

Abstract
The first oral treatment for relapsing multiple sclerosis, the nonselective sphingosine-1-phosphate receptor (S1PR) modulator fingolimod, led to identification of a pivotal role of sphingosine-1-phosphate and one of its five known receptors, S1P1R, in regulation of lymphocyte trafficking in multiple sclerosis. Modulation of S1P3R, initially thought to cause some of fingolimod’s side effects, prompted the search for novel compounds with high selectivity for S1P1R. Ponesimod is an orally active, selective S1P1R modulator that causes dose-dependent sequestration of lymphocytes in lymphoid organs. In contrast to the long half-life/slow elimination of fingolimod, ponesimod is eliminated within 1 week of discontinuation and its pharmacological effects are rapidly reversible. Clinical data in multiple sclerosis have shown a dose-dependent therapeutic effect of ponesimod and defined 20 mg as a daily dose with desired efficacy, and acceptable safety and tolerability. Phase II clinical data have also shown therapeutic efficacy of ponesimod in psoriasis. These findings have increased our understanding of psoriasis pathogenesis and suggest clinical utility of S1P1R modulation for treatment of various immune-mediated disorders. A gradual dose titration regimen was found to minimize the cardiac effects associated with initiation of ponesimod treatment. Selectivity for S1P1R, rapid onset and reversibility of pharmacological effects, and an optimized titration regimen differentiate ponesimod from fingolimod, and may lead to better safety and tolerability. Ponesimod is currently in phase III clinical development to assess efficacy and safety in relapsing multiple sclerosis. A phase II study is also ongoing to investigate the potential utility of ponesimod in chronic graft versus host disease.
Keywords: autoimmune disease, lymphocyte migration, multiple sclerosis, psoriasis, transplantation
Biology and pharmacology of sphingosine-1-phosphate receptor 1
The past decades have witnessed major advances in the treatment of autoimmune and chronic inflammatory diseases. A plethora of novel therapies targeting specific molecules involved in the inflammatory or immune system activation cascades have become available. These have significantly increased our understanding of disease pathogenesis and improved the management of immune-mediated disorders. However, most of the targeted therapies are biological drugs which need to be injected, are eliminated slowly (e.g. over several weeks) and can lose efficacy or tolerability due to their potential immunogenicity. In an attempt to overcome these hurdles, pharmaceutical research has made considerable efforts to develop novel oral targeted therapies for autoimmune and chronic inflammatory diseases.
Sphingosine-1-phosphate receptor 1 (S1P1R) is one of five known G protein-coupled receptors with nanomolar affinity for the lysophospholipid sphingosine-1-phosphate (S1P), which is generated through physiologic metabolism of the cell membrane constituent sphingomyelin by all cells [Brinkmann, 2007]. S1P receptors, including S1P1R, are widely expressed in many tissues [Chun et al. 2010]. S1P1R expression on lymphocytes controls their egress from thymus and secondary lymphoid organs [Cyster and Schwab, 2012]. Lymphocyte egress requires a gradient of S1P concentration, which is established by a high S1P concentration in blood and lymph compared with a low concentration in the interstitial fluid of lymphoid organs [Grigorova et al. 2009].
Synthetic S1P1 receptor modulators disrupt the interaction of the physiologic S1P ligand with S1P1R by promoting initial activation followed by sustained internalization and desensitization of S1P1R [Hla and Brinkmann, 2011; Pinschewer et al. 2011]. Experiments conducted in animal models of transplant rejection, multiple sclerosis, lupus erythematosus, arthritis and inflammatory bowel disease with the first-generation, nonselective S1P receptor modulator, fingolimod, have demonstrated the potential efficacy of this mode of action across several immune-mediated chronic inflammatory conditions [Brinkmann, 2007]. Fingolimod is a structural analog of sphingosine that is phosphorylated in the body by a sphingosine kinase to generate the bioactive form of the drug, fingolimod phosphate, which binds to multiple S1P receptors [Brinkmann, 2007]. Clinical trials in multiple sclerosis (MS) have confirmed the efficacy of fingolimod in relapsing MS, but not in primary progressive disease, and led to the approval of the first oral medication for the treatment of relapsing forms of MS in 2010 [Kappos et al. 2010].
The mechanism of action of fingolimod has increased our understanding of MS pathogenesis. T and B cells, but not natural killer (NK) cells, express functional S1P1R and are affected by fingolimod [Cyster and Schwab, 2012]. Furthermore, S1P1R is differentially expressed and regulated in functionally distinct subsets of lymphocytes and fingolimod has been shown to predominantly affect naïve T cells and central memory T cells (TCM) while sparing effector memory T cells (TEM), and terminally differentiated effector T cells (TE) in patients with relapsing MS [Mehling et al. 2008, 2011]. This has raised the possibility that, at least in MS, retention of TCM cells, which include pro-inflammatory T helper 17 (Th17) cells, by fingolimod may prevent their accumulation in the cerebrospinal fluid (CSF) and subsequent differentiation to TE cells in the central nervous system (CNS) [Hla and Brinkmann, 2011]. The effects of S1P1R modulation on B cells are less well defined. Recent data from patients with relapsing MS have shown predominant reduction of memory B cells and recently activated memory B cells (CD38int-high) in peripheral blood after treatment with fingolimod [Claes et al. 2014; Nakamura et al. 2014]. As memory B cells are implicated in the pathogenesis of MS and other autoimmune diseases, these observations suggest another potential mechanism underlying the therapeutic effects of S1P1R modulators.
Astrocytes, microglia, oligodendrocytes and neurons express various S1P receptors including S1P1R, S1P3R and S1P5R. Fingolimod has been shown to penetrate the CNS tissues and in vitro studies have shown activation of astrocytes and oligodendrocytes by fingolimod [Foster et al. 2007]. Conditional deletion of S1P1R on neural cells in mice reduced the severity of experimental autoimmune encephalomyelitis (EAE) and reductions in the clinical scores were paralleled by decreased demyelination, axonal loss and astrogliosis [Choi et al. 2011]. Unfortunately, there was no beneficial effect in a recently completed, large study of fingolimod in patients with primary progressive MS [Lublin et al. 2015], suggesting that the direct effect on CNS cells alone may not be sufficient. Taken together, these data suggest the possibility of a direct beneficial effect of S1P1R modulation in the brain of patients with relapsing MS [Dev et al. 2008]; however, its contribution to efficacy relative to the immunological effects remains unclear.
Initial studies in rodents suggested that modulation of S1P3R on cardiac myocytes by fingolimod was associated with a reduction of heart rate (HR) by activation of G-protein-coupled inwardly rectifying potassium channels (GIRK) that regulate pacemaker frequency, and the shape and duration of action potentials [Koyrakh et al. 2005; Camm et al. 2014]. Modulation of S1P2R and S1P3R on myofibroblasts by fingolimod was also shown to stimulate extracellular matrix synthesis [Sobel et al. 2013]. Modulation of these receptors on vascular smooth muscle cells appeared to be associated with vasoconstriction, leading to the slight increase in blood pressure observed with fingolimod treatment [Salomone et al. 2003; Watterson et al. 2005; Hu et al. 2006; Lorenz et al. 2007; Kappos et al. 2010]. These observations raised the possibility that some side effects associated with fingolimod treatment could be avoided by more selective S1P1R modulators, thus triggering the search for novel compounds.
Currently, there are several selective S1P1R modulators in clinical development [Gonzalez-Cabrera et al. 2014; Subei and Cohen, 2015]. Here we review data and the development status of ponesimod, a selective S1P1R modulator developed by Actelion Pharmaceuticals Ltd.
Ponesimod, a selective, rapidly reversible, orally active, sphingosine-1-phosphate receptor modulator
Ponesimod (ACT-128800 (Z,Z)-5-[3-chloro-4-(2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one) is a selective, rapidly reversible, orally active, S1P1R modulator. Ponesimod emerged from the discovery of a novel class of S1P1R agonists based on the 2-imino-thiazolidin-4-one scaffold (Figure 1) [Bolli et al. 2010]. Ponesimod activates S1P1R with high potency [half maximal effective concentration (EC50) of 5.7 nM] and selectivity. Relative to the potency of S1P, the potency of ponesimod is 4.4 higher for S1P1R and 150-fold lower for S1P3R, resulting in an approximately 650-fold higher S1P1R selectivity compared with the natural ligand.
