

Abstract
Clopidogrel (INN), an oral antiplatelet drug, has been revealed to have a number of biological properties, for instance, anti-inflammation and antioxidation. Oxidative stress plays an imperative role in inflammation, diabetes mellitus, atherosclerosis, and cancer. In the present study, human aortic endothelial cells (HAECs) were employed to explore the anti-inflammatory activity of INN. INN reduced TNFα-induced reactive oxygen species (ROS) generation and time-dependently prompted the expression and activity of heme oxygenase 1 (HO-1). Cellular glutathione (GSH) levels were augmented by INN. shHO-1 blocked the INN suppression of TNFα-induced HL-60 cell adhesion. The CaMKKβ/AMPK pathway and Nrf2 transcriptional factor were implicated in the induction of HO-1 by INN. Additionally, TNFα dramatically augmented VCAM-1 expression at protein and mRNA levels. INN treatment strikingly repressed TNFα-induced expression of VCAM-1 and HL-60 cell adhesion. Compound C, an AMPK inhibitor, and shNrf2 abolished TNFα-induced expression of VCAM-1 and HL-60 cell adhesion. Our data suggest that INN diminishes TNFα-stimulated VCAM-1 expression at least in part via HO-1 induction, which is CaMKKβ/AMPK pathway-dependent.
1. Introduction
Inflammation is common for patients with cardiovascular diseases and is considered to be a sign or atherogenic response. Reactive oxygen species (ROS) such as superoxide anions, hydrogen peroxide, peroxynitrite, and hydroxyl radicals plays an important role in inflammation, leading to endothelial oxidative damage and dysfunction of the cardiovascular system. Proinflammatory cytokines, for example, tumor necrosis factor-alpha (TNFα), are able to stimulate ROS liberation [1]. Superfluous ROS production not only causes endothelial dysfunction [2] but also stimulates signaling transduction pathways implicated in augmented gene expression of inflammation-related cytokine [3]. In cardiovascular cells, the normal intracellular ROS levels rely on the proper balance between ROS formation and antioxidant defense systems.
Sophisticated interactions between leukocytes and the endothelium are involved in inflammatory reaction. The endothelial cell (EC) surface is relatively smooth and nonadhesive. In cardiovascular diseases, the interactions between the ECs and components of the blood are altered by adhesion molecules, for example, ICAM-1 and VCAM-1 [4]. The endothelial cells are activated, leading to overexpression of adhesion molecules at inflammation sites. The proinflammatory molecule TNF-α prominently elevates adhesion molecules on the endothelium [5–7]. Additionally, substantial evidence demonstrates an augmented expression of VCAM-1 in inflammatory animal models and human atherosclerotic plaques [8].
Heme oxygenase 1 (HO-1) possesses antioxidant properties [9]. Its key function is to degrade heme to iron, carbon monoxide (CO), and biliverdin [10, 11]. Biliverdin, via the cytosolic enzyme biliverdin reductase, is converted to bilirubin, which possesses antioxidant characteristics [12]. In addition, HO-1-derived CO is involved in vasoregulation and signal transduction [13, 14]. Some signaling molecules, for instance, 5′ adenosine monophosphate-activated protein kinase (AMPK), mitogen-activated protein kinases (MAPK), and PI3K/Akt, as well as transcriptional factors including activator protein 1 (AP-1) and NF-E2-related factor-2 (Nrf2), regulate HO-1 gene expression [15–18].
Clopidogrel (INN) is an oral antiplatelet drug hindering blood clots and has been discovered to possess several biological properties such as anti-inflammation, antioxidation, and antiatherosclerosis [19–21]. Anti-inflammation and antioxidation can be used as therapeutic strategies to prevent or treat cardiovascular diseases. The present study was carried out to elucidate the role of AMPK and HO-1 in the INN inhibition of TNFα-stimulated expression of VCAM-1 and the underlying mechanisms implicated.
2. Materials and Methods
2.1. Reagents
Primary human aortic endothelial cells (HAECs) and endothelial growth factors were purchased from Cambrex Bioscience (Rockland, ME). HAECs were cultured in M199 complete medium. Medium 199, gentamycin, fungizone, glutamine, collagenase, gelatin, trypsin/EDTA, penicillin, and streptomycin were purchased from Sigma-Aldrich (St. Louis, MO). Fetal bovine serum (FBS) was purchased from HyClone (Logan, UT, USA). HL-60 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 plus l-glutamine and 25 mM HEPES supplemented with 15% FBS [22]. Human TNFα, DMSO, HEPES, sodium bicarbonate, Compound C, and all other chemicals were from Sigma. STO-609 was purchased from Tocris (Ellisville, Missouri). Clopidogrel (INN) (Plavix, Sanofi-Aventis, BN.F-33565, France) stock solution (10 mM) was prepared by dissolving it in DMSO. TRIzol RNA extraction reagent and 2,7-dichlorofluorescin diacetate (H2DCF-DA) were purchased from Invitrogen (Carlsbad, CA, USA). Antibodies reacting with AMPK and phosphor-AMPK (Thr172) were obtained from Cell Signaling Technology (Danvers, MA); antibodies reacting with β-actin and Nrf2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA); anti-HO-1 and VCAM-1 antibodies were obtained from Abcam (Cambridge, MA, USA). Cell culture HAECs were cultured in medium 199 added with 20 mM HEPES, 2 mM glutamine, 20% FBS, 100 mU/L penicillin, and 100 mg/L streptomycin at 37°C. The medium was refreshed every 48 h until confluence. Preincubation with inhibitors was implemented in HEPES-HSA buffer (10 mM HEPES, 10 mM glucose, 1.5 mM CaCl2, 145 mM NaCl, 5 mM KCl, 1 mM MgSO4, and 0.25% HSA). Compound C and STO-609 were dissolved in dimethyl sulfoxide (DMSO).
