

Abstract

Antifolates are widely used to treat several diseases but are not currently used in the first-line treatment of tuberculosis, despite evidence that some of these molecules can target Mycobacterium tuberculosis (Mtb) bacilli in vitro. To identify new antifolate candidates for animal-model efficacy studies of tuberculosis, we paired knowledge and tools developed in academia with the infrastructure and chemistry resources of a large pharmaceutical company. Together we curated a focused library of 2508 potential antifolates, which were then tested for activity against live Mtb. We identified 210 primary hits, confirmed the on-target activity of potent compounds, and now report the identification and characterization of 5 hit compounds, representative of 5 different chemical scaffolds. These antifolates have potent activity against Mtb and represent good starting points for improvement that could lead to in vivo efficacy studies.
Keywords: trimetrexate, dihydrofolate reductase, antifolate, tuberculosis
Despite great advances in human medicine over the past century, tuberculosis (TB), caused by the acid-fast bacterium Mycobacterium tuberculosis (Mtb), remains the leading cause of bacterial-induced mortality worldwide. All of the current first-line TB drugs were discovered over 40 years ago, and their effectiveness continues to diminish due to the increased incidence of drug-resistant Mtb strains. Of particular concern are multidrug-resistant strains, defined as resistant to the two most effective TB drugs, isoniazid and rifampin,1 and extensively drug-resistant (XDR) strains that are additionally resistant to the fluoroquinolones and one of the second-line injectable antibiotics.2,3 Since the declaration by the World Health Organization over a decade ago that “TB represents a global health emergency”, there has been more effort from both industrial and academic laboratories to capitalize on the unprecedented advances in our understanding of the fundamental biology of TB. Unfortunately, progress has not kept pace with the problem. Of the most promising new compounds for TB chemotherapy, including the nitroimidazole PA-824,4 the 1,2-diamine SQ-109,5 and the benzothiazinone BTZ-043,6 only the diarylquinoline, Bedaquiline, has advanced to approval,7 and even there, concerns about safety and drug interaction limit its use to MDR TB patients.8 It remains critical that we develop and maintain a robust pipeline of TB drug candidates. The development of new drugs with different modes of action would complement current therapies or enable altogether new drug combinations. Shortened and better tolerated treatment regimens for both drug-sensitive and MDR/XDR TB remain an urgent unmet medical need.
One approach to identifying new lead compounds is to exploit well-validated drug targets that are not the focus of current regimens. Reduced folates participate in a variety of one-carbon transfer reactions and are required for the production of essential purines, pyrimidines, and certain amino acids.9,10 Dihydrofolate reductase (DHFR) is a critical enzyme in the folate pathway and is a clinically important target of drugs to treat cancer as well as bacterial, fungal, and parasitic infections. Antifolates are also effective antimycobacterials; dapsone is the standard chemotherapy for infections of Mycobacterium leprae (leprosy) and targets DHPS, another critical enzyme in the folate pathway.9,11 Furthermore, p-aminosalicylic acid (PAS), a second-line TB treatment, was recently shown to be a prodrug activated by the folate pathway12 and possibly inhibiting DHFR.13 This evidence highlights the antifolate pathway, and DHFR specifically, as a worthy target that has been underexplored for tuberculosis drug discovery. However, we still lack the appropriate tool compounds to investigate the potential of chemically inhibiting DHFR to treat TB.
Some clinically approved antifolates, such as methotrexate (MTX), pyrimethamine, and trimetrexate, show strong inhibition of the Mtb DHFR enzyme but poor or no activity against live Mtb.14,15 In 2002 we found that the diaminotriazine WR99210 inhibits the Mtb DHFR enzyme and is also active against live bacilli.16 We engineered Mtb with reduced DHFR activity and found it is 3–4-fold more sensitive to WR99210, confirming that it targets this enzyme in live bacteria. However, perhaps due to its acute gastrointestinal toxicity in humans,17 no further development of WR99210 for the treatment of TB infections has been carried out. Nevertheless, this confirmation that a known DHFR inhibitor can target Mtb growth suggests the potential of antifolates to treat tuberculosis and highlights the need to identify novel molecules with more potent and specific activity against Mtb DHFR, as well as lowered toxicity against mammalian cells.
RESULTS AND DISCUSSION
Unique Approach to Antifolate-Based Drug Discovery for TB
In a previous attempt to identify more promising antifolate compounds for TB, we developed an assay for the Mtb DHFR enzyme and adapted it for high-throughput screening. A screen of 32000 diverse compounds performed in collaboration with the WHO and NCDS (National Center for Drug Screening, Shanghai) yielded few promising candidates.14 However, more recently we have shown that close analogues of methotrexate target Mtb DHFR and have dramatically improved activity against bacilli in vitro.15 These recently published data15 suggested that we might explore the chemical space around previously described antifolates to discover novel lead compounds for antitubercular drugs.
In 2010 GlaxoSmithKline (GSK) launched a unique initiative to foster open drug discovery partnerships with academic researchers working on diseases of the developing world. By connecting a diverse global network of researchers to the scientific drug-discovery expertise and industrial-scale facilities housed at their R&D unit in Spain, the Tres Cantos Open Lab Foundation helps to develop drug-discovery pipelines for neglected diseases (http://www.openlabfoundation.org). GSK had previously invested in antifolate drug discovery focused on antimalarials (e.g., Malarone18). Therefore, we chose to mine the large antifolate chemical space developed at GSK for inhibitors with potent DHFR-targeting activity against live Mtb. Here we report the results of these efforts, including new compound classes that are direct inhibitors of Mtb DHFR with strong activity against live Mtb.
Curating the GSK Antifolate Library
To identify the most promising antifolate library, we designed a three-pronged strategy. First, a review of the rich antifolate literature allowed us to compile a list of compounds that have previously been characterized as folate pathway inhibitors. To get a broad range of known antifolates, we expanded the search to include inhibitors of both the eukaryotic and prokaryotic folate pathway enzymes,19,20 resulting in 36 compounds. The entire GSK screening collection of over 2 million compounds was then searched in silico for close analogues of these previously identified antifolates (Tanimoto score > 0.8). Analogue retrieval based on these 36 literature-derived antifolates yielded 150 molecules (approach A), most of which had diaminopyrimidine (DAP)-like scaffolds. In addition to antifolates from prior literature, a phenotypic screen of the malarial parasite Plasmodium falciparum previously conducted at GSK had identified another set of likely antifolates21 that was used as a second source of molecules for analogue retrieval (approach B). A P. falciparum 3D7 strain was adapted to grow in low levels of folic acid (FA) in a special FA and p-amionbenzoic acid (pABA) depleted RPMI medium until normal growth rates were observed. Compounds from the Tres Cantos anti-malarial set (TCAMS) targeting the folate pathway were identified when an increase in the IC50 was observed in standard RPMI media compared to the FA/pABA depleted media.21 Analogue retrieval performed as described earlier, but using the TCAMS antifolates, retrieved 2778 DAP analogues and a further 685 compounds with diverse chemical scaffolds (approach B). Finally, we also searched the GSK screening collection for compounds containing at least one of three core substructures commonly found in antifolates: diaminopyrimidines, pyrrolo/furodiaminopyrimidines, and biguanides (approach C). There were 2324, 5175, and 312 compounds found containing these substructures, respectively (Figure 1). There was significant overlap in the compounds retrieved from each of these subsearches, and when pooled, the compounds identified from approaches A–C resulted in a total of 5232 unique structures. In a meta analysis, 2724 of these compounds had already been shown to be inactive against H37Rv or the attenuated Mycobacterium bovis strain bacillus Calmette–Guerin (BCG).22 These were excluded from further studies. The remaining antifolate library of 2508 compounds was then retrieved from the GSK compound library, plated, and tested for inhibition of live Mtb growth.
