

Summary
Neuronal growth cone filopodia contain guidance receptors and contribute to axon guidance, however the mechanism by which the guidance cue netrin increases filopodia density is unknown. Here we demonstrate that TRIM9, an E3 ubiquitin ligase that localizes to filopodia tips and binds the netrin receptor DCC, interacts with and ubiquitinates the barbed-end polymerase VASP to modulate filopodial stability during netrin-dependent axon guidance. Studies with murine TRIM9+/+ and TRIM9−/− cortical neurons, along with a non-ubiquitinatable VASP mutant, demonstrate that TRIM9-mediated ubiquitination of VASP reduces VASP filopodial tip localization, VASP dynamics at tips, and filopodial stability. Upon netrin treatment, VASP is deubiquitinated, which promotes VASP tip localization and filopodial stability. TRIM9 deletion induces axon guidance defects in vitro and in vivo, whereas a gradient of deubiquitinase inhibition promotes axon turning in vitro. We conclude that a gradient of TRIM9-mediated ubiquitination of VASP creates a filopodial stability gradient during axon turning.
Graphical Abstract

INTRODUCTION
During embryonic development, growth cones at the tips of extending axons respond to extracellular cues to direct axon growth (Kolodkin and Tessier-Lavigne, 2011). In the mammalian cortex, the secreted guidance cue netrin-1 (netrin) and its receptor DCC (deleted in colorectal cancer) promote attractive axon guidance (Richards et al., 1997; Stein, 2001) and deficiency of the murine gene encoding netrin-1 or DCC induces cortical projection defects (Fazeli et al., 1997; Serafini et al., 1996). DCC localizes to the tips of filopodia (Shekarabi and Kennedy, 2002), bundled filamentous actin (F-actin) rich protrusions that decorate the growth cone periphery and contribute to axon guidance. Furthermore, DCC is required for netrin-dependent increases in filopodia density (Lebrand et al., 2004). In addition to guidance receptors, the filopodia tip complex contains cytoskeletal regulatory proteins that modulate filopodial growth and stability (Gupton and Gertler, 2007). Cytoskeletal dynamics contribute to the extension and turning of growth cones, but how the function of the tip complex is regulated by netrin is not known.
Ena/VASP actin regulatory proteins localize within the tip complex and are essential in netrin response, filopodial formation, neuritogenesis and axon fiber tract formation in the murine cortex (Dent et al., 2007; Kwiatkowski et al., 2007; Lebrand et al., 2004). Mammals have three Ena/VASP orthologs: Mena, VASP, and EVL (Kwiatkowski et al., 2007; Lanier et al., 1999), which promote formation of unbranched F-actin through binding and protecting the barbed end from capping and promoting polymerization (Barzik et al., 2005; Bear et al., 2002; Breitsprecher et al., 2008; Hansen and Mullins, 2010). This family is characterized by an N-terminal Ena/VASP Homology 1 (EVH1) domain that binds proteins with the sequence (D/E)FPPPPX(D/E)(D/E) (abbreviated FP4), a proline-rich (Pro) domain, and an EVH2 domain that binds monomeric and F-actin and mediates tetramerization (Krause et al., 2003). Ena/VASP function is required for netrin-dependent increases in growth cone filopodia (Lebrand et al., 2004). Ena/VASP proteins are acutely phosphorylated in response to netrin, however this phosphorylation is not detected when filopodia density increases, suggesting unidentified mechanisms regulate Ena/VASP function.
We identified murine TRIM9 as a direct binding partner of DCC that regulates cortical axon branching in response to netrin (Winkle et al., 2014). Like Ena/VASP, TRIM9 localizes to filopodia tips in cortical neurons. The interaction between TRIM9 and DCC is conserved in invertebrates, where the TRIM9 ortholog is required for netrin responses (Hao et al., 2010; Morikawa et al., 2011). Their similar localization and requirement in netrin responses suggests that TRIM9 and Ena/VASP may cooperate within filopodia in response to netrin. TRIM9 is a member of the tripartite motif (TRIM) family of E3 ubiquitin ligases, which mediate covalent linkage of ubiquitin to substrates. Ubiquitin addition can trigger proteasomal degradation or alternatively modify substrate localization, trafficking or function (Chau et al., 1989; Didier et al., 2003; Schaefer et al., 2012). Additionally, ubiquitination can be reversed by deubiquitinases (DUBs, Reyes-Turcu et al., 2009). Although TRIM9 exhibits ligase activity (Tanji et al., 2010), its substrates and the consequences of its ligase activity are unknown.
