

Abstract
The purpose of this study was to examine the toxicity of combining oncolytic adenovirus-mediated cytotoxic and interleukin 12 (IL-12) gene therapy in a preclinical model to support future phase 1 trials. One hundred and twenty C57BL/6 male mice received an intraprostatic injection of saline (n = 24) or an oncolytic adenovirus (Ad5-yCD/mutTKSR39rep-mIL12) expressing two suicide genes and mouse IL-12 (n = 96). The adenovirus was administered at three dose levels (1.3 × 106, 1.3 × 107, 1.3 × 108 vp/kg) followed by 2 weeks of 5-flurocytosine (5-FC) and gancliclovir (GCV) prodrug therapy. There were no premature deaths. Daily observations of animals did not reveal any obvious clinical problems throughout the 78-day in-life phase of the study. Animals in the highest adenovirus dose group exhibited lymphopenia and transaminitis on day 3, both of which resolved by day 17. Except for mild inflammation of the prostate and seminal vesicles, histopathology of major organs was largely unremarkable. IL-12 and interferon-gamma levels in prostate and serum peaked on day 3 and were either undetectable or returned to baseline levels by day 17. No adenoviral DNA was detected in serum in any group at any time point. The results demonstrate that local administration of an oncolytic adenovirus expressing two suicide genes and IL-12 is well tolerated and support moving this investigational approach into human trials.
Introduction
Oncolytic viral therapy is an investigational cancer treatment that has been evaluated in multiple disease settings using a variety of viral platforms including adenovirus, herpesvirus, measles virus, poxvirus, and others.1 Based on hundreds of patients treated to date, this investigational approach has demonstrated a good overall safety profile and some strategies have shown signs of efficacy in randomized trials. Despite these encouraging results, it is uncertain whether oncolytic viral therapy is sufficiently robust to be efficacious when applied as a monotherapy. A major limitation is that viruses are immunogenic, and the resulting immune response restricts viral persistence and spread, as well as the effectiveness of repeated administrations. Although this limitation is desirable from a safety standpoint, it may reduce treatment efficacy depending on whether the immune response contributes to, or detracts from, antitumor activity overall. Hence, it may be necessary to arm such viruses with therapeutic genes or combine them with standard cancer treatments, or both, to push this promising strategy over the therapeutic threshold.
For the past 20 years, we have been developing a multimodal approach that utilizes an oncolytic adenovirus platform armed with two cytotoxic genes.2–12 We have evaluated the safety and potential efficacy of this approach in five clinical trials of prostate cancer. The approach has proven to be safe when applied alone or in combination with radiotherapy and antitumor activity has been demonstrated in multiple disease settings. In a randomized phase 2 trial, combining oncolytic adenovirus-mediated cytotoxic gene therapy with contemporary dose radiotherapy resulted in a 42% relative improvement in local tumor control without diminishing the patient’s quality of life.10 These encouraging results, and others,6,12 lead us to believe that further refinement of this approach may ultimately lead to a successful treatment strategy.
Although local tumor control is important, new cancer therapies must also target metastatic disease if they are to have an impact on survival. Hence, we recently incorporated interleukin 12 (IL-12) into our therapeutic platform.13 IL-12 is a proinflammatory cytokine produced by antigen presenting cells that stimulates the innate and adaptive immune response, inhibits angiogenesis, and reverses the immune suppressive nature of the tumor milieu.14–16 Although IL-12 has exhibited significant antitumor activity in preclinical models, its performance in the clinic has been modest largely because of severe toxicity when administered systemically.17 Local administration of IL-12 may circumvent systemic toxicity without diminishing its therapeutic potential. In preparation for a phase 1 trial, we examined the toxicity of administering IL-12 intraprostatically via an oncolytic, replication-competent adenovirus.
Results
Study design
One hundred and twenty C57BL/6 male mice received an injection of saline (n = 24) or an oncolytic adenovirus (Ad5-yCD/mutTKSR39rep-mIL12) expressing two suicide genes and mouse IL-12 (n = 96). The adenovirus was injected intraprostatically at three dose levels (1.36 × 106, 1.36 × 107, 1.36 × 108 vp/kg) mimicking, on a weight basis, those to be used in future clinical trials (Table 1). The adenovirus was also injected intravenously at the highest dose level to model a worst case scenario. Two days after the adenovirus injection, animals were administered 5-flurocytosine (5-FC) + gancliclovir (GCV) prodrug therapy for 2 weeks (Figure 1). Animals were observed daily and body weights were recorded once per week. Animals (eight per group) were sacrificed at three time points representing an early time point (day 3), 1 day after completion of the prodrug therapy course (day 17), and a long-term endpoint (day 78). The following toxicological parameters were examined: complete blood counts (CBCs), liver function tests (LFT), and histopathology of 12 major organs (brain, heart, lungs, liver, spleen, kidneys, small intestines, large intestines including rectum, urinary bladder, prostate and seminal vesicles, and bone marrow) (five animals/group/time point). IL-12, and its major mediator, interferon gamma (IFN-γ), were quantified in the injected tissue and serum as was adenoviral DNA (three animals/group/time point).