Figure 1.
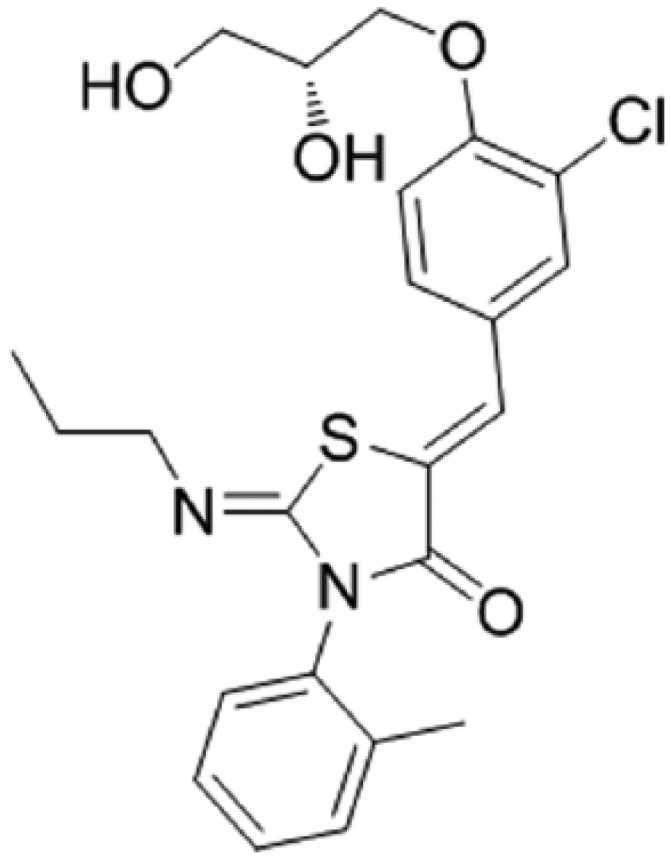
Chemical structure of ponesimod, C23H25N2O4CIS (molecular weight 460.98).
Oral administration of ponesimod has been shown to lead to a dose-dependent decrease of blood lymphocyte count in animals and humans [Piali et al. 2011; Brossard et al. 2013]. In mice with delayed-type hypersensitivity, ponesimod prevented edema formation, inflammatory cell accumulation, and cytokine release in the skin. In rats with adjuvant-induced arthritis, ponesimod prevented the increase in paw volume and joint inflammation [Piali et al. 2011]. Treatment with ponesimod of prediabetic non obese diabetic (NOD) mice, which spontaneously develop autoimmune diabetes, prevented disease development and ponesimod treatment of diabetic NOD mice induced disease remission [You et al. 2013]. In rats, ponesimod was shown to induce a dose-dependent decrease of blood lymphocyte count, which returned to baseline within 48 hours after discontinuation of dosing.
Treatment with ponesimod was able to prevent the onset and progression of EAE in mice. Ponesimod increased survival of the animals even when treatment was started after onset of EAE. Histological analyses showed that ponesimod reduced inflammation, demyelination and axonal loss in the brain, cerebellum and spinal cord of mice with EAE [Actelion Pharmaceuticals Ltd, data on file]. Tissue distribution studies of ponesimod in rats indicated that ponesimod penetrates the brain and spinal cord tissues [Actelion Pharmaceuticals Ltd, data on file]. These findings suggest that, in addition to preventing access of lymphocytes to the CNS, ponesimod may have also direct neuroprotective effects via activation of S1P1R on neural cells.
Taken together, these data showed that selective modulation of S1P1R using ponesimod is efficacious in animal models of lymphocyte-mediated tissue inflammation.
Clinical pharmacology of ponesimod
The clinical pharmacology of ponesimod has been extensively characterized in several clinical studies. To date, over a thousand subjects have been exposed to ponesimod including patients with MS or psoriasis.
The first in-human study with ponesimod was a double-blind, placebo-controlled, ascending, single-dose study in healthy male subjects who received doses of 1, 3, 8, 20, 50 and 75 mg ponesimod or placebo [Brossard et al. 2013]. Ponesimod pharmacokinetics (PK) were dose proportional with minimal effects of food intake. The median time to maximal concentration ranged from 2.0 to 4.0 hours, and ponesimod was eliminated with a mean half-life varying between 21.7 and 33.4 hours. Total lymphocyte count in peripheral blood was reduced in a dose-dependent manner reaching a maximal mean percentage (± standard deviation) reduction from baseline of 70.3 (±2.3%) and returned to normal range within 96 hours. Ponesimod reduced T and B cell counts, but not the NK cell count, and showed differential effects on T-cell subsets with a predominant reduction of circulating naïve and CD4+ T cells versus memory and CD8+ T cells while partially sparing CD4+CD25+ regulatory T (Treg) cells [D’Ambrosio et al. 2015]. Ponesimod was well tolerated. Starting with a dose of 8 mg, transient asymptomatic reduction in HR was observed.
A multiple-ascending dose study investigated the safety, tolerability, PK and pharmacodynamics (PD) of 5, 10, 20 and 40 mg once daily (o.d.) ponesimod in healthy male and female subjects [Brossard et al. 2014]. Ponesimod PK were characterized by a time to maximum concentration and an elimination halflife varying from 2.5 to 4.0 hours and 30.9 to 33.5 hours, respectively, and an accumulation of about 2.3-fold. The PK of ponesimod in male and female subjects were comparable. Ponesimod caused a dose-dependent, sustained decrease in total lymphocyte count in peripheral blood. At steady-state ponesimod plasma concentrations, the mean maximum decrease in total lymphocyte count from baseline was 47%, 59%, 74% and 81% in the 5, 10, 20 and 40 mg dose groups, respectively. Lymphocyte count returned to within the normal range within 1 week of ponesimod discontinuation. Reported adverse events (AEs) were mainly related to HR reduction, and atrioventricular (AV) conduction delays seen after the first dose and effects on pulmonary function observed mainly at the highest dose of 40 mg. Due to the occurrence of sinus bradycardia and, in some subjects, AV blocks on the first day of dosing, an uptitration regimen with a starting dose of 10 mg was introduced to reach the 40 mg dose.
An uptitration study of ponesimod to the supratherapeutic dose of 100 mg o.d. showed a plateau in mean lymphocyte count reduction of approximately 70% from baseline reached at the 40 mg dose level [Hoch et al. 2014]. A dose-dependent mean reduction in forced expiratory volume in 1 second (FEV1) from baseline was observed, with a plateau of approximately 30% reduction from baseline at 60–100 mg dose levels. Symptoms of abdominal pain, dizziness, night sweats, dyspnea and chest discomfort were more frequently reported with ponesimod than placebo. After stopping ponesimod, lymphocyte count and FEV1 returned to baseline values within 10 days. In addition, inhalation of the short acting β2 agonist, salbutamol, induced a rapid return of FEV1 close to the baseline values.
The mass balance, PK and metabolism of 14C-ponesimod were investigated in six healthy male subjects [Reyes et al. 2015]. Fecal excretion was identified as the major route of elimination of ponesimod while urinary excretion was minor. Ponesimod was extensively metabolized and 2 pharmacologically inactive metabolites, M12 and M13, were detected in the circulation corresponding to 8.1% and 25.7%, respectively, of the total drug-related exposure (AUC0–∞) in plasma.
A clinical study in Japanese and Caucasian subjects showed no relevant differences in the PK and PD of ponesimod across ethnic groups and sexes [Reyes et al. 2014b].
Similarly to fingolimod, ponesimod is teratogenic in animals and the use of effective contraception is necessary for women of child-bearing potential. A drug–drug interaction study in healthy women showed no clinically relevant PK interactions between ponesimod and combined oral contraceptives [Reyes et al. 2014a].