2.2. Western Blotting
ECs were lysed in the lysis buffer. Equivalent amounts of protein samples were separated by SDS-PAGE and then transferred to nitrocellulose membrane followed by immunoblotting with the primary antibodies including phosphor-AMPK-Thr172, AMPK, HO-1, Nrf2, PARP, and β-actin.
2.3. Real-Time PCR
Total RNA was extracted by using a QIAshredder column and RNeasy kit (Qiagen) and frozen at −80°C for further RT-PCR analysis. Primer sequences were as follows: β-actin (ATGTTTGAGACCTTCAACAC, CACGTCACACTTCATGATGG), VCAM-1 (CGTCTTGGTCAGCCCTTCCT, ACATTCATATACTCCCGCATCCTTC), and HO-1 (GGGTGATAGAAGAGGCCAAGA, AGCTCCTGCAACTCCTCAAA). cDNA (1 μL) was amplified in PCR solution (25 μL) in a Mini OpticonTM Real-Time PCR Detection System. Cycle parameters were set as 95°C for 15 min, 40 cycles of 95°C for 15 sec, 58°C for 1 min, and 72°C for 1 min.
2.4. HO-1 Activity Assay
HO-1 activity was determined as described previously [23]. The HO-1 activity in EC lysates was calculated as picomoles bilirubin produced per hour per milligram of total protein (pmol BR h-1 mg-1) and the data were expressed as fold HO-1 activity compared to control cells.
2.5. Subcellular Fractionation
Subcellular fractionation was performed with a Subcellular Protein Fractionation Kit (Thermo Fisher Scientific Inc., Rockford, IL, USA) according to the manufacturer's protocol. Nrf2 protein levels in the nuclear fractions were measured by Western analysis. Expression of PARP was utilized as loading controls for the purity of the nuclear extracts.
2.6. RNA Interference with shRNA
Lentiviral infection was implemented as described previously [24]. Two distinct sequences targeting human Nrf2 and HO-1 mRNA were selected and obtained from Sigma (St. Louis, MO, USA). The shRNA sequences were as follows: Nrf2 shRNA #1, 5′-GCTCCTACTGTGATGTGAAAT-3′, shRNA #2, 5′-CCGGCATTTCACTAAACACAA-3′; HO-1 shRNA #1, 5′-GCTGAGTTCATGAGGAACTTT-3′, shRNA #2, 5′-GCTGAGTTCATGAGGAACTTT-3′; shLuc shRNA, 5′-CAAATCACAGAATCGTCGTAT-3′. shLuc was used as a vector control.
2.7. Reactive Oxygen Species (ROS) Measurement
Intracellular ROS state was determined with the cell-permeant 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) as previously described [25]. Concisely, HAECs were grown to 50% confluence followed by serum starvation in medium 199 supplemented with 0.5% (v/v) FBS for another 24 h. The ECs were maintained in serum-free medium without phenol red for 15 min preceding being exposed to TNFα. ECs were incubated with H2DCFDA (10 μM) for 10 min and instantly observed under a confocal microscope (Leica TCS SP2).
2.8. Cellular GSH Assay
Cellular GSH assay was performed as described previously [26]. Briefly, ECs were washed two times with ice cold PBS. The homogenate was prepared in the potassium phosphate buffer (20 mM, pH 7.0) and centrifuged at 10,000 ×g for 20 min at 4°C. The supernatant was kept as the cell lysate. The protein was measured with a BCA Protein Assay Kit. Cell lysates (100 μL) were incubated with 5% TCA (150 μL) and centrifuged at 5000 ×g for 10 min at 4°C. The cell lysate was incubated with 0.4 M Tris buffer and 0.01 M DTNB. After incubation at room temperature for 5 min, the intracellular GSH production was measured with a microplate reader at 412 nm (Model 680, Bio-Rad).
2.9. Monocyte Adhesion Assay
The monocyte adhesion assay was executed as described by Chen et al. [27].
2.10. Statistical Analysis
The results were expressed as mean ± SD. Data were analyzed with one-way analysis of variance and Fisher's protected least significant difference test. P < 0.05 was considered as the level of significance.
3. Results
3.1. Effect of Clopidogrel (INN) on TNFα-Induced ROS Formation in HAECs
To determine whether INN decreases TNFα-stimulated ROS formation, we pretreated ECs with 10 μmol/L INN for 24 h and then treated ECs with 1 ng/mL TNFα for another 20 min. As illustrated in Figure 1, TNFα promoted ROS liberation at 20 min, and pretreatment with 10 μmol/L INN dramatically repressed this ROS liberation. N-Acetylcysteine (NAC) is a potent antioxidant and used as a positive control.
Figure 1.