Here we show that TRIM9, which lacks an FP4 motif, exhibits a distinct mode of direct interaction with the EVH1 domain of Ena/VASP proteins. We find that VASP is ubiquitinated in the presence of TRIM9, but not Mena or EVL, and that VASP is deubiquitinated upon netrin stimulation. Although neither TRIM9 nor netrin treatment altered the stability of VASP protein, they differentially altered VASP localization and mobility at filopodia tips. Inhibition of DUB activity or expression of a non-ubiquitinatable VASP mutant supports the hypothesis that TRIM9-mediated ubiquitination alters VASP localization, filopodial stability, and filopodia density. We show that deletion of TRIM9 disrupts attractive axon turning within a netrin gradient, whereas a gradient of DUB inhibition, and thus of ubiquitination, was sufficient to repulse axons. We propose that TRIM9 function and a netrin gradient create a gradient of VASP ubiquitination across the growth cone, and thus spatial differences in filopodia stability and density that promote extension toward netrin.
RESULTS
Identification of TRIM9 as a novel Ena/VASP interaction partner
A yeast two-hybrid screen using an embryonic mouse brain cDNA library and EVL as bait identified four independent clones containing sequences corresponding to amino acids 45-532 of TRIM9. TRIM proteins share a conserved N-terminal TRIM motif with an E3 ubiquitin ligase RING domain, 1-2 BBox domains, and a coiled-coil (CC) domain that mediates homo- and hetero-multimerization (Fig1A). In TRIM9, the TRIM motif is followed by a COS box, fibronectin type III (FN3) and SPRY domains (Short and Cox, 2006). Since the SPRY domain of TRIM9 directly interacts with DCC (Winkle et al., 2014), the interaction between Ena/VASP and TRIM9 may link netrin to filopodia.
Figure 1. TRIM9 is a brain-enriched Ena/VASP interaction partner.
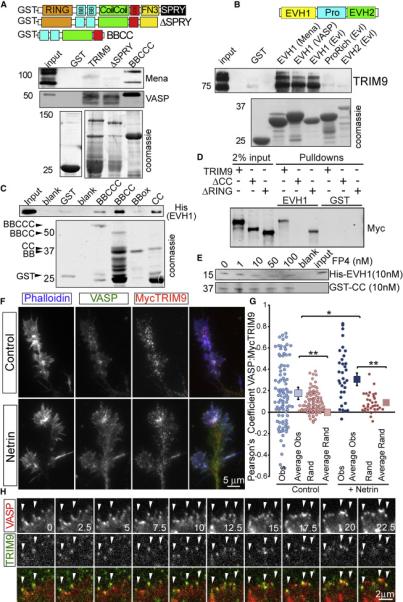
A-B) Binding assays with purified GST fusion proteins incubated in E15.5 brain lysate. Coomassie stained gels of recombinant proteins shown in lower panels. A) GST-BBox-coiled-coil-COS (BBCCC) of TRIM9 precipitates endogenous Mena and VASP. B) The GST-EVH1 domain of Mena, VASP and EVL interacts with endogenous TRIM9. C) In vitro binding assay showing that GST-Coiled Coil (CC) domain of TRIM9 directly binds His-EVH1 domain of VASP. D) Binding assay showing that GST-EVH1 precipitates Myc-TRIM9 and Myc-TRIM9ΔRING from cell lysate, but not Myc-TRIM9ΔCC. E) A 10-fold excess of FP4 containing peptide does not block the CC-EVH1 interaction. F) Axonal growth cones of control and netrin-treated cortical neurons stained for VASP (green), MycTRIM9 (red) and phalloidin (blue). G) Quantification of Pearson's correlation coefficient within filopodia (Obs=Observed measurements, Rand=pixels from one image randomized). Squares represent means +/− 95% CI. H) Montage of TIRF images of GFP-VASP (red) and mCherry-TRIM9 (green). Arrowheads denote filopodia tips in which TRIM9 and VASP colocalize, time in seconds. (See also Movies S1-3, FigS1)