A thorough QT study indicated a mild prolongation of QTc interval by ponesimod with the largest mean increase of 6.9 s and 9.1 ms for doses of 40 and 100 mg, respectively [Hoch et al. 2015a]. Based on concentration-effect analysis, QTc prolongation caused by 20 mg, currently the highest therapeutic dose, was predicted to be a mean increase of 4.4 ms [90% confidence interval (CI): 2.9–5.9 ms], which is below the level of clinical concern.
Based on the data obtained in healthy subjects, a PK/PD model of the effect of ponesimod on peripheral blood lymphocyte count has been developed to identify doses to be tested in subsequent studies in patients [Krause et al. 2014].
The PK and PD of ponesimod were also investigated in the psoriasis and MS phase II studies (Table 1). PK and PD characteristics of ponesimod in patients with psoriasis or MS were found to be similar to those observed in healthy subjects.
Table 1.
Ponesimod phase II and III clinical studies (completed and ongoing).
| Study number* | Phase status | Population | Ponesimod dose | Comparator | Treatment duration | Patients enrolled (n) | Main objectives |
|---|---|---|---|---|---|---|---|
|
Psoriasis
NCT00852670 |
Phase IIa Completed | Moderate-to-severe chronic plaque psoriasis | 20 mg o.d. | Placebo | 6 weeks | 66 | Efficacy, safety, tolerability of short-term ponesimod treatment |
|
Psoriasis
NCT01208090 |
Phase IIb Completed | Moderate-to-severe chronic plaque psoriasis | 20 or 40 mg o.d. | Placebo | 16−28 weeks | 326 | Efficacy, safety, tolerability of induction and maintenance treatment with ponesimod |
|
Multiple sclerosis
NCT01006265 |
Phase IIb (core) Completed | Relapsing remitting multiple sclerosis | 10, 20 or 40 mg o.d. | Placebo | 24 weeks | 464 | Efficacy, safety, tolerability of ponesimod treatment |
|
Multiple sclerosis
NCT01093326 |
Phase IIb (extension) Ongoing | Relapsing remitting multiple sclerosis | Treatment period 1: 10, 20 or 40 mg o.d. Treatment period 2: 10 or 20 mg o.d. (40 mg switched to 10 or 20 mg) |
None | Up to 528 weeks | 353 | Safety, tolerability, efficacy of long-term ponesimod treatment |
|
Multiple sclerosis
NCT02425644 |
Phase III Ongoing | Relapsing multiple sclerosis | 20 mg o.d. | Teriflunomide 14 mg o.d. | 108 weeks | 1100 (planned) | Efficacy, safety, tolerability of ponesimod treatment |
|
Chronic graft versus host disease
NCT02461134 |
Phase II Ongoing | Symptomatic moderate or severe chronic graft versus host disease inadequately responding to first or second line therapy | Up 20 mg o.d. (single-arm, intra-patient dose-escalation with 5, 10, and 20 mg) | None | 24 weeks | 30 (planned) | Biological activity, safety, tolerability of ponesimod treatment |
Identifier on ClinicalTrials.gov.
o.d., once daily.
Efficacy and safety of ponesimod in MS
Efficacy in MS
A double-blind, placebo-controlled, dose-finding phase IIb study evaluated the efficacy, safety and tolerability of ponesimod in the treatment of relapsing remitting MS [ClinicalTrials.gov identifier: NCT01006265] (Table 1) [Olsson et al. 2014]. A total of 464 patients were randomized across 94 centers in 23 countries (in Europe, Australia, Canada and USA) between October 2009 and November 2010 to receive ponesimod 10, 20 and 40 mg o.d. or placebo for 24 weeks. Baseline demographic and disease characteristics of patients were well balanced across groups.
The primary efficacy endpoint of cumulative number of new gadolinium-enhancing (Gd+) lesions per patient detected on T1-weighted magnetic resonance imaging (MRI) scans from weeks 12 to 24 was significantly and dose-dependently reduced by 43%, 83% and 77% with ponesimod 10, 20 and 40 mg, respectively, compared with placebo (Table 2). Although the study was not designed to show an effect on relapses, the rate of relapses was reduced by ponesimod treatment. The annualized relapse rate (ARR) up to week 24 was approximately 0.33, 0.42 and 0.25 in the 10, 20 and 40 mg ponesimod groups, respectively, compared with 0.525 in the placebo group. Peripheral blood lymphocyte count was rapidly reduced with ponesimod treatment in a dose-dependent manner. Mean reductions from baseline to week 24 were 50%, 65% and 69% for ponesimod 10, 20 and 40 mg, respectively, compared with 3% in the placebo group. Patients completing the 24 weeks of treatment in the phase II core study were eligible to enter a dose-blinded extension with ponesimod 10, 20 and 40 mg.
Table 2.
Main efficacy results of ponesimod phase II studies.
| Endpoints | Placebo | Ponesimod 10 mg | Ponesimod 20 mg | Ponesimod 40 mg |
|---|---|---|---|---|
| Phase IIb study in relapsing remitting multiple sclerosis | ||||
| Mean (SD) cumulative number of new T1 Gd+ lesions detected by MRI at week 12–24$ |
n = 110 6.2 (13.42) |
n = 88 3.5 (7.27) |
n = 98 1.1 (1.96) |
n = 93 1.4 (3.24) |
| Treatment effect ratio versus placebo (95% CI) | - | 0.566 (0.337–0.952) 0.0318 |
0.170 (0.100–0.289) <0.0001 |
0.226 (0.133–0.384) |
| p value | <0.0001 | |||
| Annualized relapse rate up to week 24* |
n = 121 0.525 (0.358–0.770) |
n = 108 0.332 (0.198–0.557) |
n = 114 0.417 (0.266–0.653) |
n = 119 0.251 (0.141–0.446) |
| Treatment effect ratio versus placebo (95% CI) | – | 0.632 (0.332–1.202) |
0.793 (0.440–1.432) |
0.478 (0.240–0.954) |
| p value | – | 0.1619 | 0.4420 | 0.0363 |
| Phase IIb study in moderate-to-severe chronic plaque psoriasis | ||||
| % PASI75 at week 16* |
n = 67 13.4 |
Not tested |
n = 126 46.0 |
n = 133 48.1 |
| p value | <0.0001 | <0.0001 | ||
| % PGA score 0 or 1 at week 16* |
n = 67 4.5 |
Not tested |
n = 126 27.8 |
n = 133 32.3 |
| p value | <0.0001 | <0.0001 | ||
| DLQI score mean change (SD) at week 16* |
n = 67 −2·5 (±5·94) |
Not tested |
n = 126 −5·9 (±5·96) |
n = 133 −6·2 (±6·41) |
| p value | 0.0003 | 0.0004 | ||
All treated analysis set.
Per protocol analysis set.
CI, confidence interval; DLQI, Dermatology Life Quality Index; Gd, gadolinium; MRI, magnetic resonance imaging; PASI, Psoriasis Area and Severity Index; PGA, physician global assessment; SD, standard deviation.
A total of 353 patients, out of the 393 who completed the core study, entered the long-term extension trial [ClinicalTrials.gov identifier: NCT01093326] (Table 1). Patients who received active treatment during the initial 24 weeks continued treatment with the same dose of ponesimod, whereas patients who received placebo were randomized in a 1:1:1 ratio to receive either 10, 20 or 40 mg ponesimod. Following completion and analysis of results from the placebo-controlled phase IIb core study, patients receiving 40 mg ponesimod in the extension study were switched to 10 or 20 mg ponesimod in a 1:1 ratio, while patients who received 10 or 20 mg ponesimod continued with the same dose. An interim analysis of the extension study was performed to help in planning for the phase 3 studies [Pozzilli et al. 2013]. At the cutoff date of this interim analysis, treatment was ongoing in 309 patients and the mean treatment duration in all treatment groups ranged between 99.5 and 119.7 weeks for the combined core plus extension phase II study.
The ARR in patients continuously treated with ponesimod 10, 20 or 40 mg in the core and extension studies was 0.24, 0.21 and 0.14, respectively, suggesting a dose-dependent reduction of relapse frequency with ponesimod. Additional analysis of ARR by treatment periods showed decreasing relapse rates with time and a clearer dose-dependent effect on relapses in the second year of treatment. Consistent with a dose-dependent reduction of disease activity by ponesimod, the number of total T1 Gd+ lesions and the number of new or enlarging T2 lesions detected by MRI after 72 weeks of treatment were consistently lower in patients receiving 20 and 40 mg compared with those receiving 10 mg ponesimod. Overall, the available phase II data indicated a sustained dose-dependent reduction in the number of brain inflammatory lesions and reduced rates of relapses with ponesimod. The lymphocyte reducing effect and the efficacy of ponesimod reached a plateau with 20 mg o.d.