Table 1. Study cohorts.
| Group | Number | Route of administration | Adenovirus dose vp (vp/kg) | Human equivalent dosea vp (vp/kg) |
|---|---|---|---|---|
| 1 | 24 | Intraprostatic | 3 × 106 (1.28 × 108) | 1 × 1010 (1.25 × 108) |
| 2 | 24 | Intraprostatic | 3 × 107 (1.28 × 109) | 1 × 1011 (1.25 × 109) |
| 3 | 24 | Intraprostatic | 3 × 108 (1.28 × 1010) | 1 × 1012 (1.25 × 1010) |
| 4 | 24 | Intravenous | 3 × 108 (1.28 × 1010) | 1 × 1012 (1.25 × 1010) |
| 5 | 24 | Intraprostatic | Saline | NA |
Assuming 80 kg (175 lbs.) human. Mean weight of 120 mice at the time of adenovirus injection was 23.4 g.
Figure 1.
Study design. Mice received an injection of saline or Ad5-yCD/mutTKSR39rep-mIL12 on day 1 as described in the text. Animals that were administered the adenovirus also received 5-FC + GCV prodrug therapy for 2 weeks beginning on day 3. Eight animals in each group were sacrificed on days 3, 17, and 78 and CBC, LFT, and histopathology of 12 major organs were examined (five mice/group/time point). IL-12, IFN-γ and adenoviral DNA were quantified in the injected tissue and serum (three mice/group/time point).
General observations
There were no premature deaths. Daily observation of animals did not reveal any obvious clinical problems throughout the 78-day in-life phase of the study. Body weights increased steadily over time for all groups and there was no statistical difference in body weights among the groups at any point of the study (data not shown).
CBC and LFT
CBC and LFT are shown in Figure 2 and the results are summarized in Table 2. Except for lymphocytes (LYMP), alanine aminotransferase (ALT), alkaline phosphatase (ALKP), and albumin (ALB), the blood chemistries of the investigational groups (groups 1–4) did not differ significantly from controls (group 5). On day 3, group 3 (highest Ad dose, intraprostatic) and group 4 (highest Ad dose, intravenous) exhibited lymphopenia. Group 4 also exhibited transaminitis (ALT). These events were expected and have been observed in human trials with oncolytic adenoviruses,2–8 as well as trials involving IL-12.17–21
Figure 2.

CBC and LFT Each data point represents the mean of five animals ± the standard deviation. WBC, white blood cells; RBC, red blood cells; HGB, hemoglobin; ALKP, alkaline phosphatase; ALT, alanine aminotransferase, AST, aspartate aminotransferease. Group 1, 3 × 106 vp intraprostatic; group 2, 3 × 107 vp intraprostatic; group 3, 3 × 108 vp intraprostatic; group 4, 3 × 108 vp intravenous; group 5, saline intraprostatic.
Table 2. Clinical chemistries.
| Parameter | Group affected | Values | P value |
|---|---|---|---|
| All investigational groups (groups 1–4) versus control (group 5) | |||
| Day 3 | |||
| LYMP | group 3 < group 5 | 2.22 ± 0.36 versus 3.84 ± 0.52 | 0.030 |
| group 4 < group 5 | 2.05 ± 0.62 versus 3.84 ± 0.52 | 0.014 | |
| ALT | group 4 > group 5 | 29 ± 6 versus 13 ± 3 | < 0.001 |
| Day 17 | |||
| LYMP | group 4 < group 5 | 2.54 ± 1.20 versus 4.20 ± 0.42 | 0.021 |
| ALKP | group 4 < group 5 | 34 ± 16 versus 76 ± 8 | < 0.001 |
| Day 78 | |||
| ALB | group 4 > group 5 | 1.7 ± 0.1 versus 1.4 ± 0.1 | 0.032 |
| Highest dose intraprostatic (group 3) versus highest dose intravenous (group 4) | |||
| Day 3 | |||
| ALT | group 3 < group 4 | 14 ± 3 versus 29 ± 6 | < 0.001 |
| Day 17 | |||
| LYMP | group 3 > group 4 | 4.32 ± 0.61 versus 2.54 ± 1.20 | 0.012 |
| ALKP | group 3 > group 4 | 73 ± 14 versus 34 ± 16 | 0.001 |
| Day 78 | |||
| ALT | group 3 < group 4 | 13 ± 2 versus 20 ± 3 | 0.025 |
| ALB | group 3 < group 4 | 1.4 ± 0.1 versus 1.7 ± 0.1 | 0.002 |
ALB, albumin; ALT, alanine aminotransferase; ALKP, alkaline phosphatase; LYMP, lymphocytes.
