

Abstract
Background
Glutathione S-transferases (GSTM1, GSTT1, and GSTP1) and methylenetetrahydrofolate reductase (MTHFR) are important enzymes for protection against oxidative stress. In addition, MTHFR has an essential role in DNA synthesis, repair, and methylation. Their polymorphisms have been implicated in the pathogenesis of ulcerative colitis (UC). The aim of the present study was to investigate the role of selected polymorphisms in these genes in the development of UC in the Moldavian population.
Methods
In a case-control study including 128 UC patients and 136 healthy individuals, GSTM1 and GSTT1 genotypes (polymorphic deletions) were determined using multiplex polymerase chain reaction (PCR). The GSTP1 rs1695 (Ile105Val), MTHFR rs1801133 (C677T), and MTHFR rs1801131 (A1298C) polymorphisms were studied with restriction fragment length polymorphism (RFLP) analysis. Genotype–phenotype correlations were examined using logistic regression analysis.
Results
None of the genotypes, either alone or in combination, showed a strong association with UC. The case-only sub-phenotypic association analysis showed an association of the MTHFR rs1801133 polymorphism with the extent of UC under co-dominant (p corrected = 0.040) and recessive (p corrected = 0.020; OR = 0.15; CI = 0.04–0.63) genetic models. Also, an association between the MTHFR rs1801131 polymorphism and the severity of UC was reported for the over-dominant model (p corrected = 0.023; coefficient = 0.32; 95% CI = 0.10–0.54).
Conclusion
The GST and MTHFR genotypes do not seem to be a relevant risk factor for UC in our sample. There was, however, evidence that variants in MTHFR may influence the clinical features in UC patients. Additional larger studies investigating the relationship between GST and MTHFR polymorphisms and UC are required.
Abbreviations: UC, Ulcerative colitis; IBD, Inflammatory bowel disease; GST, Glutathione S-transferase; MTHFR, Methylenetetrahydrofolate reductase; SAM, S-adenosyl methionine; n, Total number; SNP, Single nucleotide polymorphism; PCR, Polymerase chain reaction; RFLP, Restriction fragment length polymorphism; HWE, Hardy–Weinberg equilibrium
Keywords: Genetic polymorphism, Ulcerative colitis, Susceptibility, Methylenetetrahydrofolate reductase, Glutathione S-transferases, Moldavian population
Highlights
-
•
Polymorphisms in the GST and MTHFR genes are not associated with UC in Moldavian population.
-
•
The combined GST and MTHFR genotypes showed no clear association with UC.
-
•
Polymorphisms in the MTHFR gene may affect clinical features in UC patients.
1. Introduction
Ulcerative colitis (UC) is a chronic inflammatory condition of the large intestine, which along with Crohn's disease comprises the major part of the inflammatory bowel diseases (Abraham and Cho, 2009). Inflammatory bowel diseases (IBD) and specifically UC are not evenly distributed throughout the world, with North America and western/northern Europe having the highest rate (100–250 per 100,000 population for UC) (Burisch and Munkholm, 2015). In the Republic of Moldova, as in most other eastern/southeastern European countries, UC appears to be much less common (20–25 per 100,000 population in Moldavia, unpublished data), although continuous increase in the disease incidence over the past years has been reported (Burisch and Munkholm, 2015). The precise etiology of UC remains unclear. Several mechanisms related to immunological, genetic, toxic, and infection abnormalities are implicated in the pathogenesis of UC (Ananthakrishnan, 2015).
Oxidative stress due to overproduction of reactive oxygen species (ROS) and decreased efficiency of antioxidant defenses has been considered to be a common pathogenic factor in UC and its complications. ROS overproduction has been shown in the inflamed mucosa of UC patients and in experimental animal models of IBD (reviewed by Rezaie et al., 2007). Excessive amounts of ROS can destroy biomolecules such as lipids, proteins, and DNA, leading to cellular stress and endothelial dysfunction in UC patients (Valko et al., 2007, Piechota-Polanczyk and Fichna, 2014). Production of oxidants and free radicals can also facilitate activation of signaling events that mediate expression of inflammatory genes as well as genes regulating cell division, differentiation, and apoptosis (Valko et al., 2007). It has been suggested that the impaired prooxidant and antioxidant system in UC patients may contribute to the disease process (Pravda, 2005). The human glutathione S-transferases (GSTs) are well-known oxidative stress-related detoxification enzymes. Located mainly in the cytosol, GST enzymes catalyze the conjugation of electrophilic substrates to glutathione, thus facilitating detoxification and further metabolization and excretion (Hayes et al., 2005). They also play an important role in peroxidase and isomerase activities (Sheehan et al., 2001). Several classes of GSTs have been identified, with GSTT1, M1, and P1 being the most well-characterized forms. Polymorphisms within these genes either decrease or abolish GST enzyme activity (Strange et al., 2000). Thus, a functionally significant A to G transition in exon 5 of the GSTP1 gene (A313G, rs1695), which results in replacing isoleucine with valine (Ile105Val), substantially diminishes GSTP1 enzyme activity. By contrast, homozygous whole gene deletions of GSTT1 or GSTM1 cause a lack of the respective enzyme function. These GST genes polymorphisms have been linked to inflammation and immune processes in a number of reports (Bekris et al., 2005, Aguilera et al., 2004, Babushok et al., 2013, Liang et al., 2013, Živković et al., 2013, Ding et al., 2014).
It is known that homocysteine (Hcy) induces oxidative stress (Loscalzo, 1996). Hyperhomocysteinemia is common among UC patients, and elevated Hcy levels are associated with deep vein thrombosis and thromboembolic disease in UC patients (Peyrin-Biroulet et al., 2007, Akbulut et al., 2010). Besides its prooxidant properties, Hcy is also known to play a role in epigenetic gene regulation being directly involved in the DNA methylation process (Peyrin-Biroulet et al., 2007). Noteworthy, excessive DNA methylation is closely associated with the inflammatory state of the colon in UC (Karatzas et al., 2014). The 5,10-methylenetetrahydrofolate reductase (MTHFR) is a key regulatory enzyme in folate and Hcy metabolism (Finkelstein, 1998). It converts 5,10-methylenetetrahydrofolate (a derivative of folic acid) irreversibly to 5-methyltetrahydrofolate, which donates its methyl group to Hcy in the generation of methionine. Methionine is in turn converted to S-adenosylmethionine (SAM), the methyl donor in DNA methylation. Two single nucleotide polymorphisms (SNPs) in the MTHFR gene, rs1801133 (C677T, Ala222Val) and rs1801131 (A1298C, Glu429Ala), have been associated with reduced enzyme activity, elevated Hcy levels, and reduced methionine/SAM supply for methylation (Frosst et al., 1995, Weisberg et al., 1998). Numerous epidemiologic and experimental studies have shown that genetic polymorphisms in the MTHFR gene may be related to various pathological states, including colorectal cancer and autoimmune diseases (Zhao et al., 2013, Mavragani et al., 2007, Afeltra et al., 2005, Foffa et al., 2009, Mao et al., 2010).
