

Abstract
Here, we sequenced and functionally annotated the long reads (1–2 kb) cDNAs library of an infratentorial ependymoma tumor tissue on PacBio RSII by Iso-Seq protocol using SMRT technology. 577 MB, data was generated from the brain tissues of ependymoma tumor patient, producing 1,19,313 high-quality reads assembled into 19,878 contigs using Celera assembler followed by Quiver pipelines, which produced 2952 unique protein accessions in the nr protein database and 307 KEGG pathways.
Additionally, when we compared GO terms of second and third level with alternative splicing data obtained through HTA Array2.0. We identified four and twelve transcript cluster IDs in Level-2 and Level-3 scores respectively with alternative splicing index predicting mainly the major pathways of hallmarks of cancer. Out of these transcript cluster IDs only transcript cluster IDs of gene PNMT, SNN and LAMB1 showed Reads Per Kilobase of exon model per Million mapped reads (RPKM) values at gene-level expression (GE) and transcript-level (TE) track. Most importantly, brain-specific genes–—PNMT, SNN and LAMB1 show their involvement in Ependymoma.
Keywords: Ependymoma, Isoseq, PacBio, Annotation
Highlights
-
•
Produced 2952 unique protein accessions in the nr protein database
-
•
Identified 307 KEGG-pathways with 442 enzyme-codes for 1172 unique-sequences
-
•
Identified transcript cluster-IDs in Level-2 and -3 GO-terms with alternative splicing index
-
•
Involvement of PNMT, SNN and LAMB1-brain specific genes in Ependymoma
1. Introduction
Ependymoma arise from ependymal cells which line the ventricles of the brain and center of the spinal cord. They are soft, grayish or red tumors with cyst or mineral calcifications and are infratentorial or supratentorial in origin. They are both slow and fast growing tumors. Young adults and children show its predominance, in whom they account for approximately 10% of all brain tumors (Yao et al., 2011).
Histopathologically they have been classified into four sub-groups and World Health Organization (WHO) has classified into three grades GradeI, II and III. Myxopapillary- and sub-ependymomas are slow growing (WHO-Grade-I) tumors occurring in the lower part of the spinal column and near ventricles of the brain respectively. Ependymomas and anaplastic ependymomas are fast growing WHO-Grade-II and III tumors occurring in posterior fossa regions (Palm et al., 2009).
Microarray based-transcript profiles of ependymoma have correlated with clinicohistologic characteristics. These studies have showed association of Notch and Sonic Hedgehog pathways to intracranial ependymomas, and homeobox-containing (HOX) family genes to extracranial ependymomas (Korshunov et al., 2003, Modena et al., 2006, Taylor et al., 2005). Some reports have differentiated inra- versus extra-cranial ependymomas and normal tissue versus posterior fossa tumors on the basis of transcripts (Lukashova-v Zangen et al., 2007, Suarez-Merino et al., 2005). Additionally, dysregulation of developmental pathways were identified in anaplastic ependymomas (Palm et al., 2009).
However, ependymoma molecular characterization has been done to large extent but then also tumor-specific molecules are needed across diverse areas of oncology for use in early detection, diagnosis, prognosis and therapy. To address this issue, we performed Isoseq sequencing on histopathologically, immunohistochemically and molecularly classified Ependymoma (WHO grade-II), (Singh et al., 2015) a posterior fossa tumor from a 2 year-old-male with informed consent. We identified four and twelve transcript cluster IDs (HTA array 2.0) in Level-2 and Level-3 GO terms assignment. Further, when correlated these transcript cluster IDs with gene-level expression (GE) and transcript-level (TE) track data obtained through RNA Seq analysis, only gene PNMT, SNN and LAMB1 showed RPKM of 26.24929, 36.68109 and 3.376959 respectively and transcripts PNMT_1, SNN_1, LAMB1_3, LAMB1_1 showed RPKM 28.29116, 36.68109, 2.838438 and 0.93592 respectively.
2. Methodology
Total RNA extracted (Chomczynski and Sacchi, 1987) from tumor tissue (ependymoma) with RIN (RNA Integrity Number) > 8.0 was used for library preparation. The library was constructed as per Clontech SMARTer-PCR cDNA Synthesis Sample Preparation Guide” (Zhu et al., 2001). 1–2 KB libraries were selected on 0.8% Agarose Gel, purified, end repaired and blunt end SMARTbell adapters were ligated. The libraries were quantified using Qubit (Invitrogen) and validated for quality by running a LabChipGX (Caliper Life Sciences) subsequently sequencing was done in 8-well-SMART Cell v3 in PacBioRSII.
