

Abstract
Although the heavy metals cadmium (Cd) and lead (Pb) are known environmental health concerns, their long-term impacts on gut ecology and susceptibility to gastrointestinal autoimmune diseases have not been extensively investigated. We sought to determine whether subchronic oral exposure to Cd or Pb is a risk factor for the development and progression of inflammatory bowel disease (IBD). Mice were exposed to various doses of CdCl2 or PbCl2 in drinking water for 1, 4 or 6 weeks prior to infection with Salmonella, the induction of colitis with dextran sodium sulfate (DSS) or trinitrobenzene sulfonic acid (TNBS). In human cell-based models, exposure to Cd and Pb is associated with reduced transepithelial electric resistance and changes in bacteria-induced cytokine responses. Although 1- and 6-week exposures did not have clear effects on the response to Salmonella infectious challenges, 1-week short-term treatments with CdCl2 tended to enhance intestinal inflammation in mice. Unexpectedly, subchronic exposure to Cd and (to a lesser extent) Pb significantly mitigated some of the symptoms of DSS-induced colitis and reduced the severity of TNBS colitis in a dose-dependent manner. The possible adaptive and immunosuppressive mechanisms by which heavy metals might reduce intestinal inflammation are explored and discussed.
Genome-wide association studies have highlighted a huge number of susceptibility genes linked to chronic immune diseases, such inflammatory bowel disease (IBD). However, epidemiological studies have shown that the worldwide increase in the incidence of Crohn’s disease (CD) and ulcerative colitis (UC) over the last few decades is mainly due to environmental factors1. Thus, the etiology and pathogenesis of IBD are clearly related to changes in lifestyle, diet and pollution levels. This is probably the case in late-onset IBD (i.e. diagnosed in adulthood), associated with more exposure time elapsed2. Rappaport and Smith and Lioy and Rappaport3,4 have extended the initial concept of the individual “exposome” to the whole set of chemicals that enter the body. This encompasses many exogenous factors, such as the diet, dietary pollutants, xenobiotics and endogenous influencing features (e.g. oxidative stress, inflammation) and the microbiome. These factors interact and change over time, and so it is difficult to estimate the causality of environmental signatures. However, it is important to determine the levels of exposure that are likely to tune the many integrated biologic effects (particularly in IBD) and the extent to which putative risk factors might interfere and interact with the host’s adaptive processes.
Only a few clear environmental risk factors for IBD have been identified to date (smoking, appendectomy and early antibiotic use), although discrepancies (and even opposing effects) have been reported as a function of age and the disease subtype5,6. For example, smoking is positively correlated with the incidence of CD but negatively correlated with the incidence of UC - suggesting that the net effects of myriads of chemicals in cigarette smoke are complex and hard to predict. Indeed, the generation of reactive oxygen species and cadmium (Cd) accumulation may differentially alter the interplay between immune function, oxidative stress and microbiota function in the body as a whole an in the gut in particular. The direct impact of environmental pollutants on gut ecology and subsequent intestinal inflammatory events has not been extensively addressed. It has been shown that the xenobiotic perfluorooctanoic acid was associated with UC in highly exposed workers, although one cannot rule out the possibility whereby the inflammatory state in UC subjects is responsible for increased intestinal absorption of the molecule7. It has also been suggested that aluminum is a potent environmental factor for CD induction8. The metal’s harmful effects on gut homeostasis have been well established using several in vitro and murine models of colitis9.
Lead (Pb) and Cd are two widespread, non-essential, heavy-metal pollutants of environmental health concern in both industrialized and developing countries10,11. Humans can be exposed to Cd and/or Pb from a variety of sources resulting from past and ongoing anthropogenic activities (industrial emissions, car exhaust fumes, fossil fuel combustion, metallurgy and sea pollution). In particular, smoking and dietary intake (through contaminated cereals, fish, shellfish and drinking water) are the major sources of Cd/Pb entry into the gastrointestinal tract12. It is also noteworthy that a large proportion of inhaled Cd ends up in the gastrointestinal tract as a result of mucociliary clearance13. Heavy metal exposure is harmful because of acute poisoning on one hand and long-term toxicity (due to accumulation) on the other. However, little is known about the impact of subchronic exposure to Cd and Pb on susceptibility to colitis. We therefore sought to determine whether these two heavy metals are potential risk factors for the development and progression of IBD.
In previous works, we characterized the impact of oral exposure to Cd and Pb salts on basal gut physiology and observed (i) time- and dose-dependent dissemination of ingested metals through the blood and target organs, (ii) specific transcriptional signatures in the small intestine and colon and (iii) a clear impact of these metals on intestinal microbial communities14,15,16. However, the complexity of the various toxicological and other endpoints and markers (concomitant anemia, coexisting pro- and anti-inflammatory mediators, oxidative markers, and primary immunosuppressive and possible counter-regulatory adaptive responses) prevented us from predicting or speculating on the heavy metals’ respective effects on gut vulnerability and subsequent intestinal immune responses. After having studied some of these aspects in vitro in human-cell-based models, we designed the present study in order to evaluate the influence of oral exposure to Cd and Pb on susceptibility to gut inflammation in a model of Salmonella typhimurium infection and two different murine models of colitis.
Results
The impact of Cd and Pb on the integrity of a human enterocyte monolayer.
Although the Caco-2 cell line is derived from a human colon cancer, it shows certain traits of small intestinal cells. In contrast, the T84 cell line is characteristic of human colonocytes. In order to examine the possible impact of each heavy metal on gut functionality, subtoxic doses of CdCl2 or PbCl2 were applied to the apical compartment of these two differentiated human intestinal epithelial cell lines. Barrier integrity was evaluated by monitoring the TEER for 24 h and then (after thorough washing) for a further 48 h. Figure 1A,B show that treatment with Cd salts altered the TEER in a concentration-dependent manner for both cell lines, with the higher dose fully abolishing the resistance. After removal of the metal by washing, monolayers exposed to the lower dose rapidly recovered and returned to baseline TEER values (and slightly exceeded for Caco-2, in fact), whereas monolayers exposed to the higher dose never recovered. In contrast, Pb did not appear to affect the TEER, suggesting that Pb does not mediate epithelial damage. Taken as a whole, these in vitro data demonstrate that heavy metals have element-specific, time- and potentially reversible dose-related effects on monolayer permeability.
Figure 1. Impact of CdCl2 or PbCl2 on epithelial layer integrity.