The roles of the GSTM1, GSTT1, GSTP1, and MTHFR genotypes in susceptibility to UC have been discussed by some investigators, but no consistent conclusions have yet been made. Their effect on the particular clinical phenotypes also remains to be clarified. Here we explored the role of these genotypes in the development of UC in Moldavian population.
2. Materials and methods
2.1. Samples
This case-control study comprises 128 unrelated UC patients, recruited at the Department of Gastroenterology, Republican Clinical Hospital, Moldova. The diagnosis of UC was based on standard clinical, endoscopic, and histological criteria (Lennard-Jones, 1989). The distribution of UC lesions was defined according to the Montreal classification (Satsangi et al., 2006). The subjects with UC were also classified using three additional clinical categories: (i) disease severity as assessed by the modified Truelove–Witts disease activity index (Dignass et al., 2012); (ii) UC-related outcomes—i.e. strictures, lead pipe colon, malignancy, steroid-dependency, and colectomy: absent versus present; and (iii) disease relapse rate: infrequent (≤ 1/year) versus frequent (≥ 2/year). All patients were Caucasians of European descent. The control population consisted of 136 unrelated and ethnically matched healthy individuals who had no history of autoimmune or oncological disease. Information on smoking habits was collected from both patients and healthy controls. According to smoking habit, cases were divided into 3 groups: current smokers, ex-smokers, and never smokers, and controls were classified as current smokers and current non-smokers. The lack of correspondence between cases and controls in smoking definitions is due to the lack of information on former smoking status in controls. Therefore, ex-smoker and never-smoker UC patients were combined in one group to make them comparable with healthy controls in the case-control study. The combination is further justified by findings that both the never- and ex-smokers have an increased risk of UC compared with current smokers, as reported previously (Lakatos et al., 2013). The demographics and clinical features of the study population are depicted in Table 1. EDTA anti-coagulated venous blood samples were collected from all participants, and genomic DNA was extracted from peripheral blood leukocytes using a standard salting out method (Miller et al., 1988). The local ethics committee approved the study, and informed written consent was received from all subjects.
Table 1.
Characteristics of the study population.
| Characteristic | UC (n = 128) n (%) | Healthy controls (n = 136) n (%) |
|---|---|---|
| Sex | ||
| Female | 59 (46.1) | 52 (38.2) |
| Male | 69 (53.9) | 84 (61.8) |
| Smoking | ||
| Never | 98 (76.6) | NA |
| Former | 16 (12.5) | NA |
| Current | 14 (10.9) | 48 (35.3) |
| Age at recruitment, median ± S.D. (years) | 41.4 ± 13.7 | 45.9 ± 10.8 |
| Age at diagnosis, median ± S.D. (years) | 36.7 ± 13.4 | |
| Extent of disease | ||
| Distal colitis | 53 (41.4) | |
| Left-sided colitis | 39 (30.5) | |
| Pancolitis | 36 (28.1) | |
| Severity | ||
| Mild | 41 (32.0) | |
| Intermediate | 60 (46.9) | |
| Severe | 27 (21.1) | |
| Negative UC-related outcomes | ||
| Absent | 73 (57.9) | |
| Present | 53 (42.1) | |
| Relapse rate | ||
| Infrequent | 67 (53.2) | |
| Frequent | 59 (46.8) |
NA, not available.
2.2. Genotyping
GSTM1 and GSTT1 genotypes were determined using multiplex polymerase chain reaction (PCR). Three sets of primers were used to amplify a 434-bp sequence of the GSTT1 gene (Zheng et al., 2001), a 267-bp fragment of the GSTM1 gene (Tujague et al., 2006), and a 212-bp segment of the human albumin gene as an internal amplification control (Tujague et al., 2006). The PCR reactions were carried out in 20 μL containing 5 pmol of each primer, 100 ng genomic DNA, 1.5 mmol/L MgCl2, 200 μmol/L dNTPs, and 0.5 unit of Taq DNA polymerase in the buffer provided by the manufacturer. Amplification was performed in a Bio-Rad T100 Thermal Cycler (Bio-Rad, Hercules, California) for the PCR reaction. The amplification conditions consisted of an initial melting step 95 °C for 5 min, followed by 35 cycles of 95 °C for 30 s, 57 °C for 30 s, and 72 °C for 40 s; and a final elongation step of 72 °C for 5 min. Resulting fragments were visualized using ethidium bromide staining and 3% agarose gel electrophoresis (Fig. 1). MTHFR rs1801133 (C677T, Ala222Val), MTHFR rs1801131 (A1298C, Glu429Ala), and GSTP1 rs1695 (A313G, Ile105Val) were determined by polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) with HinfI, MboII, and Alw26I restriction endonucleases, respectively, as reported elsewhere (Garte et al., 2007, Eloualid et al., 2012). The digestion fragment sizes for the MTHFR rs1801133 genotypes were a single 198-bp band for CC; 198, 175, and 23 bp (3 fragments) for CT; and 175 and 23 bp (2 fragments) for TT. For rs1801131 genotypes, the fragments were 56, 31, 30, 28, and 18 bp (5 fragments) for AA; 84, 56, 31, 30, 28, and 18 bp (6 fragments) for AC, and 84, 31, 30 and 18 bp (4 fragments) for CC. For the GSTP1 rs1695 polymorphism, AA homozygotes had two fragments (329 bp and 104 bp), AG heterozygotes demonstrated four DNA bands (329, 222, 107, and 104 bp) and GG homozygotes showed three fragments (222, 107, and 104 bp). Genotyping errors were excluded by random re-genotyping of the respective loci and therefore, all polymorphisms were included into the association analysis.
Fig. 1.