The isolated RNA of ependymoma tumor was further compared to normal control i.e. posterior fossa tissue from the normal trauma patient. The tissues were obtained after informed consent and the study was ethically approved by the Institutional Ethics Committee of the King George's Medical University. The detailed procedure of obtaining CEL files through Affymetrix HTA 2.0 arrays using 500 ng of total RNA according to the manufacturer's recommendations (Affymetrix, Santa Clara, Calif) has been previously documented by Singh et al. (Singh et al., 2015). The CEL files generated by these arrays were converted into rma-gene-ful.chp and rma-alt-splice-dabg.chp files through Affymetrix Expression Console™ Software (version 1.3) and the data was analyzed through Transcriptome Analysis Console v3.0.
The generated raw sequences were processed for quality control checks including filtering of high-quality reads based on the score value, removal of primer/adaptor sequence reads and trimming of read-length using Celera assembler followed by Quiver (Chin et al., 2013). Putative function of the assembled contigs was deduced by using them as queries against the SwissProt and the non-redundant (nr)-protein database in the BLASTX-program using a cut off E-value was set at 1e-6 and only the top gene-ID and name were initially assigned to each contig. Gene ontology (GO) annotation analysis was performed in Blast2GO (http://www.blast2go.org/) version 2.5.0 (Conesa et al., 2005) for the assignment of gene ontology terms. After gene-ID mapping, GO term assignment, annotation augmentation and generic GO-slim process the final annotation file was produced and the results were categorized with respect to Biological Process, Molecular Function, and Cellular Component at level 2.
Pathway analyses of unique sequences were carried out based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database using the online KEGG Automatic Annotation Server (KAAS) (http://www.genome.jp/tools/kaas/) by using Bi-directional Best Hit (BBH) method (Moriya et al., 2007). Enzyme commission (EC) numbers were obtained and were putatively mapped for protein sequences to a specific biochemical pathway.
We also analyzed our clean data through RNA-seq tool (CLC Genomics Workbench-version) to produce mRNA expression levels for genes and/or transcripts, from samples of ependymoma-(m)RNA reads. This has been done by mapping the reads to reference genome (Homo sapiens-hg18) that includes gene annotations and (optionally) mRNA annotations. The total length of reference sequence was 3,080,436,051 including 36,693 genes and 49,495 transcripts. RNA seq report is generated for the query sequences giving the information of read sequences, reference sequences, total no of genes and transcripts in reference sequence, graph showing the number of transcripts per gene, graph showing the number of exons per transcript and graph showing the distribution of transcript length. Further, the mapped results were output as a reads track at the gene and transcript level showing expression values at the gene and transcript level, respectively in the form of unique count, total count and reads per kilobase per million (RPKM). Importantly, the maximum number of hits for a read parameter was set for 1 to consider uniquely mapped reads. The number of reads and reads per million were determined using the CLC mapping program.
3. Results and discussion
Using Celera assembler followed by Quiver, a total of 1,19,313 raw sequence reads with maximum length of 5580 bp length were generated encompassing about 577 MB of sequence data in fastq format. A total of 19,878 contigs (with a length of 500–3000 bp) had significant BLASTX hit corresponding to 2952 unique protein accessions in the nr protein database (Table 1 and Fig. 1). Gene ontology (GO) analysis of these 2952 unique proteins resulted in a total of 50,596 GO terms including 27,484 (54.32%) biological process terms, 10,518 (20.78%) molecular function terms and 12,594 (24.89%) cellular component terms. Among the biological process category 4592, 3980 and 3662 genes were related to cellular processes (GO:0008150), single-organism process (GO:0008150) and metabolic process respectively; a significant numbers of genes were also identified from biological regulation (2961), response to stimulus (2136), localization (1748), cellular component organization or biogenesis (1616), multicellular organismal process (1615), developmental process (1492) and signaling (1402) sub-categories. Similarly under molecular function category, 3724 genes were involved in the binding process (GO: 0005488) and 1676 genes in the catalytic activity (GO: 0003824); whereas under the cellular component category, 4925 genes from cell (GO: 0005623), and 3570 genes corresponded to organelle (GO:0043226), 1768 genes corresponded to macromolecular complex (GO:) and 1692 genes corresponded to membrane (GO:) were the most represented categories (Fig. 2).
Table 1.