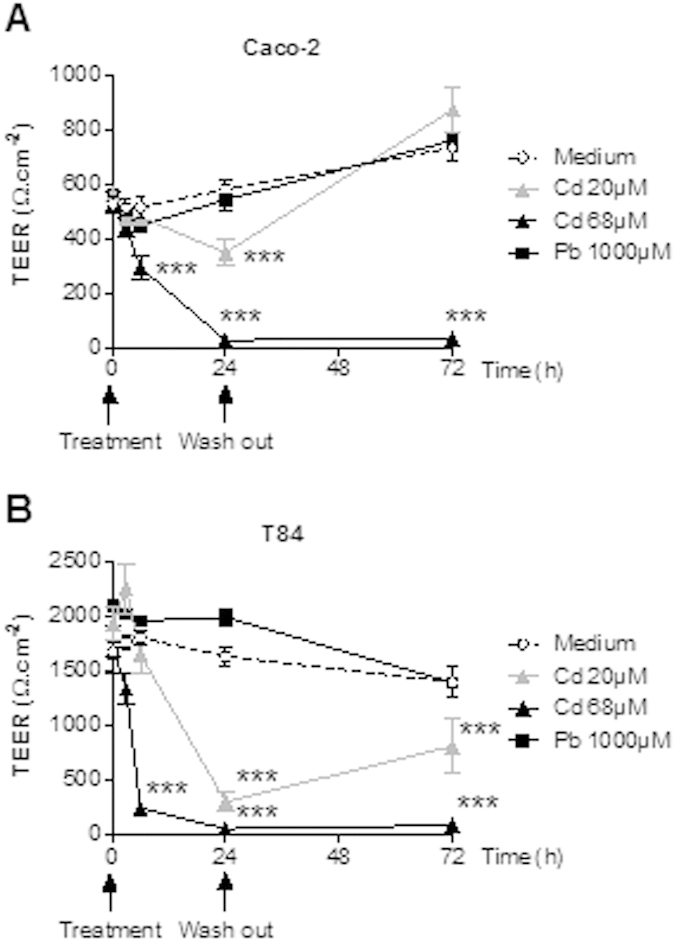
Transepithelial electric resistance (TEER) was measured for differentiated Caco-2 (A) and T84 (B) cells exposed for 24 hours, washed thoroughly and then monitored for a further 48 hours. Data are expressed as the mean ± SEM, n = 3; ***p < 0.001.
The impact of Cd and Pb on human immune cell responses
The immunotoxic properties (including both immunosuppression and immunostimulation) of heavy metals have been studied extensively in animal and in vitro models. We used human PBMCs to evaluate the immunomodulatory effects (cytokine release) of a 24 h exposure to non-toxic doses of CdCl2 and PbCl2 in the absence and presence of concomitant bacterial stimulation (Fig. 2A,B). Whereas exposure to the metals alone did not modulate basal cytokine release, concomitant incubation with various types of bacteria was associated with significantly lower levels of IL-10 or IL-12p70 release with 20 μM Cd and the absence of release for 68 μM Cd. Pb (100 μM) has a smaller, less consistent effect on immune responses. These data suggest that heavy metals have potential immunomodulatory effects on immunocompetent cells. However, this type of in vitro model cannot fully mimic the integrated physiology of the gut; preclinical models are more reliable for investigating dynamic and adaptive responses.
Figure 2. Effect of CdCl2 or PbCl2 on cytokine release by bacteria-stimulated human Peripheral blood mononuclear cells (PBMCs).
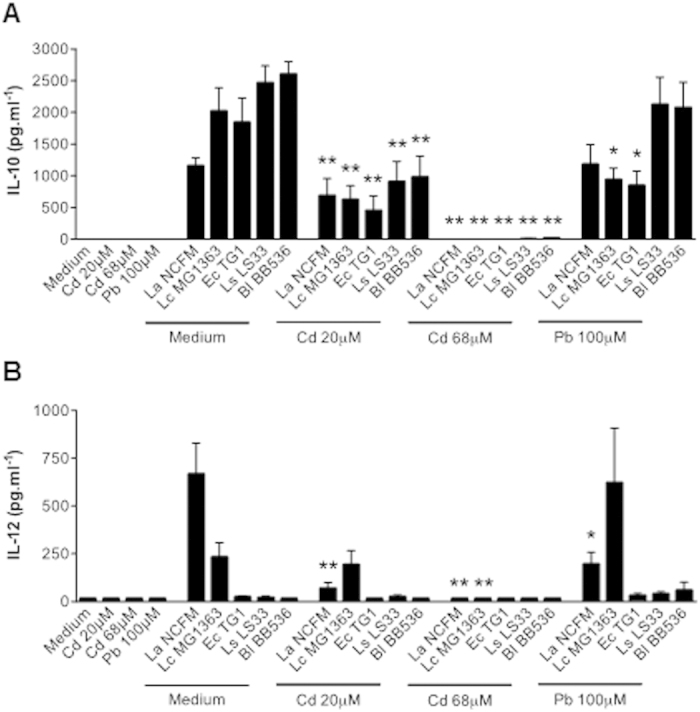
Supernatant levels of IL-10 (A) and IL-12 (B) released by bacteria-stimulated and control human PBMCs in the presence and absence of CdCl2 (20 and 68 μM) or PbCl2 (100 μM) for 24 hours. A set of bacteria Data are expressed as the mean ± SEM pg.mL−1, n = 4 donors; *p < 0.05, **p < 0.01. Metals were simultaneously added to PBMCs with respectively five bacterial strains, a set of microorganisms known for their immune stimulatory properties at various ranges of cytokine levels (Lactobacillus acidophilus NCFM, Lactococcus lactis MG1363, Escherichia coli TG1, Lactobacillus salivarius LS33 and Bifidobacterium longum BB536)55. While heavy metal salts do not induce any pro- or anti-inflammatory cytokines at baseline, they can dose-dependently affect immune responses and cytokine levels when other external stimuli occur simultaneously.
The impact of short-term or subchronic exposure to Cd and Pb salts on an oral infectious challenge with S. typhimurium
We first investigated the impact of either short-term ingestion (for 1 week) or subchronic ingestion (for 4 or 6 weeks) of Cd or Pb on the overall innate immune status of the murine intestine by studying survival following an oral challenge with the pathogen S. typhimurium mice. Neither Cd (20 ppm in drinking water, i.e. 2.5 mg.d−1.kg−1) nor Pb (100 ppm, i.e. 12.5 mg.d−1.kg−1) significantly modified the survival outcomes in any of these situations (Fig. 3A–C, p > 0.1). Thes results show that quite high doses of these heavy metals had a marginal impact on gastrointestinal and systemic antibacterial immune responses
Figure 3. Impact of the CdCl2 or PbCl2 exposure time on mice survival following an oral Salmonella challenge.