Safety in MS
In the phase II core study, ponesimod was generally safe and well tolerated. The majority of AEs were of mild or moderate intensity and the proportion of patients who had at least 1 treatment-emergent AE was similar across the ponesimod groups (73.9–77.2%) compared with the placebo group (74.4%). During the 24 weeks’ treatment period, 6.5%, 6.1% and 2.5% of the patients in the ponesimod 10, 20 and 40 mg, respectively, reported at least 1 serious adverse event (SAE) compared with 4.1% in the placebo group. The proportion of patients who prematurely discontinued study treatment due to AEs in the ponesimod 10, 20 and 40 mg groups was 11.1%, 5.3% and 13.4%, respectively, compared with 2.5% in the placebo group. Treatment-emergent AEs reported with higher incidence in ponesimod groups were anxiety, cough, dyspnea, influenza, insomnia, peripheral edema, dizziness and increased alanine transaminase (ALT). The incidence of dyspnea and peripheral edema appeared to be dose-related and had an early onset.
The interim analysis from the phase II extension study did not identify new or unexpected safety signals associated with longer exposure to ponesimod.
In the phase II core study, all patients in the ponesimod groups received 10 mg on the first day and were uptitrated to 20 or 40 mg after 1 and 2 weeks, respectively. After the first dose of 10 mg ponesimod, there was a transient reduction in HR that reached a maximum of approximately 16 beats per minute (bpm), compared with 4 bpm on placebo, from baseline at 2–3 hours post dose and returned to pre dose values 6 hours post dose. The effect of subsequent uptitration to 20 and 40 mg on HR was negligible. AEs of bradycardia and second degree AV block Mobitz Type I were reported in 2% and 0.9%, respectively, of patients receiving ponesimod 10 mg on day 1.
The rate of infections was not increased in the ponesimod groups compared with the placebo group in the phase II core study. Lymphocyte count reduction was dose-dependent, sustained with ponesimod treatment, and fully reversible within 1 week of treatment discontinuation.
There was a dose-dependent decrease in FEV1 with ponesimod treatment. In the phase II core study, the mean percentage change in FEV1 from baseline to week 24 was −0.6%, −5.2%, −6.0% and −10.3% in the placebo and ponesimod 10, 20 and 40 mg groups, respectively. In the subgroup of patients who discontinued treatment prematurely or did not enter the extension study, FEV1 returned to baseline values within 1 week of treatment discontinuation. There was also a decrease in forced vital capacity (FVC) with ponesimod treatment, but this was smaller than the decrease seen for FEV1 and also reversible, suggesting a functional obstructive change induced by ponesimod. In the phase II core study, the proportion of patients with respiratory AEs, consisting mainly of mild or moderate dyspnea, was higher in the ponesimod than in the placebo group (placebo, 6.6%; ponesimod 10 mg, 9.3%; ponesimod 20 mg, 16.7%; ponesimod 40 mg, 31.9%).
There were increases in liver transaminases with ponesimod treatment. In the phase II core study, the proportion of patients with a >3-fold increase in ALT above the upper limit of the normal range was 2.8%, 4.5% and 4.2% in the ponesimod 10, 20 and 40 mg arms, respectively, compared with none in the placebo group. These increases were asymptomatic, not associated with bilirubin elevations, and were reversible after treatment discontinuation or even upon continued treatment.
There were four cases of macular edema (one on placebo and three on ponesimod) reported in the phase II core study, but only one (on ponesimod) was confirmed while a second confirmed case occurred in the extension study. Both confirmed cases occurred within the first 3 months of initiating ponesimod treatment and resolved without sequelae after discontinuation.
Current assessment of ponesimod benefit-risk in MS
Based on the efficacy and safety data available from the phase II core and extension studies, ponesimod appears to have a favorable benefit–risk profile for the treatment of patients with relapsing MS. Oral treatment with ponesimod significantly and dose-dependently reduced the number of inflammatory lesions in the brain and was associated with a numerical reduction of relapse rates. Although comparison across different studies has several limitations and should be interpreted with caution, the magnitude of treatment effects with ponesimod in MS appears overall comparable with that associated with fingolimod treatment [Kappos et al. 2006, 2010]. However, in contrast to fingolimod, a clear dose–response relationship was identified for ponesimod, reaching a plateau of PD effect and therapeutic efficacy with 20 mg o.d. dosing.
Ponesimod was generally well tolerated at doses of 10 and 20 mg, while the 40 mg dose was associated with an increased incidence of AEs leading to treatment discontinuation, particularly in relation to respiratory function effects and symptoms of dyspnea. The 20 mg dose appears to provide an optimal benefit–risk balance for treatment of MS patients with ponesimod, but more data are needed from the ongoing phase II extension study and from other studies to provide compelling evidence. First-dose cardiac effects observed with ponesimod appeared of similar nature to those described with fingolimod [Kappos et al. 2010], but in more recent studies with ponesimod an optimized uptitration regimen is used to minimize these effects [Hoch et al. 2015b]. The S1P1R selectivity of ponesimod may provide safety advantages over fingolimod, but these may need long-term observation to become apparent.
A distinctive feature of ponesimod in comparison with fingolimod is its shorter halflife and corresponding rapid reversibility of effects on the immune system. Lymphocyte count in peripheral blood returned to the normal range within 1 week after stopping ponesimod compared with 1–2 months after stopping fingolimod. The quicker elimination of ponesimod may be an advantage in managing serious or opportunistic infections, it may be beneficial in case of vaccination or pregnancy, and it may also help to prevent sequelae or complications in case of AEs such as macular edema, pulmonary function changes and liver enzyme elevations. A phase III study is currently ongoing to establish the benefit–risk of ponesimod in patients with relapsing MS [ClinicalTrials.gov identifier: NCT02425644] (Table 1).
Efficacy and safety of ponesimod in psoriasis
Efficacy in psoriasis
The rationale to apply ponesimod in psoriasis is based on the concept that psoriasis is a T-cell mediated inflammatory skin disease. Blocking lymphocyte recirculation from secondary lymphoid organs therefore appeared a promising and novel therapeutic approach to improve psoriasis by reducing the recruitment of pathogenic T cells into the skin.
The first study of ponesimod in psoriasis was a small proof-of-concept, randomized, double-blind, placebo-controlled study in 66 patients with moderate- to-severe chronic plaque psoriasis treated with o.d. 20 mg ponesimod or placebo [ClinicalTrials.gov identifier: NCT00852670] (Table 1). The study showed ponesimod was generally well tolerated and might provide a benefit to these patients.
A double-blind, randomized, placebo-controlled, dose-finding phase IIb study evaluated the efficacy, safety and tolerability of ponesimod for the treatment of moderate-to-severe chronic plaque psoriasis [ClinicalTrials.gov identifier: NCT01208090] (Table 1) [Vaclavkova et al. 2014]. A total of 326 patients were randomized across 58 centers in 15 countries between September 2010 and October 2012 to receive o.d. ponesimod 20 or 40 mg, or placebo for 16 weeks. Patients with at least a partial improvement at 16 weeks continued ponesimod or were switched to placebo until week 28. Baseline demographic and disease characteristics of patients were well balanced across groups and consistent with a population of patients in need of systemic psoriasis treatment.
The primary efficacy endpoint of a reduction in Psoriasis Area and Severity Index (PASI) score of at least 75% from baseline (PASI75) at week 16 was met in 46.0% and 48.1% of patients in the 20 and 40 mg ponesimod groups, respectively, compared with 13.5% in the placebo group (Table 2). At week 16, physician global assessment (PGA) scores of 0 or 1 were observed in 27.8% and 32.3% of patients in the 20 and 40 mg ponesimod groups, respectively, compared with 4.5% in the placebo group. A PASI90 score at week 16 was met in 3.0%, 14.3% and 24.8% of patients in the placebo, ponesimod 20 and 40 mg groups, respectively. Improvements in quality of life were also reported more frequently by patients receiving ponesimod than placebo.