The lymphopenia observed in group 3 (highest Ad dose, intraprostatic) on day 3 resolved by day 17. In contrast, the lymphopenia observed in group 4 (highest Ad dose, intravenous) took longer to resolve; however, it was within normal limits by the end of the study (day 78). The transaminitis observed in group 4 (highest Ad dose, intravenous) on day 3 resolved by day 17.
Group 4 (highest Ad dose, intravenous) also exhibited decreased ALKP on day 17 and increased ALB on day 78. The former event was likely because of hemolysis of the blood samples.22 The basis for the slight increase in ALB on day 78 is unknown but is not considered clinically significant, as all other liver enzymes were within normal limits.
IL-12 and IFN-γ levels in injected tissue and serum
IL-12 and IFN-γ were quantified in prostate and serum at each of the three time points. IL-12 levels in prostate increased with the adenovirus dose and reached 1,328 pg/g in the highest dose (group 3) on day 3 (Table 3). At this time point, IL-12 levels in prostate were 15-fold higher than that of serum. Peak IL-12 levels in serum (90.3 pg/g or pg/ml) were well below the range (6,000–20,000 pg/ml) reported in humans that were administered IL-12 systemically at the maximum tolerated dose of 500 ng/kg.17,18 On days 17 and 78, IL-12 was undetectable in prostate and within the range of controls (group 5) in serum.
Table 3. IL-12 and IFN-γ levels in injected tissue and serum.
| Group |
Injected tissue |
Seruma |
||||
|---|---|---|---|---|---|---|
|
Day |
Day |
|||||
| 3 | 17 | 78 | 3 | 17 | 78 | |
| IL-12 (pg/g) | ||||||
| 1 | 0b | 0 | 0 | 7.2 ± 1.3 | 2.8 ± 2.8 | 2.1 ± 3.4 |
| 2 | 14.7 ± 7.1 | 0 | 0 | 6.3 ± 3.4 | 2.3 ± 2.2 | 1.7 ± 1.6 |
| 3 | 1,328 ± 59 | 0 | 0 | 90.3 ± 19.7 | 1.9 ± 1.7 | 1.9 ± 3.4 |
| 4 | Ad injected intravenously-see serum |
6.9 ± 9.9 | 11.2 ± 19.4 | 1.8 ± 2.5 | ||
| 5 | 0 | 0 | 0 | 3.8 ± 3.6 | 1.9 ± 3.4 | 2.9 ± 2.5 |
| IFN-γ (pg/g) | ||||||
| 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | 174.2 ± 26 | 0 | 0 | 0 | 0 | 0 |
| 4 | Ad injected intravenously-see serum |
0 | 0 | 0 | ||
| 5 | 0 | 0 | 0 | 0 | 0 | 0 |
Assumes density of mouse serum is 1.024 g/ml. bAll values of zero were below the limit of detection of the ELISA (7.8 pg/ml for both IL-12 and IFN-γ).
IFN-γ, which mediates IL-12’s antitumor activity and toxicity, was detectable only in prostate at the highest adenovirus dose on day 3 (Table 3). It was undetectable in the prostate on days 17 and 78. No IFN-γ was detected in serum at any time point.
Adenoviral DNA in injected tissue and blood
Ad5-yCD/mutTKSR39rep-mIL12 DNA was quantified by polymerase chain reaction (PCR) in both injected tissue and blood. Ad5-yCD/mutTKSR39rep-mIL12 DNA was detected in the prostate of animals injected intraprostatically and increased with adenovirus dose (not shown). No Ad5-yCD/mutTKSR39rep-mIL12 DNA was detected in blood at any time point.
Histopathology of major organs
Twelve organs were subjected to histopathological analysis at each of the three time points. Except for prostate and seminal vesicles, urinary bladder, liver, spleen, and bone marrow, there were no notable changes (from controls) throughout the life of the study. As expected, animals that were administered the adenovirus intraprostatically (groups 1–3) exhibited acute inflammation of the prostate and seminal vesicles on day 3 that increased with adenovirus dose (Table 4). Some of this inflammation could be attributed to the injection procedure itself as animals in the control group (group 5) also exhibited acute inflammation. The inflammation became chronic at the latter time points and essentially resolved (equal to controls) by the end of the study. Focal, mixed cell infiltrates without necrosis were observed in liver on day 3 and, as expected, was greatest in animals that received the adenovirus intravenously (group 4). Such infiltrates were less frequent in animals that received the adenovirus intraprostatically (groups 1–3) and all resolved (equal to controls) by the end of the study. Mixed cell inflammation and hematopoietic cell proliferation were observed in spleen on day 17 of all groups administered the adenovirus (groups 1–4). These events did not correlate with adenovirus dose and resolved by the end of the study. Bone marrow hypercellularity was also observed on day 17 in animals receiving the adenovirus intraprostatically (but not intravenously). Much like the spleenic events, these events did not correlate with adenovirus dose and resolved by the end of the study.