Figure 1.

Antifolate library curation. Approaches A and B (shown in the left panel) used analogue searches and approach C (right-hand panel) used substructure searches to identify compounds from the GSK screening collection. In each query the number of retrieved analogues is shown in parentheses and bold.
Screening against Live Mtb Identified Compounds with Antifolate-Mediated Inhibitory Activity
Our screening approach is outlined in Scheme 1. Compounds were tested at a single concentration of 20 µM for inhibition of H37Rv growth under aerobic conditions. The results of duplicate experiments showed good correlation (results from the two experiments are plotted on the x- and y-axes in Figure 2a). The screen had a high hit rate, and 75% growth inhibition (as an average of duplicate experiments; data points shown in red in Figure 2a) was set as a primary cutoff when compounds were selected for dose–response testing. Experiments performed in the same high-throughput format confirmed dose–response activity and provided us with a preliminary value of 50% inhibition (IC50) for each compound (Figure 2b). As expected, there was an inverse correlation between the percent inhibition in the primary screen and the IC50 calculated from the dose–response data: 210 of the 459 compounds retested were considered primary hits, with clear dose–response activity and preliminary IC50 values of <10 µM against Mtb.
Scheme 1.

Screening Flowchart: Overview of the steps Taken during This Studya
aThe total number of compounds that matched the defined cutoffs at each stage and progressed to the next level of work is shown in italicized bold font.
Figure 2.

Sreening antifolates against Mtb. (a) 2724 compounds were tested at 20 µM against live H37Rv. The percent inhibition measured in two separate assays is plotted on the x- and y-axes for each compound. (b) Active inhibitors were assayed at 11 concentrations ranging from 100 to 0.098 µM, and the concentration required for 50% growth inhibition (IC50) was calculated. Average activity of duplicate experiments from (a) was plotted on the x-axis, and calculated IC50 values were plotted on the y-axis). Compounds with IC50 values under 10 µM are in the red-shaded area of the graph. (c) Growth inhibition curves are shown for compounds from two of the five chemotypes identified in the screen, namely, WR99210 (left graph) and DQn-1 (right graph). Compounds were assayed against wild-type Mtb (H37Rv shown as solid circles) and Mtb engineered to artificially overexpress DHFR (DHFR-oe shown as clear circles). Data shown are the average of duplicate experiments.
For each hit, we next determined the minimum concentration required to inhibit growth (MIC90) in culture. We also tested whether the inhibitory activity was mediated through DHFR, using genetically manipulated strains that had been desensitized to DHFR inhibition.14,15 We had previously constructed an integrating DNA plasmid (pMRN1) containing a wild-type copy of the Mtb dfrA gene under the control of a strong constitutive promoter (MOP), which resulted in an 8-fold increase in the levels of dfrA transcript.14,15 The DHFR-overexpressing strain of Mtb (DHFR-oe) is less sensitive to compounds that target the folate pathway when compared to the parent strain.14,15 This assay cannot exclude potential involvement of other targets; however, compounds are considered to have on-target activity if the MIC90 increases by at least 4-fold in the DHFR-oe compared with H37Rv. Of the 210 compounds evaluated, 17 showed at least a 4-fold increase in the MIC90 activity against the DHFR-oe strain, suggesting that their potency against live Mtb was at least partially due to inhibition of DHFR. The inhibition curves for two of these compounds are shown in Figure 2c, where we observed a shift to lower inhibition when the compounds were tested in cells overexpressing DHFR. This same pattern was observed with all 17 compounds, and they were considered confirmed hits from this screen: highly potent compounds with MIC90 of <20 µM in H37Rv and at least 4-fold decreased potency in cells overexpressing DHFR.
Chemotypes Identified
These 17 hit compounds could be divided into five subgroups on the basis of their chemotype. All 17 compounds are shown in Table 1. One of the chemical families, represented by WR99210, has been well studied in the literature, and over 150 analogues were present in the curated antifolate library. Eleven of the 17 confirmed hits belonged to this chemotype (chemotype A). The remaining six hits (belonging to four other chemical subgroups) were novel structures but all contained the traditional DAP scaffold. Three of the chemical families (chemotypes B, C, and D, exemplified by PDP-1, PQD-1, and DPt-1) were also well represented in the curated antifolate library; however, many of these did not meet our criteria for a hit (MIC90 < 20 µM). PDP-1 (chemotype B), containing a pyrido diamino pyrimidine moiety, had 25 close analogues that were tested in our primary H37Rv screen. PQD-1 (chemotype C), a pyrroloquinazolinediamine, had 15 analogues in the curated antifolate library and was previously reported as one of the most potent antimalarial agents,23 although to the best of our knowledge this is the first time its activity against live Mtb has been reported. DPt-1 (chemotype D), a diaminopteridine, had a total of 12 analogues in our screening library, of which DPt-1 was the most potent in its class, but not as potent as some of the hits belonging to other chemotypes found in this work (see Table 1). Finally, DQn-1 (chemotype E) contained a 2,4-diaminoquinazoline core and had a structure similar to that of trimetrexate. Trimetrexate is a well-known antifolate with modest activity against Mtb.15 However, DQn-1, in which the methyl group on the quinazoline ring in trimetrexate is replaced with a chlorine and the trimethoxyphenyl group is substituted with a para carboxyphenyl (Table 1), was very potent against the DHFR enzyme and was especially interesting as it showed the most potent activity against live Mtb (MIC90 = 0.03 µM; Table 1).
Table 1.