The PASI score started to improve 3 weeks after initiation of ponesimod treatment and continued to do so during the induction period. PASI scores improved further after 16 weeks in those patients who continued ponesimod treatment until the end of the study, while patients began to lose efficacy approximately 4 weeks after switching to placebo. Peripheral blood lymphocyte count was rapidly reduced with ponesimod treatment to a similar extent as seen in patients with MS. Mean reductions of lymphocyte count from baseline to week 16 were around 2%, 56% and 65%, for the placebo, ponesimod 20 and 40 mg groups, respectively. Lymphocyte count remained stable in patients who continued ponesimod treatment up to week 28. In patients who switched to placebo, lymphocyte count recovered quickly to roughly baseline levels after stopping ponesimod.
The loss of clinical benefits in patients who switched from ponesimod to placebo was gradual, starting approximately 4 weeks after stopping ponesimod treatment and it was delayed compared with the restoration of lymphocyte count in peripheral blood. This delay suggests that pathogenic lymphocytes need to return to the circulation, enter the skin, and become activated to trigger the formation of new plaques. These observations also support the notion that a continuous recruitment of T cells from lymphoid organs rather than a persistent activation of a priori skin-resident T cells is required to induce and maintain disease activity in psoriasis [Diluvio et al. 2006; Gaide et al. 2015].
Assessment of the patients’ perception of joint pain in a subgroup of patients (n = 44) who had psoriatic arthritis suggested that ponesimod treatment may also have a beneficial effect on joint involvement in psoriatic arthritis.
Safety in psoriasis
Ponesimod appeared generally well tolerated in patients with psoriasis.
In the dose-finding phase IIb study, most AEs were of mild or moderate intensity. SAEs were reported in 1.5%, 4.0% and 3.8% patients in the placebo, ponesimod 20 and 40 mg groups, respectively. The proportion of patients who prematurely discontinued study treatment due to AEs in the ponesimod 20 and 40 mg groups was 7.9% and 10.5%, respectively, compared with 1.5% in the placebo group. Treatment-emergent AEs reported with higher incidence in ponesimod groups included dyspnea, dizziness, increased ALT or aspartate transaminase (AST), and bradycardia.
All patients in the ponesimod groups received 10 mg on the first day and were later uptitrated to 20 or 40 mg. After the first dose of 10 mg ponesimod, there was a transient reduction in HR that reached a maximum of approximately 13 bpm (compared with 2 bpm on placebo) from baseline at 2–3 hours post dose and returned close to pre dose values 6 hours post dose. As seen in the MS phase II study, the effect on HR of subsequent uptitration to 20 and 40 mg was negligible. AEs of bradycardia and second-degree AV block were reported more frequently in the ponesimod groups, almost exclusively on day 1.
The rate of infections was similar across all treatment groups. Lymphocyte count reduction was dose-dependent, sustained with ponesimod treatment, and fully reversible after 1 or 2 weeks after treatment discontinuation.
There was an early, dose-dependent decrease in FEV1 with ponesimod treatment. The mean change in FEV1 from baseline to week 8 was 0.1%, −5.1% and −10.1% in the placebo and ponesimod 20 and 40 mg groups, respectively. As seen in MS, the reduction of FVC with ponesimod was less pronounced compared with FEV1. For patients who switched at week 16 from ponesimod to placebo, the FEV1 returned to baseline values within 2 weeks of ponesimod discontinuation. The proportion of patients who reported an AE of dyspnea was higher in the ponesimod groups compared with placebo (placebo, 1.5%; ponesimod 20 mg, 11.1%; ponesimod 40 mg, 26.3%).
There were increases in aminotransferases with ponesimod treatment. The proportion of patients with >3-fold increase in aminotransferases above the upper limit of the normal range was 11.3% and 8.4% in the ponesimod 20 and 40 mg groups, respectively, compared with 3.1% in the placebo group. These increases were asymptomatic, not associated with bilirubin elevations, and were reversible after treatment discontinuation or even on continued treatment.
There was one confirmed case of macular edema which occurred within the first 3 months of initiating ponesimod treatment and resolved shortly after treatment discontinuation without sequelae.
Current assessment of ponesimod benefit-risk in psoriasis
Phase II study results indicated that ponesimod may be efficacious for treatment of moderate-to-severe chronic plaque psoriasis. Although the efficacy seen with ponesimod cannot be directly compared with that achieved in other studies, it appears similar to that of the tumor necrosis factor-α (TNF-α) antagonist, etanercept, and better than that of methotrexate and the phosphodiesterase 4 inhibitor apremilast, an oral small molecule recently approved for treatment of psoriasis and psoriatic arthritis [Kavanaugh et al. 2014; Papp et al. 2015]. Both ponesimod doses showed a similar level of efficacy. Thus it remains to be determined whether doses lower than 20 mg could also be effective.
In the population enrolled in the phase II study, which excluded psoriasis patients with some common cardiovascular or metabolic comorbidities, ponesimod was generally well tolerated, particularly at the dose of 20 mg.
First-dose cardiac effects with ponesimod were of a similar magnitude to those seen in the MS phase II trial. As for MS patients, an optimized titration regimen might be used to minimize these effects and improve the benefit–risk balance in this population.
The magnitude of lymphocyte count reduction, pulmonary function effects and incidence of infections observed with ponesimod in psoriasis patients were also similar to those seen in patients with MS. Aminotransferase elevations were apparently more frequent in patients with psoriasis, but were also not associated with any sign or symptoms of potential hepatotoxicity.
The finding that ponesimod may provide a benefit to skin as well as joint disease in patients with psoriatic arthritis suggests potentially broad applications of ponesimod for the treatment of various immune-mediated diseases in which T cells play a predominant pathogenic role. Further studies will be needed to better characterize the benefit–risk profile of ponesimod in psoriasis.
A gradual dose titration regimen to minimize first-dose effects of ponesimod
In humans, activation of S1P1 receptors on cardiomyocytes during initiation of treatment with an S1P1R modulator leads to transient HR decreases and infrequently AV conduction delays [Peters and Alewijnse, 2007; Camm et al. 2014]. These effects initially observed with the nonselective S1PR modulator fingolimod were later found to occur also with selective S1P1R modulators in humans. Acute occurrence of bradycardia and, in some cases, AV blocks seen after the initial fingolimod intake, requires electrocardiography (ECG) monitoring for at least 6 hours after administration of the first dose [Novartis, 2015]. HR decrease with fingolimod is maximal approximately 6 hours after the first dose and diminishes with the second and subsequent doses. With continued dosing of fingolimod, S1P1R becomes internalized and unable to signal on cardiomyocytes and thus the HR gradually returns to baseline within 1 month of chronic treatment [Novartis, 2015].
Clinical data from over a thousand subjects treated with ponesimod have consistently shown a dose-dependent HR reduction following first-dose administration. This effect is transient and due to initial activation of S1P1R by ponesimod before internalization and desensitization of the receptor. The desensitization phenomenon observed for the effect on HR and AV conduction suggested that the effect could be mitigated by initiating treatment with a low dose followed by uptitration to the target dose. A study in guinea pigs, a preclinical model in which S1P1R regulates AV conduction, had shown that a progressive dose titration regimen could significantly reduce the effects of ponesimod on AV conduction [Rey et al. 2013]. On the basis of these observations, different uptitration regimens were investigated to minimize the first-dose effects of ponesimod on HR and AV conduction in humans.
An uptitration regimen of ponesimod o.d. starting with 10 mg for 7 days, followed by 20 mg for 7 days, then followed by 40 mg was initially investigated in phase I studies [Brossard et al. 2014] and later implemented in the phase II studies of psoriasis and MS. This uptitration regimen was able to reduce acute changes in HR and AV conduction but bradycardia and second degree AV blocks, mostly asymptomatic, were still observed in a small proportion of subjects after first-dose administration and required monitoring for a period of 6 hours.