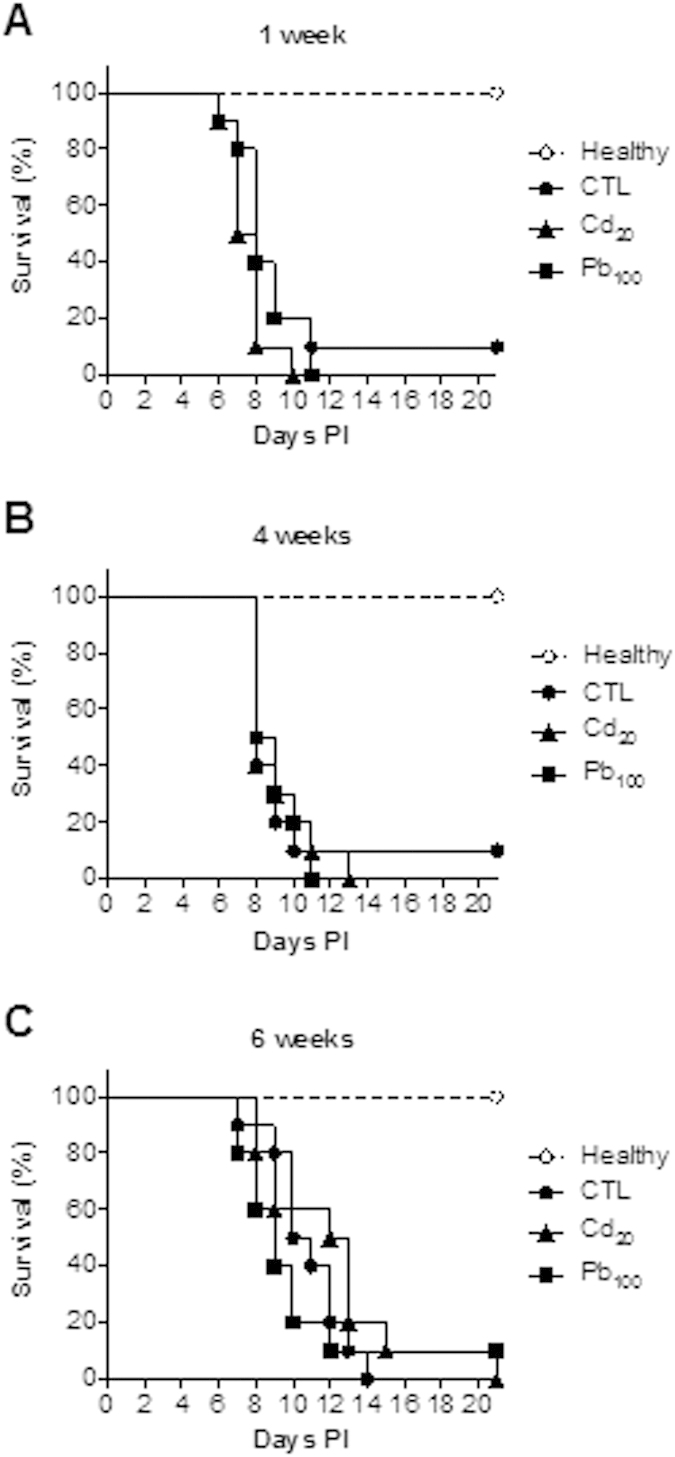
Survival curves are provided for three weeks post-infection (PI) after 1, 4 and 6 weeks of exposure to Cd or Pb (A–C), n = 10 mice per group. Intergroup differences were analyzed in a log rank test.
Short-term exposure to Cd salts accentuates the symptoms of acute DSS- and TNBS-induced colitis
The main objective of our study was to mimic realistic environmental exposure conditions and characterize the long-term effects of Cd and Pb salts on susceptibility to colitis. We first sought to determine the effect of a short period of heavy metal exposure on body weight loss (a major inflammation-associated sign). In an initial experiment, a week of daily exposure to either very low-dose (5 μg.kg−1) or high-dose (2 mg.kg−1) CdCl2 was associated with weight loss in mice subsequently exposed to DSS-related (p < 0.01 vs. controls, Fig. 4A). We then assessed the effect of very low-dose (5 μg.kg−1) or high-doses (2 and 10 mg.kg−1) CdCl2 in a model of healing following colitis induced by DSS (Fig. 4B). In DSS control animals (C2), DSS interruption led to a recovery phase with a weight regain. In mice exposed to the low dose 5 μg.kg−1 Cd, body weight recovery was slower and only partial. Mice exposed to the higher doses kept losing weight after DSS cessation until the ethical defined endpoint (i.e. 20%) was reached, thus prompting us to euthanize the animals at day 9.
Figure 4. The effect of short-term oral exposure to various doses of CdCl2 on the development of chemically-induced colitis in murine models.

(A) The change in body weight over the course of the DSS experiment. (B) The change in body weight recorded over the course of the DSS experiment and after replacement with tap water. Data are quoted as the mean ± SEM, n = 8 mice per condition. (C) The change in body weight recorded over the course of the TNBS experiment. (D) Macroscopic evaluation of colonic inflammation (the Wallace score); *p < 0.05, **p < 0.01, *0.05 < p < 0.1, compared with C2 (the colitis control).
When assessing the consequences of 5 days of Cd exposure on the severity of TNBS-induced colitis, only the higher dose (10 mg.kg−1) significantly aggravated the body weight loss (p < 0.05, Fig. 4C) and the score for macroscopic colon injury (p < 0.01, Fig. 4D). The differences with lower doses were not significant (0.05 < p < 0.1). Taken as a whole, these data suggest that acute exposure to Cd had a noticeable impact by exacerbating inflammation and delaying remodeling and tissue repair pathways.
Subchronic exposure to Cd and Pb salts does not have a marked effect on DSS-induced colitis
Ingestion of CdCl2 (5 and 20 ppm, corresponding respectively to 0.5 and 2.5 mg.d−1.kg−1) or PbCl2 (100 ppm, i.e. 12.5 mg.d−1.kg−1) in drinking water for 6 weeks did not significantly impact the main clinical outcomes in mice with DSS-induced colitis, such as body weight loss (Fig. 5A) and SAA protein levels (Fig. 5B). However, despite the absence of obvious changes in the signs of disease, the mice’s external appearance (e.g. the sheen of the fur) and behavior (e.g. searching activity) were clearly better in the metal-exposed groups. The Cd- and Pb-associated relief of colitis symptoms was confirmed histologically, with less epithelial damage in the metal-exposed groups (Fig. 5C). Accordingly, we observed lower transcriptional activity of pro-inflammatory genes (such Il6, Il-β, Tnfa, Nos2 and Cox2) in colon biopsies (Fig. 5D); this suggests that both metals had a clear, positive influence on components of the local inflammatory response. Interestingly, mRNA expression of IL-10 (a regulatory, adaptive cytokine often induced during inflammatory events) was not modified by exposure to heavy metal salts. mRNA expression of the Pparg and Zo-1 genes was unaffected or slightly induced, depending on the dose of heavy metal.