Table 4. Notable histopathological findings.
| Group |
Day |
||
|---|---|---|---|
| 3 | 17 | 78 | |
| 1 | Prostate and SV, 2.0 | Prostate and SV, 0.6 | Prostate and SV, 0.2 |
| Liver, 0.4 | Liver, 0.3 | ||
| Bladder, 0 | |||
| Liver, 0 | Spleen, 0.4 | Spleen, 0 | |
| Bladder, 0 | Bone marrow, 1.6 | Bone marrow, 0 | |
| 2 | Prostate and SV, 2.4 | Prostate and SV, 0.7 | Prostate and SV, 0.4 |
| Liver, 0.8 | Liver, 0.3 | ||
| Bladder, 0 | |||
| Liver, 0.4 | Spleen, 0.3 | Spleen, 0.2 | |
| Bladder, 0 | Bone marrow, 1.2 | Bone marrow, 0 | |
| 3 | Prostate and SV, 2.6 | Prostate and SV, 0.8 | Prostate and SV, 0.2 |
| Liver, 0.4 | |||
| Bladder, 0 | Liver, 0.4 | ||
| Liver, 0.2 | Spleen, 0.3 | Spleen, 0.2 | |
| Bladder, 0.4 | Bone marrow, 1.4 | Bone marrow, 0 | |
| 4 | Prostate and SV, 1.2 | Prostate and SV, 0.1 | Prostate and SV, 0 |
| Liver, 0.2 | Liver, 0.3 | ||
| Bladder, 0 | |||
| Liver, 1.0 | Spleen, 0.6 | Spleen, 0 | |
| Bladder, 0 | Bone marrow, 0 | Bone marrow, 0 | |
| 5 | Prostate and SV, 2.0 | Prostate and SV, 0.1 | Prostate and SV, 0.2 |
| Liver, 0.6 | |||
| Bladder, 0 | Liver, 0.3 | ||
| Liver, 0.4 | Spleen, 0.1 | Spleen, 0.2 | |
| Bladder, 0 | Bone marrow, 1.0 | Bone marrow, 0 | |
SV, seminal vesicles. Numbers represent the mean histopathological score of five animals with 1 representing minimal change and 5 representing severe change. The following pathologies were noted: prostate, seminal vesicles, and bladder—acute inflammation (day 3) that became chronic (or resolved) at the latter time points; liver—focal, mixed cell infiltrate without necrosis; spleen—mixed cell inflammation and hematopoietic cell proliferation; bone marrow—hypercellularity.
Comparison of IL-12 gene expression and biological effects in mouse versus human tissue
A limitation of the mouse model is that human adenoviruses do not produce a significant viral burst in mouse cells, although they do support adenoviral DNA replication.22 To extrapolate our findings in the mouse to the human setting, we (i) quantified IL-12 gene expression in mouse and human prostate epithelial cells following infection with Ad5-yCD/mutTKSR39rep-IL12 and (ii) compared the biological potency of mouse versus human IL-12.
Normal and malignant mouse and human prostate epithelial cells were infected with adenovirus expressing either mouse (Ad5-yCD/mutTKSR39rep-mIL12) or human (Ad5-yCD/mutTKSR39rep-hIL12) IL-12, and IL-12 production was quantified at multiple times thereafter. In all cell types, IL-12 expression increased with time (Figure 3). Seventy-two hours after infection, the amount of IL-12 produced was approximately two- (normal) to fourfold (malignant) greater in human versus mouse cells. This assay measures together the relative infectivity, DNA replication, and viral transgene expression in human versus mouse cells. Thus, for a given adenovirus dose (vp/kg), we expect that the human prostate will generate about two- to fourfold more IL-12 than the mouse prostate.
Figure 3.