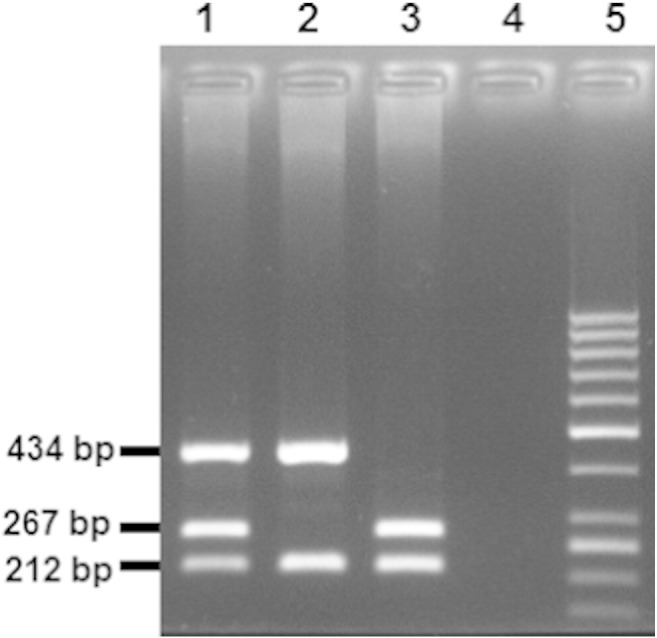
Electrophoresis of the products of the multiple PCR. Presence of 267 and 434-bp fragments indicates GSTM1 and GSTT1 wild-type (non-null) genotypes, respectively. The 212-bp band corresponding to a fragment of the human albumin gene, which serves as a positive control for the PCR. Lane 5: molecular weight marker (GeneRuler 50 bp DNA Ladder, Thermo Fisher Scientific); lane 1: GSTM1 and GSTT1 non-null genotype; lane 2: GSTM1 null/GSTT1 non-null genotype; lane 3: GSTM1 non-null/GSTT1 null genotype; lane 4: negative PCR control.
2.3. Statistical analysis
Comparison of demographical parameters between cases and controls was performed using Student's t-test for continuous variables and the χ2 test for categorical data. Deviation from Hardy–Weinberg equilibrium (HWE) was assessed by the Fisher exact test. The linkage disequilibrium between the two polymorphisms in MTHFR gene was examined using D′ and r2 coefficients. Genotype frequencies in the case and control groups were compared by logistic regression with adjustment for sex, age at investigation, and current smoking status under the additive, dominant, recessive, co-dominant, and over-dominant genetic models. Logistic regression model was also constructed to determine the effect of genetic polymorphisms on the clinical phenotypes of UC (disease extent, relapse, severity, complications, and age at onset). The odds ratios (OR) with their corresponding 95% confidence intervals (CI) and p-values were calculated as measures of associations. p-values < 0.05 were considered as significant. The Bonferroni correction for multiple comparisons was applied where appropriate, and the threshold was calculated as 0.05/5 = 0.01. For significant p-values, Fisher exact test was additionally calculated as suggested in (Lettre et al., 2007). The Fisher test is particularly relevant when one of the alleles is rare, and together with pooling the genotypes according to the genetic model, the statistical significance can be estimated more adequately (Lewis, 2002). Combined effects of polymorphisms on the disease risk were analyzed using logistic regression for all possible combinations of loci under three genetic models: dominant, recessive, and over-dominant. In the dominant model, the rare homozygous variant was combined with the heterozygous, while in the recessive model, the rare homozygous genotype was considered alone. In the over-dominant model, the heterozygous and both homozygous genotypes together were encoded using two dummy variables. Therefore, tests for genotype combinations had nine degrees of freedom for pairwise combinations of MTHFR rs1801133, rs1801131, and GSTP1 rs1695 polymorphisms (three for each SNP), six degrees of freedom for combinations of MTHFR rs1801133, rs1801131, and GSTP1 rs1695 polymorphisms with GSTT1 and GSTM1 loci, and a four-degree of freedom for combined GSTT1 and GSTM1 genotypes. Accordingly, significant p-values were corrected using the Bonferroni method by multiplying by factors 9, 6, or 4. Power of the study was calculated post-hoc assuming the following variables: significance (type 1 error) 0.05, genetic effects (odds ratio [OR]) 1.5 and 2.0, and disease prevalence of 0.00025; allele frequencies were those found in control population. The statistical tests were performed with SNPStats program and E-Views software (IHS Global Inc.).
3. Results
There were significant differences in age and smoking status (current smokers vs. non-smokers) distributions between the case and control groups (p = 0.00322 and p = 3.0E-6, respectively; Table 1). Although there was no significant difference in sex ratio between the two groups (p = 0.168462), their matching on gender was imperfect: the cases had a higher percentage of female (46.1%) than the controls (38.2%) (Table 1). All three of these demographic variables were included as covariates in all subsequent multivariate regression analyses.
Table 2 summarizes the distribution of MTHFR and GST genotypes and allele frequencies in the cohort of 128 UC patients and in 136 healthy controls. The observed frequencies of the studied polymorphisms were in a range of values observed in other Caucasian–European populations. Allelic distributions of the investigated SNPs were in accordance with Hardy–Weinberg equilibrium (HWE) in both groups except for GSTP1 rs1695, which showed a slight deviation from HWE in controls (p = 0.026). Linkage disequilibrium analysis showed strong LD (pairwise D′ = 0.969) between the two MTHFR loci at nucleotide positions 677 (rs1801133) and 1298 (rs1801131). However, due to a weak correlation (r2 = 0.205), the two SNPs cannot substitute each other and were analyzed in an independent manner.
Table 2.