Figure 5. Impact of 6 weeks of oral exposure to CdCl2 or PbCl2 on DSS-induced colitis in mice.

(A) The mean change in body weight over the course of the experiment. (B) Serum levels of SAA protein (in μg.mL−1). (C) Representative photomicrographs of the histological features of May-Grünwald-Giemsa-stained colon sections (bar = 200 μm). (D) mRNA expression levels of specific genes in the colon, relative to that of Actb. Data are quoted as the mean ± SEM, n = 8 mice per condition; *p < 0.05, **p < 0.01, ***p < 0.001, *0.05 < p < 0.1, compared with C2 (the colitis control).
Subchronic exposure to Cd and Pb salts has protective effects in TNBS-induced colitis.
Six weeks of oral exposure of CdCl2 (20 ppm and 100 ppm) and PbCl2 (100 ppm) in drinking water was associated with significantly lower TNBS-induced colonic damage in terms of body weight loss (Fig. 6A), macroscopic lesion (Fig. 6B), histological scores (Fig. 6C) and necrosis and epithelial loss (Fig. 6D). Levels of general markers of inflammation were also drastically lower following long-term exposure to Cd and/or Pb (Fig. 6E,F). These observations were consistent with the transcriptional signatures in inflamed colons (Fig. 6G) (although dependent on the doses and the metal in question). Indeed, the TNBS-induced upregulation of inflammatory genes (such as the immune-related Il6, Il1b and Tnfa and oxidative-stress-related genes (such as Nos2 and Cox2) was less marked or down-regulated in metal-exposed groups. We also observed greater induction of transcripts of Foxo4, an endogenous inhibitor of NfκB17 that is downregulated by TNBS. The expression of certain genes related to remodeling and anti-inflammatory or anti-oxidant functions are sometimes actively induced in an inflammatory context and thus reflect the adaptive response of the mucosa. This is the case for Tgfb (which was upregulated by both Cd and Pb), whereas Il10, Hmox1 and the metallothioneins (MTs) Mt1 and Mt2 were less expressed in metal-exposed animals with inflammation.
Figure 6. Impact of 6 weeks of oral exposure to CdCl2 or PbCl2 on TNBS-induced colitis in mice.

(A) The mean change in body weight over the course of the experiment. (B) Macroscopic evaluation of colonic inflammation (the Wallace score). (C) Histopathological evaluation of colonic inflammation (the Ameho score). (D) Representative photomicrographs of histological features of May-Grünwald-Giemsa-stained colon sections (bar: 100 μm). (E,F) Serum levels of IL-6 and SAA protein. (G) mRNA expression levels of specific genes in the colon, relative to that of Actb. Data are quoted as the mean ± SEM, n = 10 mice per condition; *p < 0.05, **p < 0.01, ***p < 0.001, *0.05 < p < 0.1, compared with C2 (the colitis control).
Discussion
It is well known that both short-term and chronic oral exposure to heavy metals can induce (i) breaches in the gut barrier, (ii) local and systemic immunosuppressive effects and (iii) the genesis of a pro-oxidant environment. This context may contribute to create “the perfect storm” to favor chronic intestinal disease. However, our present results show that subchronic exposure to Cd or Pb salts in drinking water may have a beneficial effect in mice by mediating protection against experimentally-induced acute colitis. This phenomenon may involve several factors inside and outside the intestine.
At baseline (without colitis), it is known that Cd induces substantial levels of MTs in all parts of the small intestine and in the colon15, but this cannot be sufficient to explain all the anti-inflammatory effects (in terms of threshold or time of impregnation). We thus hypothesized that adaptive processes are also involved. Based on our previous experiments15, we can state that no induction of SAA neither IL-6 was induced per se by the metals and thus, we cannot claim that Cd or Pb salts are pro-inflammatory.
We first confirmed that Cd (but not Pb) altered the TEER in an in vitro reconstituted epithelium system. Our findings also suggest that Cd might be associated with a reversible reduction in epithelial permeability in vivo. Furthermore, Cd and Pb may lower immune responsiveness because both heavy metals reduced bacteria-elicited cytokine release (but not baseline release). Given that pro-and anti-inflammatory pathways were affected in similar ways, it is hard to predict whether the inflammatory response will be exacerbated or dampened. However, Cd- and Pb-exposed mice were no more susceptible than controls to S. typhimurium, suggesting that the metals only have a moderate impact on intestinal innate immunity in vivo. Nevertheless, these harmful effects may account for the exacerbation of experimentally induced colitis observed in mice after short-term exposure. The alleviation of colitis after longer, continuous exposure to Cd and Pb may involve other mechanisms.
Maintaining homeostasis of the intestinal mucosa involves adaptive, counter-regulatory processes that might be preferentially induced by longer exposure to toxic heavy metals. Boirivant et al.18 showed that breaches in the intestinal barrier (caused by ethanol and another tight junction disrupter) are necessary for efficient, protective homeostatic regulatory T-cell responses to mucosal inflammation. Likewise, it has been reported that superoxide and NO have divergent roles in intestinal inflammation19, and that physiologic reactive oxygen species (ROS) signaling regulates homeostatic processes20. It has also be established that exogenous bacteria can prevent intestinal NF-kB activation by increasing epithelial levels of ROS21, whereas lactobacilli use H2O2 to activate PPARg in epithelial cells and thus modulate inflammation22. Although massive oxidative stress contributes to failure of the intestinal barrier, a moderate increase in intracellular ROS concentrations paradoxically affords protection and prevents intestinal barrier dysfunction via the upregulation of oxidative defense mechanisms23. We recently investigated the impact of subchronic Pb and Cd exposure on gut ecology, with a view to developing a more comprehensive view of the consequences of environmental exposure at baseline (i.e. before the induction of colitis)14,15,16. We found that the mRNA expression of inducible enzymes coded by genes such as Nos2 and Gpx2 did not change significantly (or was even somewhat lower) in the colon of metal-exposed mice. In contrast, transcription of Mt1 and Mt2 in the duodenum and the colon was upregulated. Furthermore, a significant, consistent rise in the levels of Cyp1a1 mRNA was measured in all parts of the intestine of exposed animals, and Hmox1 was clearly induced in the distal ileum. All of these genes (either separately or conjointly) may contribute to increased levels of anti-oxidants and may thus further limit colitis. Heavy-metal-induced genes have multiple functions, and - at least in the gut - chronic contamination by xenobiotics may paradoxically exert some beneficial or protective features. Indeed, the MTs are known to exert anti-inflammatory effects both inside the gut24,25 and outside the gut26,27 and to protect against rheumatoid arthritis and colitis28. Several mechanisms are involved, including oxygen radical scavenging and immunomodulation. However, the MTs’ role during disease onset and progression in animal models of colitis and IBD is not yet clear29. Similarly, activation of the aryl hydrocarbon receptor (AhR) pathway (induced in colitis, along with Cyp1a1) ameliorates DSS-induced colitis in mice30,31. Other examples of the modulation of mucosal immune responses by environmental factors involve the AhR and the estrogen receptor32,33; this reinforces evidence in favor of an estrogen-like role of Cd in the intestine34 and related anti-inflammatory role in the gut35. There are abundant literature data on the protective role of Hmox1 in clinical and experimental colitis36,37,38 and mechanisms might also involve Cd-induced protective autophagy39. In the context of the present study, specific gene-by-gene inactivation could be used to determine the respective roles in Cd-mediated protection but one can legitimately hypothesize that several genes act together.