Statistics of transcriptome assembly.
| Numbers of raw sequence reads | 1, 19, 313 |
|---|---|
| File size of raw data | 577 MB |
| Number of contigs generated | 19,878 |
| Size | 500–3000 bp |
| Unique protein accessions in the nr protein database | 2952 |
Fig. 1.
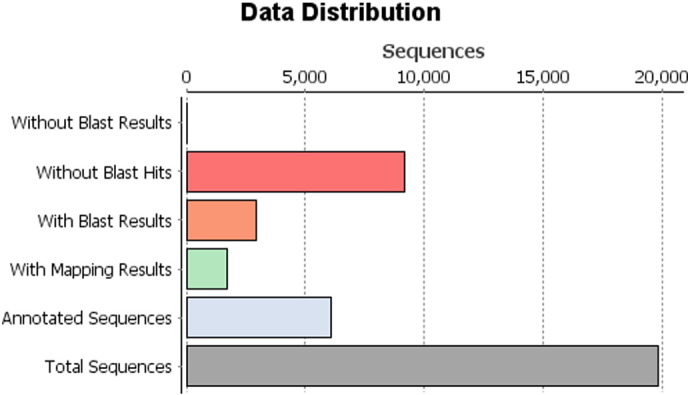
Data distribution in the non-redundant protein database.
Fig. 2.

Percentages of functionally annotated transcripts assigned with GO terms according to level 2 categories: GO-terms were categorized at level 2 under three main categories; (upper to lower): Biological process, Cellular component and Molecular function.
Enzyme commission (EC) numbers were assigned with 442 enzyme codes for 1172 unique sequences (Fig. 3). 307 KEGG pathways were represented by 2952 unique transcripts (Supplementary Table 1) including cancer-related (KO052000: 39 transcripts coding 35 genes) (Fig. 4), transcriptional misregulation (KO0502: 35 transcripts coding 21 genes), microRNA (KO0520622: 22 transcripts coding 18 genes) and proteoglycans in cancer (28 transcripts coding 26 genes) pathways.
Fig. 3.

Enzyme Code Distribution: EC numbers were assigned with 442 enzyme codes for 1172 unique sequences.
Fig. 4.

Pathways in Cancer: 39 transcripts coding 35 genes were mapped in infratentorial ependymoma tumor tissue library (KEGG databases).
Statistical distribution showed majority of BLASTX-hit sequences of ependymoma transcript produced similarities with Homo sapiens followed by other species like Macaca fasicularis and Macaca mulatta (Fig. 5).
Fig. 5.

Statistical distribution of Top Hit Species in the non-redundant protein database.
The nucleotide sequence of the infratentorial ependymoma has been deposited in NCBI/SRA under accession no. SUB1031314.
Additionally, when we screened GO terms of second and third level in alternative splicing data obtained through HTA Array2.0. The identified transcript clusters showed linear gene fold change based on the average of all the eligible exons Probe Specific Regions (PSRs) intensities in that gene between ependymoma and control samples. We identified four and twelve transcript cluster IDs in Level-2 and Level-3 respectively with alternative splicing index Supplementary Table 2. The four transcript clusters of Level-2 of HMCN2, CES1P1, LILRB4 and SRRD genes owed to various biological processes like metabolic process, response to stimulus, immune system process and rhythmic process. While twelve transcript clusters of Level-3 of IL2RG, GAL3ST2, ANTXR2, ULBP2, LAMB1, PON3, TDH, IFITM5, TPH2, HDGFRP3, SNN and PNMT genes owed to reproductive process, response to external stimulus, response to biotic stimulus, biosynthetic process, antigen processing and presentation, methylation, circadian rhythm, immune response, cell proliferation, response to stress, cellular metabolic process and cell adhesion. The various isoforms of the identified transcript clusters of level-2 and level-3 has been depicted in Supplementary Table 3 and Supplementary Figs. 1 (a–d) and 2 (a–l).
Supplementary Fig. 1.




Transcript Clusters of Level-2 GO terms as per alternative splicing data obtained through HTA Array2.0. a) TC16000460.hg.1, b) TC09002937.hg.1, c) TC19000882.hg.1, d) TC22000170.hg.1.
Supplementary Fig. 2.












Transcript Clusters of Level-3 GO terms as per alternative splicing data obtained through HTA Array2.0. a) TC04001328.hg.1, b) TC07003364.hg.1, c) TC11003502.hg.1, d) TC02005017.hg.1, e) TC06001088.hg.1, f) TC17000479.hg.1, g) TC12000629.hg.1, h) TC0X001121.hg.1, i) TC15001766.hg.1, j) TC16000165.hg.1, k) TC08002637.hg.1, l) TC07001747.hg.1.