TRIM9 binds and colocalizes with Ena/VASP proteins in cortical neurons
To confirm the interaction between TRIM9 and Ena/VASP predicted by yeast two hybrid, we incubated GST-tagged TRIM9 variants in embryonic mouse brain lysate (Fig1A). A GST-tagged protein containing the BBox-CC-COS domains of TRIM9 (BBCCC) bound endogenous Mena and VASP, whereas GST did not. GST-TRIM9 and a variant lacking the SPRY domain (ΔSPRY) failed to precipitate Mena or VASP. Even though TRIM9 lacks an FP4 motif, GST-EVH1 domains of all three Ena/VASP members precipitated endogenous TRIM9 from embryonic brain lysate, but GST-Pro or GST-EVH2 did not (Fig1B). This pattern of binding may suggest conformational changes or post-translational modifications in TRIM9 or Ena/VASP proteins are required to permit binding, or that terminal domains of the proteins modulate binding. However, direct binding assays demonstrated that GST-BBCCC was able to precipitate His-EVH1 (Fig1C). This interaction was maintained with GST-BBCC and GST-CC, but not GST-BBox, indicating that the CC domain is the minimal binding region of TRIM9. We confirmed this observation with precipitation of Myc-TRIM9 variants expressed in HEK293 cells (Fig1D). GST-EVH1 precipitated Myc-TRIM9 and Myc-TRIM9 lacking the RING domain (TRIM9ΔRING) but not Myc-TRIM9 lacking the CC motif (TRIM9ΔCC). Tes and Abi are the only identified EVH1 interacting proteins that lack an FP4 motif (Boëda et al., 2011; Chen et al., 2014). Unlike Tes and Abi, which exhibit competitive binding with FP4, His-EVH1 continued to interact with GSTCC in the presence of a 10-fold excess of FP4 (Fig1E). Although these biochemical assays confirm the direct binding predicted by the yeast-two hybrid screen, we were unable to co-immunoprecipitate endogenous TRIM9 and VASP, suggesting this interaction may be regulated or transient in vivo.
TRIM9 and Ena/VASP localize to the tips of filopodia, which decorate the periphery of neuronal growth cones (Lanier et al., 1999; Winkle et al., 2014). TRIM9ΔCC fails to localize to filopodia tips (Winkle et al., 2014), suggesting the interaction with Ena/VASP proteins may be required for TRIM9 tip localization. Immunocytochemistry of endogenous VASP (Fig1F) or Mena (Fig1SA) with Myc-tagged TRIM9 in embryonic mouse cortical neurons revealed limited colocalization within filopodia. The observed colocalization coefficient was lower than typically reported (Fig1G, Obs ~0.2), but greater than measured following pixel randomization (Rand, p<.05), suggesting only a subset of proteins colocalized within filopodia. Live-cell imaging via total internal reflection fluorescence (TIRF) microscopy revealed transient colocalization of mCherry-TRIM9 and GFP-VASP at filopodia tips (Fig1H, MovieS1). Transient interaction often occurs between E3 ligases and substrates. Consistent with this, a mutant lacking the ligase domain (TRIM9ΔRING) exhibited prominent colocalization with GFP-VASP (MovieS2). GFP-Mena also colocalized with TRIM9 in dynamic filopodia (FigS1C, MovieS3), although Mena lacked distinct tip localization.