The low levels of colonic and blood iron associated with the chronic ingestion of Cd14 might also partly explain the Cd-associated alleviation of colitis – perhaps via a drop in oxidative stress and other indirect mechanisms40,41.
Lastly, specific modification of the gut microbiota might also account for how heavy metals can protect against or promote colitis1,9,42. Our previous studies have shown that Cd and Pb can affect the structure of microbiome and change the bacterial community, leading among other things to a lower proportion of Lachnospiracea and a higher proportion of Turicibacter16. We hypothesize that adaptation of the microbiota to a heavy-metal environment drives the differential responses observed in the present study. For example, Turicibacter have been detected in the ileal pouch of a patient with ulcerative colitis43 and in human appendicitis44, whereas low levels of this genus were observed in dogs with idiopathic IBD45. Interestingly, high levels of Turicibacter were observed in colitis-resistant CD8 knock-out mice, where the genus is potentially involved in the anti-inflammatory phenotype46. Furthermore, the fact that IBD is associated with a reduction in the diversity of taxonomic clusters and specific groups (i.e., Lachnospiraceae) suggests that bacterial groups might have an important role in gastrointestinal health. However, further research is required to evaluate the functional changes associated with this type of intestinal dysbiosis. Although human IBD and experimental colitis in mice are clearly associated with specific shifts in intestinal microbiota composition, the extent to which these microbiota dynamics are indicative of health or disease is not clear47. Oral exposure to Cd and Pb may generate bidirectional, adaptive interactions between the host and the microbiota. Again, we are not yet able to discriminate between causes and causality or to state that inflammation-associated enterotypes or strains can promote or protect against colitis. It remains difficult to differentiate between the effects of isolated host factors, microbial factors and host-microbiota interactions on intestinal homeostasis and toxicological responsiveness.
In line with our present results, Ansari et al.48 showed that 3 weeks of exposure to low-dose CdCl2 (5 ppm) during the initiation phase of a collagen-induced arthritis model in rats reduced disease progression, whereas exposure to a higher dose (50 ppm) exacerbated the severity. This recent work confirms that Cd salts potentially have a collateral, beneficial effect on a chronic inflammatory disease through stimulation of anti-oxidant pathways and downregulation of NfκB and pro-inflammatory cytokine pathways. Beneficial effects for other metals of health concern have already been reported, although they depend on element speciation and administration route. In mice, for example, sodium arsenite was associated with a reduction in the severity of DSS-induced experimental colitis49 and arsenic trioxide had anti-inflammatory consequences in TNBS-induced colitis50. Likewise, preclinical studies using cobalt51, lithium52, strontium53 and uranium54 also found a clear anti-inflammatory potential. Lastly, some preliminary data strongly suggest that mice exposed to methylmercury for 3 weeks are resistant to DSS-associated inflammation (Body-Malapel and Vignal, unpublished data). Taken as a whole, these data reinforce the notion whereby specific doses and durations of exposure to dietary or contaminant heavy metals may variously elicit neutral, pro- or anti-inflammatory phenomena via a number of interrelated pathways.
Lastly, some of the potentially “positive” side effects of heavy metals based on the host’s intestinal adaptome might not underestimate toxicogenic effects at other sites (such as nephrotoxicity) or harmful long-term consequences (such as colon tumorigenesis).
Conclusion
Our present observations provide insights into the Cd- and Pb-mediated modulation of inflammatory processes and emphasize that clinical outcomes vary considerably as a function of dose and exposure time. This may also apply to other xenobiotics involved in the onset of IBD, especially when the body is exposed to mixtures of metals or other dietary and environmental risk factors. Several factors can influence the subtle pro-/anti-oxidant balance and subsequent immune responses by either disrupting or (as a sort of “collateral benefit”) reinforcing homeostatic mechanisms. Additional epidemiological and toxicological studies are needed to determine the integrated role of the environmental exposome in gut ecotoxicology and thus identify risk factors, key regulators and novel therapeutic approaches in IBD and other immune-related disorders. This research must take account of the host’s complex responses and interplay between (i) the exposome, (ii) the adaptome and (iii) the emerging techniques in microbiome science (including metagenomics and metabolomics), in order to better manage environmental factors for human health.
Methods
Materials
Chemicals and reagents were purchased from Sigma-Aldrich Chemical (St Quentin Fallavier, France), unless otherwise stated.
Cell lines and in vitro transepithelial electric resistance (TEER) measurements
Human intestinal epithelial HTB37 (Caco-2) and CCL248 (T84) cells (purchased from the American Type Tissue Collection) were seeded (2.105 cells.cm−2) on Transwell polycarbonate cell culture inserts with pore size of 3 μm (Corning Costar) and cultivated for up to 21 days in appropriate media. TEER was measured using the Millicel-ERS epithelial volt-ohmmeter (Millipore) and expressed in Ω.cm−2 as the mean of triplicate wells. Subtoxic doses (as previously determined in MTT cell viability assays) of CdCl2 (20 μM and 68 μM) and PbCl2 (1000 μM) were applied to the apical compartments. TEER was monitored for 24 h and then (after intensive washing of the cells) for a further 48 h.
In vitro immunomodulation assays
Peripheral blood mononuclear cells (PBMCs) were isolated from the blood of four healthy donors as already described55. Nontoxic doses of CdCl2 (final concentration: 20 or 68 μM) or PbCl2 (100 μM) were added in the absence or presence of 10 μl of thawed bacterial suspensions (in 20% glycerol in PBS, with a bacterium-to-cell ratio of approximately 10:1). Cytokine release (human IL-10 and IL-12p70) was measured in an ELISA using pairs of antibodies (BD PharmingenTM). All experimental protocols were approved by our institution committees (INSERM, CNRS and Institut Pasteur de Lille). Blood sampling from healthy informed donors were done upon approved agreement of volunteers (signed consents) by authorized staff in accordance with the abovementioned committees, i.e. (INSERM, CNRS and Institut Pasteur de Lille).