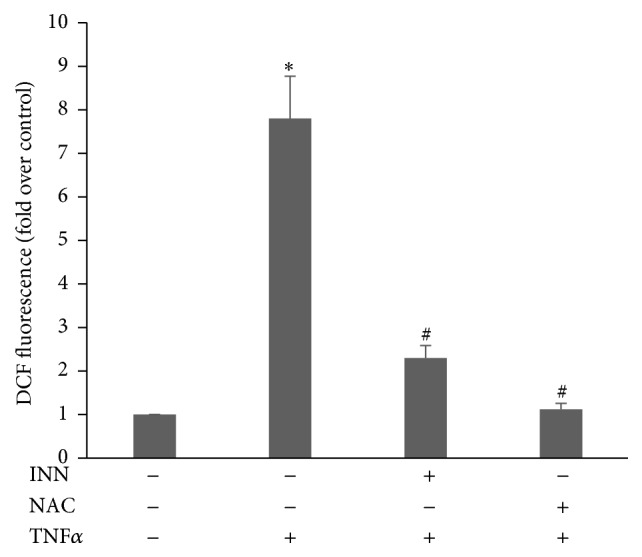
Effect of Clopidogrel (INN) on TNFα-stimulated ROS liberation in HAEC. ECs were pretreated with 10 μM INN for 24 h or 1 mM NAC for 2 h and treated with 10 mM H2DCFDA for 15 min prior to being challenged with TNFα (1 ng/mL) for another 20 min. Cells incubated with 0.1% DMSO for 24 h were used as a control (CON). ROS levels were assayed with a BioTek Fluorescence Plate Reader. The mean DCF value of control is set as “1.” The relative fluorescence intensity is presented as mean ± SD. ∗ P < 0.05 (n = 3, Student's t-test).
3.2. INN Enhances HO-1 Expression and Hinders TNFα-Stimulated HL-60 Adhesion
Since HO-1 has a potent cytoprotective effect against cellular oxidative stress [28, 29], we sought to investigate the effect of INN on HO-1 expression. HAECs were incubated with 10 μM INN for various time periods. As illustrated in Figures 2(a), 2(b), and 2(c), INN treatment time-dependently promoted expression of HO-1 at mRNA and protein levels as well as its activity. Furthermore, the role of HO-1 in the suppression of HL-60 adhesion by INN was determined with HO-1 shRNA knockdown study. ECs were transfected with shHO-1, followed by incubation with 10 μM INN for 24 h and then ECs were stimulated with TNFα for another 6 h. Knockdown of HO-1 was confirmed by Western blot (Figure 2(d)). As indicated in Figure 2(e), shHO-1 lessened the INN suppression of HL-60 adhesion. Together, our data suggest that HO-1 plays an imperative role in the suppression of TNFα-stimulated HL-60 adhesion by INN.
Figure 2.

The role of HO-1 in INN suppression of TNFα-triggered HL-60 cell adhesion. ECs were incubated with 10 μM INN for the indicated time points. Aliquots of lysate (50 μg) were subjected to Western analysis for HO-1 protein expression (a). Total RNA was extracted from ECs and underwent real-time PCR with specific primers for HO-1 and β-actin (b). HO-1 activity was measured three times in each treatment group as mentioned in Methods. Data are expressed as fold increase of HO-1 activity versus control (c). ECs transfected with shHO-1 were incubated with 10 μM INN for 24 h prior to being stimulated with 1 ng/mL TNFα for additional 6 h (d). (e) ECs were incubated with 1 ng/mL TNFα in the presence or absence of INN for 8 h. Adhesion assay was performed as described in Materials and Methods. Values are means ± SD (n = 4). ∗ P < 0.05 versus control; # P < 0.05 versus TNFα.
3.3. The CaMKKβ/AMPK Pathway Mediates INN-Induced HO-1 Expression
Previous studies [30] suggest that AMPK modulates the antioxidant status of cardiovascular ECs by upregulating expression of genes implicated in antioxidant defense, for example, manganese superoxide dismutase, catalase, and thioredoxin. To determine the signaling pathways implicated in the INN-mediated HO-1 induction, we examined the phosphorylation of AMPK. ECs were incubated with INN (10 μM) for different time periods. As illustrated in Figure 3(a), AMPK was activated by INN. However, INN did not change AMPK protein expression. To further substantiate the role of the CaMKKβ/AMPK pathway, their specific inhibitors, STO-609 and Compound C, were used. As shown in Figures 3(b), 3(c), and 3(d), STO-609 and Compound C attenuated INN-triggered HO-1 expression and activity. As indicated in Figure 3(e), the INN-stimulated elevation of cellular GSH content was repressed by STO-609 and Compound C. Our findings demonstrate that INN induces HO-1 expression and increases GSH synthesis through the CaMKKβ/AMPK pathway.
Figure 3.

