

Abstract
Although transcriptional induction of stress genes constitutes a major cellular defense program against a variety of stressors, posttranslational control directly regulating the activities of preexisting stress proteins provides a faster-acting alternative response. We propose that posttranslational control is a general adaptive mechanism operating in many stress pathways. Here with the aid of computational models, we first show that posttranslational control fulfills two roles: (1) handling small, transient stresses quickly and (2) stabilizing the negative feedback transcriptional network. We then review the posttranslational control pathways for major stress responses—oxidative stress, metal stress, hyperosmotic stress, DNA damage, heat shock, and hypoxia. Posttranslational regulation of stress protein activities occurs by reversible covalent modifications, allosteric or non-allosteric enzymatic regulations, and physically induced protein structural changes. Acting in feedback or feedforward networks, posttranslational control may establish a threshold level of cellular stress. Sub-threshold stresses are handled adequately by posttranslational control without invoking gene transcription. With supra-threshold stress levels, cellular homeostasis cannot be maintained and transcriptional induction of stress genes and other gene programs, eg, those regulating cell metabolism, proliferation, and apoptosis, takes place. The loss of homeostasis with consequent changes in cellular function may lead to adverse cellular outcomes. Overall, posttranslational and transcriptional control pathways constitute a stratified cellular defense system, handling stresses coherently across time and intensity. As cell-based assays become a focus for chemical testing anchored on toxicity pathways, examination of proteomic and metabolomic changes as a result of posttranslational control occurring in the absence of transcriptomic alterations deserves more attention.
Keywords: posttranslational, transcriptional, feedback, stress, pathway
Stress response pathways are characterized by a common structure, including sensors, transcription factors, and kinase/phosphatase transducers (Simmons et al., 2009). All of the so-called canonical pathways—oxidative stress, metal stress, hyperosmotic stress, DNA damage, heat shock, hypoxia, endoplasmic reticulum stress, and inflammation—have these components. If these pathways serve as the major repertoire by which cells deal with increasing stress levels, transcriptional activation of existing, or new gene networks should be expected to regulate low levels of stress, limiting adverse cellular responses. Over the past few years, we have examined concentration–responses for multiple compounds with differing DNA-damage mechanisms in relation to the ability of cells to initiate functional responses. To our surprise (Clewell et al., 2014; Sun et al., 2014), all measured functional responses associated with changes in p53 pathway proteins, cell cycle arrest, apoptosis, etc., were less sensitive than was micronuclei formation, a marker of irreversible damage (Fig. 1A). At chemical concentrations less than those causing increases in micronuclei and altered gene expression, we consistently found significant changes in the rates and extent of formation of DNA-repair centers (DRCs). They appear and resolve quickly at low levels of genotoxic stress (Figs. 1B and 1C). These DRCs form as DNA damage leads to phosphorylation of preexisting proteins required for repair that then come together and co-localize at sites of damage, independent of transcriptional induction (Neumaier et al., 2012). Our analysis of the DNA damage response led us to consider how prevalent posttranslational control might be for controlling other stress pathways in response to low-level stressors. Here, we undertake a more detailed review of posttranslational processes involved in the control of these canonical stress pathways to assess their importance for responses in in vitro cell systems.
FIG. 1.

BMD evaluation and DRC formation in DNA damage response. A, BMD estimates for various in vitro endpoints in HT-1080 cells after 24-h treatments with 3 compounds respectively, ETP (etoposide), QUE (quercetin), and MMS (methylmethane sulfonate) capable of causing DNA damage—measured as micronuclei formation. Although activation of gene transcription program was expected to moderate stress responses and prevent micronuclei formation, micronuclei were nevertheless the most sensitive endpoint measured in relation to their BMD. The data are adapted from a published table (Clewell et al., 2014). B–D, Dynamic behaviors of DRCs in response to various levels of genotoxic damage by radiomimic chemical neocarzinostatin (NCS) in HT-1080 cells. B, Images of DRC foci detected by TP53BP1 antibody at various times following treatment with 5 or 25 ng/ml NCS. C, Dynamics of quantified TP53BP1 foci/cell in response to various concentrations of NCS treatment. Foci form quickly but only resolve relatively quickly with low NCS concentrations whereas high NCS concentrations lead to more persistent unresolved foci beyond 24 h with potential adverse cellular outcomes. D, TP53BP1 foci/cell remaining at 24 h in (B) (solid line, NOEL = 5 ng/ml) and cumulative (total) TP53BP1 foci/cell produced during the first 24 h (dashed line, NOEL = 0.5 ng/ml). The data were originally reported in Sun et al. (2014).
An organism’s ability to cope with and adapt to adverse external conditions and to maintain internal stability is implemented at multiple levels of biological hierarchy, ie, in whole bodies, organs, tissues, and cells. Adaptation is especially important for unicellular species living in frequently changing, unpredictable surroundings. In multicellular organisms such as mammals, the extracellular microenvironment is relatively stable as a result of whole-body homeostatic regulation, yet the capability of stress response is still largely intact and essential at the cellular level. Many environmental and industrial chemicals cause cellular stress responses, and a good understanding of how cells resist chemical perturbations is important for developing toxicity tests relevant to human health safety assessment and for interpreting the results of these tests.
A suite of intracellular biochemical control networks underpin adaptive cellular stress responses and homeostasis. There is a consistent topological scheme to these networks, emerging across various types of stresses and species. These control networks are primarily organized in the form of negative feedback and/or incoherent feedforward loops (Fig. 2) (Zhang and Andersen, 2007). The stress response pathways underlying these control networks can be activated by transcriptional, translational, and posttranslational processes. The most well studied of these is the transcriptionally mediated program, where the stress first activates a master transcription factor, followed by the induction of a suite of stress genes by the transcription factor to help restore homeostasis (Fig. 2, solid lines). Each of the stress response pathways—including pathways responding to oxidative stress, metal stress, hyperosmotic stress, DNA damage, heat shock, and hypoxia—has its own set of sensor molecules, master transcription factors, and key stress genes (Simmons et al., 2009). These canonical, transcriptionally mediated response programs are essential for survival under normal or stressed conditions as evidenced by embryonic lethality or compromised viability of animals with deletions of the master transcription factors or sensor proteins (Simmons et al., 2009). Many studies have explored the design principles of cellular stress response networks from a control engineering perspective, including activation of the master transcription factors, transcriptional induction of stress genes, and complex formation of functional stress proteins (El-Samad et al., 2005; Muzzey et al., 2009; Zhang and Andersen, 2007; Zhang et al., 2010a).
FIG. 2.

Cellular stress response to external stressor (S) may involve both transcriptional (outer solid lines) and posttranslational control (inner dotted lines) of the cellular state (Y). The transcriptional induction of stress genes (g) is activated by transcription factor (T) either through feedback (bottom arm) or feedforward (top arm). Posttranslational control bypasses the slow-acting transcriptional loops by regulating the activities of preexisting molecules of stress proteins (G) through covalent modifications or other fast mechanisms (dotted lines). Pointed arrows denote activation and blunted arrows denote inhibition.
Although transcriptionally mediated gene induction responds in the face of large perturbations, posttranslational regulation of stress proteins also contributes to controlling cellular stress. Perturbed cellular states, such as altered redox potential, metal concentrations, and cell volume, or the perturbing stressors themselves, may directly alter preexisting stress proteins (Fig. 2, dotted lines). These posttranslational alterations may occur in a variety of forms, including reversible covalent modifications (such as phosphorylation, methylation, acetylation, oxidation, ubiquitination, and sumoylation), allosteric or non-allosteric regulation (such as inhibition of an enzyme by its end product) or physically induced protein structural changes (such as by high temperature or mechanical forces) (Schaber et al., 2012; Winter and Jakob, 2004; Zhang et al., 2010a). These alternations of stress proteins lead to changes in their activities to mitigate perturbations elicited by the stress and restore cellular homeostasis. Importantly, these posttranslational control programs operate quickly without requiring transcriptional activation.
Thus, cells have evolved two parallel pathways: the transcriptionally dependent canonical pathways that increase the abundance of stress proteins, and the transcriptionally independent posttranslational pathways that increase the activity of stress proteins. This raises the following questions. Why do cells need two separate ways to handle stresses; what different roles do the two play; and how are they coordinated to operate across time and stress intensity? In this article, we first use simple computational modeling to show why transcriptional control alone is inadequate to control stress across all time scales and stressor levels, while on the other hand, rapid posttranslational control effectively handles transient and low-level stresses and stabilizes the intrinsic transcriptional network. We then review posttranslational control processes for various stress responses, including oxidative stress, metal stress, hyperosmotic stress, DNA damage, heat shock, and hypoxia, using examples from both mammalian and unicellular organisms. As we gain a better understanding of the posttranslational regulation of stress pathway functions, it will be possible to use these cellular behaviors to identify points-of-departure for dose response modeling and to transform design of in vitro testing system for these stress pathways.
ROLES OF POSTTRANSLATIONAL CONTROL IN STRESS RESPONSES
Transcriptional stress control is unlikely to be sufficient for robust adaptation, due primarily to the time required to go from transcriptional activation to completed synthesis of new gene products. These processes require several hours or even days before there are meaningful increases in protein abundance. In addition, some transcriptional stress responses may require several levels of response in a cascade, further delaying the appearance of new gene products. For stresses lasting for a short period of time but occurring frequently, the transcriptional program would not be activated in time to counteract the transient changes in stressor intensity. To illustrate this issue we calculated expected responses for a transcriptional feedback model (Fig. 3A, see details in Supplementary Material and the SBML models). For transient but recurring stresses, where each episode lasts from minutes to about an hour, there is hardly any transcriptional induction of the stress protein (G). As a result, the cellular state (Y) varies dramatically, rising and falling along with the stressor S (Fig. 3C). In contrast, when there is a feedback path for posttranslational activation of the stress protein (Fig. 3B), the stress-handling capacity of the cell increases quickly, in the order of seconds to minutes, as seen with the rapid rise of the active form of G (Fig. 3D). As a result, the cellular state Y is only perturbed minimally, displaying very limited fluctuations. Posttranslational control also helps curb the initial impact to the cellular state at the onset of a high-level, persistent stress (simulation not shown). In this way cells gain time, in the absence of otherwise severe damage, to launch the slower-acting transcriptional program for handling the chronic stress.