TRIM9 ubiquitinates VASP but not Mena or EVL
We first used HEK293 cells to determine if Ena/VASP proteins were ubiquitinated in the presence of TRIM9; however endogenous TRIM9 complicated these experiments. Therefore we generated TRIM9−/− cells via CRISPR/Cas genome editing (Fig2A). Clone 3 contained mutations in both TRIM9 alleles and had no detectable TRIM9, whereas clone 1 exhibited partial expression (Fig2A). We expressed HA-DCC, GFP-VASP and FLAG-ubiquitin in 293-WT (TRIM9+/+) and clone 3 (TRIM9−/−) cells. Upon immunoprecipitating GFP-VASP, in addition to the expected 75kDa band, we observed a ~25kDa heavier GFP band that co-migrated with ubiquitin in TRIM9+/+ cells, indicative of VASP ubiquitination (Fig2B). Either netrin stimulation or deletion of TRIM9 reduced GFP-VASP ubiquitination (p<.05), suggesting TRIM9 ubiquitinated VASP, and this modification was lost in the presence of netrin. The netrin-dependent decrease in VASP ubiquitination was not observed in cells lacking HA-DCC (Fig S2A), indicating that loss of ubiquitination occurred downstream of DCC. Although Ena/VASP proteins are often functionally redundant, we did not detect ubiquitination of GFP-Mena or GFP-EVL (Fig S2B-C). To confirm ubiquitination of VASP was relevant to cortical neurons, we performed similar experiments by immunoprecipitating endogenous VASP and blotting for ubiquitin. A higher molecular weight VASP+ band that co-migrated with ubiquitin was similarly lost upon netrin treatment or deletion of TRIM9 (Fig2C). Although ubiquitination can decrease substrate half-life via proteasomal-degradation, we observed no change in VASP protein in embryonic cortical neurons cultured from TRIM9+/+ or TRIM9−/− mice (Fig2D). Furthermore neither netrin stimulation nor proteasome inhibition altered VASP protein levels. Without evidence for degradation of VASP, we hypothesized that VASP was deubiquitinated in the presence of netrin. To test this, we inhibited DUB activity with PR-619, a cell permeable inhibitor of DUBs that increases levels of ubiquitinated proteins with little inhibitory effect on other proteases (Altun et al., 2011; Seiberlich et al., 2012). DUB inhibition blocked the netrin-dependent reduction in VASP ubiquitination in cortical neurons (Fig2E). Based on bioinformatic predictions and conservation across VASP orthologs, we mutated single lysines to arginine within VASP to identify ubiquitination sites, but this approach failed to reduce VASP ubiquitination (FigS2D). Since E3 ligases can promiscuously ubiquitinate lysine residues on substrates (Rodriguez et al., 2000), we combined nine lysine mutations based on bioinformatic analysis and mass spectrometry studies (Danielsen et al., 2011). This mutant (VASP K-R) exhibited significantly reduced ubiquitination (Fig2F). Together these results suggest that TRIM9 specifically ubiquitinates VASP and that VASP is deubiquitinated upon netrin treatment. E3 ligases and their substrates typically interact transiently (Kim et al., 2015); this may account for the low, transient colocalization (Fig1G) of these proteins and their inability to co-immunoprecipitate.
Figure 2. VASP ubiquitination occurs in the presence of TRIM9 and is lost following netrin treatment.
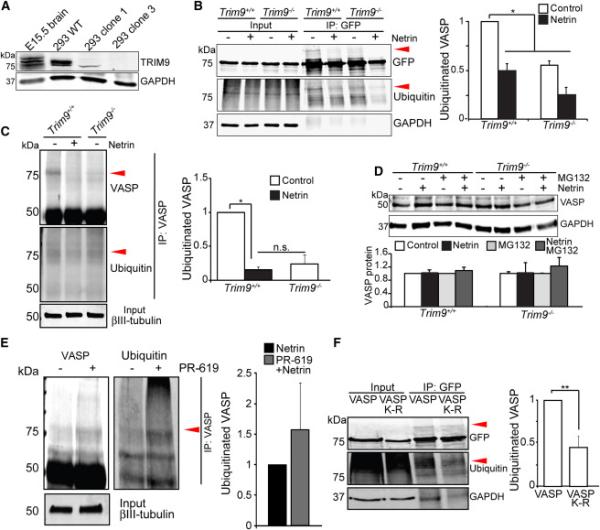