Primary Screen Hit Confirmation
| Chemotype | Structure | MIC90(µM) | Enzyme IC50(nM) | ||
|---|---|---|---|---|---|
| Hit | wt | DHFR-oe | Mtb | Human | |
| A. WR99210(WR) | |||||
| WR99210 | 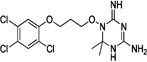 |
1–2 | 15–30 | 14a | 9.0 |
| WR-1 | 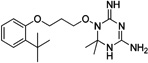 |
0.3 | 2.0 | 10.1 | 8.7 |
| WR-2 | 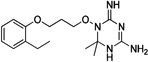 |
0.5 | 2.0 | 5.6 | 3.6 |
| WR-3 | 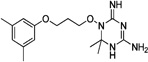 |
1.0 | 15.6 | nd | nd |
| WR-4 | 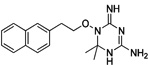 |
1.0 | 11.7 | nd | nd |
| WR-5 | 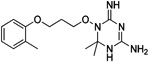 |
0.5 | 15.6 | nd | nd |
| WR-6 | 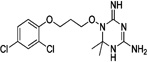 |
0.7 | 11.7 | nd | nd |
| WR-7 | 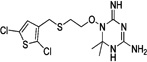 |
0.5 | >2 | nd | nd |
| WR-8 | 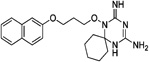 |
0.5 | 2.0 | nd | nd |
| WR-9 | 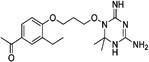 |
0.5 | >2 | nd | nd |
| WR-10 | 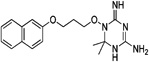 |
2.0 | 23.4 | nd | nd |
| B. Pyrido diamine Pyrimidine (PDP) | |||||
| PDP-1 | 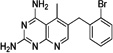 |
1.3 | 30.0 | 8.2 | 6.7 |
| C. Pyrroloquinazolinediamine (PQD | |||||
| PQD-1 | 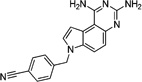 |
0.5 | >2 | 10.1 | 6.8 |
| PQD-2 | 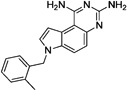 |
1.0 | 15.6 | nd | nd |
| D. Diaminopteridine (Dpt) | |||||
| Dpt-1 | 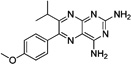 |
2.9 | 250.0 | 54.5 | 382.1 |
| Dpt-2 | 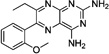 |
15.6 | >250 | 70.5 | 3844.4 |
| E. Diaminoquinazoline (DQn) | |||||
| DQn-1 | 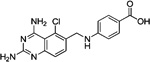 |
0.03 | 1.0 | 8.7 | 7.6 |
| Trimetrexatea,b | 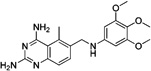 |
6.3 | >100 | 17.0 | nd |
Data from Nixon et al.15
The compound was not identified in the screen.
Antitubercular Antifolates Directly Inhibit DHFR Enzyme Activity
Each of the representative molecules of the five different chemotypes showed decreased activity against Mtb with artificially elevated levels of DHFR (Table 1), suggesting that they targeted DHFR in our cell-based assay. To test directly whether the molecules target DHFR, we assayed their ability to inhibit the purified enzyme. We were able to test most of the 17 hits, and at least 1 in each chemotype, and confirmed that they directly inhibited the Mtb DHFR enzyme. Their potency varied, although most of the hits displayed similar levels of inhibition against the human DHFR (Table 1). The exception was DPt-1 that, although not as active an inhibitor of Mtb growth, showed selectivity for the Mtb DHFR enzyme with ~7-fold lower activity against the human enzyme (Table 1).
Postscreening Cytotoxicity and Preliminary in Vivo Characterization of Hits
The potential of a compound in animal studies is determined by a variety of factors, including potency, in vivo safety profile, and pharmacokinetic properties. To identify the best hits for progression to in vivo studies, we first evaluated their in vitro safety and DMPK profile (Tables 2 and 3). The cytotoxicity of the selected molecules was assessed by measuring the concentration of compound resulting in 50% inhibition (Tox50) of HepG2, a mammalian cell line. Furthermore, we measured compound activity against intracellular Mtb growth in macrophages (THP-1 cells) and evaluated their potential for drug-induced human hepatotoxicity using an in vitro high-content cell-based assay (Cell Health or CH assay).24 Briefly, nuclear area, cell number, and plasma membrane permeability were determined in HepG2 human hepatocytes cultured in 96-well plates. Human cytotoxicity potential was defined as 80% sensitivity and 90% specificity when cells were treated with 30 times the maximal efficacious concentration of a compound. We assessed parameters of compound quality: the property forecast index (PFI), an approximate predictor of the lipophilicity and solubility of the compound;25 ligand efficiency (LE);26 and lipophilic ligand efficiency (LLE).27 Furthermore, aqueous solubility was measured by chemiluminescent nitrogen detection (CLND),25 and occasionally for promising hits solubility was also tested in biorelevant gastrointestinal media to simulate the fasted and fed states in vivo.28 For most compounds an in vitro evaluation of metabolic stability was conducted, measuring the rate of clearance in mouse microsomes. Finally, with one compound preliminary DMPK studies were carried out in mice. When taken together these results allowed us to determine the potential for further progression of our hits for in vivo efficacy studies to validate DHFR as a new target for TB treatment.
Table 2.
In Vitro Characterization of Representative Compounds
| compound | enzyme IC50 (nM) |
MIC90 (µM) |
Tox50 (µM) |
selectivity index (Tox50/MIC90) | intracell MIC90 (µM) | cell healtha (µM) | ||
|---|---|---|---|---|---|---|---|---|
| Mtb | human | wt | DHFR-oe | HepG2 | ||||
| WR99210 | 14a | 9.0 | 1–2 | 15–30 | 40 | 20 | 9.7 | 10/19/25 |
| PDP-1 | 8.2 | 6.7 | 1.25 | 30 | 40 | 32 | 1.2 | ndc |
| PQD-1 | 7.1 | 6.8 | 0.50 | >2 | 10 | 20 | 0.7 | nd |
| DPt-1 | 49.1 | 382.1 | 2–8 | 250 | >50 | >17 | 28 | 200/200/200 |
| DQn-1 | 6.2 | 7.6 | 0.03 | 1.0 | >50 | >1000 | 0.05 | 200/200/200 |
| trimetrexateb | 17 | nd | 6.25 | >100 | nd | nd | nd | nd |
Nuclear morphology/membrane permeability/mitochondrial potential.
Data from Nixon et al.15
nd, not determined.
Table 3.