This shows that identified transcripts in 1–2 Kb fragments of ependymoma sequence show differential regulation at gene level and alternative splicing index on comparison with normal sample. Most of the proteins predicted include the major pathways of hallmarks of cancer (Hanahan and Weinberg, 2011).
Through RNA-seq analysis (mapping statistics) we identified that out of 4, 69, and 559 fragments, 1, 83, and 302 were counted and 2, 86, and 257 were uncounted fragments. In counted fragments 1, 77, 550 fragments uniquely mapped to reference human genome (hg18) and 5,752 non-specifically mapped (Supplementary Table 4). Further, counted fragments were divided into different types like reads that mapped completely to exon; reads that mapped across an exon junction and reads that completely mapped to introns.
The gene-level expression (GE) track holds information about counts, length as annotated and expression values for each gene involved in Ependymoma on using one reference sequence per transcript (Supplementary Table 5a). Total number of genes identified were 20, 274. The expression values are expressed as total count (sum of total number of reads mapped to the gene and total number of reads mapped to the reference sequence (un-annotated references)), unique count (total number of reads mapped to the gene) and normalized RPKM (Reads Per Kilobase of exon model per Million mapped reads). It also shows unique (number of reads that match uniquely to the gene) and total gene reads (number of reads that match uniquely to the gene and reads that map to more positions in the reference other than the gene), annotated (based on mRNA annotations) and detected transcripts (no of annotated transcripts to which reads have been assigned). Similarly, it also expresses unique exonic reads (number of reads that match uniquely to the exons including across exon–exon junctions) and total exon reads (number of reads mapped to this gene that fall entirely within an exon or in exon–exon or exon–intron junctions) accompanied with unique exon–exon (reads that uniquely match across an exon–exon junction of the gene) and total exon–exon reads (reads that uniquely match across an exon–exon junction of the gene as well as multiple matches that were assigned to an exon–exon junction of this gene). Besides exonic, unique intron–exon (reads that uniquely match across an intron–exon junction of the gene) and total intron–exon reads (maps across an intron–exon boundary of the gene) are also depicted in the GE track (Supplementary Table 5a).
Each row in the transcript-level expression (TE) track table corresponds to an mRNA annotation in the mRNA track used as reference (Supplementary Table 5b). It holds information about counts i.e. 11, 202 transcripts for 20, 274 genes, length as annotated and expression values as RPKM and relative RPKM (RPKM for the transcript divided by the maximum of the RPKM values among all transcripts of the same gene) for each transcripts involved in Ependymoma (Supplementary Table 5b). The track also depicts information about unique (number of reads in the mapping for the gene that are uniquely assignable to the transcript) and total transcript reads (‘Unique transcript read's plus the remaining (non-unique) transcript reads which are assigned randomly to one of the transcripts to which they match), annotated (number of transcripts based on the mRNA annotations on the reference) and detected transcripts (no of annotated transcripts to which reads have been assigned).
Importantly, genes that mapped distinctly at gene level (GE track) did not mapped uniquely to particular transcript i.e. were non-specific at transcript level (TE track), hence suggestive of transcript isoforms of the particular gene involved in Ependymoma (Supplementary Table 5a and b).
The identified four and twelve transcript cluster IDs (HTA array 2.0) in Level-2 and Level-3 GO terms when correlated with GE and TE track data obtained through RNA Seq, only gene PNMT, SNN and LAMB1 showed RPKM of 26.24929, 36.68109 and 3.376959 respectively and transcripts PNMT_1, SNN_1, LAMB1_3, LAMB1_1 showed RPKM 28.29116, 36.68109, 2.838438 and 0.93592 respectively (Supplementary Table 6a and b). Involvement of Phenylethanolamine N-Methyltransferase (PNMT) has been reported in neuroendocrine differentiation (Yang et al., 2013) and its differential regulation has also been reported in neuroblastoma (Castro-Vega et al., 2015a), ependymoma (Castro-Vega et al., 2015a), paraganglioma (Castro-Vega et al., 2015, Castro-Vega et al., 2015) and different subgroups of breast cancer (Dai et al., 2014). SNN (Stannin), a protein coding gene associated with the disease-cavernous hemangioma, a collection of small blood vessels (capillaries) in the central nervous system (CNS) shows its mRNA expression in brain tissue (SNN Gene Cards). Laminin B1 (LAMB1), involved in the organization of the laminar architecture of cerebral cortex. It is most likely also required for the integrity of the basement membrane/glia limitans that serves as an anchor point for the end feet of radial glial cells and as a physical barrier to migrating neurons. Radial glial cells play a central role in cerebral cortical development, where they act both as the proliferative unit of the cerebral cortex and a scaffold for neurons migrating toward the gial surface (LAMB1 gene cards). LAMB1 has also been identified in Diffuse intrinsic pontine glioma (DIPG) a highly morbid form of pediatric brainstem glioma (Saratsis et al., 2014). As per the gene cards the expression images based on data from Illumina Human BodyMap show mRNA expression of all the three genes in brain tissue as well as cerebellum. Hence, after streamlining the data through both HTA array 2.0 and Isoseq analysis, we report the involvement of brain-specific genes—PNMT, SNN and LAMB1 in Ependymoma.