Association tests for single polymorphisms.
| Polymorphism | Genotype/Allele | Controls n (%) | Cases n (%) | Model | p-value⁎ |
|---|---|---|---|---|---|
| MTHFR rs1801133 | CC | 70 (51.5) | 59 (46.1) | Codominant | 0.63 |
| CT | 52 (38.2) | 55 (43) | Dominant | 0.35 | |
| TT | 14 (10.3) | 14 (10.9) | Recessive | 0.87 | |
| C | 192 (70.6) | 173 (67.6) | Overdominant | 0.39 | |
| T | 80 (29.4) | 83 (32.4) | Additive | 0.44 | |
| MTHFR rs1801131 | AA | 66 (48.5) | 52 (41.3) | Codominant | 0.27 |
| AC | 59 (43.4) | 57 (45.2) | Dominant | 0.14 | |
| CC | 11 (8.1) | 17 (13.5) | Recessive | 0.27 | |
| A | 191 (70.2) | 161 (63.9) | Overdominant | 0.43 | |
| C | 81 (29.8) | 91 (36.1) | Additive | 0.11 | |
| GSTP1 rs1695 | AA | 60 (44.4) | 62 (49.2) | Codominant | 0.29 |
| AG | 68 (50.4) | 53 (42.1) | Dominant | 0.42 | |
| GG | 7 (5.2) | 11 (8.7) | Recessive | 0.27 | |
| A | 188 (69.6) | 177 (70.2) | Overdominant | 0.18 | |
| G | 82 (30.4) | 75 (29.8) | Additive | 0.83 | |
| GSTT1 | Present | 111 (81.6) | 108 (84.4) | 0.77 | |
| Null | 25 (18.4) | 20 (15.6) | |||
| GSTM1 | Present | 61 (44.9) | 52 (40.6) | 0.65 | |
| Null | 75 (55.1) | 76 (59.4) |
The p-values were obtained from logistic regression with co-dominant, dominant, recessive, over-dominant and additive models, and adjusted for sex, age and current smoking status.
No significant differences were observed in the frequencies of the MTHFR and GST genotypes and alleles between UC subjects and controls (Table 2). The lack of association persisted after stratification by gender or smoking status (data not shown). In pairwise combined analyses of MTHFR rs1801133 and rs1801131, GSTP1 rs1695, GSTM1, and GSTT1 gene polymorphisms, only five different genotype combinations were significantly associated with UC (Table 3). However, the significance was eliminated after applying a Bonferroni correction (p corrected > 0.05). No other combinatorial genotypes reached a nominal significance (data not shown).
Table 3.
Pairwise genotype - genotype interaction effects on UC risk revealed by logistic regression under dominant, recessive and over-dominant genetic models. Only associations with a nominal p-value p < 0.05 are shown.
| 1st locus | Genotype | 2nd locus | Genotype | Controls n (%) | Cases n (%) | OR (95% CI)a | p-valuea | p-Value corrected |
|---|---|---|---|---|---|---|---|---|
| MTHFR rs1801133 | CT | MTHFR rs1801131 | AC | 22 (16.2%) | 32 (25.4%) | 2.18 (1.12–4.21) | 0.019 | 0.171 |
| MTHFR rs1801133 | CT + TT | MTHFR rs1801131 | AC | 23 (16.9%) | 32 (25.4%) | 2.03 (1.06–3.89) | 0.03 | 0.27 |
| MTHFR rs1801131 | AA | GSTP1 rs1695 | AG | 32 (23.7%) | 17 (13.6%) | 0.45 (0.23–0.90) | 0.021 | 0.189 |
| MTHFR rs1801131 | AA | GSTP1 rs1695 | AG + GG | 34 (25.2%) | 21 (16.8%) | 0.51 (0.27–0.98) | 0.04 | 0.36 |
| GSTP1 rs1695 | AG | GSTM1 | Present | 34 (25.2%) | 19 (15.1%) | 0.50 (0.26–0.98) | 0.039 | 0.234 |
OR, odds ratio; CI, confidence interval.
Adjusted for sex, age at investigation and smoking.
In addition, we also performed a detailed genotype–phenotype analysis of GST and MTHFR variants in UC patients. There was no association of these polymorphisms with age of onset, complications, and relapse rate (data not shown). Also, no association was observed between GSTM1 and GSTT1 genes and disease spread or severity (data not shown). We showed that the AC genotype of MTHFR rs1801131 was significantly associated with increased severity in the over-dominant genetic model (coefficient = 0.32; 95% CI = 0.10–0.54; p = 0.0046; Table 4). Both the co-dominant genetic model (p = 0.018) and the dominant genetic model (coefficient = 0.26; 95% CI = 0.04–0.48; p = 0.022) also obtained significant results (Table 4). However, only the p-value under the over-dominant model remained significant after Bonferroni correction for multiple tests was applied (p corrected = 0.023). We also report a significant association between MTHFR rs1801133 polymorphism and disease extent (co-dominant model: p = 0.008; Table 5). This was primarily due to a significantly higher frequency of the TT genotype in patients with distal colitis than in patients with more extensive UC types (recessive model: OR = 0.15; CI = 0.04–0.63; p = 0.0041). The values were still significant after Bonferroni correction (p corrected = 0.040 and p corrected = 0.020 for co-dominant and recessive models, respectively; Table 5). Furthermore, the associations were validated by Fisher exact test (p corrected = 0.0375 and p corrected = 0.0185 for co-dominant and recessive models, respectively; Table 5).
Table 4.
Effect of MTHFR polymorphism rs1801131 on severity of UC, analyzed by logistic regression.
| Genotype | Severity grade |
Severity mean (s.e.) | LR coefficient (95% CI)a | p-Valuea | p-Value correcteda | ||
|---|---|---|---|---|---|---|---|
| 1. (n = 41) | 2. (n = 59) | 3. (n = 26) | |||||
| AA | 21 (51.2%) | 24 (40.7%) | 7 (26.9%) | 1.73 (0.1) | 0 (Reference) | 0.018 | 0.09 |
| AC | 13 (31.7%) | 28 (47.5%) | 16 (61.5%) | 2.05 (0.1) | 0.33 (0.10–0.56) | ||
| CC | 7 (17.1%) | 7 (11.9%) | 3 (11.5%) | 1.76 (0.18) | 0.04 (− 0.30–0.38) | ||
| AA | 21 (51.2%) | 24 (40.7%) | 7 (26.9%) | 1.73 (0.1) | 0 (Reference) | 0.022 | 0.11 |
| AC + CC | 20 (48.8%) | 35 (59.3%) | 19 (73.1%) | 1.99 (0.08) | 0.26 (0.04–0.48) | ||
| AA + AC | 1.9 (0.07) | 0 (Reference) | 0.43 | - | |||
| CC | 7 (17.1%) | 7 (11.9%) | 3 (11.5%) | 1.76 (0.18) | − 0.13 (− 0.46–0.19) | ||
| AA + CC | 28 (68.3%) | 31 (52.6%) | 10 (38.4%) | 1.74 ± 0.08 | 0 (Reference) | 0.0046 | 0.023 |
| AC | 13 (31.7%) | 28 (47.5%) | 16 (61.5%) | 2.05 ± 0.1 | 0.32 (0.10–0.54) | ||
1 = mild, 2 = moderate, 3 = severe.
aAdjusted for sex, age at investigation and smoking.