Physicochemical Properties and Initial DMPK of Representative Compounds
| compound | mol wt (g/mol) | cLogP | LE | LLE | PFI | CLND solubility (µM) | in vitro clearance in mouse microsomes (mg/mL·g) |
|---|---|---|---|---|---|---|---|
| WR99210 | 432.14 | 4.00 | 0.46 | 4.00 | 4.9 | 373 | <0.5 |
| PDP-1 | 380.67 | 3.51 | 0.52 | 4.49 | 8.1 | 52 | >30 |
| PQD-1 | 314.34 | 2.53 | 0.46 | 5.47 | 5.8 | 12 | <0.5 |
| DPt-1 | 310.35 | 2.68 | 0.42 | 4.32 | 6.6 | 30 | nd |
| DQn-1 | 343.77 | 2.83 | 0.46 | 5.17 | 3.1 | 401 | <0.5 |
As expected from previously published work, WR99210 showed some toxicity against HepG2 cells and furthermore had a poor hepatotoxicity profile in the Cell Health Assay (Table 2). PDP-1 showed very low metabolic stability and was also very lipophilic, with a very high PFI and poor LLE values (see Table 3). Its predicted poor solubility was confirmed when tested experimentally in aqueous solution (Table 3) as well as in a variety of biorelevant media (data not shown). PQD-1 showed some toxicity in vitro (Table 2) and was insoluble as measured by CLND (Table 3), excluding it from further in vivo progression. DPt-1 showed moderate whole cell potency, no cytotoxicity in vitro, and an initial clean profile regarding hepatotoxicity (CH > 200 µM) but was 10-fold less potent at inhibiting Mtb growing within THP-1 cells compared to its extracellular activity (Table 2). Furthermore, it showed poor LLE and was also only moderately soluble, which did not make it an ideal candidate for further in vivo progression (Table 3). In comparison, DQn-1, the most potent compound of the 17 confirmed hits, showed promising results for progression, with high whole cell potency values of ~50 nM in both the intra-and extracellular assays (Table 2). Furthermore, it showed no cytotoxicity against HepG2 cells, with a selectivity index (SI) >1000 and an initial clean profile regarding hepatotoxicity (CH > 200 µM). Its compound quality parameters were suitable (Table 3): PFI was <6, and both efficiency indicators, LE and LLE, were in the appropriate range for progression. DQn-1 had a high solubility profile in both the aqueous CLND assay and biorrelevant media once formulated. In vitro DMPK studies showed that it was metabolically stable, as it was not cleared rapidly in mouse microsomal stability assays (Table 3). Furthermore, preliminary assessment of drug–drug interactions revealed no inhibition of the CYP3A isoform29 (CYP P450 3A4VR and 3A4VG values were >50 µM), although other cytochrome P450 isoforms remain to be tested. In preliminary evaluations of the in vitro safety pharmacology profile30 for DQn-1, no relevant flags were observed that might have indicated potentially undesirable off-target activity for this hit.
These promising data encouraged us to evaluate the in vivo profile of DQn-1. Initial pharmacokinetic parameters were considered adequate to progress to in vivo efficacy experiments. However, multiday dosing in mice revealed toxicity (low responsiveness to stimuli) >10 mg/kg (data not shown). A subsequent experiment limited to low doses of DQn-1 failed to detect any significant reduction in Mtb burden after 8 days of treatment (Supporting Information). Therefore, although its in vitro and DMPK properties matched the profile we were seeking to develop a suitable lead for progression, it was not possible to pursue this series further at this time.
Scope of DHFR Inhibitors for the Treatment of TB
Inhibitors of DHFR have been used to treat a variety of diseases but could be underutilized as antimicrobial chemicals against Mtb. There are currently several potent inhibitors of the Mtb enzyme commercially available, most notably methotrexate, which has poor to no activity against live Mtb. More recent evidence suggests that slight modifications to known antifolates can dramatically alter the potency of these molecules against live Mtb.15 Our objective was to identify new antifolates that are selective for Mtb DHFR and potent inhibitors of in vitro Mtb growth with adequate physicochemical properties and safety profiles that might be suitable for testing in an animal model of infection.
GSK’s Open Lab Foundation initiative provided an excellent collaborative platform for this project, embracing an innovative partnership between academic laboratories, nonprofit organizations, and a major pharmaceutical company. Together we were able to combine biochemical, genetic, and microbiological assays and tools developed in an academic setting with deep industry-based knowledge of drug design and development to identify and characterize novel antimycobacterial antifolates. This project also benefitted from GSK’s extensive work with antifolate chemistry. The prior malaria-driven expansion of antifolates in the GSK library allowed us to search within the antifolate chemical space for close analogues with strong antimycobacterial activity. When we mined the GSK screening collection for likely antifolates, we identified a large library of compounds that had not previously been tested for antimycobacterial activity. Furthermore, we were able to identify chemotypes with very potent activity against in vitro Mtb growth despite similarity to previously described antifolates that were not active against Mtb, confirming that this chemical space was indeed underexplored.
Some years ago potency of compounds and the “rule of five” physical property guidelines for drug permeability27,31,32 were in general the standard analysis considered for developmental selection of hits in drug discovery. However, current experience indicates that maximizing compound quality from the very beginning is a better strategy for lowering the risk of problems in progression related to drug absorption, distribution, metabolism, and excretion (ADME) and drug safety.30,33,34 With an eye toward developing scaffolds that can be progressed rapidly into tolerated antibiotics, preliminary analyses beyond the “rule of five” (e.g., PFI, aqueous solubility, ligand efficiency (LE), and lipophilic ligand efficiency (LLE)) have become increasingly relevant to reducing the attrition rate in drug discovery. Furthermore, specific physicochemical features may be required of antimycobacterial drugs due to the unique architecture of the mycobacteria cell wall that affects the permeability of the compounds across membranes. Sufficient potency may require a certain structure, complexity, and lipophilicity that could be in conflict with conventional metrics of drug-like space.35 For that reason our medicinal chemistry decisions to progress the hits found in this work were based on the identification of good starting points for lead characterization; compounds with high lipophilicity (PFI and cLogP), low solubility, inadequate LE and LLE values, and other issues such as cytotoxicity were considered unsuitable for progression.
One class of hits from our screen, represented by WR99210 (Table 1), was previously described as an antifolate with antimycobacterial activity, but with some toxicity issues.22 Our preliminary SAR analysis of chemotype A (WR99210 analogues) indicates that a wide variety of structural modifications around the phenoxy group and the length of the alkoxy chain maintain similar optimal whole cell potencies. Although our screen identified numerous analogues in this class, none had improved properties, discouraging further exploration of this family at this time. The remaining four chemotypes we identified were not previously described as antimycobacterials (Table 1). Chemotypes B–D lacked enough representation to allow any preliminary SAR analyses. However, there is an opportunity to expand the chemistry work in these series to generate a larger data set to draw some conclusions about their SAR. PDP-1 was the only potent compound in chemotype B (Table 1), and most of the other analogues had similar predicted lipophilic and solubility values (data not shown). For chemotype C cytotoxicity evaluation of different analogues confirmed the acute toxicity profile of the series. In chemotype D, DPt-1 was the most potent among its analogues and was the only chemotype that showed some selectivity for the Mtb DHFR enzyme, ~7 times more potent an inhibitor of the mycobacterial compared with the human DHFR enzyme (Table 1). This selectivity is encouraging, although its moderate whole cell potency, poor LLE, and low solubility excluded it from further progression at this time. However, given its in vitro selectivity, it may provide a promising starting point for further chemistry that might improve its drug profile.