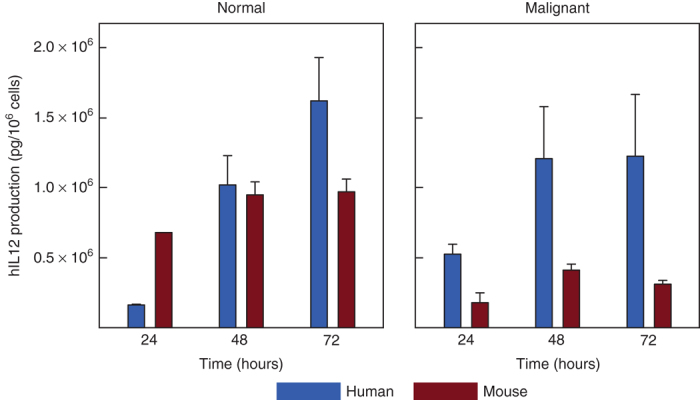
IL-12 production in mouse versus human prostate epithelial cells. Normal and malignant mouse and human prostate epithelial cells were infected with Ad5-yCD/mutTKSR39rep-IL12 and IL-12 production was measured by ELISA at various times thereafter as indicated. The bars represent the mean of three independent determinations ± SD. Primary cultures of mouse (C57BL/6) and human prostate epithelial cells were used for normal cells. TRAMP-C2 and DU145 cells were used for malignant mouse and human prostate epithelial cells, respectively. Shown are results with Ad5-yCD/mutTKSR39rep-hIL12 (expressing human IL-12). Ad5-yCD/mutTKSR39rep-mIL12 (expressing mouse IL-12) gave essentially identical results.
Finally, we compared the relative biological potency of mouse versus human IL-12. Since mouse IL-12 binds efficiently to the human IL-12 receptor (but not vice versa), we used human peripheral blood mononuclear cells (PBMCs) as targets to limit the number of variables in the study (i.e., the source of IL-12) to one. Human PBMCs were incubated with increasing amounts of IL-12 produced from Ad5-yCD/mutTKSR39rep-mIL12 or Ad5-yCD/mutTKSR39rep-hIL12 and IFN-γ production and cell proliferation were measured 72 hours later. Over all IL-12 concentrations examined (0–1,000 pg/ml), the biological potency of mouse and human IL-12 differed by less than twofold (Figure 4). At the IL-12 concentration detected in mouse serum on day 3 (90.3 pg/ml, see Table 3), IFN-γ production and cell proliferation were essentially identical. We conclude that the biological potency of mouse and human IL-12 is similar.
Figure 4.

Relative biological potency of mouse versus human IL-12. Human PBMCs were incubated with increasing amounts of IL-12 produced from Ad5-yCD/mutTKSR39rep-mIL12 (expressing mouse IL-12) or Ad5-yCD/mutTKSR39rep-hIL12 (expressing human IL-12) and IFN-γ production (left) and cell proliferation (right) were measured 72 hours later. For human IL-12, both laboratory grade and clinical grade adenovirus was examined. Each data point represents the mean of three independent determinations with SD <5%. IFN-γ production with no added IL-12 was 3.2 pg/ml.
Discussion
IL-12 has been investigated as a potential cancer treatment for ~15 years. Although it has exhibited significant antitumor activity in preclinical models, its activity in the clinic has been modest (<11% tumor response rate) largely because of severe toxicity when administered systemically. In an attempt to circumvent this limitation, we and others have examined the merit of administering IL-12 locally (ref. 13, for reviews see refs. 14–16). Intratumoral administration of Ad5-yCD/mutTKSR39rep-mIL12 improved both local and metastatic tumor control in a preclinical model of prostate cancer. These effects were accompanied by increased natural killer and cytotoxic T lymphocyte activity, both of which were required for IL-12’s antitumor effects. We report here that intraprostatic administration of Ad5-yCD/mutTKSR39rep-mIL12 at doses to be used in human trials (on a vp/kg basis) was associated with low systemic toxicity. Although animals in the highest adenovirus dose group developed transaminitis, lymphopenia, and inflammation of the prostate/seminal vesicles, these adverse events were expected, low grade, and transient. Overall, our preclinical data support moving this investigational therapy into the clinic.
A limitation of the mouse model is that mouse cells do not support the productive replication of human adenoviruses (i.e., they do not generate a significant viral burst). Hence, when examining the toxicity of oncolytic adenoviruses, the mouse model may underestimate the toxicity that will occur in humans. Although other animal species (e.g., Syrian hamster) support the productive replication of human adenoviruses containing a wild-type E1 region,24 we have found that the replication of 55 kDa E1B-deleted adenoviruses, such as Ad5-yCD/mutTKSR39rep-mIL12, is markedly attenuated (by ~100-fold) in the Syrian hamster relative to wild-type Ad5 (unpublished results). Because we have demonstrated the safety of administering oncolytic adenoviruses without IL-12 to humans in five clinical trials of prostate cancer,4–10 our major concern here was not the toxicity of the oncolytic adenovirus and/or suicide gene therapy, but the increased toxicity because of the addition of IL-12. Owing to the fact that human IL-12 is not active in the mouse,25 we could not use an adenovirus expressing human hIL-12 (i.e., Ad5-yCD/mutTKSR39rep-hIL12) for these studies and, instead, elected to use an adenovirus expressing mouse IL-12 (i.e., Ad5-yCD/mutTKSR39rep-mIL12). However, the biological activity of mouse IL-12 in the Syrian hamster is not known. Therefore, we chose to conduct these studies in the mouse so that the host would match the variant of IL-12 contained in injected adenovirus. We acknowledge that the viral burst obtained in human cells may heighten the immune response and exacerbate toxicity when combined with IL-12. Hence, patients in our upcoming clinical studies may exhibit greater inflammation of the prostate and other immune-related toxicities than what was observed here in the mouse studies. We demonstrate that human prostate epithelial cells express only approximately two- (normal) to fourfold (malignant) more IL-12 than mouse prostate epithelial cells following infection with an oncolytic adenovirus expressing IL-12. This modest difference is likely attributable to the fact that mouse prostate epithelial cells are readily infectable by human adenovirus and support viral DNA replication (although they do not generate a viral burst).23 We also demonstrate that mouse IL-12 has roughly the same biological potency (on a mass basis) as human IL-12. Thus, when considering the toxicity because of the addition of IL-12, we believe the mouse model is as good as any and is likely to recapitulate many of the events that will occur in humans.