A) Immunoblot for TRIM9 and GAPDH in E15.5 brain lysates, HEK293 lysates: TRIM9+/+ (WT), TRIM9+/− (clone 1) and TRIM9−/− (clone 3). B) Input and GFP-VASP immunoprecipitation (IP) from ubiquitination assay immunoblotted (IB) for GFP, ubiquitin and GAPDH. Shown are GFP-VASP (75kDa) in TRIM9+/+ and TRIM9−/− cell lysates and ~25kDa heavier VASP band present in TRIM9+/+ lysate that co-migrates with ubiquitin (red arrowheads). Plot shows quantification of VASP ubiquitination. C) Endogenous VASP ubiquitination in TRIM9+/+ and TRIM9−/− neurons at 2DIV. A higher molecular weight VASP+ band (red arrowhead) that co-migrates with ubiquitin is seen in TRIM9+/+ cortical neurons. D) Endogenous VASP in TRIM9+/+ and TRIM9−/− control neurons or neurons treated with MG132 and/or 600 ng/ml netrin. IB for VASP (50kDa) and GAPDH. No difference in VASP protein levels detected between genotypes or treatment conditions. E) Endogenous VASP ubiquitination in TRIM9+/+ cortical neurons treated with netrin or netrin and 4 μM PR-619. Elevated ubiquitination is observed upon PR-619 treatment. F) Ubiquitination assay and quantification of wildtype VASP and VASP K-R mutant. The VASP K-R mutant exhibits a reduction in the ~25kDa heavier VASP band that co-migrates with ubiquitin (red arrowheads). All error bars represent SEM. (See also FigS2)
Deletion of TRIM9 disrupts growth cone filopodia, netrin response and VASP localization
To determine if TRIM9 regulated filopodia, we compared murine axonal growth cones from TRIM9+/+ and TRIM9−/− embryos (Fig3A). This revealed that TRIM9−/− growth cones exhibited increased area, and increased filopodia number, density and length (Fig3B-E, p<.01). Since we previously showed that TRIM9 directly interacts with DCC and was important for response to netrin (Winkle et al., 2014), we investigated growth cone response to netrin stimulation. Following netrin treatment, TRIM9+/+ growth cones increased filopodia number, density and length (Fig3B-E, p<.01), whereas TRIM9−/− growth cones (p<.01) were netrin insensitive. Pre-incubation with an antibody that recognizes an epitope within the extracellular domain of DCC impeded netrin-dependent increases in filopodia density (FigS3A, p=0.93), as previously observed (Lebrand et al., 2004). This suggests that netrin stimulated increases in filopodia are DCC-dependent. However this antibody did not change filopodia density in TRIM9−/− growth cones (FigS3B), suggesting TRIM9 regulates filopodia density downstream of DCC. To determine if VASP localization was sensitive to netrin treatment or TRIM9, we measured VASP intensity relative to phalloidin along filopodia. In TRIM9+/+ neurons, VASP localized throughout the growth cone and accumulated at filopodia tips (Fig3F). Netrin treatment or deletion of TRIM9 increased this tip ratio more than two fold (p<.01). Expression of MycTRIM9 in TRIM9−/− neurons reduced elevated tip localization and rescued netrin-dependent increases in VASP tip accumulation (Fig3GH, p<.01). MycTRIM9 also accumulated at filopodia tips, but decreased in response to netrin (Fig3I, p<.01). Although Mena and EVL accumulated at filopodia tips, both were insensitive to netrin treatment or deletion of TRIM9 (FigS3C-F). Thus TRIM9 negatively regulates VASP localization to filopodia tips, whereas netrin promotes VASP tip localization.
Figure 3. Deletion of TRIM9 increases growth cone size and filopodia density and alters VASP localization to filopodia tips.