LR, logistic regression; CI, confidence interval.
p-values above 1.0 after Bonferroni correction are not shown.
Table 5.
Effect of MTHFR polymorphism rs1801133 on extent of UC, analyzed by logistic regression and Fisher exact test.
| Genotype | Distal colitis (n = 53) | Extended colitisa (n = 75) | OR (95% CI)b | p-Value log. Reg.b | p-value log. reg. correctedb | p-Value isher test | p-Value fisher test corrected |
|---|---|---|---|---|---|---|---|
| CC | 24 (45.3%) | 35 (46.7%) | 1 (Reference) | 0.008 | 0.040 | 0.0075 | 0.0375 |
| CT | 18 (34%) | 37 (49.3%) | 1.69 (0.71–4.02) | ||||
| TT | 11 (20.8) | 3 (4%) | 0.19 (0.04–0.83) | ||||
| CC | 24 (45.3%) | 35 (46.7%) | 1 (Reference) | 0.91 | - | > 0.9999 | - |
| CT + TT | 29 (54.7%) | 40 (53.3%) | 1.05 (0.48–2.28) | ||||
| CC + CT | 42 (79.3%) | 72 (96%) | 1 (Reference) | 0.0041 | 0.020 | 0.0037 | 0.0185 |
| TT | 11 (20.8) | 3 (4%) | 0.15 (0.04–0.63) | ||||
| CC + TT | 35 (66%) | 38 (50.7%) | 1 (Reference) | 0.05 | 0.25 | 0.0729 | 0.3645 |
| CT | 18 (34%) | 37 (49.3%) | 2.28 (1.00–5.21) |
OR, odds ratio; CI, confidence interval.
p-values above 1.0 after Bonferroni correction are not shown.
Left-sided UC + Pancolitis.
Adjusted for sex, age, at investigation and smoking.
With regard to the power analysis in the combined sample, our sample set was estimated to have enough power (> 89%) to detect moderate high-risk alleles (OR = 2) but limited (49–64%) for moderate low-risk alleles (OR = 1.5). The power was even smaller in the stratified and combinatorial analyses and this was a limitation of this study.
4. Discussion
Wide evidence suggests that oxidative stress is involved in UC pathogenesis (Rezaie et al., 2007, Valko et al., 2007, Piechota-Polanczyk and Fichna, 2014). The enzymes glutathione S-transferases T1, M1, and P1, and methylenetetrahydrofolate reductase (MTHFR) are implicated in the antioxidant defenses (Hayes et al., 2005, Raza, 2011, Hoffman, 2011). The analysis of their variation has been widely used in the field of cancer and inflammatory genetics. Their role in the pathogenesis of UC has been also proposed but has not been extensively studied. The present study was designed to investigate the contribution of GSTT1, GSTM1, GSTP1, and MTHFR genetic polymorphisms to the risk and pathogenesis of UC in Moldavian population. To the best of our knowledge, this is the first report on GSTT1, GSTM1, GSTP1, and MTHFR genes in UC patients from eastern–southeastern Europe.
We did not observe significant associations between any studied polymorphisms and UC. Furthermore, no Bonferroni-corrected significant associations of combined genotypes were found, implying that these combinations are probably not synergistic in their effect on UC risk in Moldavian population. These findings were not totally unexpected. Indeed, previous studies on the association between the same polymorphisms and susceptibility to UC have reported conflicting results. Thus, an association of MTHFR rs1801133 genotypes with UC has been reported in Ireland (Mahmud et al., 1999), Denmark (Nielsen et al., 2000), and Portugal (Magro et al., 2003), but not in China (Chen et al., 2005, Chen et al., 2008, Jiang et al., 2012), the UK (Herrlinger et al., 2005), Italy (Vecchi et al., 2000, Papa et al., 2001), Turkey (Yilmaz et al., 2006), or Morocco (Senhaji et al., 2013). For the MTHFR rs1801131 polymorphism, positive association results were obtained in studies from Southeast China (Jiang et al., 2012) and Turkey (Yilmaz et al., 2006) but were not confirmed on samples from Central China (Chen et al., 2005, Chen et al., 2008) and the UK (Herrlinger et al., 2005). Regarding GSTT1/M1 genes, both homozygous GSTT1 and GSTM1 deletion polymorphisms were shown to be associated with UC in Central China (Ye et al., 2011), northern India (Mittal et al., 2007), and in Turkish population (Buyukgoze et al., 2013). Conversely, studies in Denmark (Ernst et al., 2010) and Holland (Broekman et al., 2014) as well as in Israeli Jews (Karban et al., 2011) failed to demonstrate any relationship between GSTT1/M1 loci and the risk of UC. Likewise, the GSTP1 rs1695 polymorphism showed significant association with UC in Central China (Ye et al., 2011) and no association in Denmark (Ernst et al., 2010). The observed discrepancy between individual studies may be due to the relatively small number of patients used in different studies. In addition, the impact of these polymorphisms on the risk of UC may also vary from population to population because of differences in genetic backgrounds as well as environmental and nutritional factors. For example, folate supplementation can efficiently reduce plasma Hcy level and may therefore affect UC susceptibility. A study by Chen et al. (2008) suggested a link between interethnic difference in folate consumption and association of polymorphisms in the MTHFR gene with UC. Interestingly, Moldavia, along with Kosovo, is the only European country that mandates fortification of wheat products with folic acid (Food Fortification Initiative, 2015). This background folic acid supplementation could potentially smooth possible effects of MTHFR polymorphisms on UC and partially explain the lack of association in the Moldavian population. Consideration of additional information on the dietary habits of participated individuals and controlling for the markers of folate status (i.e. folic acid, Hcy, vitamin B12) would certainly help to clarify the role of MTHFR variants in the development of UC and, therefore, are desired for further in-depth studies of UC.