FIG. 3.
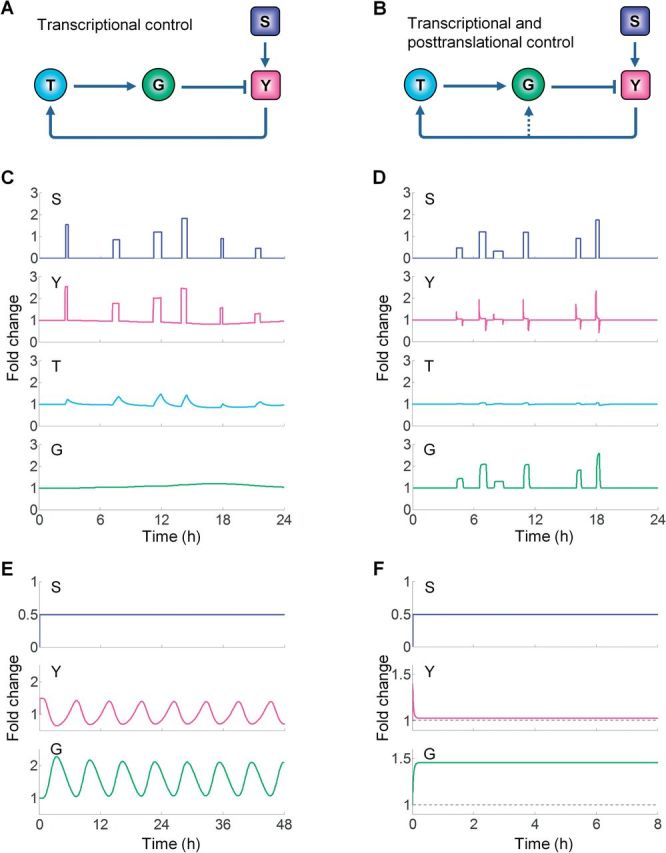
Simulations comparing the different dynamic behaviors in the absence (A) versus presence (B) of posttranslational control. Posttranslational control allows effective handling of transient stresses. C, Transcriptionally mediated induction of the stress protein G cannot keep up with the rapidly changing stressor S, resulting in nearly unmitigated consequences on the cellular state variable, Y. D, Posttranslational activation of stress protein G results in more rapid upregulation of anti-stress activity, occurring almost in sync with the changing stressor S. The cellular state Y shows very brief increases at the initiation of the square wave for stressor S and then brief decreases with the cessation of the square wave input. Posttranslational control stabilizes the transcriptionally mediated negative feedback circuit, reducing pathway oscillations. E, A transcriptionally mediated negative feedback circuit with high amplification is prone to oscillation. F, Adding a fast-acting posttranslational pathway eliminates the time delay and damps out or eliminates oscillation. See Supplementary Material and the accompanying SBML model files for model details.
The second limitation with transcriptional control for stress responses is the potential instability of negative feedback networks. Negative feedback is the primary network motif for perturbation resistance and homeostasis; however, it is also the predominant network structure underlying biochemical oscillation (Novak and Tyson, 2008). Negative feedback loops oscillate when (1) there is a long time delay in signaling through the loop and (2) a high loop gain, ie, there is significant signal amplification within the feedback loop. The sequence of events from transcriptional factor activation to stress protein synthesis introduces significant time delays. Moreover, negative feedback circuits need a high loop gain to achieve near-perfect stress adaptation, which requires multiple ultrasensitive motifs (Zhang and Andersen, 2007). High loop gains can be achieved with ultrasensitive motifs embedded within the feedback loop, which strongly amplify the signal representing the altered cellular state, leading to a high induction of stress proteins (Zhang et al., 2013). Our simulations illustrated that sustained oscillations readily arise with transcriptionally mediated feedback containing ultrasensitive gene induction (Fig. 3E). Feedback circuits may also produce damped oscillations that prolong the time to reach adaptation. Due to its faster time scale, the posttranslational feedback loop ‘short-circuits’ the transcriptional feedback loop, drastically shortening the time delay in signaling. In this way, the feedback loop oscillations either become much smaller or disappear entirely (Fig. 3F).
Another factor limiting the ability of transcriptionally mediated control to effectively handle small stresses is that many stress genes have a constitutive (basal) expression level, independent of that driven by their master transcription factors. With this basal expression, the sensitivity of gene expression to activated transcription factors would be small (in percentage-change terms), resulting in insufficient induction of stress genes, especially when responding to low-level stresses where the extent of transcription factor activation is still small (Zhang and Andersen, 2007). Insufficient increases in stress gene expression would lead to incomplete counteracting of and only partial adaptation to even small stresses. In contrast, posttranslational processes, such as reversible covalent modification of preexisting stress proteins by kinases or other types of enzymes, can provide strong signal amplification to overcome the sub-sensitivity in the transcriptional response to small stresses (Goldbeter and Koshland, 1981).
Taken together, these limitations imply that by itself transcriptionally mediated negative feedback control does not serve to maintain robust homeostasis for handling small intermittent changes in cellular stress or more significant prolonged stresses with quick adaptation. Inclusion of a feedback path of posttranslational activation of stress proteins qualitatively changes the dynamic behaviors of the transcriptionally mediated feedback circuit and overcomes the drawbacks of transcriptional control (Schaber et al., 2012). In the following section, we look at various posttranslational mechanisms that are at work with specific cellular stress pathways (summarized in Supplementary Table S1).
POSTTRANSLATIONAL CONTROL OF STRESS RESPONSES
Oxidative stress response
During basal metabolism, reactive oxygen species (ROS), mainly superoxide anion (O2−·), hydrogen peroxide (H2O2), and hydroxyl radical (OH·), are constantly produced primarily in the mitochondrion (Turrens, 2003). To limit the adverse effects of ROS on nucleic acids, proteins and lipids, cells contain a system of endogenous antioxidants, including specific enzymes and small scavenging molecules (Nguyen et al., 2003). The enzymes include superoxide dismutase, glutathione peroxidase, catalase, glutaredoxin, peroxiredoxin, thioredoxin, thioredoxin reductase (TrxR), glutathione reductase (GR), glutamate cysteine ligase (GCL), and glucose-6-phosphate dehydrogenase. The latter three enzymes are responsible for the synthesis of glutathione (GSH) and reducing agents such as NADPH. Transcriptional induction of these endogenous antioxidant enzymes, mediated by redox sensor Keap1 and transcription factor Nrf2, serves as long-term responses to oxidant stress (Motohashi and Yamamoto, 2004). In contrast, rapid fluctuations in oxidative stress require fast, redox-sensitive changes in the cellular antioxidant capacity, which occur through reversible posttranslational modifications on the thiol group of the cysteine residues in antioxidant enzymes (Zhang et al., 2010a). Common types of modification include sulfenation, s-glutathionylation, and disulfide bond formation (Jacob et al., 2012).
Under oxidative stress, GSH depletion disrupts cellular redox homeostasis. Limiting GSH depletion is crucial to the cell’s ability to handle these oxidative perturbations. Synthesis of GSH, a tripeptide, relies on two enzymatic reactions. In the first, GCL covalently attaches glutamate to cysteine. GCL is a heterodimer containing two subunits, GCL catalytic (GCLC) and GCL modulatory (GCLM). Although oxidative or electrophilic stress transcriptionally increases the expression of these subunits, they also undergo direct oxidative covalent modification by reactive species, resulting in rapid alterations in their catalytic activity absent any changes in the enzyme’s abundance (Ochi, 1995, 1996). The reversible process of association and dissociation between the GCLC and GCLM subunits is sensitive to redox potential changes: an oxidative intracellular environment promotes association and thus formation of the holoenzyme (Fraser et al., 2002; Huang et al., 1993; Krejsa et al., 2010; Seelig et al., 1984; Tu and Anders, 1998). The redox-regulation of the holoenzyme activity and heterodimerization is likely to ensue from cysteine modifications in the subunit proteins (Fraser et al., 2003; Tu and Anders, 1998). For example, cysteine 553 of GCLC can be covalently modified by low-concentrations of 4-hydroxy-2-nonenal, significantly increasing GCLC’s enzymatic activity (Backos et al., 2011). Another way to rapidly boost GSH production is through feedback inhibition of its own synthesis. GSH binds to the same site as glutamate on GCLC, competitively inhibiting GCLC activity (Richman and Meister, 1975). Together, these posttranslational mechanisms enhance GCL activity to help restore the GSH level very quickly in the face of any decrease in its concentration (Fig. 4A, a and b).
FIG. 4.
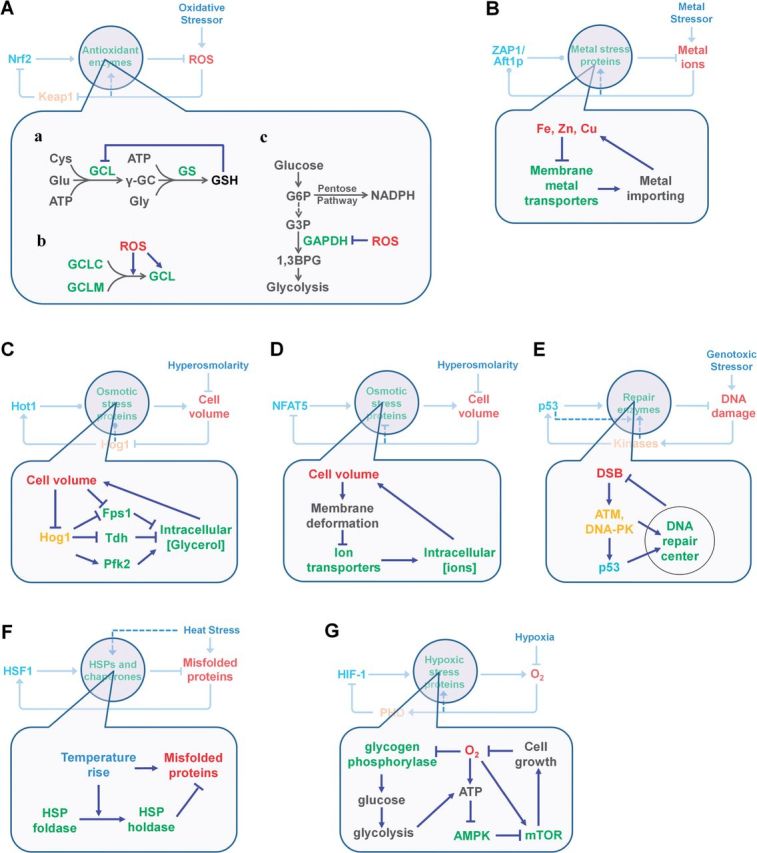
Posttranslational control processes in stress response pathways. A, Oxidative stress response, B, Yeast metal stress response, C, Yeast hyperosmotic stress response, D, Mammalian hyperosmotic stress response, E, DNA damage response, F, Heat shock response, and G, hypoxic response. Denotations of colored arrow heads: pointed, activation; blunted, inhibition; dotted, activation or inhibition. Refer to the text for details of these processes.