CaMKKβ/AMPK is involved in INN-induced HO-1 expression and GSH synthesis. ECs were incubated with 10 μM INN for various time periods. (a) Total protein was subjected to Western analysis for p-AMPK and AMPK expression. ECs were treated with 10 μM STO-609 and Compound C for 1 h, followed by incubation with 10 μM INN for another 16 h. Aliquots of cell lysate underwent immunoblotting analysis for HO-1 expression (b). Total RNA was extracted and underwent RT-PCR with specific primers for HO-1 and β-actin (c). HO-1 activity was measured three times in each treatment group as mentioned in Methods. Data are expressed as fold increase of HO-1 activity versus control (d). ECs were treated with 10 μM STO-609 and Compound C for 1 h, followed by incubation with 10 μM INN for another 24 h. GSH content was detected as mentioned in Methods (e). The results represent means ± SD (n = 3). ∗ P < 0.05 versus control; # P < 0.05 versus INN.
3.4. Nrf2 Is Activated by INN and Implicated in the HO-1 Induction
To illuminate the downstream signaling pathway of CaMKKβ/AMPK for the stimulation of HO-1 by INN, we examined Nrf2 nuclear translocation. ECs were incubated with 10 μM INN for the indicated time. As indicated in Figure 4(a), Nrf2 nuclear translocation was augmented as early as 8 h and continued until 24 h after INN treatment. Furthermore, Nrf2 nuclear translocation was repressed in ECs pretreated with the AMPK inhibitor Compound C for 1 h before being challenged with 10 μM INN. Our results suggest that the CaMKKβ/AMPK pathway is implicated in INN-stimulated Nrf2 nuclear translocation. To further determine the role of Nrf2 in INN-stimulated HO-1 expression, ECs were transfected with shNrf2 and shLuc. As illustrated in Figures 4(b) and 4(c), knockdown of Nrf2 eliminated INN-induced HO-1 expression. Together, our results demonstrate that Nrf2 is a transcriptional factor responsible for INN-stimulated HO-1 expression.
Figure 4.

Nrf2 are implicated in HO-1 expression induced by INN. (a) ECs were treated with 10 μM Compound C for 1 h followed by incubation with 10 μM INN for the indicated time. Aliquots of cell lysate underwent immunoblotting analysis. ∗ P < 0.05 versus control; # P < 0.05 versus INN (24 h). (b) ECs were transfected with shLuc or shNrf2 and then treated with 10 μM INN for 16 h. Aliquots of cell lysate underwent immunoblotting analysis. (c) Total RNA was extracted from ECs and was used to analyze HO-1 and β-actin mRNA by using RT-PCR with their specific primers. Data are expressed as means ± SD (n = 3). ∗ P < 0.05 versus control; # P < 0.05 versus INN/shLuc.
3.5. AMPK and Nrf2 Are Responsible for INN Inhibition of TNFα-Stimulated VCAM-1 Expression and Monocyte Adhesion in HAEC
Next, we sought to determine whether INN hinders TNFα-induced VCAM-1 activation. As indicated in Figure 5, TNFα significantly increased expression of VCAM-1 and HL-60 cell adhesion. INN treatment dramatically suppressed TNFα-induced VCAM-1 expression at mRNA and protein levels as well as HL-60 cell adhesion. To further identify the role of AMPK and Nrf2 in the TNFα-stimulated VCAM-1 expression, we then used Compound C to inhibit AMPK and shNrf2 to knock down Nrf2 expression. As illustrated in Figure 5, Compound C and shNrf2 abolished TNFα-triggered VCAM-1 expression and HL-60 cell adhesion. Together, these results suggest that AMPK and Nrf2 mediate INN inhibition of TNFα-stimulated VCAM-1 expression and monocyte adhesion in HAEC.
Figure 5.

Effect of INN alone or in combination with AMPK or Nrf2 inhibition on TNFα-stimulated VCAM-1 expression and monocyte adhesion in HAEC. (a) ECs were treated with 1 ng/mL TNFα with or without DMSO (vehicle), INN, or INN + Compound C (Comp C) for VCAM-1, p-AMPK, and AMPK expression measurement. (d) ECs transfected with shLuc or shNrf2 were incubated with 1 ng/mL TNFα in the presence or absence of INN for 8 h for VCAM-1 expression measurement. Aliquots of cell lysate underwent immunoblotting analysis. (b and e) Total RNA was extracted and underwent RT-PCR with specific primers for VCAM-1. (c and f) Adhesion assay was carried out as described in Methods. Results are expressed as means ± SD (n = 3). ∗ P < 0.05 versus control; # P < 0.05 versus TNFα; § P < 0.05 versus TNFα/INN.
4. Discussion
Improvement of intracellular antioxidant ability is thought to diminish the risk of oxidative stress-induced diseases. Clopidogrel (INN) is an oral antiplatelet drug used to impede blood clots in coronary artery disease (CAD). Previous studies have shown that INN has anti-inflammation and antioxidation activities [19–21]. Recent work has demonstrated that INN augments nitric oxide (NO) and prostacyclin production in endothelial cells [31]. In addition, INN impedes CD40 ligand both in vitro and in vivo [32], stimulating HO-1 expression [33]. These findings suggest that INN can preserve endothelial function via a mechanism independent of its antiplatelet activity. More importantly, McClung et al. suggest that INN protects against diabetes-induced vascular damage and reduces circulating endothelial cells (CECs) in type-2 diabetes patients [34]. The effect of INN was to improve vascular function, protect against oxidative stress, and inhibit apoptosis in patients with type-2 diabetes. This involved an increase in the expression of both phosphorylated Akt and AMPK. In the present study, we revealed that INN repressed TNFα-stimulated ROS liberation, VCAM-1 expression, and cell adhesion and that this effect was related to the increase in HO-1 expression and GSH content via the CaMKKβ/AMPK/Nrf2 pathway.