Based on the sustained S1P1R desensitization induced by ponesimod, PK/PD modeling was used to design new uptitration regimens with the objective to minimize the acute effects of ponesimod on HR and AV conduction. An early study comparing three different uptitration regimens of ponesimod in healthy subjects suggested that a dose around 2.5 mg might be sufficient to start inducing S1P1R desensitization while causing a very small first-dose effect [Scherz et al. 2015]. The study also investigated the duration of S1P1R desensitization upon 1–3 days interruption of a maintenance dosing regimen of 20 mg o.d. The first-dose cardiac effects seen upon re-initiation with 20 mg were small even after 3 days interruption, thus indicating that S1P1R desensitization was sustained for at least 3 days after stopping treatment.
A more recent study in 32 healthy male and female subjects compared a novel gradual uptitration regimen of ponesimod to the uptitration regimen used in phase II studies [Hoch et al. 2015b]. The novel uptitration regimen of ponesimod consisted of a starting dose of 2 mg given for 2 days, followed by administration of 3 and 4 mg for 2 days at each dose, followed by 5, 6, 7, 8 and 9 mg for 1 day at each dose, 10 mg for 2 days and then a 20 mg maintenance dose. This gradual uptitration regimen was able to minimize the effects of ponesimod on HR and AV conduction. Mean decrease of HR from baseline following the first dose was smaller with the gradual uptitration compared with the previous uptitration (a decrease of 6 versus 12 bpm). The effects on HR and AV conduction were smaller during the entire 2-week regimen with the novel uptitration compared with the previous one. There were no SAEs or AEs leading to treatment discontinuation and no AEs of bradycardia or AV blocks reported with the gradual regimen. Dizziness and headache AEs were less frequently reported with the new regimen and fewer subjects had occurrences of a marked HR decrease with the novel uptitration.
Overall, the novel, gradual uptitration was better than the previous one to minimize the effects of ponesimod on HR and AV conduction. This gradual uptitration regimen is currently implemented in ponesimod clinical studies. Data from the ongoing studies will help to determine whether first-dose monitoring of patients on site may be reduced or even avoided with the novel uptitration regimen.
Future of ponesimod for the treatment of MS and other lymphocyte-driven diseases
Development in MS
The extensive clinical experience gained with ponesimod allowed the selection of an optimized uptitration dosing regimen to minimize first-dose cardiac effects and of a maintenance dose of 20 mg o.d. to achieve the desired level of therapeutic efficacy for phase III studies.
A multinational, randomized, double-blind, active-control phase III study of ponesimod in relapsing MS, OPTIMUM [ClinicalTrials.gov identifier: NCT02425644) (Table 1), has recently been started. The OPTIMUM study is evaluating the efficacy and safety of ponesimod in patients with relapsing MS. This study will randomize approximately 1100 patients in a 1:1 ratio to receive 20 mg ponesimod or 14 mg teriflunomide orally o.d. for 2 years. The primary objective of the trial is to assess whether ponesimod is superior to teriflunomide in reducing the ARR over 108 weeks. Secondary endpoints include time to 12-week confirmed disability accumulation, time to first confirmed relapse, cumulative number of combined unique active lesions of the brain, and percent change in brain volume from baseline to week 108.
The study is enrolling adult male and female subjects aged 18–55 years with an established diagnosis of relapsing MS, as defined by the 2010 revision of the McDonald Diagnostic criteria, with relapsing course from onset (i.e. relapsing remitting MS or secondary progressive MS with superimposed relapses). Patients are required to have active disease based on at least 1 MS attack within 12 months or 2 MS attacks within 24 months or at least 1 Gd+ lesion in the brain or spinal cord on an MRI performed within 6 months of study entry. The trial permits inclusion of patients with secondary progressive MS with superimposed relapses up to a maximum of 15%. This will allow the evaluation of the benefit–risk profile of ponesimod in this group of patients, who have often not been included in pivotal trials of medications approved for treatment of relapsing MS.
This study will be the first to compare the efficacy and safety of two oral treatments in relapsing MS patients. Teriflunomide is a first-line oral medication, approved for the treatment of patients with relapsing MS in 2013 [O’Connor et al. 2011, 2013; Freedman, 2013]. Both ponesimod and teriflunomide are given orally o.d., which facilitates blinding and improves patient compliance in the clinical trial.
The OPTIMUM study has a number of unique features. Nonconventional MRI techniques of magnetization transfer ratio (MTR) and double inversion recovery are implemented at selected centers to investigate the effect of ponesimod on grey matter lesions and to measure the changes in myelin density in normal appearing white matter and focal lesions, respectively. A patient-reported outcome measure of fatigue in relapsing MS, developed according to FDA guidance [Hudgens, 2015], is being used for the first time to measure the effect of ponesimod on fatigue-related symptoms. A purpose-designed e-diary is used to enhance and monitor treatment compliance of patients in the trial.
A substudy of OPTIMUM will aim to collect patients’ preferences and value judgments for selected treatment outcomes via a purpose-designed questionnaire. The substudy has a target of approximately 360 patients and the data collected will be analyzed using the Measuring Attractiveness by a Categorical Based Evaluation Technique (MACBETH) [Bana e Costa and Vansnick, 1999]. This approach, implemented for the first time in an interventional MS trial, may help to shape future healthcare decisionmaking based on patient preferences [Wilson et al. 2014].
Patients who will complete the 108 weeks of treatment in the OPTIMUM study will have the possibility to enter an open-label, long-term extension study in which all patients will receive 20 mg o.d. ponesimod. The main objectives of this extension study will be to evaluate the long-term safety and tolerability, and the effect on disease control of ponesimod as well as the effects of re-initiating ponesimod treatment after interruption and of switching from teriflunomide to ponesimod treatment.
Finally, the long-term efficacy, safety and tolerability of 10 and 20 mg ponesimod will continue to be evaluated in the long-term dose-blinded, phase II extension study.
These and potentially other studies are expected to support the benefit–risk assessment of ponesimod for the treatment of patients with relapsing forms of MS.
Development in chronic graft versus host disease
Chronic graft versus host disease (GVHD) is a pleomorphic syndrome with autoimmune-like features, which represents the most serious and common long-term complication of allogeneic hematopoetic stem cell transplantation (HSCT). The prognosis of patients with chronic GVHD is poor, especially for glucocorticoid refractory disease [Lee and Flowers, 2008; Martin et al. 2011]. The pathophysiology of chronic GVHD is complex and involves both autoreactive and alloreactive T and B lymphocytes [Wolff et al. 2011]. Fingolimod has shown beneficial effects in an animal model of chronic GVHD [Huu et al. 2013], suggesting a potential role of S1P1R modulators in the treatment of this condition.
Based on these premises, a phase II study has recently been initiated with ponesimod in patients with chronic GVHD. This is a prospective, multi-center, open-label, intra-subject dose-escalation study [ClinicalTrials.gov identifier: NCT02461134] (Table 1). The study is designed to investigate the biological activity, safety, tolerability and PK of ponesimod in subjects with symptomatic moderate or severe chronic GVHD inadequately responding to first- or second-line therapy.
Approximately 30 subjects will be enrolled to receive ponesimod in escalating doses of 5, 10 and 20 mg over the core treatment period of 24 weeks. The study will be conducted at approximately 10 sites in the USA. Subjects successfully completing the initial 24 weeks of the study have the possibility to restart ponesimod for an extension treatment course of 96 weeks should their chronic GVHD progress within a defined timeframe after stopping ponesimod.
Patients with chronic GVHD may be particularly fragile due to the underlying causes of and procedures associated with HSCT, which include treatment with multiple cytotoxic and immunosuppressive medications. An important goal of this study is to investigate the safety and tolerability of ponesimod in this patient population. Equally important is to explore the effects of ponesimod on the newly reconstituted immune system of these patients and to investigate its potential beneficial effects on the disease.