NADPH, a key reducing agent in antioxidant defense, contributes many reactions, including GR-catalyzed conversion of GSSG to GSH and TrxR-catalyzed reduction of thioredoxin. These reactions convert NADPH to NADP. The recycling of NADP back to NADPH relies primarily on the pentose phosphate pathway, a branch of glucose metabolism that competes with the glycolysis pathway for carbon flux. Both pathways use a common substrate, glucose-6-phosphate (G6P). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a rate-limiting enzyme in the glycolysis pathway, and some other glycolytic enzymes including enolase, are redox-sensitive in both mammalian and yeast cells (Brodie and Reed, 1990; Schuppe-Koistinen et al., 1994; Shenton and Grant, 2003). Upon exposure to H2O2 or other oxidative chemicals, these enzymes can be reversibly S-thiolated, resulting in their inhibition. In this way, oxidative stress causes rapid inhibition of GAPDH, decreasing flux of glucose metabolism through glycolysis, redirecting G6P to the pentose pathway and producing more NADPH to support various antioxidant reactions utilizing NADPH (Fig. 4A, c) (Grant, 2008; Ralser et al., 2007). Other enzymes also control NADPH production and consumption (Ying, 2008). Malate dehydrogenase catalyzes a reaction that produces NADPH. The inter-converting enzyme pyridine nucleotide transhydrogenase and enzymes such as those involved in NADH production, isocitrate dehydrogenase, can regulate the equilibrium between NADPH and NADP. Any oxidative stress-induced posttranslational changes affecting the activities of these enzymes—oxidation, phosphorylation, and oligomerization—would quickly regulate NADPH/NADH homeostasis and the ability of the cell to cope with oxidative stress.
The posttranslational control mechanisms for oxidative stress may also operate with reductive stressors, which could also result in adverse outcomes if left unchecked (Brewer et al., 2013). The enzymatic activities that regulate the intracellular redox potential can be either enhanced or suppressed depending on the direction of the redox potential change. In this way, the cellular redox state remains within a narrow range regardless of the presence of oxidative or reductive stress.
Metal stress response
Cellular stress response from exposures to high concentrations of metals, especially heavy metals such as zinc (Zn), copper (Cu), and cadmium (Cd), is regulated at both transcriptional and posttranslational levels. Zn can directly bind to and activate transcription factor MTF-1, which induces a suite of stress genes responsible for metal homeostasis (Gunther et al., 2012). Among them, cysteine-rich metallothionein sequesters Zn and other heavy metals, reducing their free concentrations in the cytosol. Metal transporters, such as slc30a1 and slc30a2 encoding ZnT-1 and ZnT-2 exporters, pump Zn out of the cell. In contrast, slc39a10 encoding Zip10 importer is transcriptionally repressed by MTF-1, reducing Zn uptake. MTF-1 is activated by metals other than Zn, such as Cu and Cd, through competitive binding to metallothionein. At high concentrations, Cu and Cd replace Zn associated with metallothionein, freeing up Zn to activate MTF-1.
Posttranslational control of metal stress is so far best studied in yeast. It generally involves a mechanism of stress-elicited downregulation of cell membrane metal importers (Fig. 4B). In yeast, Ctr1p is the rate-limiting transporter for Cu uptake and is stable under low-copper conditions. It was found that higher concentrations of Cu specifically induce degradation of Ctr1p within an hour, and the downregulation is due to increased proteolysis rather than Ctr1p internalization (Ooi et al., 1996). This posttranslational mechanism allows a relatively quick shutdown of Cu uptake, and along with induced repression of Ctr1p gene transcription, it prevents Cu overload. Similar to the Cu transporter, Zn transporters ZRT1 and ZRT2, in addition to being transcriptionally repressed via inhibition of transcription activator ZAP1 by Zn, are also subject to stress-induced stability regulation. Exposure of yeast cells to high Zn concentrations first induces ubiquitination of ZRT1 (Gitan and Eide, 2000). Ubiquitinated ZRT1 is prone to increased internalization, causing a rapid loss of membrane ZRT1, as evidenced by a decrease of ZRT1 half-life from >150 to 39 min under Zn stress (Gitan et al., 1998). The internalized ZRT1 is later degraded by vacuolar protease.
Yeast iron transporter, comprising the multicopper oxidase Fet3p and transmembrane-permease Ftr1p, is regulated by iron both transcriptionally via iron-sensing Aft1p and posttranslationally. Similar to ZRT1, exposure to high iron concentrations leads to internalization and degradation by vacuolar protease of both Fet3p and Ftr1p (Felice et al., 2005). The internalization of Ftr1p, not Fet3p, requires ubiquitination. Interestingly, the direct trigger of Ftr1p internalization is not the high iron concentration per se in either the extracellular or intracellular space; rather it requires the active transportation of iron through the permease. In Arabidopsis, iron-regulated transporter 1 is also subject to increased turnover by iron through possible ubiquitination of lysine residues in an intracellular loop of the transporter protein (Kerkeb et al., 2008).
The regulation of iron concentrations in mammalian cells primarily involves posttranscriptional processes, including alterations in translation and stabilization of mRNAs for the key genes involved in iron homeostasis (Anderson et al., 2012). At low iron concentrations, the iron regulatory protein 1 (IRP1) lacking the [4Fe-4S] cluster has a high affinity for the iron-response elements located in the 5′ or 3′ untranslated region of several mRNAs encoding proteins ferritin, ferroportin, and transferrin receptor (Haile et al., 1992). IRP1 binding to these mRNAs hinders translation of ferritin and ferroportin and stabilizes transferrin receptor mRNA (Binder et al., 1994; Gray and Hentze, 1994; McKie et al., 2000). At high iron concentrations, formation of the [4Fe-4S] cluster in IRP1 causes dissociation of IRP1 from target mRNAs. Ferritin and ferroportin are then translated at higher rates and transferrin receptor, whose mRNA becomes less stable, at a lower rate. Higher levels of ferritin and ferroportin bind and export more iron molecules, respectively. The lower levels of transferrin receptor reduces cellular iron uptake. Together, these processes reduce free concentrations of iron in the cell to maintain homeostasis.
Hyperosmotic stress response
High osmolality in the extracellular space causes immediate water loss and shrinkage of the cell, a physical stress that has to be handled swiftly. Cells deal with hyperosmotic stress by increasing the intracellular concentrations of osmolytes, which would pull water back into the cell. For the adaptive osmotic stress response, yeast cells are the best studied model system (Fig. 4C). The canonical pathway involves the following sequence of events: signal transduction from the membrane sensor proteins Sln1 and Sho1 to Hog1, a yeast MAPK homolog, translocation of Hog1 into the nucleus, phosphorylation of transcription factors such as Hot1 by Hog1, and transcriptional regulation of stress genes including Gpd1, which increases the synthesis of glycerol, the main osmolyte used by yeast (Miermont et al., 2011). However, an increasing number of studies have begun to show that this transcriptional control scheme may not be as crucial as originally thought. In contrast, fast posttranslational control of glycerol production (Fig. 5) appears to be essential, and sufficient in many cases, especially for surviving mild to moderate hyperosmotic stresses (Mettetal et al., 2008).
FIG. 5.

Schematic illustration of Hog1-mediated posttranslational and transcriptional hyperosmotic stress response pathway. At low stress levels, activated Hog1 posttranslationally alters the activities of a suite of enzymes involved in glycerol metabolism (Pfk2, Tdh, Gpd1, Fps1, etc.), resulting in increased production and decreased exportation of intracellular glycerol. At high stress levels, Hog1 activates transcription factor Hot1, which transcriptionally regulates the expression of the enzymes above, resulting in alterations in their abundance, which helps increase intracellular glycerol concentration on a longer time scale. Denotations of line colors: blue, posttranslational control; red, transcriptional control; green, common pathway shared by both posttranslational and transcriptional control. Denotations of colored arrow heads: pointed, activation; blunted, inhibition; dotted, activation or inhibition depending on target.
By blocking Hog1 from entering the nucleus or tethering it to the plasma membrane, Westfall et al. (2008) showed that while these yeast cells lack apparent Hog1-initiated stress gene induction, they could withstand hyperosmotic stress as well as wide-type cells do. Subsequent studies further confirmed that increased glycerol production flux in wild-type yeast cells is for the most part not due to de novo synthesis of related enzymes such as GPD1 and GPP, even though their expression levels do increase under hyperosmotic stress (Bouwman et al., 2011). Direct Hog1-mediated metabolic regulation of glycerol production seems to play a predominant role. The increased glycerol production can be partially explained by posttranslational events such as phosphorylation of Tdh by Hog1 (Westfall et al., 2008). Tdh is an enzyme isoform of GAPDH which converts glyceraldehyde-3-phosphate (GA3P) to 1,3-biphosphoglycerate, diverting GA3P away from being used for the glycerol-synthesizing branch. Phosphorylation of GAPDH by Hog1 inhibits GAPDH activity, making more GA3P available for glycerol synthesis and leading to its rapid accumulation in the cell. In addition, phosphorylation and activation of 6-phosphofructo-2-kinase (Pfk2) directly or indirectly by Hog1 also contributes to glycerol accumulation (Dihazi et al., 2004). Pfk2 catalyzes a metabolic reaction that produces fructose 2,6-bisphosphate, which is a signaling molecule that activates 6-phosphofructo-1-kinase. The latter increases the supply of 1,6-bisphosphate as a substrate precursor for glycerol synthesis. Reduced clearance of intracellular glycerol also appears to be important and can be mediated posttranslationally. One of the main ways glycerol leaves cells is through the membrane transporter Fps1. Hog1 regulates Fps1 directly or indirectly through phosphorylation, resulting in its closure or loss from the cell membrane (Beese et al., 2009; Lee et al., 2013; Mollapour and Piper, 2007). Independent of Hog1, the mechanical force generated by decreased cell volume or turgor pressure may directly close Fps1 as well, producing the fastest adaptive response among the mechanisms discussed so far (Luyten et al., 1995; Schaber et al., 2012). Both Hog1-dependent and independent closure of Fps1, along with its increased production, lead to fast accumulation of glycerol in the cell, which quickly restores cell volume.