Finally, DQn-1 is a particularly interesting novel hit; not only was it the most potent of our hits in vitro, it is also a close analog of the FDA approved trimetrexate (Table 1). Trimetrexate is a well-known nonclassical antifolate analogue of methotrexate (MTX) that has modest activity against live Mtb (MIC = 6.25 µM15). In comparison, DQn-1 had an MIC90 value of 0.03 µM with similar potency against intracellular Mtb growth in activated macrophages (Table 2). We found only one other compound in the GSK compound library belonging to this chemotype, an extremely close analogue in which the carboxyphenyl group was in the meta position, which was about 10-fold less potent against in vitro Mtb growth. The reasons for these differences are unclear, but could reflect differential uptake by Mtb or differential engagement with the DHFR active site.
We have reported above that DQn-1 had a very promising in vitro profile and high drug-like quality. Also, when progressed to in vivo DMPK studies its exposure and bioavailability were considered sufficient to be administered orally (Supporting Information). However, a lack of linearity was observed between the doses administered (Table S1). In addition, multiday dosing revealed toxicity >10 mg/kg (data not shown), and lower doses were not efficacious in reducing bacterial burden (Supporting Information). As noted earlier, no other analogue to DQn-1 endowed with whole-cell potency in the nanomolar range was identified, and to the best of our knowledge there are no other compounds in this series currently available as promising alternatives for in vivo evaluation. However, the scaffold for DQn-1 is ripe for improvement, following closer evaluation of toxicity and pharmacokinetics.
Of the five representative tool compounds identified in our study, none had a profile that allowed us to complete full in vivo characterization. However, additional analogues within these chemotypes could be further explored. The 3-D structures of both the Mtb and human DHFR have been solved both alone and in complex with a variety of inhibitors.36,37 These structures have been valuable in the development of inhibitors with higher specificity for Mtb DHFR38 and will be useful in modifying new scaffolds for improved selectivity. Although resources did not permit us to pursue further chemistry at this time, it is possible that chemical modifications of these families could improve their physicochemical and DMPK properties. We might then be able to identify compounds that are both highly effective in animal models of TB and endowed with the selectivity and safety profile needed to proceed into clinical development.
In summary, our approach was a unique collaboration between an academic laboratory and a large pharmaceutical company that has delivered potent hits of different chemical scaffolds that may serve as starting points for further development into lead compounds suitable for in vivo efficacy animal model studies. Although many of the hits did not provide much scope for immediate work because of poor selectivity, toxicity, or DMPK profiles, their properties could be improved with chemical modifications. Importantly, this work demonstrated that antifolates targeting DHFR have not been exhaustively mined for potent hits against Mtb and that continued efforts could identify antifolates to be introduced into the TB drug discovery pipeline.
METHODS
Cheminformatic Generation of the Antifolate Library
The curation of the antifolate library used a series of analoueg searches employing 2D topological fingerprints and Tanimoto similarities, as well as substructure searches using SMARTS queries (SMARTS Theory Manual, Daylight Chemical Information Systems, Santa Fe, NM,USA). Both types of calculations were run using in-house applications employing the ChemAxon toolkit (http://www.chemaxon.com).
Mtb Low-Throughput Microplate Alamar Blue Assay (MABA)
Mtb strain H37Rv was grown at 37 °C in Middlebrook 7H9 broth supplemented with 0.025% Tween 80 and 10% ADC or on Middlebrook 7H10 plates supplemented with 10% OADC. Plates (96-well flat-bottom polystyrene microtiter plates) were prefilled with 95 µL of Middlebrook 7H9 medium. Into this was added 5 µL of neat DMSO or compound dilution made in DMSO. For each compound 10 2-fold dilutions were prepared, and each dilution series was added to one row across the plate in columns 1–10. In each plate were plated seven test compounds in rows A–G along with a dilution series of isoniazid (INH; 2-fold dilutions starting at 160 µg/mL) as a control in row H. In each plate columns 11 and 12 were used for blank and growth controls (5 µL of rifampicin or 5 µL of neat DMSO was added to columns 11 and 12, respectively, for blank and growth controls). Fresh H37Rv cultures at logarithmic growth were diluted to approximately 1 × 107 CFU/mL by measuring the optical density of the culture and then further diluted 1:100 in Middlebrook 7H9 medium to a final inoculum of 1 × 105 CFU/mL. All 96 wells in the plate were inoculated with 100 µL of the inoculum, sealed, placed in a closed box to prevent drying out, and incubated without shaking at 37 °C for 6 days. A resazurin solution was prepared using resazurin tablets (Resazurin Tablets for Milk Testing ref 330884Y VWR International Ltd.) dissolved in sterile phosphate-buffered saline (PBS). Twenty-five microliters of this solution was added to each well, and after a further 48 h of incubation at 37 °C, fluorescence was measured (Spectramax M5Molecular Devices, excitation at 530 nm, emission at 590 nm, cutoff at 570 nm).
Mtb High-Throughput Screen
For screening of compounds at a single concentration 384-well polystyrene flat-bottom black plates with clear bottoms were prefilled with 500 nL of neat DMSO or compound dissolved in DMSO (neat DMSO was plated in column 6, 0.5 mg/mL rifampicin in column 18, and 1 mM test compound in all remaining wells). H37Rv inoculum of 1 × 105 CFU/mL was prepared as above, and 25 µL was plated in each well of the plate using a liquid-handling machine (Multidrop from Thermo Scientific). The final concentrations were 20 µM for test compounds and 0.5 µg/mL for rifampicin. Plates were sealed, placed in a closed box to prevent drying out, and incubated at 37 °C without shaking for 7 days. The ATP-dependent fluorescent reagent (Bactiter Glo from Promega) was prepared according to the manufacturer’s instructions, and 10 µL of the solution was added to each well using the Multidrop liquid-handling equipment. Plates were incubated for 30 min at room temperature (covered in foil), and then luminescence was measured (Spectramax M5Molecular Devices). The percent inhibition of Mtb growth in each well was calculated on the basis of the growth and blank controls in columns 6 and 18, respectively.