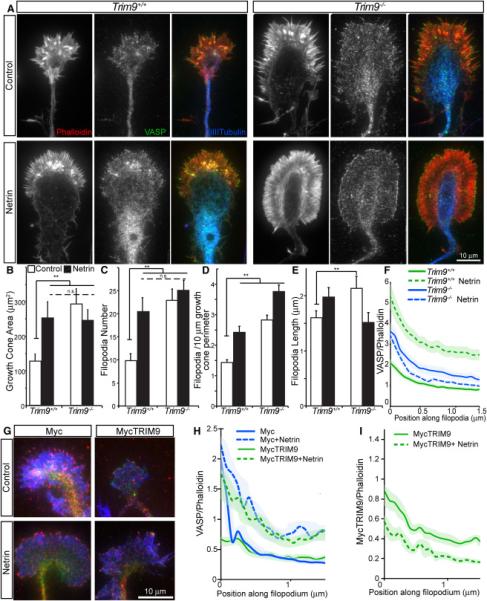
A-F) Images and quantification of axonal growth cones from control and netrin-treated TRIM9+/+ and TRIM9−/− neurons, stained for phalloidin (red, left), VASP (green, middle) and βIII tubulin (blue, merge). Quantification of B) growth cone area +/−SEM, C) growth cone filopodia number +/−SEM, D) density of growth cone filopodia +/−SEM, E) filopodia length +/−SEM, and F) VASP fluorescence intensity relative to phalloidin +/− 95% CI from the tip of filopodia into growth cone. G-I) Images and quantification of TRIM9−/− growth cones stained for VASP (red), phalloidin (blue), and Myc (green, Myc or MycTRIM9). H) VASP fluorescence intensity normalized to phalloidin +/− 95% CI from filopodia tip into growth cone. Expression of TRIM9 rescues VASP localization. I) MycTRIM9 fluorescence intensity normalized to phalloidin +/− 95% CI from filopodia tip into growth cone. (See also FigS3)
TRIM9 regulates filopodia density through VASP
To ensure that excess filopodia in TRIM9−/− growth cones required barbed end polymerization and thus were conventional, we treated neurons with a low dose of the F-actin capping drug cytochalasin D (100 nM CytoD, FigS4A). This blocks filopodia without disrupting growth cone extension (Dent and Kalil, 2001; Dent et al., 2007). As expected, netrin-dependent increases in TRIM9+/+ filopodia and excess filopodia in TRIM9−/− growth cones were blocked by CytoD (FigS4B, p<.005). To further test the specificity of TRIM9 function in netrin and DCC-dependent filopodia increases, neurons were treated with Fibroblast growth factor 2 (FGF-2), an attractive guidance cue that functions through a distinct signaling pathway (Webber et al., 2005; Zechel et al., 2010). FGF2 increased filopodia density in both TRIM9+/+ and TRIM9−/− growth cones (p<.05, Fig4AB). Expression of MycTRIM9 in TRIM9−/− neurons restored filopodia density and rescued netrin sensitivity (Fig4EF, p<.005). Expression of a mutant of TRIM9 that lacks the DCC binding domain, Myc-TRIM9ΔSPRY, reduced elevated filopodia, but failed to rescue netrin sensitivity (Fig4F, p<.005), although FGF-2 sensitivity remained intact (p<.001, Fig4AB). Together these results suggest that TRIM9 functions specifically downstream of netrin, and that the interaction between TRIM9 and DCC is required for increases in filopodia density specifically in response to netrin.
Figure 4. TRIM9 constrains filopodia density through VASP.
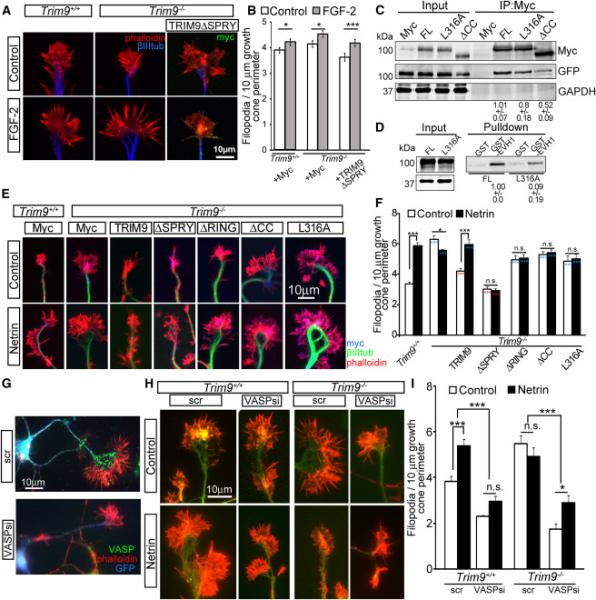
A-B) Images and quantification of filopodia +/−SEM in axonal growth cones from control and FGF2-treated TRIM9+/+ and TRIM9−/− cortical neurons expressing Myc or MycTRIM9ΔSPRY, stained for Myc (green), βIII tubulin (blue) and phalloidin (red). C) TRIM9 dimerization assay IB for GFP-TRIM9 (IB:GFP) co-IP with Myc-TRIM9 variants (IP:Myc, IB:Myc). GAPDH is loading control. Numbers indicate levels of coIP GFP-TRIM9 +/−SEM. D) Binding assay immunoblot showing MycTRIM9 and TRIM9-L316A interaction with GST-EVH1. Numbers denote relative levels of MycTRIM9 variant precipitated by GST-EVH1 beads +/−SEM. E-F) Images and quantification of