We further investigated whether GST and MTHFR polymorphisms are associated with certain phenotypic characteristics in UC patients. We found that the MTHFR rs1801131 AC heterozygote was more frequently associated with severe clinical subtypes of UC, whereas AA homozygous wild type had an inverse correlation, indicating a possible role of the MTHFR gene polymorphism on the severity of the disease. However, no positive correlation has been shown for the homozygous CC genotype, although, given its lowest enzymatic activity (van der Put et al., 1998), a stronger effect for CC than for AC genotype was expected. This lack of association could be due to chance because of small sample size and low occurence of genotype CC in the groups. Yet the possibility of over-dominant inheritance of MTHFR rs1801131 polymorphism (‘heterozygous advantage’) cannot be ruled out. Indeed, although no ‘heterozygous advantage’ has been reported for MTHFR polymorphisms in UC, it has been previously described for other human diseases (Li et al., 2014, Hubacek et al., 2015). Further research is needed to confirm the causal effect of MTHFR on the severity of UC.
Finally, we observed a significant association between the MTHFR rs1801133 polymorphism and the extent of inflammation. Patients with the TT genotype achieved significantly higher protection against more extensive disease (left-sided UC and pancolitis) than patients with CT or CC genotypes. This finding contradicts the oxidative stress hypothesis of UC and the previous reports on correlation between MTHFR rs1801133 and UC extension (Chen et al., 2008, Jiang et al., 2012, Senhaji et al., 2013, Vecchi et al., 2000). The differential results could be due to multifunctional properties of methylenetetrahydrofolate reductase, again modulated by population and nutritional factors. Indeed, the MTHFR rs1801133 TT genotype is known to reduce enzyme activity (~ 30% of normal) (Frosst et al., 1995), and therefore, its carriers may be especially susceptible to Hcy-induced diseases, particularly in the presence of folate deficiency. On the other hand, MTHFR has a crucial role in regulating cellular methylation and gene expression (Chen et al., 2001, Friso et al., 2002, Lu, 2013). A potential biochemical explanation for the inverse association between UC and the MTHFR rs1801133 TT genotype is that low MTHFR activity results in lower production of SAM (Schwahn and Rozen, 2001). Eventually, this may lead to diminished spreading of hypermethylation and inflammation. The above scenario on the protective effect of the MTHFR rs1801133 TT genotype appears to be more suitable for countries with a sufficient total folate intake, like Moldavia, where the overproduction of SAM conditioned by nutrition has to be balanced by intrinsic regulatory factors. The relevance of the inverse association between the MTHFR rs1801133 TT genotype and UC is further supported by association studies on colorectal cancer and Graves' disease (Mao et al., 2010, Zhao et al., 2013) that are etiologically related to UC.
In conclusion, the present study has shown that GSTT1, GSTM1, GSTP1, and MTHFR genetic variants, alone or combined, have no significant influence on the primary risk of having UC in Moldavian population. However, our results suggest that MTHFR genotypes may affect the spread and severity of UC. The main limitation of our study is its small sample size, which is partly due to the low incidence of UC in Moldavia. It also does not consider the nutritional status of participants as assessed by serum Hcy and folic acid levels. Hence, further work using a larger population and studying additional folate and oxidative stress-related genes taking into account the gene–gene and gene–environment interactions is needed to refine the present results. Such knowledge may have important implications for prevention and management of UC.
Conflict of interest statement
The authors declare no conflicts of interest.
Acknowledgments
The authors thank Nina Perlug for her help with laboratory work. The authors also thank Sabine Hoffjan for her valuable comments and suggestions on the manuscript. We are likewise grateful to medical staff, UC patients, and healthy donors for their participation in this study. The study was partially supported by the Academy of Sciences of Moldova (grant no. 09.819.09.01F).
References
- Abraham C., Cho J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afeltra A. Thrombosis in systemic lupus erythematosus: congenital and acquired risk factors. Arthritis Rheum. 2005;53:452–459. doi: 10.1002/art.21172. [DOI] [PubMed] [Google Scholar]
- Aguilera I., Sousa J.M., Gavilán F., Bernardos A., Wichmann I., Nuñez-Roldán A. Glutathione S-transferase T1 mismatch constitutes a risk factor for de novo immune hepatitis after liver transplantation. Liver Transpl. 2004;10:1166–1172. doi: 10.1002/lt.20209. [DOI] [PubMed] [Google Scholar]
- Akbulut S., Altiparmak E., Topal F., Ozaslan E., Kucukazman M., Yonem O. Increased levels of homocysteine in patients with ulcerative colitis. World J. Gastroenterol. 2010;16:2411–2416. doi: 10.3748/wjg.v16.i19.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthakrishnan A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015;12:205–217. doi: 10.1038/nrgastro.2015.34. [DOI] [PubMed] [Google Scholar]
- Babushok D.V. Common polymorphic deletion of glutathione S-transferase theta predisposes to acquired aplastic anemia: independent cohort and meta-analysis of 609 patients. Am. J. Hematol. 2013;88:862–867. doi: 10.1002/ajh.23521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekris L.M. Glutathione-s-transferase M1 and T1 polymorphisms and associations with type 1 diabetes age-at-onset. Autoimmunity. 2005;38:567–575. doi: 10.1080/08916930500407238. [DOI] [PubMed] [Google Scholar]
- Broekman M.M. GST theta null genotype is associated with an increased risk for ulcerative colitis: a case–control study and meta-analysis of GST Mu and GST theta polymorphisms in inflammatory bowel disease. J. Hum. Genet. 2014;59:575–580. doi: 10.1038/jhg.2014.77. [DOI] [PubMed] [Google Scholar]
- Burisch J., Munkholm P. The epidemiology of inflammatory bowel disease. Scand. J. Gastroenterol. 2015;50:942–951. doi: 10.3109/00365521.2015.1014407. [DOI] [PubMed] [Google Scholar]