Dose–response studies of compounds in high-throughput format were very similar to the single-concentration studies described above, except that for each compound an 11-point 2-fold serial dilution was performed, beginning at a stock of 10 mM, and 250 nL of each dilution was preplated into the 384-well assay plate. The dilution series was plated across the plate in a given row either in columns 1–5 and 7–12 or in columns 13–17 and 19–24, allowing for a total of 32 dose–response curves in a single 384-well plate. After the addition of 25 µL of H37Rv inoculum, the final concentrations ranged from 100 to 0.098 µM for test compounds.
Intracellular Assay of Mtb Clearance
Mycobacterial Preparation
Prior to the day of THP-1 cell infection, the Mtb subcultures were prepared for an OD600 of 0.6–1.0 at infection. The subcultures were pelleted from 160 mL (in five 50 mL sterile tubes) of mycobacterial cultures by centrifugation at 3500 rpm for 10 min. The bacterial pellets were aseptically dispersed (60 s of shaking with 10 glass beads; diameter = 3 mm), and 6 mL of RPMI-1640 was added to each tube for 5 min. The 5 mL of supernatant from each was pooled and centrifuged at 1500 rpm for 8 min. The OD600 of the supernatant containing the dispersed bacterial suspension was measured at 1:3 or 1:4 dilutions in RPMI-1640 with 0.05% Tween 80. The multiplicity of infection of the THP-1 cells was calculated on the basis that an OD600 of 0.1 represents 1.0 × 107 CFU/mL.
THP-1 Cell Preparation
THP-1 cells were maintained in suspension in RPMI-1640 containing 10% fetal bovine serum, 1 mM pyruvate, and 2 mM l-glutamine (RPMI complete medium), at 37 °C in 5% CO2 at 95% relative humidity. At subconfluence (5 × 105 cell/mL), the THP-1 cells were infected in a cell roller bottle for 4 h with a multiplicity of infection of 1.0. The excess bacteria were removed by washing the cells five times in RPMI-1640 (1500 rpm for 5 min). Following the final resuspension in RPMI complete medium, 100 µL/well (ca. 50000 cells/well) of the infected cells was dispensed into each well of 96-well white plates, and the serially diluted antimycobacterial agents (at a further 1:3 dilution) were added to each well. The DMSO was maintained under 0.5% by 1:400 dilution in culture medium. After 5 days, the luminescence was measured using Steady-Glo Promega kits in a Victor 1420 system, according to the manufacturer’s instructions. The data were processed using Excel and Grafit software. The MIC90 values were calculated from the dose-response curves, using nonlinear regression analysis.
THP1 Cytotoxicity Assay
THP-1 cell line (ATCC TIB-202) was cultured in RPMI-1640 medium supplied with 10% FBS, 1 mM sodium pyruvate, and 2 mM l-glutamine and incubated at 37 °C, 5% CO2, and 95% humidity. Cells were harvested by centrifugation (1500 rpm for 5 min at room temperature), resuspended in growth medium supplied with 5% FBS, and counted using a cell counter. A suspension with 50000 cells/mL was prepared using the same medium specification, and then 50 µL of this suspension was transferred to each well of a black 96-well microplate with clear bottom tissue culture treated with collagen type 1 (except in column 11, where only 50 µL of the growth medium was added). A serial dilution in DMSO of the tested compound (concentration = 0.1–50 µM) was made in a V-bottom 96-well cell culture plate. Then, using a flat-bottom 96-well cell culture plate, 2.0 µL of each drug dilution was added to 200 µL of RPMI-1640 medium supplemented with 5% FBS, 1 mM sodium pyruvate, and 2 mM l-glutamine. At column 12, only DMSO was added, and the final concentration was 0.5%. Serial dilution of doxorubicin was used as an internal control of the assay. In column 11 only medium was added. Then 50 µL of the medium containing the dilutions of the compounds/DMSO was added to the plate containing the cells. Each compound serial dilution was added in two replicates. The plate was then incubated for 48 h at 37 °C, 5% CO2, and 95% humidity.
After 2 days of incubation, the solubility of the compounds was checked. Then 100 µL of reconstituted substrate solution of an ATP monitoring assay kit was added per well. Then the plate was shaken for 1 min, and the luminescence was measured using a microplate reader (1420 Multilabel HTS counter Victor Wallac). The cytotoxicity was estimated using a sigmoidal dose–response curve, plotting percentage of growth inhibition versus log of compound concentration using GraphPad software. The Tox50 value corresponds to the concentration of the compound necessary to inhibit 50% of cell growth.
Biochemical Assay of Mtb and Human DHFR
Measurement of DHFR activity was performed essentially as described previously.14 Recombinant Mtb DHFR was cloned and isolated from Escherichia coli as previously described,14 and recombinant human DHFR was purchased from Sigma (catalog no. D6566). The concentration required for 50% inhibition of enzyme activity (IC50) was determined in a biochemical fluorescence intensity assay. Assays were conducted in 384-well flat-bottomed plates in a final volume of 40 µL containing 20 µM dihydrofolate, 50 µM NADPH, 150 mM KCl, and either 0.5 µg/mL Mtb DHFR or 1.2 µg/mL human DHFR, all in 40.7 mM HEPES (pH 7.8) buffer. Substrate-free reactions were used as a negative control for activity, and 2.5% final concentration of DMSO was used as a positive control for activity. Enzyme activity was determined by measuring the rate of substrate depletion; the cofactor NADPH has an absorbance spectrum at 340 nm and is oxidized by DHFR into NADP+, which has no absorbance at this wavelength. The rate of NADPH depletion was measured by monitoring the absorbance at 340 nm and the Vmax for each curve was calculated in Spectramax Pro. The percent activity was calculated as (Vmax(x) − Vmax(−))/(Vmax(+) − Vmax(−)). IC50 values were calculated with nonlinear curve fitting in Graphpad Prism.
HepG2 Cytotoxicity Assays
The day before the subculturing of the plates, the HepG2 (HB-8065) cell line was cultured in essential minimum Eagle’s medium supplemented with 5% fetal calf serum and 2 mM l-glutamine. On the day of the assay, the cells (10000 cells/well) were seeded into clear-bottomed, black 96-well collagen-coated microplates (Becton Dickinson), including 100 µL of culture medium controls. Stock solutions for each test substance were prepared in 100% DMSO. Ten serial 1:2 dilutions in medium were carried out for each test compound, with a final 1:200 dilution, in a final concentration of 0.5% DMSO. Resazurin tablets (Merck) were dissolved in phosphate-buffer saline at 0.0042%.