The following are the supplementary data related to this article.
KEGG mappings.
a. Biological Processes related to GO terms.
b. GO terms of second level in alternative splicing data obtained through HTA Array2.0.
c. GO terms of second level in alternative splicing data obtained through HTA Array2.0.
a. Gene-level expression (GE) track holding information about counts, length as annotated and expression values for each gene involved in Ependymoma on using one reference sequence per transcript.
b. Transcript-level expression (TE) track holding information about counts, length as annotated and expression values as RPKM and relative RPKM (RPKM for the transcript divided by the maximum of the RPKM values among all transcripts of the same gene) for each transcripts involved in Ependymoma.
Various isoforms of the identified transcript clusters of level-2 and level-3.
a. Correlation between HTA array-based transcript cluster ID identified in GO-terms level-3 and gene expression (GE) track data. b. Correlation between HTA array-based transcript cluster ID identified in GO-terms level-3 and transcript-level expression (TE) track data.
RNA Seq Report.
Author contributions
Conceived and designed the experiments: NS, DKG and RK. Samples were provided by: BKO and CS; Histopathological and immunohistochemical details: provided by MG. Sample Collection and nucleic acid isolation: RC and AM; Performed and Analyzed the experiments: NS and DKS. Wrote the paper and discussion: NS. Contributed reagents/materials/analysis tools: RK. Reviewed the paper: BKO, CS, DKG and RK.
Competing interests
None of the authors have any competing interests.
Acknowledgements
We are grateful to Mr. Prasad Bajaj, of Xcelris Genomics, India and Dr. Paras Yadav, Mr. Rakshit Choudhary of Imperial Life Sciences, Gurgaon, Haryana for their technical assistance.
References
- Yao Y., Mack S.C., Taylor M.D. Molecular genetics of ependymoma. Chin. J. Cancer. 2011;30(10):669–681. doi: 10.5732/cjc.011.10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm T., Figarella-Branger D., Chapon F., Lacroix C., Gray F., Scaravilli F., Ellison D.W., Salmon I., Vikkula M., Godfraind C. Expression profiling of ependymomas unravels localization and tumor grade-specific tumorigenesis. Cancer. 2009;115(17):3955–3968. doi: 10.1002/cncr.24476. [DOI] [PubMed] [Google Scholar]
- Korshunov A., Neben K., Wrobel G., Tews B., Benner A., Hahn M., Golanov A., Lichter P. Gene expression patterns in ependymomas correlate with tumor location, grade, and patient age. Am. J. Pathol. 2003 Nov;163(5):1721–1727. doi: 10.1016/S0002-9440(10)63530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modena P., Lualdi E., Facchinetti F., Veltman J., Reid J.F., Minardi S., Janssen I., Giangaspero F., Forni M., Finocchiaro G., Genitori L., Giordano F., Riccardi R., Schoenmakers E.F., Massimino M., Sozzi G. Identification of tumor-specific molecular signatures in intracranial ependymoma and association with clinical characteristics. J. Clin. Oncol. 2006;24(33):5223–5233. doi: 10.1200/JCO.2006.06.3701. [DOI] [PubMed] [Google Scholar]
- Taylor M.D., Poppleton H., Fuller C., Su X., Liu Y., Jensen P., Magdaleno S., Dalton J., Calabrese C., Board J., Macdonald T., Rutka J., Guha A., Gajjar A., Curran T., Gilbertson R.J. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8(4):323–335. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Lukashova-v Zangen I., Kneitz S., Monoranu C.M., Rutkowski S., Hinkes B., Vince G.H., Huang B., Roggendorf W. Ependymoma gene expression profiles associated with histological subtype, proliferation, and patient survival. Acta Neuropathol. 2007;113(3):325–337. doi: 10.1007/s00401-006-0190-5. Epub 2007 Jan 31. [DOI] [PubMed] [Google Scholar]