filopodia +/−SEM in axonal growth cones from control and netrin-treated TRIM9+/+ and TRIM9−/− neurons stained for Myc (blue: Myc, Myc-TRIM9 or TRIM9 mutants), βIII tubulin (green) and phalloidin (red). *denotes significance compared to TRIM9+/+ and *denotes significance compared to TRIM9−/−. G) Representative images of TRIM9−/− neurons transfected with scrambled (scr) or VASP siRNA, along with GFP to identify transfected cells. GFP (blue), VASP immunostaining (green), phalloiding (red). H-I) Images and quantification of axonal growth cone filopodia +/−SEM from control and netrin-treated TRIM9+/+ and TRIM9−/− cortical neurons transfected with scramble (scr) or VASP siRNA (VASPsi), stained for GFP (green) and phalloidin (red). (See also FigS4)
TRIM9ΔRING or TRIM9ΔCC expression did not reduce the excess filopodia of TRIM9−/− growth cones, suggesting ligase activity and interaction with Ena/VASP proteins were essential to constrain filopodia density. Because the CC domain also mediates dimerization of TRIM proteins and the interaction between SNAP25 and TRIM9 (Li et al., 2001; Short et al., 2002; Winkle et al., 2014), we wanted to confirm that aberrant filopodia in TRIM9−/− neurons were dependent upon Ena/VASP function. To address whether TRIM9 dimerization influenced filopodia density, we attempted to design a TRIM9 variant incapable of dimerization by mutating Leu316 within the CC domain to Ala (L316A). However, similar amounts of GFP-TRIM9 co-immunoprecipitated with MycTRIM9L316A as with MycTRIM9 (Fig4C), indicating this mutation did not block dimerization, as a similar mutation does in TRIM25 (Sanchez et al., 2014). However the L316A mutant exhibited reduced binding to GST-EVH1 (Fig4D) and did not reduce aberrant filopodia in TRIM9−/− neurons (Fig4EF), further supporting the conclusion that TRIM9 interaction with Ena/VASP proteins was necessary for constraining filopodia density and TRIM9 dimerization was not sufficient.
To determine if SNAP25 function was required for the filopodia responses, we used Botulinum neurotoxin A (BoNTA) to cleave SNAP25 and block SNAP25-mediated exocytosis (Williamson et al., 1996). BoNTA treatment had no effect on filopodia density (FigS4CD). In cortical neurons, genetic loss of Ena/VASP members blocks filopodia formation, and this is phenocopied by the mis-localization of Ena/VASP proteins with a mitochondrial targeting construct attached to an FP4 motif (FP4Mito, Dent et al., 2007; Gupton and Gertler, 2010; Lebrand et al., 2004). Whereas control EGFP-AP4Mito expressing TRIM9+/+ and TRIM9−/− neurons were unaffected, EGFP-FP4Mito expression reduced TRIM9−/− growth cone filopodia (FigS4EF, p<.005), indicating the filopodia in TRIM9−/− growth cones required Ena/VASP function. Since this approach sequesters all Ena/VASP proteins, we used siRNA to specifically deplete VASP. Knockdown was confirmed by a reduction in VASP immunostaining (Fig4G). VASP siRNA, but not a scrambled control (scr) blocked netrin-dependent increases in filopodia density in TRIM9+/+ growth cones and reduced elevated filopodia density in TRIM9−/− growth cones (Fig4HI, p<.001).