TNFα is a powerful proinflammatory cytokine secreted by various innate immune cells, predominantly activated macrophages, as well as neutrophils, mast cells, and eosinophils cells [35–39]. In macrophages, inflammatory stimuli result in TNFα synthesis and release by constitutive exocytosis [38]. The plasma levels of TNFα are augmented in some pathologies, such as cancer, atherosclerosis, rheumatoid arthritis, and preeclampsia [40]. Several studies have revealed that TNFα, via activation of NFκB, induces expression of proinflammatory cytokines and adhesion molecules, for example, IL-6 and VCAM-1 [41, 42]. In the current study, we revealed that TNFα increased ROS production, VCAM-1 expression, and cell adhesion. Intriguingly, these effects of TNFα were abolished by INN.
Heme oxygenase or haem oxygenase (HO) is a stress-inducible enzyme. Increasing evidence supports that it protects against many chronic diseases such as cardiovascular diseases, hypertension, diabetes mellitus, and neurological disorders [43]. Using systemic manipulation of either HO-1 expression or activity, some studies suggested that HO-1 plays a vital role in atherosclerosis initiation and development. HO-1-null mice show a noteworthy increase in plasma lipid hydroperoxides [44]. Similarly, rabbits treated with SnPP, an HO-1 inhibitor, exhibit a significant lipid deposits in abdominal aortic plaques [45]. Contrarily, HO-1 induction alleviated the oxLDL-induced formation of foam cells [46]. Taken together, these studies substantiated the anti-inflammatory or antiatherosclerotic activity of HO-1.
The early step in atherogenesis is endothelial dysfunction resulting in several compensatory responses that change the vascular homeostasis [47]. Proinflammatory stimuli such as a diet rich in saturated fat, obesity, hypercholesterolemia, and hyperglycemia cause the expression of adhesion molecules, for example, vascular cell adhesion molecule-1 (VCAM-1) and P-selectin in endothelium, and these molecules facilitate the attachment of monocytes and lymphocyte [48, 49]. In addition, turbulent flow resulting from an unfavorable serum lipid profile probably leads to overexpression of adhesion molecules in endothelial cells triggering atherosclerosis. Animals fed a proatherogenic diet quickly overexpress VCAM-1 [50]. Overexpression of VCAM-1 enhances recruitment of monocytes to endothelial injury locations; succeeding liberation of monocyte chemoattractant protein-1 (MCP-1) by leukocytes amplifies the inflammatory cascade through recruiting other leukocytes, stimulating leukocytes in the media, and initiating recruitment and proliferation of smooth muscle cells [51]. Here, we demonstrate that suppression of VCAM-1 expression by INN reduced HL-60 cell adhesion to TNFα-stimulated HAECs. Nevertheless, this inhibition was eliminated by AMPK inhibitor Compound C, shHO-1, and shNrf2 (Figures 2(d), 5(c), and 5(f)). These data demonstrate the contribution of AMPK, Nrf2, and HO-1 to the suppression of HL-60 cell adhesion by INN.
Nrf2 is an imperative transcriptional factor implicated in cellular anti-inflammatory action and related to the induction of HO-1 and glutathione S-transferase [52, 53]. Nrf2 interacts with its cytosolic inhibitor Keap1 under basal circumstances. In response to stress, Nrf2 is released from Keap1 and successively translocates to the nucleus, activating its target gene transcription via ARE [54]. In the current study, INN augmented Nrf2 nuclear translocation, and silencing Nrf2 abolished the INN induction of HO-1. These results indicate that this transcriptional factor is indispensable for INN-induced HO-1 expression. Nrf2 activation is controlled by several kinases, including JNK, p38, ERK, and PI3K/Akt [55]. In the current study, we demonstrated that INN activates CaMKKβ and AMPK. By using specific inhibitors of CaMKKβ and AMPK, we revealed that the CaMKKβ/AMPK pathway is implicated in the INN-triggered HO-1 induction.
In summary, we have revealed that INN hinders TNFα-induced ROS formation, expression of VCAM-1, and HL-60 cell adhesion by upregulating HO-1 gene expression and elevating GSH levels through the CaMKKβ/AMPK/Nrf2 pathway (Figure 6). The antioxidant and anti-inflammatory characteristic of INN is thought to protect against oxidative stress-induced diseases including arthrosclerosis.
Figure 6.

Proposed diagram summarizing the suppression of TNFα-induced inflammation by INN via upregulating HO-1 expression through the CaMKKβ/AMPK/Nrf2 pathway and repressing ROS formation, VCAM-1 expression, and monocyte adhesion. In summary, we have revealed that INN hinders TNFα-induced ROS formation, expression of VCAM-1, and HL-60 cell adhesion by upregulating HO-1 gene expression and elevating GSH levels through the CaMKKβ/AMPK/Nrf2 pathway.
Acknowledgments
This study was supported by Chinese National Natural Science Foundation (no. 81202733) to Huabing Yang and grants from the Specialized Research Fund for the Doctoral Program of Higher Education (20123156120002) to Shiliu Tian.