- Suarez-Merino B., Hubank M., Revesz T., Harkness W., Hayward R., Thompson D., Darling J.L., Thomas D.G., Warr T.J. Microarray analysis of pediatric ependymoma identifies a cluster of 112 candidate genes including four transcripts at 22q12.1–q13.3. Neuro-Oncology. 2005;7(1):20–31. doi: 10.1215/S1152851704000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N., Sahu D.K., Mishra A., Agarwal P., Goel M.M., Chandra A., Singh S.K., Srivastava C., Ojha B.K., Gupta D.K., Kant R. Multiomics approach showing genome-wide copy number alterations and differential gene expression in different types of North-Indian pediatric brain tumors. Gene. 2015 doi: 10.1016/j.gene.2015.09.078. (ii: S0378-1119(15)01187–7. [Epub ahead of print]) [DOI] [PubMed] [Google Scholar]
- Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162(1):156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Zhu Y.Y., Machleder E.M., Chenchik A., Li R., Siebert P.M. Reverse transcriptase template switching: a SMART™ approach for full-length cDNA library construction. Biotechniques. 2001;30:892–897. doi: 10.2144/01304pf02. [DOI] [PubMed] [Google Scholar]
- Chin C.S., Alexander D.H., Marks P., Klammer A.A., Drake J., Heiner C., Clum A., Copeland A., Huddleston J., Eichler E.E., Turner S.W., Korlach J. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods. 2013;10(6):563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- Conesa A., Gotz S., Garcia-Gomez J.M., Terol J., Talon M., Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Moriya Y., Itoh M., Okuda S., Yoshizawa A.C., Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:182–185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Yang M., Soga T., Pollard P.J. Oncometabolites: linking altered metabolism with cancer. J. Clin. Invest. 2013;123(9):3652–3658. doi: 10.1172/JCI67228. (Epub 2013 Sep 3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Vega L.J., Lepoutre-Lussey C., Gimenez-Roqueplo A.P., Favier J. Rethinking pheochromocytomas and paragangliomas from a genomic perspective. Oncogene. 2015 doi: 10.1038/onc.2015.172. ([Epub ahead of print]) [DOI] [PubMed] [Google Scholar]
- Castro-Vega L.J., Letouzé E., Burnichon N., Buffet A., Disderot P.H., Khalifa E., Loriot C., Elarouci N., Morin A., Menara M., Lepoutre-Lussey C., Badoual C., Sibony M., Dousset B., Libé R., Zinzindohoue F., Plouin P.F., Bertherat J., Amar L., de Reyniès A., Favier J., Gimenez-Roqueplo A.P. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat. Commun. 2015;6044 doi: 10.1038/ncomms7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X., Chen A., Bai Z. Integrative investigation on breast cancer in ER, PR and HER2-defined subgroups using mRNA and miRNA expression profiling. Sci. Rep. 2014;4:6566. doi: 10.1038/srep06566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saratsis A.M., Kambhampati M., Snyder K., Yadavilli S., Devaney J.M., Harmon B., Hall J., Raabe E.H., An P., Weingart M., Rood B.R., Magge S.N., TJ M.D., Packer R.J., Nazarian J. Comparative multidimensional molecular analyses of pediatric diffuse intrinsic pontine glioma reveals distinct molecular subtypes. Acta Neuropathol. 2014;27(6):881–895. doi: 10.1007/s00401-013-1218-2. (Epub 2013 Dec 3) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
KEGG mappings.
a. Biological Processes related to GO terms.
b. GO terms of second level in alternative splicing data obtained through HTA Array2.0.
c. GO terms of second level in alternative splicing data obtained through HTA Array2.0.
a. Gene-level expression (GE) track holding information about counts, length as annotated and expression values for each gene involved in Ependymoma on using one reference sequence per transcript.
b. Transcript-level expression (TE) track holding information about counts, length as annotated and expression values as RPKM and relative RPKM (RPKM for the transcript divided by the maximum of the RPKM values among all transcripts of the same gene) for each transcripts involved in Ependymoma.
Various isoforms of the identified transcript clusters of level-2 and level-3.
a. Correlation between HTA array-based transcript cluster ID identified in GO-terms level-3 and gene expression (GE) track data. b. Correlation between HTA array-based transcript cluster ID identified in GO-terms level-3 and transcript-level expression (TE) track data.
RNA Seq Report.