After a 24 h incubation at 37 °C in 5% CO2 at 95% relative humidity, 150 µL of culture medium containing the appropriate test compound concentrations was added to the cells, in duplicates; 150 µL of 0.5% DMSO was also added as the blank control. Following a 48 h incubation at 37 °C in 5% CO2 at 95% relative humidity, the medium was removed from these samples, and Resazurin solution was added to each well, followed by incubation for a further 1.5 h. The fluorescence was then measured at an excitation wavelength of 515 nm and an emission wavelength of 590 nm, in a microplate reader (1420 Multilabel HTS counter, Victor 2; Wallac). The fluorescence from each well was corrected by background subtraction (mean of the blank controls). Inhibition is expressed as the means of the duplicates, each relative to the mean of the DMSO controls wells. The data were processed using Excel and GraphPad software analysis. The Tox50 values were calculated from the sigmoidal dose–response (variable slope) curves, using nonlinear regression analysis. We ensured that all human biological samples used for these in vitro studies were collected and transferred in accordance with all applicable national laws including those that relate to ethical standards.
Microsomal Fraction Stability Experimental Procedure
Pooled mouse liver microsomes were purchased from Xenotech. Microsomes (final protein concentration = 0.5 mg/ mL), MgCl2 (final concentration = 5 mM), and test compound (final substrate concentration = 0.5 µM; final DMSO concentration = 0.5%) in 0.1 M phosphate buffer (pH 7.4) were pre-incubated at 37 °C prior to the addition of NADPH (final concentration = 1 mM) to initiate the reaction. The final incubation volume was 600 µL. All incubations were performed singularly for each test compound. Each compound was incubated for 30 min, and samples (90 µL) of incubate were taken at 0, 3, 6, 9, 12, 15, 18, 21, 24, 27, and 30 min. The reactions were stopped by the addition of sample to 200 µL of acetonitrile/methanol (3:1) containing an internal standard. The terminated samples were centrifuged at 3700 rpm for 15 min at 4 °C to precipitate the protein. Quantitative analysis was performed following protein precipitation; the samples were analyzed using specific LC-MS/MS conditions. Data analysis was performed from a plot of ln peak area ratio (compound peak area/internal standard peak area) against time, and the gradient of the line was determined. Subsequently, half-life and intrinsic clearance were calculated using the equations
where V = incubation volume mL/g microsomal protein.
Cytochrome P450 (CYP450)
The P450 inhibition profiles of the DHFR inhibitors were determined as previously described by Kajbaf et al39.
In Vivo Assays
All experimental work with animals was performed by following the 3R principles, replacing, reducing, and refining animal testing, and took place in our Association for the Assessment of Laboratory Animal Care International (AAALAC-1)-accredited facilities under the control and approval of the Tres Cantos GSK local ethics committee. All animal studies were ethically reviewed and carried out in accordance with European Directive 2010/63/EU and the GSK Policy on the Care, Welfare and Treatment of Animals.
Murine DMPK Assays
The compound was assayed at 5 and 500 mg/kg (po) in mice. For this pharmacokinetic studies CD1 male mice of 25–30 g weight were used (n = 3 mice per oral dose). Experimental compound was administered by oral gavage at 5 and 500 mg/kg single dose at a volume of 10 mL/ kg (n = 3 mice per oral dose). All mice received treatment in the fed state. Compound was administered as 1% methyl cellulose suspension. Peripheral total blood was the compartment chosen for the establishment of compound concentrations: aliquots of 20 µL of blood were taken from the caudal vein at 15, 30, and 45 min and 1, 2, 4, 8, 24, and 48 h. LC-MS was used as the analytical method of choice for the establishment of compound concentration in blood with a sensitivity of LLQ = 1–5 ng/mL in 20 µL of blood. The noncompartmental data analysis (NCA) was performed with WinNonlin Phoenix 6.3 (Pharsight, Certara LP), and supplementary analysis was performed with GraphPad Prism 6 (GraphPad Software, Inc.).
Acute Murine Model of Intratracheal Mtb Infection
Mice
C57BL/6 specific pathogen free (spf) 6–8-week-old female mice were obtained from Harlan Iberica (St. Feliu de Codines, Spain). They were shipped in suitable travel conditions, with the corresponding certificate of health and origin, and were allowed to acclimate for 1 week. All of the animals were kept under controlled conditions in a P3 high-security facility with sterile food and water ad libitum.
Infection and Antibiotic Administration
The experimental design has been previously described.40 In brief, mice were intratracheally infected with 100,000 CFU/mouse (Mtb H37Rv strain). Products were administered once a day for 8 consecutive days starting 1 day after infection. Moxifloxacin was provided by Sequoia Research Products (United Kingdom) and diluted in 20% (2-hydroxypropyl)-b-cyclodextrin (Sigma-Aldrich). DQn-1 was formulated as a suspension in 1% methyl cellulose (Sigma, Madrid, Spain), and three mice were tested per dose at 5 and 10 mg/kg.
CFU Determination
Lungs were harvested on day 9 after infection, 24 h after the eighth administration. All lung lobes were aseptically removed, homogenized, and frozen. Special care was taken not to include hiliar lymph nodes at the time of removal of the lung in order to not artificially increase the CFU value. The number of viable bacteria in lung homogenates was measured by plating serial dilutions on nutrient Middlebrook 7H11 agar (Biomedics s.l., Madrid, Spain) supplemented with 10% OADC and 0.4% activated charcoal. CFUs were counted after incubation for 18 days at 37 °C.
Animal Health
Mice were supervised every day under a protocol paying attention to weight loss, apparent good health (bristled hair and wounded skin), and behavior (signs of aggressiveness or isolation). Mice showing clear signs of toxicity were sacrificed and excluded from the analysis. Animals were euthanized by CO2 inhalation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Reiling Liao for expert technical help. The research leading to these results has received funding from the Tres Cantos Open Lab Foundation (program code TC027).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.5b00063.
Results of the in vivo DMPK studies and in vivo efficacy study with DQn-1 (PDF)
Author Contributions
A.K., R.R.G., R.M.G., and P.T. performed all of the in vitro experiments; F.O., E.J., and J.R. performed all of the in vivo experiments; A.G. and R.C.G. performed the chemical analyses of hits; G.C. and A.G. did the in silico curation of the compound library; A.K., A.G., E.P., D.C., L.B., and D.R.S. designed the study and analyzed the data; A.K., A.G., and D.R.S. prepared the manuscript.
The authors declare no competing financial interest.
Contributor Information
Ana Guardia, Email: ana.a.guardia@gsk.com.
David R. Sherman, Email: david.sherman@cidresearch.org.