Conflict of Interests
The authors declare no conflict of interests.
References
- 1.Kim Y. S., Ahn Y., Hong M. H., et al. Curcumin attenuates inflammatory responses of TNF-α-stimulated human endothelial cells. Journal of Cardiovascular Pharmacology. 2007;50(1):41–49. doi: 10.1097/fjc.0b013e31805559b9. [DOI] [PubMed] [Google Scholar]
- 2.Shi Y., Vanhoutte P. M. Reactive oxygen-derived free radicals are key to the endothelial dysfunction of diabetes. Journal of diabetes. 2009;1(3):151–162. doi: 10.1111/j.1753-0407.2009.00030.x. [DOI] [PubMed] [Google Scholar]
- 3.Mitra S., Abraham E. Participation of superoxide in neutrophil activation and cytokine production. Biochimica et Biophysica Acta—Molecular Basis of Disease. 2006;1762(8):732–741. doi: 10.1016/j.bbadis.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 4.Gimbrone M. A., Jr., Nagel T., Topper J. N. Biomechanical activation: an emerging paradigm in endothelial adhesion biology. Journal of Clinical Investigation. 1997;100(11, supplement):S61–S65. [PubMed] [Google Scholar]
- 5.Morisaki N., Takahashi K., Shiina R., et al. Platelet-derived growth factor is a potent stimulator of expression of intercellular adhesion molecule-1 in human arterial smooth muscle cells. Biochemical and Biophysical Research Communications. 1994;200(1):612–618. doi: 10.1006/bbrc.1994.1492. [DOI] [PubMed] [Google Scholar]
- 6.Read M. A., Neish A. S., Luscinskas F. W., Palombella V. J., Maniatis T., Collins T. The proteasome pathway is required for cytokine-induced endothelial-leukocyte adhesion molecule expression. Immunity. 1995;2(5):493–506. doi: 10.1016/1074-7613(95)90030-6. [DOI] [PubMed] [Google Scholar]
- 7.Rothlein R., Czajkowski M., O'Neill M. M., Marlin S. D., Mainolfi E., Merluzzi V. J. Induction of intercellular adhesion molecule 1 on primary and continuous cell lines by pro-inflammatory cytokines: regulation by pharmacologic agents and neutralizing antibodies. Journal of Immunology. 1988;141(5):1665–1669. [PubMed] [Google Scholar]
- 8.Cybulsky M. I., Gimbrone M. A., Jr. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251(4995):788–791. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- 9.Choi A. M. K., Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. American Journal of Respiratory Cell and Molecular Biology. 1996;15(1):9–19. doi: 10.1165/ajrcmb.15.1.8679227. [DOI] [PubMed] [Google Scholar]
- 10.Maines M. D., Kappas A. Cobalt induction of hepatic heme oxygenase, with evidence that cytochrome P-450 is not essential for this enzyme activity. Proceedings of the National Academy of Sciences of the United States of America. 1974;71(11):4293–4297. doi: 10.1073/pnas.71.11.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maines M. Carbon monoxide and nitric oxide homology: differential modulation of heme oxygenases in brain and detection of protein and activity. Methods in Enzymology. 1996;268:473–488. doi: 10.1016/s0076-6879(96)68049-5. [DOI] [PubMed] [Google Scholar]
- 12.Stocker R., Yamamoto Y., McDonagh A. F., Glazer A. N., Ames B. N. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235(4792):1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 13.Motterlini R., Gonzales A., Foresti R., Clark J. E., Green C. J., Winslow R. M. Heme oxygenase-1-derived carbon monoxide contributes to the suppression of acute hypertensive responses in vivo. Circulation Research. 1998;83(5):568–577. doi: 10.1161/01.RES.83.5.568. [DOI] [PubMed] [Google Scholar]
- 14.Sammut I. A., Foresti R., Clark J. E., et al. Carbon monoxide is a major contributor to the regulation of vascular tone in aortas expressing high levels of haeme oxygenase-1. British Journal of Pharmacology. 1998;125(7):1437–1444. doi: 10.1038/sj.bjp.0702212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X.-M., Peyton K. J., Shebib A. R., Wang H., Korthuis R. J., Durante W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. The American Journal of Physiology—Heart and Circulatory Physiology. 2011;300(1):H84–H93. doi: 10.1152/ajpheart.00749.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang C.-S., Lii C.-K., Lin A.-H., et al. Protection by chrysin, apigenin, and luteolin against oxidative stress is mediated by the Nrf2-dependent up-regulation of heme oxygenase 1 and glutamate cysteine ligase in rat primary hepatocytes. Archives of Toxicology. 2013;87(1):167–178. doi: 10.1007/s00204-012-0913-4. [DOI] [PubMed] [Google Scholar]
- 17.Yu A.-L., Lu C.-Y., Wang T.-S., et al. Induction of heme oxygenase 1 and inhibition of tumor necrosis factor alpha-induced intercellular adhesion molecule expression by andrographolide in EA.hy926 cells. Journal of Agricultural and Food Chemistry. 2010;58(13):7641–7648. doi: 10.1021/jf101353c. [DOI] [PubMed] [Google Scholar]