The results of a computational model of the yeast Hog1 pathway based on an ensemble modeling approach incorporating existing literature also supported the importance of fast posttranslational control (Schaber et al., 2012). It concluded that (1) the main adaptation mechanism is through posttranslational activation of glycerol production rather than through transcriptional gene induction; this posttranslational feedback pathway is tonically active at unstressed conditions (Macia et al., 2009). (2) A secondary posttranslational mechanism is glycerol channel closure mediated through Hog1 or directly driven by turgor pressure changes, leading to glycerol retention. (3) Transcriptional response is still activated but at a later time, and is more relevant in response to severe stresses (Mettetal et al., 2008). It may also serve to reset Hog1 activity to pre-stress levels and replenish the pool of stress proteins most of which have been posttranslationally modified, thus readying cells for still rapid response to future hyperosmotic stresses. (4) Fast posttranslational feedback control of glycerol production and of upstream components such as Sln1 or Sho1 helps stabilize the transcriptional feedback circuit, reducing the likelihood of oscillation.
Although less frequent, osmotic stress is relevant to mammalian cells in a number of circumstances, including cells in the renal medulla where antidiuresis constantly occurs, and metabolically active cells such as lymphocytes in the thymus, where rapid clonal expansion causes quick consumption of intracellular nutrients, leading to lower intracellular osmotic pressures. Osmotic stress responses could also be important for intestinal and skin cells which may experience rapid osmotic changes in the extracellular environment. In mammalian cells, an immediate response to hyperosmolarity, which occurs in a matter of seconds to minutes, is the so-called ‘regulatory volume increase’ (RVI). RVI is mediated by rapid changes in the activities of preexisting membrane ion transporters such as Na+–K+–2Cl- cotransporter, Na+/H+ exchanger, and Cl−/HCO3− exchanger (Hoffmann et al., 2009), which allow accumulation of ions in the cell, temporarily boosting the intracellular osmolarity (Fig. 4D). Moderate RVI occurs in isolated guinea-pig tracheal epithelial cells exposed to hyperosmotic solutions (Fedan et al., 2013), suggesting that the airway epithelium may rely on RVI to counteract cell volume shrinkage in the presence of inhaled hyperosmotic aerosols. In response to hypoosmotic stress ‘regulatory volume decrease’ (RVD) occurs. In this regard, cystic fibrosis transmembrane conductance regulator (CFTR), a cell membrane Cl− transporter, besides being regulated by protein kinases, also appears to serve as a mechanosensitive gating channel (Zhang et al., 2010b). CFTR may play a role in RVD: its deletion impairs fast cell volume regulation in response to hypotonic challenges (Valverde et al., 1995). Overall, dysregulation of transmembrane electrolyte and fluid transfer via ion transporters may lead to pathological conditions such as those that occur in cystic fibrosis patients who often possess mutations in the CFTR gene (Guggino, 1999).
RVI is usually followed by accumulation of non-ionic osmolytes through transcriptional induction of pertinent stress genes, which increase the uptake and synthesis of sorbitol, myoinositol, neutral amino acids, and their derivatives (Burg et al., 1997; Yancey et al., 1982). The master transcription factor activating the transcriptional program is TonEBP/NFAT5 (Miyakawa et al., 1999). It appears that what triggers the activation of TonEBP/NFAT5 is the increase in the intracellular ionic concentrations due to initial cell volume shrinkage and importation of Na+, K+, and Cl− as part of the RVI process rather than the mechanical force of cell volume shrinkage (Rodgaard et al., 2008). It is unclear whether mammalian cells possess a posttranslational pathway homologous to the yeast Hog1 pathway.
DNA damage response
The DNA damage response to genotoxic insults is an essential stress pathway crucial to the maintenance of genome integrity. Although there is a large literature base on tumor suppressor protein p53-induced transcriptional events leading to inducible DNA repair, cell cycle arrest, and apoptosis, rapid formation of DRCs allows DNA damage to activate organization of various proteins including p53, p53-binding protein 1 (TP53BP1), and γ-H2AX at the sites of damage (Al Rashid et al., 2005; Neumaier et al., 2012). Formation of these DRCs appears to involve phosphorylation of preexisting proteins that then aggregate to the sites of damage. Large-scale proteomic studies found over 700 protein substrates are phosphorylated by ATM or ATR in response to ionizing radiation (Matsuoka et al., 2007). Many of them take part in mismatch repair, excision repair, cross-link repair, and homologous recombination repair, indicating that posttranslational enhancement of the cellular DNA repair capacity is a possible damage-control mechanism. Upon appearance of double strand breaks (DSBs), phosphorylated p53 moves to the damage foci, where localized γ-H2AX, phosphorylated ATM, DNA-PK, scaffold proteins, and repair proteins may participate to form DRCs (Al Rashid et al., 2005). These observations suggest a direct, transcription-independent role of p53 in DSB repair (Fig. 4E).
Rapid formation/resolution of DRCs suggests that existing repair and accessory proteins work to repair these lesions, in addition to transcriptional upregulation of p53-dependent genes at higher levels of damage (Lisby and Rothstein, 2004; Neumaier et al., 2012). In our laboratories at The Hamner, we have conducted concentration–response evaluations of multiple biomarkers after treating HT-1080 cells with chemicals causing different types of DNA damage (Clewell et al., 2014; Sun et al., 2014). The response curve for the formation of micronuclei, a biomarker of cellular adversity, had a lower benchmark concentration than for any other measures, with the exception of formation of DRCs. DRCs form quickly after exposure and at low levels of damage they resolve quickly as well (Fig. 1). The architecture of the stress controlling pathways for DNA damage has characteristics similar to the HOG pathway. In each case there is a phosphorylated intermediate (p53 and Hog1, respectively) that plays a dual role: altering activities of key enzymes and acting as a transcriptional regulator at higher levels of cellular perturbation.
Sumoylation of DNA repair proteins has emerged as an important posttranslational modification mechanism that quickly upregulates cellular DNA repair capacity (Sarangi and Zhao, 2015). In normal human skin fibroblasts UV irradiation induces rapid sumoylation of XPC, a protein that participates in the early stage of nucleotide excision repair (Wang et al., 2005). Sumoylation appears to stabilize the XPC protein, preventing it from ubiquitination-mediated degradation. In yeast, sumoylation of endonuclease Rad1 takes place at the site of DNA damage following exposure to UV irradiation or some genotoxicants (Sarangi et al., 2014). Sumoylation accelerates the dissociation of Rad1 from DNA substrates after completion of nucleotide cleavage, promoting efficient proceeding to subsequent repair steps. Rad52, a DNA repair protein involved in repair via homologous recombination, becomes sumoylated in response to DNA DSBs, a process believed to stabilize Rad52 from degradation (Sacher et al., 2006).
Heat shock response
At the cellular level, heat shock due to temperature rise causes significant disruption to the proteasome, resulting in protein unfolding, misfolding, and aggregation. To handle heat-induced disruption, cells possess an intricate set of heat shock proteins (HSPs), including primarily molecular chaperones, co-chaperones, and small HSPs (Priya et al., 2013; Richter et al., 2010). These proteins are transcriptionally upregulated by heat shock through activated transcription factors, such as HSF1 in eukaryotic cells and σ32 in prokaryotic cells. HSPs function both as holdases to sequester misfolded proteins, preventing them from aggregating, and as foldases to help fold misfolded molecules back to their native 3D configurations (Richter et al., 2010). As a stress pathway involving both feedback and feedforward regulations, the heat shock response, especially the transcriptionally mediated pathway, has been closely examined from a systems control perspective (El-Samad et al., 2005; Guisbert et al., 2008). Below we describe the posttranslational aspect of the heat shock pathway.
Although heat can disrupt the structures of biomacromolecules, thermally induced structural changes are also exploited by nature to sense temperature shift as a feedforward signal to kick-start heat shock defense (Schumann, 2012). A number of HSPs can undergo heat-induced conformation changes with consequent alterations in their chaperone activities. These changes generally make chaperones function as more efficient holdases, which capture non-native protein intermediates generated acutely due to heat shock (Fig. 4F). This way, the futile, ATP-consuming repair/damage cycle—refolding and releasing protein substrates which are only to be misfolded again if the heat stress persists—is prevented (Winter and Jakob, 2004).
Most small HSPs exist as large oligomers containing up to 50 monomeric subunits at normal temperatures (Haslbeck, 2002). They primarily function as chaperones preventing non-native protein from forming non-functional aggregates (Jakob et al., 1993). When exposed to heat, some small HSPs such as Hsp26 in yeast, a spherical 24mer (Bentley et al., 1992) dissociate reversibly into smaller oligomers, such as dimers (Franzmann et al., 2008; Haslbeck et al., 1999; Stromer et al., 2003). As a result of dissociation, more hydrophobic surface of Hsp26 is exposed, allowing for more high-affinity binding to misfolded or unfolded protein substrates. The augmented holdase capacity prevents non-natively structured proteins from assembling into large aggregates and prepares cells for refolding, after stress recedes, with the help of large ATP-utilizing HSPs such as Hsp70. Another example of heat-induced de-oligomerization of small HSPs is the dodecameric Hsp16.9 in wheat, which disassembles into high-affinity dimers for binding to protein substrates (Winter and Jakob, 2004).