Ad5-yCD/mutTKSR39rep-mIL12 (and Ad5-yCD/mutTKSR39rep-hIL12) has the same viral tropism as the three oncolytic adenoviruses we have studied earlier. Because the biodistribution of these adenoviruses was examined in preclinical models and humans,8,9,22 there was no need to examine the biodistribution of Ad5-yCD/mutTKSR39rep-mIL12 here. Using an oncolytic adenovirus (Ad5-yCD/mutTKSR39rep-hNIS) similar to Ad5-yCD/mutTKSR39rep-hIL12 that can be imaged noninvasively, we demonstrated earlier in humans that the vast majority of the injected adenovirus remains in the prostate following intraprostatic administration.8,9 No extraprostatic gene expression was detected in 18 patients treated, 6 of whom were administered 5 × 1012 vp (6.25 × 1010 vp/kg) or five times the highest dose (on a weight basis) used here. Our human imaging results must be interpreted cautiously owing to the insensitivity of the imaging procedures (lower limit of detection is ~3 × 1011 vp in humans) and the fact that roughly half of patients develop low grade flu-like symptoms and transaminitis. We believe these latter events are because of a low level of adenovirus dissemination beyond the prostate gland. Because systemic administration of IL-12 can result in severe toxicity,17 it will be important to limit extraprostatic dissemination of Ad5-yCD/mutTKSR39rep-hIL12 in future trials. We believe this can be accomplished by carefully controlling the volume, dose, and placement of the adenovirus deposits within the prostate gland.
In summary, we have demonstrated in a preclinical model that intraprostatic administration of an oncolytic adenovirus expressing two cytotoxic genes and IL-12 at doses to be used in humans is associated with low toxicity. These results, and others,13 support moving this investigational treatment into the clinic for further evaluation.
Materials and Methods
Adenovirus and cell lines
Ad5-yCD/mutTKSR39rep-mIL12 has been described earlier.13 In brief, it is a 55 kDa E1B-deleted replication-competent adenovirus that contains a yeast cytosine deaminase (yCD)/mutant SR39 herpes simplex virus thymidine kinase (mutTKSR39) fusion gene in the E1 region and a single-chain murine IL-12 gene in the E3 region. Both the yCD/mutTKSR39 fusion gene and IL-12 genes are under the transcriptional control of the human cytomegalovirus promoter. Ad5-yCD/mutTKSR39rep-mIL12 was propagated in human embryonic kidney (HEK) 293 cells and was free of contaminating wild-type adenovirus based on PCR. The viral particle (vp) to plaque-forming unit ratio of the Ad5-yCD/mutTKSR39rep-mIL12 preparation used in the toxicology study was 18. HEK 293, DU145, TRAMP-C2, and primary human prostate epithelial cells (PCS-440-010) were obtained from the American Type Culture Collection (Manassas, VA). Mouse (C57BL/6) primary prostate epithelial cells (C57-6038) were obtained from Cell Biologics, Inc (Chicago, IL). All cell lines were maintained in medium recommended by the supplier.
Toxicology study
One hundred and twenty C57BL/6 male mice (22–24 g) were administered an intraprostatic injection of saline (n = 24) or Ad5-yCD/mutTKSR39rep-mIL12 (n = 72) at one of three dose levels (1.3 × 106, 1.3 × 107, 1.3 × 108 vp/kg) on day 1. To model a worst case scenario, 24 mice were administered the adenovirus intravenously at the highest dose level (1.3 × 108 vp/kg). The volume of the adenovirus injection was 10 µl. Animals that were administered adenovirus also received daily intraperitoneal injections (1 ml) of 5-FC (500 mg/kg) and GCV (30 mg/kg) for 2 weeks beginning on day 3.