- 18.Levy S., Jaiswal A. K., Forman H. J. The role of c-Jun phosphorylation in EpRE activation of phase II genes. Free Radical Biology & Medicine. 2009;47(8):1172–1179. doi: 10.1016/j.freeradbiomed.2009.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hadi N. R., Mohammad B. I., Ajeena I. M., Sahib H. H. Antiatherosclerotic potential of clopidogrel: antioxidant and anti-inflammatory approaches. BioMed Research International. 2013;2013:10. doi: 10.1155/2013/790263.790263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graff J., Harder S., Wahl O., Scheuermann E.-H., Gossmann J. Anti-inflammatory effects of clopidogrel intake in renal transplant patients: effects on platelet-leukocyte interactions, platelet CD40 ligand expression, and proinflammatory biomarkers. Clinical Pharmacology and Therapeutics. 2005;78(5):468–476. doi: 10.1016/j.clpt.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 21.Klinkhardt U., Bauersachs R., Adams J., Graff J., Lindhoff-Last E., Harder S. Clopidogrel but not aspirin reduces P-selectin expression and formation of platelet-leukocyte aggregates in patients with atherosclerotic vascular disease. Clinical Pharmacology and Therapeutics. 2003;73(3):232–241. doi: 10.1067/mcp.2003.13. [DOI] [PubMed] [Google Scholar]
- 22.Millius A., Weiner O. D. Chemotaxis in neutrophil-like HL-60 cells. Methods in Molecular Biology. 2009;571:167–177. doi: 10.1007/978-1-60761-198-1_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Visner G. A., Lu F., Zhou H., Liu J., Kazemfar K., Agarwal A. Rapamycin induces heme oxygenase-1 in human pulmonary vascular cells: implications in the antiproliferative response to rapamycin. Circulation. 2003;107(6):911–916. doi: 10.1161/01.cir.0000048191.75585.60. [DOI] [PubMed] [Google Scholar]
- 24.Tang S.-C., Wu M.-F., Wong R.-H., et al. Epigenetic mechanisms for silencing glutathione S-transferase m2 expression by hypermethylated specificity protein 1 binding in lung cancer. Cancer. 2011;117(14):3209–3221. doi: 10.1002/cncr.25875. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y.-C., Lii C.-K., Wei Y.-L., et al. Docosahexaenoic acid inhibition of inflammation is partially via cross-talk between Nrf2/heme oxygenase 1 and IKK/NF-κB pathways. The Journal of Nutritional Biochemistry. 2013;24(1):204–212. doi: 10.1016/j.jnutbio.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Lu C.-Y., Yang Y.-C., Li C.-C., Liu K.-L., Lii C.-K., Chen H.-W. Andrographolide inhibits TNFα-induced ICAM-1 expression via suppression of NADPH oxidase activation and induction of HO-1 and GCLM expression through the PI3K/Akt/Nrf2 and PI3K/Akt/AP-1 pathways in human endothelial cells. Biochemical Pharmacology. 2014;91(1):40–50. doi: 10.1016/j.bcp.2014.06.024. [DOI] [PubMed] [Google Scholar]
- 27.Chen H.-W., Lin A.-H., Chu H.-C., et al. Inhibition of TNF-alpha-induced inflammation by andrographolide via down-regulation of the PI3K/Akt signaling pathway. Journal of Natural Products. 2011;74(11):2408–2413. doi: 10.1021/np200631v. [DOI] [PubMed] [Google Scholar]
- 28.Kang J., Choi I., Han M., et al. The cytoprotective effect of petalonia binghamiae methanol extract against oxidative stress in C2C12 myoblasts: mediation by upregulation of heme oxygenase-1 and nuclear factor-erythroid 2 related factor 2. Marine Drugs. 2015;13(5):2666–2679. doi: 10.3390/md13052666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scapagnini G., Butterfield D. A., Colombrita C., Sultana R., Pascale A., Calabrese V. Ethyl ferulate, a lipophilic polyphenol, induces HO-1 and protects rat neurons against oxidative stress. Antioxidants and Redox Signaling. 2004;6(5):811–818. doi: 10.1089/ars.2004.6.811. [DOI] [PubMed] [Google Scholar]
- 30.Colombo S. L., Moncada S. AMPKalpha1 regulates the antioxidant status of vascular endothelial cells. The Biochemical Journal. 2009;421(2):163–169. doi: 10.1042/bj20090613. [DOI] [PubMed] [Google Scholar]
- 31.Ziemianin B., Olszanecki R., Uracz W., Marcinkiewicz E., Gryglewski R. J. Thienopyridines: effects on cultured endothelial cells. Journal of Physiology and Pharmacology. 1999;50(4):597–604. [PubMed] [Google Scholar]
- 32.Vishnevetsky D., Kiyanista V. A., Gandhi P. J. CD40 ligand: a novel in the fight against cardiovascular disease. The Annals of Pharmacotherapy. 2004;38(9):1500–1508. doi: 10.1345/aph.1d611. [DOI] [PubMed] [Google Scholar]
- 33.Hancock W. W., Buelow R., Sayegh M. H., Turka L. A. Antibody-induced transplant arteriosclerosis is prevented by graft expression of anti-oxidant and anti-apoptotic genes. Nature Medicine. 1998;4(12):1392–1396. doi: 10.1038/3982. [DOI] [PubMed] [Google Scholar]