In Escherichia coli, the major chaperone system contains DnaK, DnaJ, and GrpE, which are also subject to heat-induced function shift from primarily a foldase to holdase. The thermo-sensor is the co-chaperone GrpE, a nucleotide exchange factor. GrpE is a homodimer comprising two monomeric subunits that are held together by interactions between two α-helices of the monomers. At normal temperatures, unfolding protein substrates are first captured by DnaJ and then presented to ATP-bound DnaK (an Hsp70), which hydrolyzes into ADP-bound DnaK that has a high affinity for the DnaJ and protein substrate complex. As a nucleotide exchange factor, GrpE then comes in and replaces the ADP to ATP in DnaK, which diminishes its affinity for the protein substrate, allowing the refolded substrate to be released (Winter and Jakob, 2004). At high temperatures, the pairing α-helices of the two monomeric subunits of GrpE partially ‘melt’, a structural change that suppresses the nucleotide exchange activity of GrpE. As a result, DnaK is left in the high-affinity state, holding protein substrates as long as the heat stress persists (Grimshaw et al., 2001, 2003). This way, protein substrates are prevented from being released prematurely into the cytosol under conditions not favoring protein refolding, and futile ATP/ADP cycles of DnaK are avoided (Siegenthaler et al., 2004). Similarly, the second major HSP system in E. coli, GroEL/GroES, also shifts from a foldase to holdase position under heat stress due to potentially heat-induced structural alterations that affect interactions between the two proteins (Goloubinoff et al., 1997; Llorca et al., 1998). Another temperature-regulated HSP in E. coli is DegP. Its function can shift from being a chaperone to a protease as temperature increases. This way, protein substrates that are severely damaged and impossible to refold can be degraded directly (Spiess et al., 1999).
Hypoxic stress response
Hypoxia, a lowering of the partial oxygen pressure in the extracellular fluid, disrupts energy homeostasis of cells. The hypoxic response involves alterations of a number of metabolic pathways, including inhibition of ATP-consuming anabolic metabolism. Under hypoxia, the master transcription factor HIF-1 is stabilized due to diminished hydroxylation of its proline residue by O2. HIF-1 then partners with ARNT to induce a suite of genes relevant in anti-hypoxic response. Besides this canonical transcriptional response, a number of proteins are targeted for posttranslational modification under hypoxia (Kumar and Klein, 2004; Kumar and Prabhakar, 2008). Those that respond acutely and immediately relevant to oxygen and energy homeostasis are discussed below (Fig. 4G).
mTOR is a serine/threonine kinase, which phosphorylates multiple target proteins promoting protein synthesis and cell growth. mTOR is inhibited under hypoxia to reduce ATP consumption and thus oxygen consumption. Inhibition of mTOR occurs through various mechanisms including transcriptional repression through HIF-1 and posttranslational regulation. Among the latter, activation of AMPK due to lowered ATP levels under hypoxia plays a major role. AMPK activates tuberous sclerosis complex-1 and -2 (TSC1/2), which functions as a GTPase that converts GTP-Rheb, a G protein, into GDP-Rheb (Liu et al., 2006). Because only GTP-Rheb can associate with and activate the mTORC1 complex that contains mTOR, conversion of GTP-Rheb into GDP-Rheb thus inhibits mTOR, shutting down cell growth-related protein synthesis. Inhibition of mTOR can also be effected through phosphorylation of mTOR binding partner, raptor, by AMPK (Gwinn et al., 2008). In addition, under hypoxia the mTORC1 pathway can be inhibited posttranslationally independent of AMPK, potentially through a heme-containing protein (Tan and Hagen, 2013), or through hypophosphorylation of mTOR or its downstream effectors involved in translation initiation (Arsham et al., 2003).
Under hypoxia, glucose metabolism switches from oxygen-consuming oxidative phosphorylation to glycolysis to maintain ATP production and reduce ROS production (Brahimi-Horn et al., 2007). Among the immediate responses, the active form of glycogen phosphorylase in the rat heart increases in response to acute hypoxia (England and Krause, 1987). This may increase the supply of glucose to glycolysis for more ATP production. In response to hypoxia, there is also a rapid increase in the adduction of soluble epoxide hydrolase (sEH) by electrophilic 15-deoxy-delta-prostaglandin J2 (15 d-PGJ2), potentially at cysteine 521. The adduction inhibits sEH activity, which normally metabolizes vasodilating epoxyeicosatrienoic acids (EET). This way, EET accumulates and vasodilation increases blood supply to the hypoxic tissue (Charles et al., 2011).
DISCUSSION
Over the past decade, we have seen the emergence of numerous activities to develop novel in vitro assays for examining cellular responses to chemicals with diverse modes of action. In the EU, the initiatives were largely developed to support 3R efforts—refinement, reduction, and replacement—for the use of laboratory animals. This program dates back to the work of Russell and Burch and their groundbreaking book on animal alternatives (Russell and Burch, 1959). The ongoing efforts to validate in vitro assays through the European Committee for the Validation of Animal Alternatives (ECVAM) have a well-defined process for determining if in vitro assays provide equivalent results with those obtained from more conventional, usually in-life studies in test animals. In the USA, there was growing frustration that the methods for assessing risks from chemicals were becoming too cumbersome, expensive, and time-consuming to provide adequate testing of the large numbers of compounds in commerce and those entering commerce each year. The US EPA pushed forward with the ToxCast program to use repurposed assays from the pharmaceutical industry to develop ‘signatures’ for the biological activities of large numbers of compounds using tools called quantitative high-throughput screening (qHTS). The 2007 NRC report, Toxicity Testing in the 21st Century: A Vision and A Strategy, noted that in vitro testing in human cells or cell lines should be the preferred approach to modern toxicity testing, providing deep biological understanding on toxicity pathways affected in human tissues across broad regions of dose and perturbation (NRC, 2007).
In categorizing expected cellular responses, the two broadest dichotomies for toxic responses are those that are receptor-mediated and those that are reactivity-mediated. In general, stress pathway responses are associated with tissue reactivity or with changes in a physical state, such as temperature or osmotic pressure that affect macromolecular integrity. Much of the focus for receptor-mediated pathways relates to alterations in gene expression by receptor-agonist complexes acting as transcriptional activators, eg, ER, AR, AhR, CAR, and PPARα. The activities of these pathways, as measured in ToxCast Phase 1 for a group of 300 plus pesticides and pesticide inerts, provided readouts on receptor binding, receptor transactivation, reporter gene activities, etc. (Dix et al., 2007). These assays provided much less information on stress pathways. Stress pathway responses are available from whole-genome microarray evaluations of in vitro and in vivo assays that show activation of canonical stress response sub-networks. These responses indicate the presence of significant perturbations that cause transcriptional activation of the pathways. In addition, there are ongoing efforts to develop multiplexed assays for stress pathway activation using binding or activation of key transcriptional components involved in stress pathway signaling. In our view, the appearance of transcriptional activation of the stress pathways concurs with adversity at the cellular level. A recent genomic study on a number of legacy chemicals demonstrated that the appearance of cancer or non-cancer apical endpoint outcomes coincided with the most sensitive transcriptional changes at comparable benchmark doses and at the same exposure time (Thomas et al., 2013). As discussed here, the dose regions below those activating gene induction through key transcription factors likely correspond to low-level stresses where adaptive control is fully at work. Examination of cellular responses at these low stressor levels will require different biomarkers than are currently available for transcription factor binding to DNA or altered gene expression. For the DNA-damage response pathway, in the adaptive region, there were increases in DRCs that resolved quickly after their formation, while at concentrations of genotoxic chemicals causing changes in p53-mediated gene expression there were more DRCs but they did not resolve as quickly. At these high concentrations, the level of micronuclei, a marker of genotoxic adversity, began to increase significantly compared with control cells (Clewell et al., 2014).
When examined in detail, the design characteristics of transcriptional feedback control are clearly not optimal for maintaining homeostasis, ie, a region of stress level where there is no increase in the controlled variable (Y in our examples) despite some increases in the stressor S (Fig. 2). Transcriptional control has time delays associated with both RNA and protein synthesis. The transcriptional feedback loops also become prone to oscillation with fluctuating levels of Y leading to fluctuating rates of transcription (ie, levels of G). These less-than-optimal characteristics of the transcriptional feedback loop are avoided with posttranslational feedback control. Many posttranslational regulations involve protein covalent modifications, as occurs with enzymes involved in oxidative stress response and in osmotic stress response. Physically induced protein structure and configuration changes are also at play for chaperones in heat shock response and for glycerol transporter and ion transporters in osmotic stress response. Although many of the posttranslational regulations discussed were studied in low-level organisms such as bacteria and yeasts, the conservation of stress responses across species suggests that they also operate in higher multicellular organisms (Kultz, 2003). Further, several of the stress pathways are well studied in mammalian systems, including oxidative stress and DNA damage. In higher organisms, inflammation, as part of the innate immune response, is also regarded by some as a stress response. It primarily requires NF-kB-mediated transcriptional events, involving multiple types of cells interacting in a local tissue. Nonetheless, some acute non-transcriptional responses also seem to occur, including the respiratory burst associated with phagocytosis in neutrophils and macrophages and release of pro-inflammatory mediators via exocytosis by neutrophils (Hampton et al., 1998; Jiang et al., 2011; Sheshachalam et al., 2014). These acute innate immune responses involving only posttranslational events may be sufficient to contain minor pathogen invasion or tissue damage.
Because posttranslational control appears to work more efficiently to ensure homeostatic adaptation, why would cells still use transcriptional activation at higher stress levels? There must be considerable values accrued to cells to invest in coordinated activation of posttranslational and transcriptional pathways to control cellular stress and maintain homeostasis. One reason may be energy efficiency. It would require a large amount of preexisting stress proteins as a reserve to allow prompt action in the face of high stresses. The maintenance of a large protein reservoir at unstressed conditions would be an energy burden to the cell, making posttranslational control energetically inefficient if used alone across a wider range of stress levels. Second, as suggested by the study of yeast osmotic stress response, transcriptional induction of stress genes may also serve to reset the posttranslational pathway to a different set-point, making it ready for future acute insults superimposed on the already elevated basal stress (Mettetal et al., 2008; Schaber et al., 2012). Lastly, various transcriptional networks can be activated in the face of high stresses to affect multiple cellular functions. The activated networks would integrate information from multiple stress pathways and coordinate signals to make decisions on cell cycle arrest, apoptosis, inflammation, and other functions.