Conclusion
Ponesimod is an orally active, selective S1P1R modulator developed by Actelion Pharmaceuticals Ltd. Ponesimod has successfully completed phase II studies in relapsing remitting MS and moderate-to-severe chronic plaque psoriasis showing therapeutic efficacy in these conditions and a potential to treat other immune-mediated disorders. The selectivity for S1P1R and the rapid onset and reversibility of pharmacological effects of ponesimod constitute important differences from the first-generation S1PR modulator fingolimod, and have the potential for an improved tolerability and safety profile. An optimized uptitration regimen for ponesimod treatment initiation can minimize the first-dose effects associated with modulation of the S1P1R on cardiomyocytes and might require a less burdensome monitoring than fingolimod. A phase III study is ongoing to establish ponesimod’s place among the available treatment options for patients with relapsing MS. A phase II study is also ongoing to investigate the potential utility of ponesimod for the treatment of patients with GVHD.
Acknowledgments
The authors would like to thank Andrea Vaclavkova, Luca Piali, Jasper Dingemanse, Alberto Gimona, Marjolaine Phan, Volker Breu and Per Nilsson at Actelion for their critical review of the manuscript.
Footnotes
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article. All ponesimod studies discussed in this review were funded by Actelion Pharmaceuticals Ltd.
Declaration of conflicting interest: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: D.D’A. is a full-time employee of, and has shares, in Actelion Pharmaceuticals Ltd. M.F. received consultancy fees from Actelion Pharmaceuticals Ltd and is an investigator in an ongoing clinical trial sponsored by Actelion Pharmaceuticals Ltd. J.P. received consultancy fees from Actelion Pharmaceuticals Ltd and has served as member of an independent Data Safety Monitoring Board in a completed clinical trial sponsored by Actelion Pharmaceuticals Ltd.
Contributor Information
Daniele D’Ambrosio, Actelion Pharmaceuticals – Global Clinical Science and Epidemiology, Gewerbestrasse 16, Basel 4056, Switzerland.
Mark S. Freedman, Multiple Sclerosis Research Clinic, University of Ottawa, Ottawa, Canada
Joerg Prinz, Dermatology, University of Munich, Munich, Germany.
References
- Bana e Costa C., Vansnick J. (1999) The Macbeth approach: basic ideas, software and an application. In: Meskens N., Roubens M. (eds), Advances in Decision Analysis. Dordrecht: Kluwer Academic Publishers, pp. 131–157. [Google Scholar]
- Bolli M., Abele S., Binkert C., Bravo R., Buchmann S., Bur D., et al. (2010) 2-imino-thiazolidin-4-one derivatives as potent, orally active S1P1 receptor agonists. J Med Chem 53: 4198–4211. [DOI] [PubMed] [Google Scholar]
- Brinkmann V. (2007) Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther 115: 84–105. [DOI] [PubMed] [Google Scholar]
- Brossard P., Derendorf H., Xu J., Maatouk H., Halabi A., Dingemanse J. (2013) Pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 receptor modulator, in the first-in-human study. Br J Clin Pharmacol 76: 888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brossard P., Scherz M., Halabi A., Maatouk H., Krause A., Dingemanse J. (2014) Multiple-dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: favorable impact of dose up-titration. J Clin Pharmacol 54: 179–188. [DOI] [PubMed] [Google Scholar]
- Camm J., Hla T., Bakshi R., Brinkmann V. (2014) Cardiac and vascular effects of fingolimod: mechanistic basis and clinical implications. Am Heart J 168: 632–644. [DOI] [PubMed] [Google Scholar]
- Choi J., Gardell S., Herr D., Rivera R., Lee C., Noguchi K., et al. (2011) FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A 108: 751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J., Hla T., Lynch K., Spiegel S., Moolenaar W. (2010) International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol Rev 62: 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes N., Dhaeze T., Fraussen J., Broux B., Van Wijmeersch B., Stinissen P., et al. (2014) Compositional changes of B and T cell subtypes during fingolimod treatment in multiple sclerosis patients: a 12-month follow-up study. PLoS One 9: e111115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster J., Schwab S. (2012) Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol 30: 69–94. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio D., Steinmann J., Brossard P., Dingemanse J. (2015) Differential effects of ponesimod, a selective S1P1 receptor modulator, on blood-circulating human T cell subpopulations. Immunopharmacol Immunotoxicol 37: 103–109. [DOI] [PubMed] [Google Scholar]
- Dev K., Mullershausen F., Mattes H., Kuhn R., Bilbe G., Hoyer D., et al. (2008) Brain sphingosine-1-phosphate receptors: implication for FTY720 in the treatment of multiple sclerosis. Pharmacol Ther 117: 77–93. [DOI] [PubMed] [Google Scholar]
- Diluvio L., Vollmer S., Besgen P., Ellwart J., Chimenti S., Prinz J. (2006) Identical TCR beta-chain rearrangements in streptococcal angina and skin lesions of patients with psoriasis vulgaris. J Immunol 176: 7104–7111. [DOI] [PubMed] [Google Scholar]
- Foster C., Howard L., Schweitzer A., Persohn E., Hiestand P., Balatoni B., et al. (2007) Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther 323: 469–475. [DOI] [PubMed] [Google Scholar]
- Freedman M. (2013) Teriflunomide in relapsing multiple sclerosis: therapeutic utility. Ther Adv Chronic Dis 4: 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaide O., Emerson R., Jiang X., Gulati N., Nizza S., Desmarais C., et al. (2015) Common clonal origin of central and resident memory T cells following skin immunization. Nat Med 21: 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Cabrera P., Brown S., Studer S., Rosen H. (2014) S1P signaling: new therapies and opportunities. F1000Prime Rep 6: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorova I., Schwab S., Phan T., Pham T., Okada T., Cyster J. (2009) Cortical sinus probing, S1P1-dependent entry and flow-based capture of egressing T cells. Nat Immunol 10: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hla T., Brinkmann V. (2011) Sphingosine 1-phosphate (S1P): physiology and the effects of S1P receptor modulation. Neurology 76: S3–S8. [DOI] [PubMed] [Google Scholar]
- Hoch M., D’Ambrosio D., Wilbraham D., Brossard P., Dingemanse J. (2014) Clinical pharmacology of ponesimod, a selective S1P1 receptor modulator, after uptitration to supratherapeutic doses in healthy subjects. Eur J Pharm Sci 63: 147–153. [DOI] [PubMed] [Google Scholar]
- Hoch M., Darpo B., Brossard P., Zhou M., Stoltz R., Dingemanse J. (2015a) Effect of ponesimod, a selective S1P1 receptor modulator, on the QT interval in healthy individuals. Basic Clin Pharmacol Toxicol 116: 429–437. [DOI] [PubMed] [Google Scholar]
- Hoch M., Vaclavkova A., Krause A., Strong A., Bush J., Dingemanse J., et al. (2015b) A novel gradual up-titration regimen mitigates the first-dose effects of ponesimod, a selective S1P1 receptor modulator. Clin Ther 37: e36–e37. [Google Scholar]
- Hu W., Mahavadi S., Huang J., Li F., Murthy K. (2006) Characterization of S1P1 and S1P2 receptor function in smooth muscle by receptor silencing and receptor protection. Am J Physiol Gastrointest Liver Physiol 291: G605–G610. [DOI] [PubMed] [Google Scholar]
- Hudgens S., Schüler R., Hunsche E., Leist T. (2015) Measurement confirmation of the fatigue symptoms and impacts questionnaire – relapsing multiple sclerosis (FSIQ RMS). Value Health 18: A26. [DOI] [PubMed] [Google Scholar]
- Huu D., Matsushita T., Jin G., Hamaguchi Y., Hasegawa M., Takehara K., et al. (2013) FTY720 ameliorates murine sclerodermatous chronic graft-versus-host disease by promoting expansion of splenic regulatory cells and inhibiting immune cell infiltration into skin. Arthritis Rheum 65: 1624–1635. [DOI] [PubMed] [Google Scholar]
- Kappos L., Antel J., Comi G., Montalban X., O’Connor P., Polman C., et al. (2006) Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med 355: 1124–1140. [DOI] [PubMed] [Google Scholar]
- Kappos L., Radue E., O’Connor P., Polman C., Hohlfeld R., Calabresi P., et al. (2010) A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 362: 387–401. [DOI] [PubMed] [Google Scholar]
- Kavanaugh A., Mease P., Gomez-Reino J., Adebajo A., Wollenhaupt J., Gladman D., et al. (2014) Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis 73: 1020–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyrakh L., Roman M., Brinkmann V., Wickman K. (2005) The heart rate decrease caused by acute FTY720 administration is mediated by the G protein-gated potassium channel I. Am J Transplant 5: 529–536. [DOI] [PubMed] [Google Scholar]
- Krause A., Brossard P., D’Ambrosio D., Dingemanse J. (2014) Population pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 receptor modulator. J Pharmacokinet Pharmacodyn 41: 261–278. [DOI] [PubMed] [Google Scholar]
- Lee S., Flowers M. (2008) Recognizing and managing chronic graft-versus-host disease. Hematology Am Soc Hematol Educ Program 2008: 134-141. [DOI] [PubMed] [Google Scholar]
- Lorenz J., Arend L., Robitz R., Paul R., Maclennan A. (2007) Vascular dysfunction in S1P2 sphingosine 1-phosphate receptor knockout mice. Am J Physiol Regul Integr Comp Physiol 292: R440–R446. [DOI] [PubMed] [Google Scholar]
- Lublin F., Miller D., Freedman M., Cree B., Wolinsky J., Weiner H. (2015) Oral fingolimod versus placebo in primary progressive multiple sclerosis: results of a large phase III, randomised trial. Lancet, in press. [DOI] [PubMed] [Google Scholar]
- Martin P., Inamoto Y., Carpenter P., Lee S., Flowers M. (2011) Treatment of chronic graft-versus-host disease: past, present and future. Korean J Hematol 46: 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehling M., Brinkmann V., Antel J., Bar-Or A., Goebels N., Vedrine C., et al. (2008) FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology 71: 1261–1267. [DOI] [PubMed] [Google Scholar]
- Mehling M., Johnson T., Antel J., Kappos L., Bar-Or A. (2011) Clinical immunology of the sphingosine 1-phosphate receptor modulator fingolimod (FTY720) in multiple sclerosis. Neurology 76: S20–S27. [DOI] [PubMed] [Google Scholar]
- Nakamura M., Matsuoka T., Chihara N., Miyake S., Sato W., Araki M., et al. (2014) Differential effects of fingolimod on B-cell populations in multiple sclerosis. Mult Scler 20: 1371–1380. [DOI] [PubMed] [Google Scholar]
- Novartis (2015) Summary of Product Characteristics. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002202/WC500104528.pdf (accessed 4 August 2015).