Animals were observed daily for any clinical problems (weight loss, listlessness, hunching, and poor grooming) and observations were documented. Body weights were recorded once per week. Animals (eight per group) were sacrificed at three time points representing an early time point (day 3), 1 day after completion of the prodrug therapy course (day 17), and a long-term endpoint (day 78). Blood was collected by single stick of the caudal vena cava, and all blood that spilled into the abdomen was collected.26 The following toxicological parameters were examined: CBCs, LFTs, and histopathology of 12 major organs (brain, heart, lungs, liver, spleen, kidneys, small intestines, large intestines including rectum, urinary bladder, prostate and seminal vesicles, and bone marrow) (five mice/group/time point). Gross observations at necropsy were recorded on study-specific necropsy forms. Tissues (except blood) were immediately fixed in 10% (v/v) neutral buffered formalin until processed for histopathology. IL-12 and IFN-γ were quantified in the injected tissue and serum by enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN) (three mice/group/time point). Adenoviral DNA in blood was quantified by PCR (three mice/group/time point). Histopathology was conducted by Dr. John Seely (Experimental Pathology Laboratories; Herdon, VA). CBC and blood chemistries were analyzed by Antech GLP (Morrisville, NC). The study was conducted under Institutional Animal Care and Use Committee (IACUC)-approved protocol 1137 and monitored by an independent study monitor.
IFN-γ production and cell proliferation assays
Human PBMCs were isolated on Histopaque 1077 gradients per the manufacturer’s protocol. PBMCs were washed twice with saline and stimulated with phytohemagglutinin-M (1.5% v/v) for 3 days in medium containing 10% fetal bovine serum. Cells were washed, counted, and incubated in 96 well round-bottomed plates (IFN-γ assays—100,000 cells/well; proliferation assays—12,500 cell/well) with increasing amounts of conditioned media from DU145 cells infected with Ad5-yCD/mutTKSR39rep-mIL12 (expressing mouse IL-12) or Ad5-yCD/mutTKSR39rep-hIL12 (expressing human IL-12). The IL-12 concentration in the conditioned media was quantified by ELISA prior to setting up the assays. Conditioned media from mock-infected cells served as a baseline control. IFN-γ production was quantified by ELISA, and cell proliferation by Vybrant MTT pulse assay (Life Technologies, Grand Island, NY), 72 hours later.
Statistical methods
Blood values of all groups were compared by ANOVA using SigmaPlot v12.3. A blood value was considered abnormal if it was statistically different (P < 0.05) than the control group (group 5) at the same time point.
Acknowledgments
The authors thank Kenneth Barton for serving as the independent study monitor. This work was supported by NIH grant CA160289 to S.O.F.
The authors declared no conflict of interest.
References
- Russell, SJ, Peng, KW and Bell, JC (2012). Oncolytic virotherapy. Nat Biotechnol 30: 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freytag, SO, Rogulski, KR, Paielli, DL, Gilbert, JD and Kim, JH (1998). A novel three-pronged approach to kill cancer cells selectively: concomitant viral, double suicide gene, and radiotherapy. Hum Gene Ther 9: 1323–1333. [DOI] [PubMed] [Google Scholar]
- Freytag, SO, Paielli, D, Wing, M, Rogulski, K, Brown, S, Kolozsvary, A et al. (2002). Efficacy and toxicity of replication-competent adenovirus-mediated double suicide gene therapy in combination with radiation therapy in an orthotopic mouse prostate cancer model. Int J Radiat Oncol Biol Phys 54: 873–885. [DOI] [PubMed] [Google Scholar]
- Freytag, SO, Khil, M, Stricker, H, Peabody, J, Menon, M, DePeralta-Venturina, M et al. (2002). Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer Res 62: 4968–4976. [PubMed] [Google Scholar]
- Freytag, SO, Stricker, H, Pegg, J, Paielli, D, Pradhan, DG, Peabody, J et al. (2003). Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res 63: 7497–7506. [PubMed] [Google Scholar]
- Freytag, SO, Stricker, H, Peabody, J, Pegg, J, Paielli, D, Movsas, B et al. (2007). Five-year follow-up of trial of replication-competent adenovirus-mediated suicide gene therapy for treatment of prostate cancer. Mol Ther 15: 636–642. [DOI] [PubMed] [Google Scholar]
- Freytag, SO, Movsas, B, Aref, I, Stricker, H, Peabody, J, Pegg, J et al. (2007). Phase I trial of replication-competent adenovirus-mediated suicide gene therapy combined with IMRT for prostate cancer. Mol Ther 15: 1016–1023. [DOI] [PubMed] [Google Scholar]