- 34.McClung J. A., Kruger A. L., Ferraris A., et al. Usefulness of clopidogrel to protect against diabetes-induced vascular damage. The American Journal of Cardiology. 2010;105(7):1014–1018. doi: 10.1016/j.amjcard.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beil W. J., Weller P. F., Peppercorn M. A., Galli S. J., Dvorak A. M. Ultrastructural immunogold localization of subcellular sites of TNF-α in colonic Crohn's disease. Journal of Leukocyte Biology. 1995;58(3):284–298. doi: 10.1002/jlb.58.3.284. [DOI] [PubMed] [Google Scholar]
- 36.Brezina R., Schramek S., Kazar J. Selection of chlortetracycline resistant strain of Coxiella burnetii . Acta Virologica. 1975;19(6, article 496) [PubMed] [Google Scholar]
- 37.Arimura H. Correlation between molecular size and interferon-inducing activity of poly I:C. Acta Virologica. 1975;19(6):457–466. [PubMed] [Google Scholar]
- 38.Wachter R. F., Briggs G. P., Pedersen C. E., Jr. Precipitation of phase I antigen of Coxiella burnetii by sodium sulfite. Acta Virologica. 1975;19(6):p. 500. [PubMed] [Google Scholar]
- 39.Ignatovich V. F. Enhancement of the antigenic activity and virulence of the vaccine strain E of Rickettsia prow azeki by passages in cell culture. Acta Virologica. 1975;19(6):481–485. [PubMed] [Google Scholar]
- 40.Share J. B. Review of drug treatment for Down's syndrome persons. American Journal of Mental Deficiency. 1976;80(4):388–393. [PubMed] [Google Scholar]
- 41.Pogodina V. V. Elizaveta Nilolaevna Levkovich-75th birthday. Acta Virologica. 1975;19(6, article 509) [PubMed] [Google Scholar]
- 42.Bland R. D., Clarke T. L., Harden L. B. Rapid infusion of sodium bicarbonate and albumin into high-risk premature infants soon after birth: a controlled, prospective trial. American Journal of Obstetrics & Gynecology. 1976;124(3):263–267. doi: 10.1016/0002-9378(76)90154-x. [DOI] [PubMed] [Google Scholar]
- 43.Abraham N. G., Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacological Reviews. 2008;60(1):79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- 44.Ishikawa K., Navab M., Lusis A. J. Vasculitis, atherosclerosis, and altered HDL composition in heme-oxygenase-1-knockout mice. International Journal of Hypertension. 2012;2012:6. doi: 10.1155/2012/948203.948203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li T., Tian H., Zhao Y., et al. Heme oxygenase-1 inhibits progression and destabilization of vulnerable plaques in a rabbit model of atherosclerosis. European Journal of Pharmacology. 2011;672(1–3):143–152. doi: 10.1016/j.ejphar.2011.09.188. [DOI] [PubMed] [Google Scholar]
- 46.Tsai J.-Y., Su K.-H., Shyue S.-K., et al. EGb761 ameliorates the formation of foam cells by regulating the expression of SR-A and ABCA1: role of haem oxygenase-1. Cardiovascular Research. 2010;88(3):415–423. doi: 10.1093/cvr/cvq226. [DOI] [PubMed] [Google Scholar]
- 47.Pyorala K., Laakso M., Uusitupa M. Diabetes and atherosclerosis: an epidemiologic view. Diabetes/Metabolism Reviews. 1987;3(2):463–524. doi: 10.1002/dmr.5610030206. [DOI] [PubMed] [Google Scholar]
- 48.Fan J., Watanabe T. Inflammatory reactions in the pathogenesis of atherosclerosis. Journal of Atherosclerosis and Thrombosis. 2003;10(2):63–71. doi: 10.5551/jat.10.63. [DOI] [PubMed] [Google Scholar]
- 49.Packard R. R. S., Libby P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clinical Chemistry. 2008;54(1):24–38. doi: 10.1373/clinchem.2007.097360. [DOI] [PubMed] [Google Scholar]
- 50.Li H., Cybulsky M. I., Gimbrone M. A., Jr., Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arteriosclerosis, Thrombosis, and Vascular Biology. 1993;13(2):197–204. doi: 10.1161/01.ATV.13.2.197. [DOI] [PubMed] [Google Scholar]
- 51.Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 52.Chen C., Kong A. N. Dietary cancer-chemopreventive compounds: from signaling and gene expression to pharmacological effects. Trends in Pharmacological Sciences. 2005;26(6):318–326. doi: 10.1016/j.tips.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 53.Shen G., Jeong W.-S., Hu R., Kong A.-N. T. Regulation of Nrf2, NF-κB, and AP-1 signaling pathways by chemopreventive agents. Antioxidants and Redox Signaling. 2005;7(11-12):1648–1663. doi: 10.1089/ars.2005.7.1648. [DOI] [PubMed] [Google Scholar]
- 54.Itoh K., Wakabayashi N., Katoh Y., et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes & Development. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Costa G., Francisco V., Lopes M. C., Cruz M. T., Batista M. T. Intracellular signaling pathways modulated by phenolic compounds: application for new anti-inflammatory drugs discovery. Current Medicinal Chemistry. 2012;19(18):2876–2900. doi: 10.2174/092986712800672049. [DOI] [PubMed] [Google Scholar]