- O’Connor P., Lublin F., Wolinsky J., Confavreux C., Comi G., Freedman M., et al. (2013) Teriflunomide reduces relapse-related neurological sequelae, hospitalizations and steroid use. J Neurol 260: 2472–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor P., Wolinsky J., Confavreux C., Comi G., Kappos L., Olsson T., et al. (2011) Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med 365: 1293–1303. [DOI] [PubMed] [Google Scholar]
- Olsson T., Boster A., Fernandez O., Freedman M., Pozzilli C., Bach D., et al. (2014) Oral ponesimod in relapsing-remitting multiple sclerosis: a randomised phase II trial. J Neurol Neurosurg Psychiatry 85: 1198–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp K., Reich K., Leonardi C., Kircik L., Chimenti S., Langley R., et al. (2015) Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol 73: 37–49. [DOI] [PubMed] [Google Scholar]
- Peters S., Alewijnse A. (2007) Sphingosine-1-phosphate signaling in the cardiovascular system. Curr Opin Pharmacol 7: 186–192. [DOI] [PubMed] [Google Scholar]
- Piali L., Froidevaux S., Hess P., Nayler O., Bolli M., Schlosser E., et al. (2011) The selective sphingosine 1-phosphate receptor 1 agonist ponesimod protects against lymphocyte-mediated tissue inflammation. J Pharmacol Exp Ther 337: 547–556. [DOI] [PubMed] [Google Scholar]
- Pinschewer D., Brinkmann V., Merkler D. (2011) Impact of sphingosine 1-phosphate modulation on immune outcomes. Neurology 76: S15–S19. [DOI] [PubMed] [Google Scholar]
- Pozzilli C., Fernandez O., Olsson T., Freedman M.S., Melanson M., Boster A., et al. (2013) Maintenance of efficacy, safety and tolerability of ponesimod in patients with relapsing remitting multiple sclerosis: phase II extension study. Poster presentation at ECTRIMS (2013) European Committee for Treatment & Research in Multiple Sclerosis, 29th Congress, 2–5 October, Copenhagen, Denmark. [Google Scholar]
- Rey M., Hess P., Clozel M., Delahaye S., Gatfield J., Nayler O., et al. (2013) Desensitization by progressive up-titration prevents first-dose effects on the heart: guinea pig study with ponesimod, a selective S1P1 receptor modulator. PLoS One 8: e74285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M., Brossard P., Chassard D., Hoch M., Dingemanse J. (2014a) Effects of ponesimod, a selective S1P1 receptor modulator, on the pharmacokinetics of a hormonal combination contraceptive. Eur J Clin Pharmacol 70: 287–293. [DOI] [PubMed] [Google Scholar]
- Reyes M., Hoch M., Brossard P., Dingemanse J. (2014b) Effects of ethnicity and sex on the pharmacokinetics and pharmacodynamics of the selective sphingosine-1-phosphate receptor 1 modulator ponesimod: a clinical study in Japanese and Caucasian subjects. Pharmacology 94: 223–229. [DOI] [PubMed] [Google Scholar]
- Reyes M., Hoch M., Brossard P., Wagner-Redeker W., Miraval T., Dingemanse J. (2015) Mass balance, pharmacokinetics and metabolism of the selective S1P1 receptor modulator ponesimod in humans. Xenobiotica 45: 139–149. [DOI] [PubMed] [Google Scholar]
- Salomone S., Yoshimura S., Reuter U., Foley M., Thomas S., Moskowitz M., et al. (2003) S1P3 receptors mediate the potent constriction of cerebral arteries by sphingosine-1-phosphate. Eur J Pharmacol 469: 125–134. [DOI] [PubMed] [Google Scholar]
- Scherz M., Brossard P., D’ Ambrosio D., Ipek M., Dingemanse J. (2015) Three different up-titration regimens of ponesimod, an S1P1 receptor modulator, in healthy subjects. J Clin Pharmacol 55: 688–697. [DOI] [PubMed] [Google Scholar]
- Sobel K., Menyhart K., Killer N., Renault B., Bauer Y., Studer R., et al. (2013) Sphingosine 1-phosphate (S1P) receptor agonists mediate pro-fibrotic responses in normal human lung fibroblasts via S1P2 and S1P3 receptors and smad-independent signaling. J Biol Chem 288: 14839–14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subei A., Cohen J. (2015) Sphingosine 1-phosphate receptor modulators in multiple sclerosis. CNS Drugs 29: 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaclavkova A., Chimenti S., Arenberger P., Hollo P., Sator P., Burcklen M., et al. (2014) Oral ponesimod in patients with chronic plaque psoriasis: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet 384: 2036–2045. [DOI] [PubMed] [Google Scholar]
- Watterson K., Ratz P., Spiegel S. (2005) The role of sphingosine-1-phosphate in smooth muscle contraction. Cell Signal 17: 289–298. [DOI] [PubMed] [Google Scholar]
- Wilson L., Loucks A., Bui C., Gipson G., Zhong L., Schwartzburg A., et al. (2014) Patient centered decision making: use of conjoint analysis to determine risk-benefit trade-offs for preference sensitive treatment choices. J Neurol Sci 344: 80–87. [DOI] [PubMed] [Google Scholar]
- Wolff D., Schleuning M., Von Harsdorf S., Bacher U., Gerbitz A., Stadler M., et al. (2011) Consensus conference on clinical practice in chronic GVHD: second-line treatment of chronic graft-versus-host disease. Biol Blood Marrow Transplant 17: 1–17. [DOI] [PubMed] [Google Scholar]
- You S., Piali L., Kuhn C., Steiner B., Sauvaget V., Valette F., et al. (2013) Therapeutic use of a selective S1P1 receptor modulator ponesimod in autoimmune diabetes. PLoS One 8: e77296. [DOI] [PMC free article] [PubMed] [Google Scholar]