Animal experiments and ethics statements
Animal experiments were performed in accordance with the guidelines issued by the Institut Pasteur de Lille’s Animal Care and Use Committee, which are based on the Amsterdam Protocol on Animal Protection and Welfare and Directive 86/609/EEC on the Protection of Animals Used for Experimental and Other Scientific Purposes, updated in the Council of Europe’s Appendix A. The animal experiments also complied with French legislation (Government Act 87-848) and the European Communities Amendment of Cruelty to Animals Act 1976. All the studies were approved by the local investigational ethics review board (Nord-Pas-de-Calais CEEA N°75, Lille, France; protocol reference numbers 192009R, 212009R, 222009R and 042011).
Short-term exposure (1 week) was performed by daily oral gavage with 5 μg.kg−1, 2 mg.kg−1 or 10 mg.kg−1 of CdCl2. Subchronic exposure consisted of 1, 4 or 6 weeks of ad libitum access to contaminated drinking water containing 20 or 100 ppm CdCl2 (corresponding to daily exposures of 2.5 and 12.5 mg.kg−1, respectively, based on a daily intake of 2.5 ml) or 100 ppm of PbCl2 (corresponding to 12.5 mg.kg−1 daily). Indeed, we retained these two ranges of optimal doses respectively, as the modulation of inflammation does not require the same intensity between each model, i.e. DSS and TNBS, based on preliminary studies (data not shown).
Salmonella typhimurium challenges
Ten- to twelve-week-old female BALB/c mice were intragastrically challenged with 5 × 105 CFU (200 μL) of an exponential culture of Salmonella enterica serovar Typhimurium strain C556 grown in LB broth and resuspended in distilled water. Animals were kept in positive-pressure cabinets, and mortality was monitored daily over a three-week period, as previously described57.
Induction of colitis with dextran sodium sulfate (DSS) and trinitrobenzene sulfonic acid (TNBS)
DSS and TNBS chemically induce colitis models of gut inflammation, sharing common features but also exhibiting specific traits. Since a single model of colitis does not resemble all the features of human IBD, we used two different models, namely trinitrobenzenesulfonic acid (TNBS)- and dextran sulphate sodium (DSS)-induced colitis, to strengthen the value of our findings. Therefore, these two models are thought to be reliable and rather complementary when studying the pathogenesis of IBD, and in this particular case, the overall inflammation inside the gut, as a general deleterious inflammatory context in rodents. Two models of chemically-induced acute colitis were implemented in 12-week-old female BALB/c, as previously described58. For the DSS model, mice (n = 8 per group) were exposed to 5% DSS molecular weight 36–50 kDa (MP Biomedicals) in their drinking water for seven consecutive days prior to necropsy. For the TNBS model (n = 10 mice per group), the colitis was triggered by the intrarectal administration of 50 μl TNBS (100 mg.kg−1) in 0.9% NaCl/ethanol (50/50 v/v). Three days after the induction of colitis, mice were euthanized and blood samples were collected immediately. The serum was separated and frozen (−20 °C). After dissection, two independent observers blindly scored the macroscopic inflammation of the colon by using the Wallace score59. The Wallace score rates macroscopic lesions on a scale from 0 to 10 based on features reflecting inflammation, such as hyperaemia (score 1 to 2), moderate to intense thickening of the bowel (score 2 to 3), and the gradual extent of 1 cm-long ulceration (score 3 to 7), according to the maximal mouse colon length. Dead mice due to over-inflammation were scored at 10, the maximal inflammatory score seen in mouse models. Following examination under microscope, tissue lesions were scored according to the Ameho criteria60. Briefly, histological findings identical to those of normal mice were scored as 0, whereas a score of 1 indicated mild mucosal and/or submucosal inflammatory infiltrate and edema, punctate mucosal erosions, and intact muscularis mucosae; the same histological findings involving 50% of the specimen were scored as 2. Prominent inflammatory infiltrate and edema, deeper areas of ulceration extending through the muscularis mucosae into the submucosa, and rare inflammatory cells invading the muscularis propria but without muscle necrosis were scored as 3, the same histological findings involving 50% of the specimen were scored as 4, extensive ulceration with coagulative necrosis with deep extension of the necrosis into the muscularis propria were scored as 5, and the same histological findings involving 50% of the specimen were scored as 6. The Wallace and Ameho scoring systems used here are the most appropriate for the TNBS model; they both report a gradual analysis of the lesions. However, they cannot be used to score the diffuse and patchy lesions occurring in DSS. Indeed, the body weight loss appears for us to be the most relevant clinical marker/symptom of inflammation as previously described58 and we decided to retain this main marker as a representative of the pathology.
For both models, distal colon samples (0.5 cm in length) were processed with RNA stabilization solution (RNA-later, Ambion, Life Technologies) and stored at −80 °C for transcriptional analysis. Samples for histologic analysis were processed for paraffin-embedding, prepared as 5 μm sections and stained with May Grünwald-Giemsa reagent to score tissue lesions according to the Ameho criteria. Blood levels of murine IL-6 and serum amyloid A (SAA) protein were measured using ELISA kits (BD Pharmingen) and (Biosource International) respectively.
Quantitative reverse-transcriptase polymerase chain reaction (RT-PCR)
Samples were homogenized using the FastPrep instrument (MP Biomedicals), total RNA was isolated using RNAspin columns (Macherey-Nagel). Reverse transcription and real-time PCR were performed with reaction kits (the High-Capacity cDNA RT Kit) and reagents (Universal PCR Master Mix, Applied Biosystems), according to the manufacturer’s instructions. PCR reactions were performed with a MX3005P machine (Stratagene, Agilent Technologies). A custom gene expression assay (TaqMan, Applied Biosystems) was used with commercially designed and validated primers (Table S1). The housekeeping gene beta actin was run as an internal control. Data were analyzed using the 2−ΔΔCt method and expressed as a fold-increase over the control group’s values.
Statistical analysis
All analyses were performed by comparing experimental groups with their respective controls in a non-parametric, one–way analysis of variance (the Mann–Whitney U-test) or a two-tailed Student’s-t-test, as appropriate (version 6.0, GraphPad Software Inc.). Survival rates after Salmonella challenge were analyzed using the log rank test. Data are presented as the mean ± SEM. Differences were judged to be statistically significant when the p-value was <0.05. However, p-values between 0.05 and 0.1 are specified to indicate trends.
