREB and AP1 signalling: implications in ADPKD cell proliferation. J Mol Med (Berl) 2012;90:1267–1282. doi: 10.1007/s00109-012-0902-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003;63:1983–1994. doi: 10.1046/j.1523-1755.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 34.Mekahli D, Parys JB, Bultynck G, Missiaen L, De Smedt H. Polycystins and cellular Ca2+ signaling. Cell Mol Life Sci. 2013;70:2697–2712. doi: 10.1007/s00018-012-1188-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aguiari G, Catizone L, Del Senno L. Multidrug therapy for polycystic kidney disease: a review and perspective. Am J Nephrol. 2013;37:175–182. doi: 10.1159/000346812. [DOI] [PubMed] [Google Scholar]
- 36.Aguiari G, Campanella M, Manzati E, Pinton P, Banzi M, Moretti S, Piva R, Rizzuto R, del Senno L. Expression of polycystin-1 C-terminal fragment enhances the ATP-induced Ca2+ release in human kidney cells. Biochem Biophys Res Commun. 2003;301:657–664. doi: 10.1016/s0006-291x(02)03011-5. [DOI] [PubMed] [Google Scholar]
- 37.Manzati E, Aguiari G, Banzi M, Manzati M, Selvatici R, Falzarano S, Maestri I, Pinton P, Rizzuto R, del Senno L. The cytoplasmic C-terminus of polycystin-1 increases cell proliferation in kidney epithelial cells through serum-activated and Ca(2+)-dependent pathway(s) Exp Cell Res. 2005;304:391–406. doi: 10.1016/j.yexcr.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 38.Puri S, Magenheimer BS, Maser RL, Ryan EM, Zien CA, Walker DD, Wallace DP, Hempson SJ, Calvet JP. Polycystin-1 activates the calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway. J Biol Chem. 2004;279:55455–55464. doi: 10.1074/jbc.M402905200. [DOI] [PubMed] [Google Scholar]
- 39.Goilav B. Apoptosis in polycystic kidney disease. Biochim Biophys Acta. 2011;1812:1272–1280. doi: 10.1016/j.bbadis.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 40.Wegierski T, Steffl D, Kopp C, Tauber R, Buchholz B, Nitschke R, Kuehn EW, Walz G, Köttgen M. TRPP2 channels regulate apoptosis through the Ca2+ concentration in the endoplasmic reticulum. EMBO J. 2009;28:490–499. doi: 10.1038/emboj.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boletta A, Qian F, Onuchic LF, Bhunia AK, Phakdeekitcharoen B, Hanaoka K, Guggino W, Monaco L, Germino GG. Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Mol Cell. 2000;6:1267–1273. doi: 10.1016/s1097-2765(00)00123-4. [DOI] [PubMed] [Google Scholar]
- 42.Boca M, Distefano G, Qian F, Bhunia AK, Germino GG, Boletta A. Polycystin-1 induces resistance to apoptosis through the phosphatidylinositol 3-kinase/Akt signaling pathway. J Am Soc Nephrol. 2006;17:637–647. doi: 10.1681/ASN.2005050534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367:2407–2418. doi: 10.1056/NEJMoa1205511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xue C, Dai B, Mei C. Long-term treatment with mammalian target of rapamycin inhibitor does not benefit patients with autosomal dominant polycystic kidney disease: a meta-analysis. Nephron Clin Pract. 2013;124:10–16. doi: 10.1159/000354398. [DOI] [PubMed] [Google Scholar]
- 45.Leuenroth SJ, Okuhara D, Shotwell JD, Markowitz GS, Yu Z, Somlo S, Crews CM. Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc Natl Acad Sci USA. 2007;104:4389–4394. doi: 10.1073/pnas.0700499104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leuenroth SJ, Bencivenga N, Igarashi P, Somlo S, Crews CM. Triptolide reduces cystogenesis in a model of ADPKD. J Am Soc Nephrol. 2008;19:1659–1662. doi: 10.1681/ASN.2008030259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitobe M, Yoshida T, Sugiura H, Shiohira S, Shimada K, Nitta K, Tsuchiya K. Clinical effects of calcium channel blockers and renin-angiotensin-aldosterone system inhibitors on changes in the estimated glomerular filtration rate in patients with polycystic kidney disease. Clin Exp Nephrol. 2010;14:573–577. doi: 10.1007/s10157-010-0329-5. [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Harris PC, Somlo S, Batlle D, Torres VE. Effect of calcium-sensing receptor activation in models of autosomal recessive or dominant polycystic kidney disease. Nephrol Dial Transplant. 2009;24:526–534. doi: 10.1093/ndt/gfn527. [DOI] [PMC free article] [PubMed] [Google Scholar]