- Barton, KN, Stricker, H, Brown, SL, Elshaikh, M, Aref, I, Lu, M et al. (2008). Phase I study of noninvasive imaging of adenovirus-mediated gene expression in the human prostate. Mol Ther 16: 1761–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, KN, Stricker, H, Elshaikh, MA, Pegg, J, Cheng, J, Zhang, Y et al. (2011). Feasibility of adenovirus-mediated hNIS gene transfer and 131I radioiodine therapy as a definitive treatment for localized prostate cancer. Mol Ther 19: 1353–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freytag, SO, Stricker, H, Lu, M, Elshaikh, M, Aref, I, Pradhan, D et al. (2014). Prospective randomized phase 2 trial of intensity modulated radiation therapy with or without oncolytic adenovirus-mediated cytotoxic gene therapy in intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys 89: 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freytag, SO, Stricker, H, Movsas, B and Kim, JH (2007). Prostate cancer gene therapy clinical trials. Mol Ther 15: 1042–1052. [DOI] [PubMed] [Google Scholar]
- Freytag S, Stricker H, Movsas B, Elshaikh M, Aref I, Barton K et al. Gene therapy of prostate cancer. In: Roth JA (ed.). Gene-Based Therapies for Cancer. Springer, New York, 2010. pp. 33–49. [Google Scholar]
- Freytag, SO, Barton, KN and Zhang, Y (2013). Efficacy of oncolytic adenovirus expressing suicide genes and interleukin-12 in preclinical model of prostate cancer. Gene Ther 20: 1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri, G (2003). Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 3: 133–146. [DOI] [PubMed] [Google Scholar]
- Del Vecchio, M, Bajetta, E, Canova, S, Lotze, MT, Wesa, A, Parmiani, G et al. (2007). Interleukin-12: biological properties and clinical application. Clin Cancer Res 13: 4677–4685. [DOI] [PubMed] [Google Scholar]
- Colombo, MP and Trinchieri, G (2002). Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev 13: 155–168. [DOI] [PubMed] [Google Scholar]
- Leonard, JP, Sherman, ML, Fisher, GL, Buchanan, LJ, Larsen, G, Atkins, MB et al. (1997). Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 90: 2541–2548. [PubMed] [Google Scholar]
- Atkins, MB, Robertson, MJ, Gordon, M, Lotze, MT, DeCoste, M, DuBois, JS et al. (1997). Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res 3: 409–417. [PubMed] [Google Scholar]
- van Herpen, CM, Looman, M, Zonneveld, M, Scharenborg, N, de Wilde, PC, van de Locht, L et al. (2004). Intratumoral administration of recombinant human interleukin 12 in head and neck squamous cell carcinoma patients elicits a T-helper 1 profile in the locoregional lymph nodes. Clin Cancer Res 10: 2626–2635. [DOI] [PubMed] [Google Scholar]
- Sangro, B, Mazzolini, G, Ruiz, J, Herraiz, M, Quiroga, J, Herrero, I et al. (2004). Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J Clin Oncol 22: 1389–1397. [DOI] [PubMed] [Google Scholar]
- Alvarez, RD, Sill, MW, Davidson, SA, Muller, CY, Bender, DP, DeBernardo, RL et al. (2014). A phase II trial of intraperitoneal EGEN-001, an IL-12 plasmid formulated with PEG-PEI-cholesterol lipopolymer in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer: a gynecologic oncology group study. Gynecol Oncol 133: 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah, H, Al-Atoom, A and Shehab, G (2012). Explanation of the decrease in alkaline phosphatase (ALKP) activity in hemolyzed blood samples from the clinical point of view: in vitro study. Jordan J Biol Sci 5: 125–128. [Google Scholar]
- Paielli, DL, Wing, MS, Rogulski, KR, Gilbert, JD, Kolozsvary, A, Kim, JH et al. (2000). Evaluation of the biodistribution, persistence, toxicity, and potential of germ-line transmission of a replication-competent human adenovirus following intraprostatic administration in the mouse. Mol Ther 1: 263–274. [DOI] [PubMed] [Google Scholar]
- Thomas, MA, Spencer, JF, La Regina, MC, Dhar, D, Tollefson, AE, Toth, K et al. (2006). Syrian hamster as a permissive immunocompetent animal model for the study of oncolytic adenovirus vectors. Cancer Res 66: 1270–1276. [DOI] [PubMed] [Google Scholar]
- Schoenhaut, DS, Chua, AO, Wolitzky, AG, Quinn, PM, Dwyer, CM, McComas, W et al. (1992). Cloning and expression of murine IL-12. J Immunol 148: 3433–3440. [PubMed] [Google Scholar]
- Schnell, MA, Hardy, C, Hawley, M, Propert, KJ and Wilson, JM (2002). Effect of blood collection technique in mice on clinical pathology parameters. Hum Gene Ther 13: 155–161. [DOI] [PubMed] [Google Scholar]