Additional Information
How to cite this article: Breton, J. et al. Does oral exposure to cadmium and lead mediate susceptibility to colitis? The dark-and-bright sides of heavy metals in gut ecology. Sci. Rep. 6, 19200; doi: 10.1038/srep19200 (2016).
Supplementary Material
Acknowledgments
The present study was funded by the French National Research Agency (grant ANR-09-CES-016 Mélodie-Reve). The Institut Pasteur de Lille receives funding from the Nord Pas de Calais Regional Council. We thank Joëlle Dewulf and Ademar Pasamoto for technical assistances.
Footnotes
Author Contributions B.F. and J.B. conceived, designed and performed the experiments, and analyzed the data and wrote the paper. C.P., C.D., C.V., M.B. and A.G. contributed to animal experiments, helped with data analysis and proofed the paper.
References
- Wang M. H. & Achkar J. P. Gene-environment interactions in inflammatory bowel disease pathogenesis. Curr. Opin. Gastroenterol. 31, 277–282 (2015). [DOI] [PubMed] [Google Scholar]
- Ruel J., Ruane D., Mehandru S., Gower-Rousseau C. & Colombel J. F. IBD across the age spectrum: is it the same disease? Nat. Rev. Gastroenterol. Hepatol. 11, 88–98 (2014). [DOI] [PubMed] [Google Scholar]
- Rappaport S. M. & Smith M. T. Epidemiology. Environment and disease risks. Science 330, 460–461 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy P. J. & Rappaport S. M. Exposure science and the exposome: an opportunity for coherence in the environmental health sciences. Environ. Health. Perspect. 119, A466–467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S. C. et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 62, 630–649 (2013). [DOI] [PubMed] [Google Scholar]
- Rogler G. & Vavricka S. Exposome in IBD: recent insights in environmental factors that influence the onset and course of IBD. Inflam. Bowel. Dis. 21, 400–408 (2015). [DOI] [PubMed] [Google Scholar]
- Steenland K., Zhao L., Winquist A. & Parks C. Ulcerative colitis and perfluorooctanoic acid (PFOA) in a highly exposed population of community residents and workers in the mid-Ohio valley. Environl. Health. Perspect. 121, 900–905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner A. Aluminum is a potential environmental factor for Crohn’s disease induction: extended hypothesis. Ann. N. Y. Acad. Sci. 1107, 329–345 (2007) [DOI] [PubMed] [Google Scholar]
- Pineton de Chambrun G. et al. Aluminum enhances inflammation and decreases mucosal healing in experimental colitis in mice. Mucosal. Immunol. 7, 589–601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarup L. & Akesson A. Current status of cadmium as an environmental health problem. Toxicol. Appl. Pharmacol. 238, 201–208 (2009). [DOI] [PubMed] [Google Scholar]
- Skerfving S. & Bergdahl I. A. Lead. Handbook on the Toxicology of Metals (Third Edition), Chapter 31, 599–643 (2007). [Google Scholar]
- Satarug S., Garrett S. H., Sens M. A. & Sens D. A. Cadmium, environmental exposure, and health outcomes. Environ. Health. Perspect. 118, 182–190 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalups R. K. & Ahmad S. Molecular handling of cadmium in transporting epithelia. Toxicol. Appl. Pharmacol. 186, 163–188 (2003). [DOI] [PubMed] [Google Scholar]
- Breton J. et al. Gut microbiota limits heavy metals burden caused by chronic oral exposure. Toxicol. Lett. 222, 132–138 (2013). [DOI] [PubMed] [Google Scholar]
- Breton J. et al. Chronic ingestion of cadmium and lead alters the bioavailability of essential and heavy metals, gene expression pathways and genotoxicity in mouse intestine. Arch. Toxicol. 87, 1787–1795 (2013). [DOI] [PubMed] [Google Scholar]
- Breton J. et al. Ecotoxicology inside the gut: impact of heavy metals on the mouse microbiome. BMC Pharmacol. Toxicol. 14, 62 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W. et al. FoxO4 inhibits NF-kappaB and protects mice against colonic injury and inflammation. Gastroenterology 137, 1403–1414 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boirivant M. et al. A transient breach in the epithelial barrier leads to regulatory T-cell generation and resistance to experimental colitis. Gastroenterology 135, 1612–1623 (2008). [DOI] [PubMed] [Google Scholar]
- Krieglstein C. F. et al. Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: divergent roles of superoxide and nitric oxide. J. Exp. Med. 194, 1207–1218 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veal E. A., Day A. M. & Morgan B. A. Hydrogen peroxide sensing and signaling. Mol. Cell 26, 1–14 (2007). [DOI] [PubMed] [Google Scholar]
- Lin P. W. et al. Lactobacillus rhamnosus blocks inflammatory signaling in vivo via reactive oxygen species generation. Free Radic. Biol. Med. 47, 1205–1211 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voltan S. et al. Lactobacillus crispatus M247-derived H2O2 acts as a signal transducing molecule activating peroxisome proliferator activated receptor-gamma in the intestinal mucosa. Gastroenterology 135, 1216–1227 (2008). [DOI] [PubMed] [Google Scholar]
- Lutgendorff F. et al. Probiotics prevent intestinal barrier dysfunction in acute pancreatitis in rats via induction of ileal mucosal glutathione biosynthesis. PloS One 4, e4512 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano H. et al. Protective role of metallothionein in acute lung injury induced by bacterial endotoxin. Thorax 59, 1057–1062 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita M. et al. Metallothionein is a crucial protective factor against Helicobacter pylori-induced gastric erosive lesions in a mouse model. Am. J. Physiol. Gastrointest. Liver. Physiol. 294, 877–884 (2008). [DOI] [PubMed] [Google Scholar]
- Takano H. et al. Cytoprotection by metallothionein against gastroduodenal mucosal injury caused by ethanol in mice. Lab. Invest. 80, 371–377 (2000). [DOI] [PubMed] [Google Scholar]
- Ashino T. et al. Effect of interleukin-6 neutralization on CYP3A11 and metallothionein-1/2 expressions in arthritic mouse liver. Eur. J. Pharmacol. 558, 199–207 (2007). [DOI] [PubMed] [Google Scholar]
- Tsuji T. et al. Role of metallothionein in murine experimental colitis. Int. J. Mol. Med. 31, 1037–46 (2013). [DOI] [PubMed] [Google Scholar]
- Devisscher L. et al. Role of metallothioneins as danger signals in the pathogenesis of colitis. Journal Pathol. 233, 89–100 (2014). [DOI] [PubMed] [Google Scholar]
- Takamura T. et al. Activation of the aryl hydrocarbon receptor pathway may ameliorate dextran sodium sulfate-induced colitis in mice. Immunol. Cell. Biol. 88, 685–689 (2010). [DOI] [PubMed] [Google Scholar]