How do cells coordinate the posttranslational and transcriptional control pathways to coherently cope with stresses? The background stress and small transient excursions in ambient conditions require constant fine tuning of the rates at which cells alleviate stress conditions. In yeast, active basal signaling from the sensor molecule Sln to Hog1 provides a tonic control mediated via posttranslational modifications of existing proteins (Macia et al., 2009). Posttranslational regulation of glycerol metabolism by a combination of Hog1-dependent and Hog1-independent processes is able to fend off mild and moderate hyperosmotic stresses (Westfall et al., 2008). Thus at low stress levels, posttranslational pathways are the main player absorbing the impact and avoiding transcriptional induction. Only at high stress levels when the posttranslational pathway is running at maximum capacity, does the transcriptional pathway become activated, inducing genes to replenish the pool of stress proteins and activate other cellular response pathways. A schematic of this type of control transition is provided (Fig. 6), capturing the differential behavior expected with increasing stressor levels culminating in transcriptional activation. In the yeast osmotic stress response, Hog1 is used as a common signaling protein for active posttranslational control of low-level stress and then for transcriptional activation of multiple genes at higher levels of stress. The p53 protein may act in a similar fashion by contributing to DRCs at low levels of DNA damage and activating transcription at higher levels of damage. One possible way for the switching from posttranslational to transcriptional control to occur is through, in network motif terminology, molecular titration (Buchler and Louis, 2008). At low stress levels the increased phosphorylated forms of Hog1 and p53 are all engaged in posttranslational processes which, through high-affinity binding, titrate Hog1 and p53 away from DNA promoter binding. Only at higher stress levels when phosphorylated Hog1 and p53 rise to levels high enough to saturate the posttranslational control processes in which they participate, do they become available for transcriptional activation. This way, a threshold level of cellular stress can be defined separating activation of transcriptional processes from posttranslational ones. Other network motif mechanisms giving rise to threshold responses at the cellular level are also possible, as we have recently discussed (Zhang et al., 2014). One possibility is integral feedback control in the Hog1 pathway which is believed to underpin perfect adaptation in the yeast osmotic stress response (Muzzey et al., 2009). Hormetic response curves may result from posttranslational control that operates in a feedforward manner, where overcompensation of the perturbed cellular state may occur at low stress levels (Kim et al., 2008).
FIG. 6.

A proposed model for coherent transition from posttranslational control to transcriptional control as stressor level increases. A, At basal condition in the absence of exogenous stressor, a small fraction of preexisting stress proteins are posttranslationally modified to cope with background/endogenous stress. B, At very low stressor levels, more preexisting stress proteins are posttranslationally modified and thus activated to maintain homeostasis. C, At slightly higher stressor levels, even more preexisting stress proteins are posttranslationally modified and activated to maintain homeostasis. D, At considerably higher stressor levels, preexisting stress proteins are exhausted in terms of posttranslational modification; the cellular state cannot be maintained at the baseline level, and transcriptional induction of stress genes and genes responsible for cell cycle arrest, apoptosis, and other functional changes starts to occur. At these stressor levels, loss of cellular homeostasis and altered cellular function/fate may lead to adverse outcomes. The dial denotes the cellular state that is perturbed and needs to be maintained.
Thus, posttranslational and transcriptional activities together cope with stresses coherently across time and across the intensity of stress. Posttranslational control allows rapid responses to acute or chronic, low-level stress by using preexisting stress proteins, while transcriptional control is initiated when dealing with higher-level stresses through de novo synthesis of more stress proteins. The stress level at which posttranslational control reaches a maximum may correspond to a threshold above which the controlled cellular state significantly deviates from the baseline level, leading to transcriptional activation of a broad suite of gene products, including both increased levels of stress proteins and other cellular function pathway proteins (Fig. 6D). This threshold may serve as a point of departure where cells transition from adaptation to stressed and to stress-related adversity. When a cell has perfectly adapted to a persistent stress, the cellular state initially perturbed (such as ROS, DNA damage, osmolarity, etc.) recovers to the pre-stress level. This adaptation requires changes in stress protein activities and/or their abundances. With reallocation of cellular resource and energy, these alterations may affect other cellular functions. For instance, upregulation of Nrf2 and antioxidant enzymes is protective against low-level oxidative stresses, but it may disrupt the physiological glucose-stimulated ROS signal that triggers insulin secretion in β-cells (Pi et al., 2010). As a result, chronic endogenous antioxidant upregulation, resulting from posttranslational and/or transcriptional control processes activated by oxidative stressors, may contribute to β-cell dysfunction and diabetes. Augmented expression of Nrf2 and antioxidant activities may promote chemoprevention, but it can also enhance the resistance of cancer cells to chemotherapy drugs (Kwak and Kensler, 2010). Therefore, whether an adaptive response is completely beneficial or may have ‘side-effects’ has to be evaluated by considering the time frame of stress exposure and its effects beyond controlling cellular homeostasis. Adaptive responses that include transcriptional changes are much more likely to have other cellular consequences.
Over the past decade, tools for assessing transcriptomic responses have improved considerably, allowing various evaluations such as estimates of Benchmark doses for enriched genes within biological pathways (Thomas et al., 2007) and for visualization of the pathways and networks affected by exposures (McMullen et al., 2014). There has been less appreciation of the ability of posttranslational activation, in the absence of transcriptional upregulation of stress genes, to achieve perfect control of low-level stressor exposures. In the context of new approaches for cell-based toxicity testing (NRC, 2007), we will need to take these posttranslational pathways into account in creating biomarkers and computational models of cellular threshold response. Biomarkers that look at transcriptional activation of canonical stress pathway signaling proteins reflect high-dose responses. In the region of adaptation, other biomarkers can provide insight into dose response and allow examining the issue of threshold response. Optimal biomarkers will differ from one stress pathway to another. In some instances, the focus will be phosphorylated protein products or specific responses such as DRC formation below levels of mutation or micronuclei formation. With others, it might be metabolomic changes in GSH below dose–time treatments that cause transcriptional activation. At this time, it is difficult to foresee approaches for multiplexing posttranslational responses across multiple stress pathways until we have better information on the non-transcriptional processes at work in dose regions reflecting adaptation. Using case studies for both p53-mediated DNA damage and GSH-depleting compounds for oxidative stress should allow better definition of the types of studies necessary to look in these adaptive regions and improve our tools for dose response below exposure levels showing transcriptional responses.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
The study was funded by a number of key sponsors of the Computational Systems Biology Pathway Modeling Program at The Hamner Institutes, including Unilever, the ExxonMobil Foundation, Dow Chemical Foundation, Dow Corning Chemical Corporation, the American Chemistry Council Long-Range Research Initiative, and NIEHS (ES016005 and P42ES04911). The authors have declared that there are no conflicts of interest.
Supplementary Material
REFERENCES
- Al Rashid S. T., Dellaire G., Cuddihy A., Jalali F., Vaid M., Coackley C., Folkard M., Xu Y., Chen B. P., Chen D. J., et al. (2005). Evidence for the direct binding of phosphorylated p53 to sites of DNA breaks in vivo. Cancer Res. 65, 10810–10821. [DOI] [PubMed] [Google Scholar]
- Anderson C. P., Shen M., Eisenstein R. S., Leibold E. A. (2012). Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 1823, 1468–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsham A. M., Howell J. J., Simon M. C. (2003). A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J. Biol. Chem. 278, 29655–29660. [DOI] [PubMed] [Google Scholar]
- Backos D. S., Fritz K. S., Roede J. R., Petersen D. R., Franklin C. C. (2011). Posttranslational modification and regulation of glutamate-cysteine ligase by the alpha,beta-unsaturated aldehyde 4-hydroxy-2-nonenal. Free Radic. Biol. Med. 50, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese S. E., Negishi T., Levin D. E. (2009). Identification of positive regulators of the yeast fps1 glycerol channel. PLoS Genet. 5, e1000738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley N. J., Fitch I. T., Tuite M. F. (1992). The small heat-shock protein Hsp26 of Saccharomyces cerevisiae assembles into a high molecular weight aggregate. Yeast 8, 95–106. [DOI] [PubMed] [Google Scholar]
- Binder R., Horowitz J. A., Basilion J. P., Koeller D. M., Klausner R. D., Harford J. B. (1994). Evidence that the pathway of transferrin receptor mRNA degradation involves an endonucleolytic cleavage within the 3' UTR and does not involve poly(A) tail shortening. Embo J. 13, 1969–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman J., Kiewiet J., Lindenbergh A., van Eunen K., Siderius M., Bakker B. M. (2011). Metabolic regulation rather than de novo enzyme synthesis dominates the osmo-adaptation of yeast. Yeast 28, 43–53. [DOI] [PubMed] [Google Scholar]
- Brahimi-Horn M. C., Chiche J., Pouyssegur J. (2007). Hypoxia signalling controls metabolic demand. Curr. Opin. Cell Biol. 19, 223–229. [DOI] [PubMed] [Google Scholar]