- Buyukgoze O., Osmanoglu N., Arslan S., Sen A. Association of the CYP1A1*2A, GSTT1 null, GSTM1 null, mEPHX*3, and XRCC1-399 genetic polymorphisms with ulcerative colitis. Int. J. Color. Dis. 2013;28:593–595. doi: 10.1007/s00384-012-1507-6. [DOI] [PubMed] [Google Scholar]
- Chen Z. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum. Mol. Genet. 2001;10:433–443. doi: 10.1093/hmg/10.5.433. [DOI] [PubMed] [Google Scholar]
- Chen M., Xia B., Rodriguez-Gueant R.M., Bigard M., Gueant J.L. Genotypes 677TT and 677CT + 1298 AC of methylenetetrahydrofolate reductase are associated with the severity of ulcerative colitis in central China. Gut. 2005;54:733–734. doi: 10.1136/gut.2004.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. Methionine synthase A2756G polymorphism may predict ulcerative colitis and methylenetetrahydrofolate reductase C677T pancolitis, in central China. BMC Med. Genet. 2008;9:78. doi: 10.1186/1471-2350-9-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignass A. Second European evidence-based consensus on the diagnosis and management of ulcerative colitis part 2: current management. J. Crohns Colitis. 2012;6:991–1030. doi: 10.1016/j.crohns.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Ding B., Sun W., Han S., Cai Y., Ren M. Polymorphisms of glutathione S-transferase M1 (GSTM1) and T1 (GSTT1) and endometriosis risk: a meta-analysis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014;183:114–120. doi: 10.1016/j.ejogrb.2014.10.032. [DOI] [PubMed] [Google Scholar]
- Eloualid A. Association of the MTHFR A1298C variant with unexplained severe male infertility. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0034111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst A. Genetic variants of glutathione S-transferases mu, theta, and pi display no susceptibility to inflammatory bowel disease in the Danish population. Scand. J. Gastroenterol. 2010;45:1068–1075. doi: 10.3109/00365521.2010.490594. [DOI] [PubMed] [Google Scholar]
- Finkelstein J.D. The metabolism of homocysteine: pathways and regulation. Eur. J. Pediatr. 1998;157:S40–S44. doi: 10.1007/pl00014300. [DOI] [PubMed] [Google Scholar]
- Foffa I., Festa P.L., Ait-Ali L., Mazzone A., Bevilacqua S., Andreassi M.G. Ascending aortic aneurysm in a patient with bicuspid aortic valve, positive history of systemic autoimmune diseases and common genetic factors: a case report. Cardiovasc. Ultrasound. 2009;7:30–34. doi: 10.1186/1476-7120-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Food Fortification Initiative Country profiles. 2015. http://www.ffinetwork.org/country_profiles/index.php (June)
- Friso S. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc. Natl. Acad. Sci. U. S. A. 2002;99:5606–5611. doi: 10.1073/pnas.062066299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frosst P. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- Garte S. Effects of metabolic genotypes on intermediary biomarkers in subjects exposed to PAHS: results from the EXPAH study. Mutat. Res. 2007;620:7–15. doi: 10.1016/j.mrfmmm.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Hayes J.D., Flanagan J.U., Jowsey I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- Herrlinger K.R., Cummings J.R., Barnardo M.C., Schwab M., Ahmad T., Jewell D.P. The pharmacogenetics of methotrexate in inflammatory bowel disease. Pharmacogenet. Genomics. 2005;15:705–711. doi: 10.1097/01.fpc.0000172242.19675.33. [DOI] [PubMed] [Google Scholar]
- Hoffman M. Hypothesis: hyperhomocysteinemia is an indicator of oxidant. Med. Hypotheses. 2011;77:1088–1093. doi: 10.1016/j.mehy.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Hubacek J.A., Rynekrova J., Kasparova D., Adamkova V., Holmes M.V., Fait T. Association of MTHFR genetic variants C677T and A1298C on predisposition to spontaneous abortion in Slavonic population. Clin. Chim. Acta. 2015;440:104–107. doi: 10.1016/j.cca.2014.11.018. [DOI] [PubMed] [Google Scholar]
- Jiang Y. Hyperhomocysteinemia and related genetic polymorphisms correlate with ulcerative colitis in Chinese Han population in Central China. Cell Biochem. Biophys. 2012;62:203–210. doi: 10.1007/s12013-011-9283-4. [DOI] [PubMed] [Google Scholar]
- Karatzas P.S., Gazouli M., Safioleas M., Mantzaris G.J. DNA methylation changes in inflammatory bowel disease. Ann. Gastroenterol. 2014;27:125–132. [PMC free article] [PubMed] [Google Scholar]
- Karban A. Non-Jewish Israeli IBD patients have significantly higher glutathione S-transferase GSTT1-null frequency. Dig. Dis. Sci. 2011;56:2081–2087. doi: 10.1007/s10620-010-1543-4. [DOI] [PubMed] [Google Scholar]
- Lakatos P.L. Is current smoking still an important environmental factor in inflammatory bowel diseases? Results from a population-based incident cohort. Inflamm. Bowel Dis. 2013;19:1010–1017. doi: 10.1097/MIB.0b013e3182802b3e. [DOI] [PubMed] [Google Scholar]
- Lennard-Jones J.E. Classification of inflammatory bowel disease. Scand. J. Gastroenterol. Suppl. 1989;170:2–6. doi: 10.3109/00365528909091339. [DOI] [PubMed] [Google Scholar]
- Lettre G., Lange C., Hirschhorn J.N. Genetic model testing and statistical power in population-based association studies of quantitative traits. Genet. Epidemiol. 2007;31:358–362. doi: 10.1002/gepi.20217. [DOI] [PubMed] [Google Scholar]
- Lewis C.M. Genetic association studies: design, analysis and interpretation. Brief. Bioinform. 2002;3:146–153. doi: 10.1093/bib/3.2.146. [DOI] [PubMed] [Google Scholar]
- Li X. Heterozygote advantage of methylenetetrahydrofolate reductase polymorphisms on clinical outcomes in advanced non-small cell lung cancer (NSCLC) patients treated with platinum-based chemotherapy. Tumour Biol. 2014;35:11159–11170. doi: 10.1007/s13277-014-2427-6. [DOI] [PubMed] [Google Scholar]
- Liang S. Significant association between asthma risk and the GSTM1 and GSTT1 deletion olymorphisms: an updated meta-analysis of case–control studies. Respirology. 2013;18:774–783. doi: 10.1111/resp.12097. [DOI] [PubMed] [Google Scholar]