- Furumatsu K. et al. A role of the aryl hydrocarbon receptor in attenuation of colitis. Dig. Dis. Sci. 56, 2532–2544 (2011). [DOI] [PubMed] [Google Scholar]
- Kluxen F. M., Diel P., Hofer N., Becker E. & Degen G. H. The metallohormone cadmium modulates AhR-associated gene expression in the small intestine of rats similar to ethinyl-estradiol. Arch. Toxicol. 87, 633–643 (2013). [DOI] [PubMed] [Google Scholar]
- Kluxen F. M., Hofer N., Kretzschmar G., Degen G. H. & Diel P. Cadmium modulates expression of aryl hydrocarbon receptor-associated genes in rat uterus by interaction with the estrogen receptor. Arch. Toxicol. 86, 591–601 (2012). [DOI] [PubMed] [Google Scholar]
- Hofer N. et al. Investigations on the estrogenic activity of the metallohormone cadmium in the rat intestine. Arch. Toxicol. 84, 541–552 (2010). [DOI] [PubMed] [Google Scholar]
- Verdu E. F., Deng Y., Bercik P. & Collins S. M. Modulatory effects of estrogen in two murine models of experimental colitis. Am. J. Physiol Gastrointest. Liv. Physiol. 283, 27–36 (2002). [DOI] [PubMed] [Google Scholar]
- Paul G. et al. Analysis of intestinal haem-oxygenase-1 (HO-1) in clinical and experimental colitis. Clin. Exp. Immunol. 140, 547–555 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga C. et al. Modulation by heme and zinc protoporphyrin of colonic heme oxygenase-1 and experimental inflammatory bowel disease in the rat. Eur. J. Pharmacol. 561, 164–171 (2007). [DOI] [PubMed] [Google Scholar]
- Chang M., Xue J., Sharma V. & Habtezion A. Protective role of hemeoxygenase-1 in gastrointestinal diseases. Cell. Molecular Life Sci. 72, 1161–1173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surolia R. et al. Heme oxygenase-1 mediated autophagy protects against pulmonary endothelial cell death and development of emphysema in cadmium treated mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, 280–92 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharshak N. et al. Escherichia coli heme oxygenase modulates host innate immune responses. Microbiol. Immunol. 59, 452–65 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua A. C. et al. Dietary iron enhances colonic inflammation and IL-6/IL-11-Stat3 signaling promoting colonic tumor development in mice. PloS One 8, e78850 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K. et al. Arsenic exposure perturbs the gut microbiome and its metabolic profile in mice: an integrated metagenomics and metabolomics analysis. Environ. Health. Perspect. 122, 28491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk A. et al. Ileal pelvic pouch microbiota from two former ulcerative colitis patients, analysed by DNA-based methods, were unstable over time and showed the presence of Clostridium perfringens. Scand. J. of Gastroenterol. 42, 973–985 (2007). [DOI] [PubMed] [Google Scholar]
- Bosshard P. P., Zbinden R. & Altwegg M. Turicibacter sanguinis gen. nov., sp. nov., a novel anaerobic, Gram-positive bacterium. Int. J. Syst. Evol. Microbiol. 52, 1263–1266 (2002). [DOI] [PubMed] [Google Scholar]
- Suchodolski J. S. et al. The fecal microbiome in dogs with acute diarrhea and idiopathic inflammatory bowel disease. PloS One 7, e51907 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley L. L Wei B., Braun J. & Borneman J. Bacteria associated with immunoregulatory cells in mice. Appl. Environ. Microbiol. 76, 936–941 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry D. et al. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J. 6, 2091–2106 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari M. M., Neha & Khan H. A. Effect of cadmium chloride exposure during the induction of collagen induced arthritis. Chem. Biological Interact. 238, 55–65 (2015). [DOI] [PubMed] [Google Scholar]
- Malago J. J. & Nondoli H. Sodium arsenite reduces severity of dextran sulfate sodium-induced ulcerative colitis in rats. J. Zhejiang Univ. Sci. B 9, 341–350 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M., Trugnan G. & Chelbi-Alix M. K. Arsenic trioxide reduces 2,4,6-trinitrobenzene sulfonic acid-induced murine colitis via nuclear factor-kappaB down-regulation and caspase-3 activation. Innate Immun. 17, 365–374 (2011). [DOI] [PubMed] [Google Scholar]
- Oh S. W. et al. Cobalt chloride attenuates oxidative stress and inflammation through NF-kappaB inhibition in human renal proximal tubular epithelial cells. J. Korean Med. Sci. 29, S139–145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneshmand A. et al. Chronic lithium administration ameliorates 2,4,6-trinitrobenzene sulfonic acid-induced colitis in rats; potential role for adenosine triphosphate sensitive potassium channels. J. Gastroenterol. Hepatol. 26, 1174–1181 (2011). [DOI] [PubMed] [Google Scholar]
- Topal F. et al. Strontium chloride: can it be a new treatment option for ulcerative colitis? BioMed. Res. Int. 2014, 530687 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dublineau I. et al. Modifications of inflammatory pathways in rat intestine following chronic ingestion of depleted uranium. Toxicol. Sci. 98, 458–468 (2007). [DOI] [PubMed] [Google Scholar]
- Foligne B. et al. Correlation between in vitro and in vivo immunomodulatory properties of lactic acid bacteria. World J. Gastroenterol. 13, 236–243 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormaeche C. E. Natural resistance to Salmonella typhimurium in different inbred mouse strains. Immunology 37, 311–318 (1979). [PMC free article] [PubMed] [Google Scholar]
- Zoumpopoulou G. et al. Lactobacillus fermentum ACA-DC 179 displays probiotic potential in vitro and protects against trinitrobenzene sulfonic acid (TNBS)-induced colitis and Salmonella infection in murine models. Int. J. Food Microbiol. 121, 18–26 (2008). [DOI] [PubMed] [Google Scholar]
- Foligne B. et al. Prevention and treatment of colitis with Lactococcus lactis secreting the immunomodulatory Yersinia LcrV protein. Gastroenterology 133, 862–874 (2007). [DOI] [PubMed] [Google Scholar]
- Wallace J. L. et al. Inhibition of leukotriene synthesis markedly accelerates healing in a rat model of inflammatory bowel disease. Gastroenterology. 96, 29–36 (1989). [DOI] [PubMed] [Google Scholar]
- Ameho C., Adjei A. A., Harrison E. K. et al. Prophylactic effect of dietary glutamine supplementation on interleukin 8 and tumour necrosis factor alpha production in trinitrobenzene sulphonic acid induced colitis. Gut 41, 487–493 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.