- Brewer A. C., Mustafi S. B., Murray T. V., Rajasekaran N. S., Benjamin I. J. (2013). Reductive stress linked to small HSPs, G6PD, and Nrf2 pathways in heart disease. Antioxid. Redox Signal. 18, 1114–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie A. E., Reed D. J. (1990). Cellular recovery of glyceraldehyde-3-phosphate dehydrogenase activity and thiol status after exposure to hydroperoxides. Arch. Biochem. Biophys. 276, 212–218. [DOI] [PubMed] [Google Scholar]
- Buchler N. E., Louis M. (2008). Molecular titration and ultrasensitivity in regulatory networks. J. Mol. Biol. 384, 1106–1119. [DOI] [PubMed] [Google Scholar]
- Burg M. B., Kwon E. D., Kultz D. (1997). Regulation of gene expression by hypertonicity. Annu. Rev. Physiol. 59, 437–455. [DOI] [PubMed] [Google Scholar]
- Charles R. L., Burgoyne J. R., Mayr M., Weldon S. M., Hubner N., Dong H., Morisseau C., Hammock B. D., Landar A., Eaton P. (2011). Redox regulation of soluble epoxide hydrolase by 15-deoxy-delta-prostaglandin J2 controls coronary hypoxic vasodilation. Circ. Res. 108, 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clewell R. A., Sun B., Adeleye Y., Carmichael P., Efremenko A., McMullen P. D., Pendse S., Trask O. J., White A., Andersen M. E. (2014). Profiling dose-dependent activation of p53-mediated signaling pathways by chemicals with distinct mechanisms of DNA damage. Toxicol. Sci. 142, 56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dihazi H., Kessler R., Eschrich K. (2004). High osmolarity glycerol (HOG) pathway-induced phosphorylation and activation of 6-phosphofructo-2-kinase are essential for glycerol accumulation and yeast cell proliferation under hyperosmotic stress. J. Biol. Chem. 279, 23961–23968. [DOI] [PubMed] [Google Scholar]
- Dix D. J., Houck K. A., Martin M. T., Richard A. M., Setzer R. W., Kavlock R. J. (2007). The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci. 95, 5–12. [DOI] [PubMed] [Google Scholar]
- El-Samad H., Kurata H., Doyle J. C., Gross C. A., Khammash M. (2005). Surviving heat shock: control strategies for robustness and performance. Proc. Natl. Acad. Sci. U.S.A. 102, 2736–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England P. J., Krause E. G. (1987). The effect of hypoxia on the phosphorylation of contractile and other proteins in perfused rat heart challenged by isoprenaline. Biomed. Biochim. Acta 46, 369–380. [PubMed] [Google Scholar]
- Fedan J. S., Thompson J. A., Ismailoglu U. B., Jing Y. (2013). Tracheal epithelium cell volume responses to hyperosmolar, isosmolar and hypoosmolar solutions: relation to epithelium-derived relaxing factor (EpDRF) effects. Front. Physiol. 4, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felice M. R., De Domenico I., Li L., Ward D. M., Bartok B., Musci G., Kaplan J. (2005). Post-transcriptional regulation of the yeast high affinity iron transport system. J. Biol. Chem. 280, 22181–22190. [DOI] [PubMed] [Google Scholar]
- Franzmann T. M., Menhorn P., Walter S., Buchner J. (2008). Activation of the chaperone Hsp26 is controlled by the rearrangement of its thermosensor domain. Mol. Cell 29, 207–216. [DOI] [PubMed] [Google Scholar]
- Fraser J. A., Kansagra P., Kotecki C., Saunders R. D., McLellan L. I. (2003). The modifier subunit of Drosophila glutamate-cysteine ligase regulates catalytic activity by covalent and noncovalent interactions and influences glutathione homeostasis in vivo. J. Biol. Chem. 278, 46369–46377. [DOI] [PubMed] [Google Scholar]
- Fraser J. A., Saunders R. D., McLellan L. I. (2002). Drosophila melanogaster glutamate-cysteine ligase activity is regulated by a modifier subunit with a mechanism of action similar to that of the mammalian form. J. Biol. Chem. 277, 1158–1165. [DOI] [PubMed] [Google Scholar]
- Gitan R. S., Eide D. J. (2000). Zinc-regulated ubiquitin conjugation signals endocytosis of the yeast ZRT1 zinc transporter. Biochem. J. 346(Pt 2), 329–336. [PMC free article] [PubMed] [Google Scholar]
- Gitan R. S., Luo H., Rodgers J., Broderius M., Eide D. (1998). Zinc-induced inactivation of the yeast ZRT1 zinc transporter occurs through endocytosis and vacuolar degradation. J. Biol. Chem. 273, 28617–28624. [DOI] [PubMed] [Google Scholar]
- Goldbeter A., Koshland D. E., Jr (1981). An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl. Acad. Sci. U.S.A. 78, 6840–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloubinoff P., Diamant S., Weiss C., Azem A. (1997). GroES binding regulates GroEL chaperonin activity under heat shock. FEBS Lett. 407, 215–219. [DOI] [PubMed] [Google Scholar]
- Grant C. M. (2008). Metabolic reconfiguration is a regulated response to oxidative stress. J. Biol. 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray N. K., Hentze M. W. (1994). Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. Embo J. 13, 3882–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimshaw J. P., Jelesarov I., Schonfeld H. J., Christen P. (2001). Reversible thermal transition in GrpE, the nucleotide exchange factor of the DnaK heat-shock system. J. Biol. Chem. 276, 6098–6104. [DOI] [PubMed] [Google Scholar]
- Grimshaw J. P., Jelesarov I., Siegenthaler R. K., Christen P. (2003). Thermosensor action of GrpE. The DnaK chaperone system at heat shock temperatures. J. Biol. Chem. 278, 19048–19053. [DOI] [PubMed] [Google Scholar]
- Guggino W. B. (1999). Cystic fibrosis and the salt controversy. Cell 96, 607–610. [DOI] [PubMed] [Google Scholar]
- Guisbert E., Yura T., Rhodius V. A., Gross C. A. (2008). Convergence of molecular, modeling, and systems approaches for an understanding of the Escherichia coli heat shock response. Microbiol. Mol. Biol. Rev. 72, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther V., Lindert U., Schaffner W. (2012). The taste of heavy metals: gene regulation by MTF-1. Biochim. Biophys. Acta 1823, 1416–1425. [DOI] [PubMed] [Google Scholar]
- Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., Shaw R. J. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haile D. J., Rouault T. A., Harford J. B., Kennedy M. C., Blondin G. A., Beinert H., Klausner R. D. (1992). Cellular regulation of the iron-responsive element binding protein: disassembly of the cubane iron-sulfur cluster results in high-affinity RNA binding. Proc. Natl. Acad. Sci. U.S.A. 89, 11735–11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton M. B., Kettle A. J., Winterbourn C. C. (1998). Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92, 3007–3017. [PubMed] [Google Scholar]
- Haslbeck M. (2002). sHsps and their role in the chaperone network. Cell. Mol. Life Sci. 59, 1649–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslbeck M., Walke S., Stromer T., Ehrnsperger M., White H. E., Chen S., Saibil H. R., Buchner J. (1999). Hsp26: a temperature-regulated chaperone. Embo J. 18, 6744–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann E. K., Lambert I. H., Pedersen S. F. (2009). Physiology of cell volume regulation in vertebrates. Physiol. Rev. 89, 193–277. [DOI] [PubMed] [Google Scholar]
- Huang C. S., Chang L. S., Anderson M. E., Meister A. (1993). Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J. Biol. Chem. 268, 19675–19680. [PubMed] [Google Scholar]
- Jacob C., Battaglia E., Burkholz T., Peng D., Bagrel D., Montenarh M. (2012). Control of oxidative posttranslational cysteine modifications: from intricate chemistry to widespread biological and medical applications. Chem. Res. Toxicol. 25, 588–604. [DOI] [PubMed] [Google Scholar]
- Jakob U., Gaestel M., Engel K., Buchner J. (1993). Small heat shock proteins are molecular chaperones. J. Biol. Chem. 268, 1517–1520. [PubMed] [Google Scholar]
- Jiang F., Zhang Y., Dusting G. J. (2011). NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 63, 218–242. [DOI] [PubMed] [Google Scholar]
- Kerkeb L., Mukherjee I., Chatterjee I., Lahner B., Salt D. E., Connolly E. L. (2008). Iron-induced turnover of the Arabidopsis IRON-REGULATED TRANSPORTER1 metal transporter requires lysine residues. Plant Physiol. 146, 1964–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Kwon Y. K., Cho K. H. (2008). The biphasic behavior of incoherent feed-forward loops in biomolecular regulatory networks. Bioessays 30, 1204–1211. [DOI] [PubMed] [Google Scholar]
- Krejsa C. M., Franklin C. C., White C. C., Ledbetter J. A., Schieven G. L., Kavanagh T. J. (2010). Rapid activation of glutamate cysteine ligase following oxidative stress. J. Biol. Chem. 285, 16116–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kultz D. (2003). Evolution of the cellular stress proteome: from monophyletic origin to ubiquitous function. J. Exp. Biol.206(Pt 18), 3119–3124. [DOI] [PubMed] [Google Scholar]
- Kumar G. K., Klein J. B. (2004). Analysis of expression and posttranslational modification of proteins during hypoxia. J. Appl. Physiol. (1985) 96, 1178–1186; discussion 1170–1172. [DOI] [PubMed] [Google Scholar]
- Kumar G. K., Prabhakar N. R. (2008). Post-translational modification of proteins during intermittent hypoxia. Respir. Physiol. Neurobiol. 164, 272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak M. K., Kensler T. W. (2010). Targeting NRF2 signaling for cancer chemoprevention. Toxicol. Appl. Pharmacol. 244, 66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Reiter W., Dohnal I., Gregori C., Beese-Sims S., Kuchler K., Ammerer G., Levin D. E. (2013). MAPK Hog1 closes the S. cerevisiae glycerol channel Fps1 by phosphorylating and displacing its positive regulators. Genes Dev. 27, 2590–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M., Rothstein R. (2004). DNA damage checkpoint and repair centers. Curr. Opin. Cell Biol. 16, 328–334. [DOI] [PubMed] [Google Scholar]
- Liu L., Cash T. P., Jones R. G., Keith B., Thompson C. B., Simon M. C. (2006). Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 21, 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorca O., Galan A., Carrascosa J. L., Muga A., Valpuesta J. M. (1998). GroEL under heat-shock. Switching from a folding to a storing function. J. Biol. Chem. 273, 32587–32594. [DOI] [PubMed] [Google Scholar]
- Luyten K., Albertyn J., Skibbe W. F., Prior B. A., Ramos J., Thevelein J. M., Hohmann S. (1995). Fps1, a yeast member of the MIP family of channel proteins, is a facilitator for glycerol uptake and efflux and is inactive under osmotic stress. Embo J. 14, 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia J., Regot S., Peeters T., Conde N., Sole R., Posas F. (2009). Dynamic signaling in the Hog1 MAPK pathway relies on high basal signal transduction. Sci. Signal. 2, ra13. [DOI] [PubMed] [Google Scholar]
- Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R., 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166. [DOI] [PubMed] [Google Scholar]