REFERENCES
- 1.Rattan A, Kalia A, Ahmad N. Multidrug-resistant Mycobacterium tuberculosis: molecular perspectives. Emerging Infect. Dis. 1998;4(2):195–209. doi: 10.3201/eid0402.980207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Becerra MC, et al. Redefining MDR-TB transmission ‘hot spots’. Int. J. Tuberc. Lung Dis. 2000;4(5):387–394. [PubMed] [Google Scholar]
- 3.WHO. Global Tuberculosis Control: WHO Report. Geneva, Switzerland: 2014. [Google Scholar]
- 4.Stover CK, et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405(6789):962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 5.Jia L, et al. Pharmacodynamics and pharmacokinetics of SQ109, a new diamine-based antitubercular drug. Br. J. Pharmacol. 2005;144(1):80–87. doi: 10.1038/sj.bjp.0705984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makarov V, et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science. 2009;324(5928):801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andries K, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis . Science. 2005;307(5707):223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 8.Provisional CDC guidelines for the use and safety monitoring of bedaquiline fumarate (Sirturo) for the treatment of multidrug-resistant tuberculosis. MMWR Recomm. Report. 2013;62(RR-09):1–12. [PubMed] [Google Scholar]
- 9.Kompis IM, Islam K, Then RL. DNA and RNA synthesis: antifolates. Chem. Rev. 2005;105(2):593–620. doi: 10.1021/cr0301144. [DOI] [PubMed] [Google Scholar]
- 10.Hitchings GH, Smith SL. Dihydrofolate reductases as targets for inhibitors. Adv. Enzyme Regul. 1980;18:349–371. doi: 10.1016/0065-2571(80)90025-4. [DOI] [PubMed] [Google Scholar]
- 11.Barr J. A short history of dapsone, or an alternative model of drug development. J. Hist. Med. Allied Sci. 2011;53(4):123–147. doi: 10.1093/jhmas/jrq068. [DOI] [PubMed] [Google Scholar]
- 12.Chakraborty S, et al. p-Aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis . Science. 2013;339(6115):88–91. doi: 10.1126/science.1228980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng J, et al. p-Aminosalicylic acid is a prodrug targeting dihydrofolate reductase in Mycobacterium tuberculosis . J. Biol. Chem. 2013;288(32):23447–23456. doi: 10.1074/jbc.M113.475798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar A. High-throughput screening and sensitized bacteria identify an M. tuberculosis dihydrofolate reductase inhibitor with whole cell activity. PLoS One. 2012;7(6):0039961. doi: 10.1371/journal.pone.0039961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nixon MR, et al. Folate pathway disruption leads to critical disruption of methionine derivatives in Mycobacterium tuberculosis . Chem. Biol. 2014;21(7):819–830. doi: 10.1016/j.chembiol.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Gerum AB, et al. Novel Saccharomyces cerevisiae screen identifies WR99210 analogues that inhibit Mycobacterium tuberculosis dihydrofolate reductase. Antimicrob. Agents Chemother. 2002;46(11):3362–3369. doi: 10.1128/AAC.46.11.3362-3369.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canfield CJ. New antimalarial under development. In: Bruce-Chevatt LJ, editor. Chemotherapy of Malaria. Vol. 27. Geneva, Switzerland: World Health Organization Monograph Series; 1986. [Google Scholar]
- 18.Malarone-GSK Product Detail Page. available from www.gsksource.com/pharma/content/gsk/source/us/en/brands/malarone.html.
- 19.Sharma M, Chauhan PM. Dihydrofolate reductase as a therapeutic target for infectious diseases: opportunities and challenges. Future Med. Chem. 2012;4(10):1335–1365. doi: 10.4155/fmc.12.68. [DOI] [PubMed] [Google Scholar]
- 20.Wright DL, Anderson AC. Antifolate agents: a patent review (2006–2010) Expert Opin. Ther. Pat. 2011;21(9):1293–1308. doi: 10.1517/13543776.2011.587804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gamo FJ, et al. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465(7296):305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 22.Ballell L, et al. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem. 2013;8(2):313–321. doi: 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin AJ, et al. Synthesis and antimalarial activity of pyrrolo[3,2-f]quinazoline-1,3-diamine derivatives, Google Patents. 2007 [Google Scholar]
- 24.O’Brien PJ, et al. High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch. Toxicol. 2006;80(9):580–604. doi: 10.1007/s00204-006-0091-3. [DOI] [PubMed] [Google Scholar]
- 25.Young RJ, et al. Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discovery Today. 2011;16(17–18):822–830. doi: 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today. 2004;9(10):430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 27.Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery. 2007;6(11):881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 28.Kostewicz ES, et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014;57:342–366. doi: 10.1016/j.ejps.2013.08.024. [DOI] [PubMed] [Google Scholar]
- 29.Ince I, et al. Developmental changes in the expression and function of cytochrome P450 3A isoforms: evidence from in vitro and in vivo investigations. Clin. Pharmacokinet. 2013;52(5):333–345. doi: 10.1007/s40262-013-0041-1. [DOI] [PubMed] [Google Scholar]
- 30.Bowes J, et al. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat. Rev. Drug Discovery. 2012;11(12):909–922. doi: 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- 31.Lipinski CA, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 32.Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technol. 2004;1(4):337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 33.Hann MM, Keseru GM. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discovery. 2012;11(5):355–365. doi: 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- 34.Arnott JA, Planey SL. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discovery. 2012;7(10):863–875. doi: 10.1517/17460441.2012.714363. [DOI] [PubMed] [Google Scholar]
- 35.O’Shea R, Moser HE. Physicochemical properties of antibacterial compounds: implications for drug discovery. J. Med. Chem. 2008;51(10):2871–2878. doi: 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- 36.Li R, et al. Three-dimensional structure of M. tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. J. Mol. Biol. 2000;295(2):307–323. doi: 10.1006/jmbi.1999.3328. [DOI] [PubMed] [Google Scholar]
- 37.Dias MV, et al. Mycobacterium tuberculosis dihydrofolate reductase reveals two conformational states and a possible low affinity mechanism to antifolate drugs. Structure. 2014;22(1):94–103. doi: 10.1016/j.str.2013.09.022. [DOI] [PubMed] [Google Scholar]
- 38.El-Hamamsy MH, et al. Structure-based design, synthesis and preliminary evaluation of selective inhibitors of dihydrofolate reductase from Mycobacterium tuberculosis . Bioorg. Med. Chem. 2007;15(13):4552–4576. doi: 10.1016/j.bmc.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 39.Kajbaf M, et al. A comparative study of the CYP450 inhibition potential of marketed drugs using two fluorescence based assay platforms routinely used in the pharmaceutical industry. Drug Metab. Lett. 2011;5(1):30–39. doi: 10.2174/187231211794455262. [DOI] [PubMed] [Google Scholar]
- 40.Rullas J, et al. Fast standardized therapeutic-efficacy assay for drug discovery against tuberculosis. Antimicrob. Agents Chemother. 2010;54(5):2262–2264. doi: 10.1128/AAC.01423-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.