- Loscalzo J. The oxidant stress of hyperhomocyst(e)inemia. J. Clin. Invest. 1996;98:5–7. doi: 10.1172/JCI118776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q. The critical importance of epigenetics in autoimmunity. J. Autoimmun. 2013;41:1–5. doi: 10.1016/j.jaut.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Magro F. High prevalence of combined thrombophilic abnormalities in patients with inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 2003;15:1157–1163. doi: 10.1097/00042737-200311000-00002. [DOI] [PubMed] [Google Scholar]
- Mahmud N. Increased prevalence of methylenetetrahydrofolate reductase C677T variant in patients with inflammatory bowel disease, and its clinical implications. Gut. 1999;45:389–394. doi: 10.1136/gut.45.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao R. Association study between methylenetetrahydrofolate reductase gene polymorphisms and graves' disease. Cell Biochem. Funct. 2010;28:585–590. doi: 10.1002/cbf.1694. [DOI] [PubMed] [Google Scholar]
- Mavragani C.P., Patronas N., Dalakas M., Moutsopoulos H.M. Ill-defined neurological syndromes with autoimmune background: a diagnostic challenge. J. Rheumatol. 2007;34:341–345. [PubMed] [Google Scholar]
- Miller S.A., Dykes D.D., Polesky H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal R.D., Manchanda P.K., Bid H.K., Ghoshal U.C. Analysis of polymorphisms of tumor necrosis factor-alpha and polymorphic xenobiotic metabolizing enzymes in inflammatory bowel disease: study from northern India. J. Gastroenterol. Hepatol. 2007;22:920–924. doi: 10.1111/j.1440-1746.2006.04538.x. [DOI] [PubMed] [Google Scholar]
- Nielsen J.N., Larsen T.B., Fredholm L., Brandslund I., Munkholm P., Hey H. Increased prevalence of methylenetetrahydrofolate reductase C677T variant in patients with IBD. Gut. 2000;47:456–457. doi: 10.1136/gut.47.3.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa A. Hyperhomocysteinemia and prevalence of polymorphisms of homocysteine metabolism-related enzymes in patients with inflammatory bowel disease. Am. J. Gastroenterol. 2001;96:2677–2682. doi: 10.1111/j.1572-0241.2001.04127.x. [DOI] [PubMed] [Google Scholar]
- Peyrin-Biroulet L. Vascular and cellular stress in inflammatory bowel disease: revisiting the role of homocysteine. Am. J. Gastroenterol. 2007;102(5):1108–1115. doi: 10.1111/j.1572-0241.2007.01170.x. [DOI] [PubMed] [Google Scholar]
- Piechota-Polanczyk A., Fichna J. Review article: the role of oxidative stress in pathogenesis and treatment of inflammatory bowel diseases. Naunyn Schmiedeberg's Arch. Pharmacol. 2014;387:605–620. doi: 10.1007/s00210-014-0985-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravda J. Radical induction theory of ulcerative colitis. World J. Gastroenterol. 2005;11:2371–2384. doi: 10.3748/wjg.v11.i16.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease. FEBS J. 2011;278:4243–4251. doi: 10.1111/j.1742-4658.2011.08358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaie A., Parker R.D., Abdollahi M. Oxidative stress and pathogenesis of inflammatory bowel disease: an epiphenomenon or the cause? Dig. Dis. Sci. 2007;52:2015–2021. doi: 10.1007/s10620-006-9622-2. [DOI] [PubMed] [Google Scholar]
- Satsangi J., Silverberg M.S., Vermeire S., Colombel J.F. The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut. 2006;55:749–753. doi: 10.1136/gut.2005.082909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwahn B., Rozen R. Polymorphisms in the methylenetetrahydrofolate reductase gene: clinical consequences. Am. J. Pharmacogenomics. 2001;1:189–201. doi: 10.2165/00129785-200101030-00004. [DOI] [PubMed] [Google Scholar]
- Senhaji N. Methylenetetrahydrofolate reductase C677T variant in Moroccan patients with inflammatory bowel disease. Gene. 2013;521:45–49. doi: 10.1016/j.gene.2013.02.046. [DOI] [PubMed] [Google Scholar]
- Sheehan D., Meade G., Foley V.M., Dowd C.A. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001;360:1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange R.C., Jones P.W., Fryer A.A. Glutathione S-transferase: genetics and role in toxicology. Toxicol. Lett. 2000;112-113:357–363. doi: 10.1016/s0378-4274(99)00230-1. [DOI] [PubMed] [Google Scholar]
- Tujague J., Bastaki M., Holland N., Balmes J.R., Tager I.B. Antioxidant intake, GSTM1 polymorphism and pulmonary function in healthy young adults. Eur. Respir. J. 2006;27:282–288. doi: 10.1183/09031936.06.00033705. [DOI] [PubMed] [Google Scholar]
- Valko M., Leibfritz D., Moncol J., Cronin M.T., Mazur M., Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- van der Put N.M. A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am. J. Hum. Genet. 1998;62:1044–1051. doi: 10.1086/301825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchi M. Inflammatory bowel diseases are not associated with major hereditary conditions predisposing to thrombosis. Dig. Dis. Sci. 2000;45:1465–1469. doi: 10.1023/a:1005541028045. [DOI] [PubMed] [Google Scholar]
- Weisberg I., Tran P., Christensen B., Sibani S., Rozen R. A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol. Genet. Metab. 1998;64:169–172. doi: 10.1006/mgme.1998.2714. [DOI] [PubMed] [Google Scholar]
- Ye X., Jiang Y., Wang H., Chen L., Yuan S., Xia B. Genetic polymorphisms of glutathione S-transferases are associated with ulcerative colitis in central China. Cell Biochem. Biophys. 2011;60:323–328. doi: 10.1007/s12013-011-9154-z. [DOI] [PubMed] [Google Scholar]
- Yilmaz S., Bayan K., Tüzün Y., Batun S., Altintaş A. A comprehensive analysis of 12 thrombophilic mutations and related parameters in patients with inflammatory bowel disease: data from Turkey. J. Thromb. Thrombolysis. 2006;22:205–212. doi: 10.1007/s11239-006-9032-5. [DOI] [PubMed] [Google Scholar]
- Zhao M., Li X., Xing C., Zhou B. Association of methylenetetrahydrofolate reductase C677T and A1298C polymorphisms with colorectal cancer risk: a meta-analysis. Biomed. Rep. 2013;1:781–791. doi: 10.3892/br.2013.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S., Ma X., Buffler P.A., Smith M.T., Wiencke J.K. Whole genome amplification increases the efficiency and validity of buccal cell genotyping in pediatric populations. Cancer Epidemiol. Biomark. Prev. 2001;10:697–700. [PubMed] [Google Scholar]
- Živković M., Životić I., Dinčić E., Stojković L., Vojinović S., Stanković A. The glutathione S-transferase T1 deletion is associated with susceptibility to multiple sclerosis. J. Neurol. Sci. 2013;334:6–9. doi: 10.1016/j.jns.2013.07.001. [DOI] [PubMed] [Google Scholar]