- McKie A. T., Marciani P., Rolfs A., Brennan K., Wehr K., Barrow D., Miret S., Bomford A., Peters T. J., Farzaneh F., et al. (2000). A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 5, 299–309. [DOI] [PubMed] [Google Scholar]
- McMullen P. D., Bhattacharya S., Woods C. G., Sun B., Yarborough K., Ross S. M., Miller M. E., McBride M. T., LeCluyse E. L., Clewell R. A., et al. (2014). A map of the PPARalpha transcription regulatory network for primary human hepatocytes. Chem. Biol. Interact. 209, 14–24. [DOI] [PubMed] [Google Scholar]
- Mettetal J. T., Muzzey D., Gomez-Uribe C., van Oudenaarden A. (2008). The frequency dependence of osmo-adaptation in Saccharomyces cerevisiae. Science 319, 482–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miermont A., Uhlendorf J., McClean M., Hersen P. (2011). The dynamical systems properties of the HOG signaling cascade. J. Signal. Transduct. 2011, 930940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa H., Woo S. K., Dahl S. C., Handler J. S., Kwon H. M. (1999). Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc. Natl. Acad. Sci. U.S.A. 96, 2538–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M., Piper P. W. (2007). Hog1 mitogen-activated protein kinase phosphorylation targets the yeast Fps1 aquaglyceroporin for endocytosis, thereby rendering cells resistant to acetic acid. Mol. Cell Biol. 27, 6446–6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H., Yamamoto M. (2004). Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends. Mol. Med. 10, 549–557. [DOI] [PubMed] [Google Scholar]
- Muzzey D., Gomez-Uribe C. A., Mettetal J. T., van Oudenaarden A. (2009). A systems-level analysis of perfect adaptation in yeast osmoregulation. Cell 138, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumaier T., Swenson J., Pham C., Polyzos A., Lo A. T., Yang P., Dyball J., Asaithamby A., Chen D. J., Bissell M. J., et al. (2012). Evidence for formation of DNA repair centers and dose-response nonlinearity in human cells. Proc. Natl. Acad. Sci. U.S.A. 109, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T., Sherratt P. J., Pickett C. B. (2003). Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 43, 233–260. [DOI] [PubMed] [Google Scholar]
- Novak B., Tyson J. J. (2008). Design principles of biochemical oscillators. Nat. Rev. Mol. Cell Biol. 9, 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NRC (2007). Toxicity Testing in the 21st Century: A Vision and a Strategy. The National Academies Press, Washington, DC. [Google Scholar]
- Ochi T. (1995). Hydrogen peroxide increases the activity of gamma-glutamylcysteine synthetase in cultured Chinese hamster V79 cells. Arch. Toxicol. 70, 96–103. [DOI] [PubMed] [Google Scholar]
- Ochi T. (1996). Menadione causes increases in the level of glutathione and in the activity of gamma-glutamylcysteine synthetase in cultured Chinese hamster V79 cells. Toxicology 112, 45–55. [DOI] [PubMed] [Google Scholar]
- Ooi C. E., Rabinovich E., Dancis A., Bonifacino J. S., Klausner R. D. (1996). Copper-dependent degradation of the Saccharomyces cerevisiae plasma membrane copper transporter Ctr1p in the apparent absence of endocytosis. Embo J. 15, 3515–3523. [PMC free article] [PubMed] [Google Scholar]
- Pi J., Zhang Q., Fu J., Woods C. G., Hou Y., Corkey B. E., Collins S., Andersen M. E. (2010). ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol. Appl. Pharmacol. 244, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya S., Sharma S. K., Goloubinoff P. (2013). Molecular chaperones as enzymes that catalytically unfold misfolded polypeptides. FEBS Lett. 587, 1981–1987. [DOI] [PubMed] [Google Scholar]
- Ralser M., Wamelink M. M., Kowald A., Gerisch B., Heeren G., Struys E. A., Klipp E., Jakobs C., Breitenbach M., Lehrach H., et al. (2007). Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol. 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman P. G., Meister A. (1975). Regulation of gamma-glutamyl-cysteine synthetase by nonallosteric feedback inhibition by glutathione. J. Biol. Chem. 250, 1422–1426. [PubMed] [Google Scholar]
- Richter K., Haslbeck M., Buchner J. (2010). The heat shock response: life on the verge of death. Mol. Cell 40, 253–266. [DOI] [PubMed] [Google Scholar]
- Rodgaard T., Schou K., Friis M. B., Hoffmann E. K. (2008). Does the intracellular ionic concentration or the cell water content (cell volume) determine the activity of TonEBP in NIH3T3 cells? Am. J. Physiol. Cell Physiol. 295, C1528–C1534. [DOI] [PubMed] [Google Scholar]
- Russell W. M. S., Burch R. L. (1959). The Principles of Humane Experimental Technique. Methuen, London. [Google Scholar]
- Sacher M., Pfander B., Hoege C., Jentsch S. (2006). Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat. Cell Biol. 8, 1284–1290. [DOI] [PubMed] [Google Scholar]
- Sarangi P., Bartosova Z., Altmannova V., Holland C., Chavdarova M., Lee S. E., Krejci L., Zhao X. (2014). Sumoylation of the Rad1 nuclease promotes DNA repair and regulates its DNA association. Nucleic Acids Res. 42, 6393–6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarangi P., Zhao X. (2015). SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem. Sci. 40, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaber J., Baltanas R., Bush A., Klipp E., Colman-Lerner A. (2012). Modelling reveals novel roles of two parallel signalling pathways and homeostatic feedbacks in yeast. Mol. Syst. Biol. 8, 622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann W. (2012). Thermosensor systems in eubacteria. Adv. Exp. Med. Biol. 739, 1–16. [DOI] [PubMed] [Google Scholar]
- Schuppe-Koistinen I., Moldeus P., Bergman T., Cotgreave I. A. (1994). S-thiolation of human endothelial cell glyceraldehyde-3-phosphate dehydrogenase after hydrogen peroxide treatment. Eur. J. Biochem. 221, 1033–1037. [DOI] [PubMed] [Google Scholar]
- Seelig G. F., Simondsen R. P., Meister A. (1984). Reversible dissociation of gamma-glutamylcysteine synthetase into two subunits. J. Biol. Chem. 259, 9345–9347. [PubMed] [Google Scholar]
- Shenton D., Grant C. M. (2003). Protein S-thiolation targets glycolysis and protein synthesis in response to oxidative stress in the yeast Saccharomyces cerevisiae. Biochem. J. 374(Pt 2), 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheshachalam A., Srivastava N., Mitchell T., Lacy P., Eitzen G. (2014). Granule protein processing and regulated secretion in neutrophils. Front. Immunol. 5, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler R. K., Grimshaw J. P., Christen P. (2004). Immediate response of the DnaK molecular chaperone system to heat shock. FEBS Lett. 562, 105–110. [DOI] [PubMed] [Google Scholar]
- Simmons S. O., Fan C. Y., Ramabhadran R. (2009). Cellular stress response pathway system as a sentinel ensemble in toxicological screening. Toxicol. Sci. 111, 202–225. [DOI] [PubMed] [Google Scholar]
- Spiess C., Beil A., Ehrmann M. (1999). A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell 97, 339–347. [DOI] [PubMed] [Google Scholar]
- Stromer T., Ehrnsperger M., Gaestel M., Buchner J. (2003). Analysis of the interaction of small heat shock proteins with unfolding proteins. J. Biol. Chem. 278, 18015–18021. [DOI] [PubMed] [Google Scholar]
- Sun B., Trask J. O., Ross S., Carmichael P. L., Adeleye Y., Andersen M. E., Clewell R. A. (2014). Evaluation of DNA repair center kinetics after treatment with chemicals causing linear and nonlinear dose-response curves for micronuclei. Phoenix, AZ, p. Abstract #1934. [Google Scholar]
- Tan C. Y., Hagen T. (2013). Post-translational regulation of mTOR complex 1 in hypoxia and reoxygenation. Cell Signal. 25, 1235–1244. [DOI] [PubMed] [Google Scholar]
- Thomas R. S., Allen B. C., Nong A., Yang L., Bermudez E., Clewell H. J., 3rd, Andersen M. E. (2007). A method to integrate benchmark dose estimates with genomic data to assess the functional effects of chemical exposure. Toxicol. Sci. 98, 240–248. [DOI] [PubMed] [Google Scholar]
- Thomas R. S., Wesselkamper S. C., Wang N. C., Zhao Q. J., Petersen D. D., Lambert J. C., Cote I., Yang L., Healy E., Black M. B., et al. (2013). Temporal concordance between apical and transcriptional points of departure for chemical risk assessment. Toxicol. Sci. 134, 180–194. [DOI] [PubMed] [Google Scholar]
- Tu Z., Anders M. W. (1998). Identification of an important cysteine residue in human glutamate-cysteine ligase catalytic subunit by site-directed mutagenesis. Biochem. J. 336(Pt 3), 675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552(Pt 2), 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde M. A., O’Brien J. A., Sepulveda F. V., Ratcliff R. A., Evans M. J., Colledge W. H. (1995). Impaired cell volume regulation in intestinal crypt epithelia of cystic fibrosis mice. Proc. Natl. Acad. Sci. U.S.A. 92, 9038–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. E., Zhu Q., Wani G., El-Mahdy M. A., Li J., Wani A. A. (2005). DNA repair factor XPC is modified by SUMO-1 and ubiquitin following UV irradiation. Nucleic Acids Res. 33, 4023–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfall P. J., Patterson J. C., Chen R. E., Thorner J. (2008). Stress resistance and signal fidelity independent of nuclear MAPK function. Proc. Natl. Acad. Sci. U.S.A. 105, 12212–12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter J., Jakob U. (2004). Beyond transcription—new mechanisms for the regulation of molecular chaperones. Crit. Rev. Biochem. Mol. Biol. 39, 297–317. [DOI] [PubMed] [Google Scholar]
- Yancey P. H., Clark M. E., Hand S. C., Bowlus R. D., Somero G. N. (1982). Living with water stress: evolution of osmolyte systems. Science 217, 1214–1222. [DOI] [PubMed] [Google Scholar]
- Ying W. (2008). NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox Signal. 10, 179–206. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Andersen M. E. (2007). Dose response relationship in anti-stress gene regulatory networks. PLoS Comput. Biol. 3, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Bhattacharya S., Andersen M. E. (2013). Ultrasensitive response motifs: basic amplifiers in molecular signalling networks. Open Biol. 3, 130031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Bhattacharya S., Conolly R. B., Clewell H. J., 3rd, Kaminski N. E., Andersen M. E. (2014). Molecular signaling network motifs provide a mechanistic basis for cellular threshold responses. Environ. Health Perspect. 122, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Pi J., Woods C. G., Andersen M. E. (2010a). A systems biology perspective on Nrf2-mediated antioxidant response. Toxicol. Appl. Pharmacol. 244, 84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. K., Wang D., Duan Y., Loy M. M., Chan H. C., Huang P. (2010b). Mechanosensitive gating of CFTR. Nat. Cell Biol. 12, 507–512. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.