Filopodia response to netrin requires VASP ubiquitination and DUB activity
Based on our observations suggesting that TRIM9-mediated ubiquitination inhibited VASP localization and reduced filopodia density, we predicted that the deubiquitination of VASP upon netrin treatment would be required for increased filopodia density. Inhibition of DUB activity with PR-619 blocked netrin-dependent increases in filopodia density in TRIM9+/+ growth cones but did not alter filopodia density in TRIM9−/− growth cones (Fig5AB). Accordingly, although expression of GFP-VASP did not alter filopodia density in TRIM9+/+ growth cones, expression of GFP-VASP K-R increased filopodia density and blocked netrin response (Fig5CD), suggesting non-ubiquitinated VASP was sufficient to increase filopodia. In contrast expression of GFPVASP or GFP-VASP-K-R in TRIM9−/− neurons did not alter filopodia density (FigS5). TIRF imaging of GFP-VASP K-R and mCherry-VASP in TRIM9+/+ neurons demonstrated that VASP K-R exhibited significantly increased filopodia tip localization compared to wildtype VASP upon PR-619 treatment (Fig5EF). Together, these data confirm that TRIM9-mediated ubiquitination inhibits VASP localization to filopodia tips and VASP ability to increase filopodia density.
Figure 5. VASP deubiquitination is required for netrin-dependent increases in filopodia density.
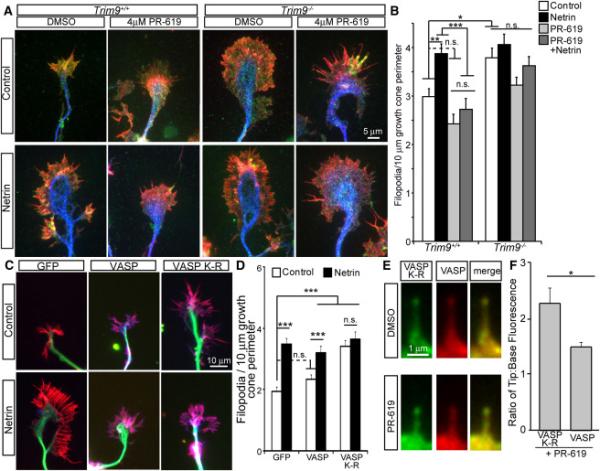
A-B) Images and quantification of filopodia +/−SEM in axonal growth cones from control, PR-619 netrin or PR-619/netrin treated TRIM9+/+ and TRIM9−/− neurons, stained for VASP (green), βIII tubulin (blue) and phalloidin (red). C-D) Images and quantification of filopodia +/−SEM in TRIM9+/+ growth cones expressing GFP, GFP-VASP or GFP-VASP K-R, stained for GFP (blue), βIII tubulin (green) and phalloidin (red). E) TRIM9+/+ filopodia containing GFP-VASP K-R (green) and mCherry-VASP (red) before and after PR-619 treatment. F) Ratio of fluorescence intensity at filopodial tip:filopodial base of GFP-VASP K-R or mCherry-VASP +/−SEM in PR-619 treated TRIM9+/+ neurons. (See also FigS5)
TRIM9 decreases filopodia stability
We hypothesized that TRIM9-mediated ubiquitination altered VASP function, which modulated filopodia density. To test this hypothesis, we performed kymography of individual filopodia (Fig6A) in TRIM9+/+ and TRIM9−/− neurons expressing mCherry (MovieS4). Although there were minor changes in retraction dynamics in the absence of TRIM9, filopodial protrusion was unaltered (FigS6A-C). Netrin treatment increased filopodia lifetimes in TRIM9+/+ growth cones (Fig6B, p<.005) and reduced the frequency of filopodia formation and loss (FigS6D, p<.05). TRIM9−/− filopodia exhibited elevated lifetimes (p<.05) and were insensitive to netrin treatment, supporting the hypothesis that TRIM9-mediated ubiquitination altered VASP function. Further, treatment with PR-619 decreased filopodial lifetime and blocked netrin response in TRIM9+/+ neurons (p<.05) without affecting TRIM9−/− filopodia. Expression of VASP K-R in the presence of endogenous VASP in TRIM9+/+ neurons produced an intermediate phenotype not significantly different from control or netrin-treated TRIM9+/+ neurons or TRIM9−/− neurons (Fig6C). Thus filopodial stability and number were decreased by TRIM9-mediated ubiquitination of VASP.
Figure 6. Ubiquitination of VASP reduces filopodia lifetime and the rate of VASP dissociation from filopodia tips.
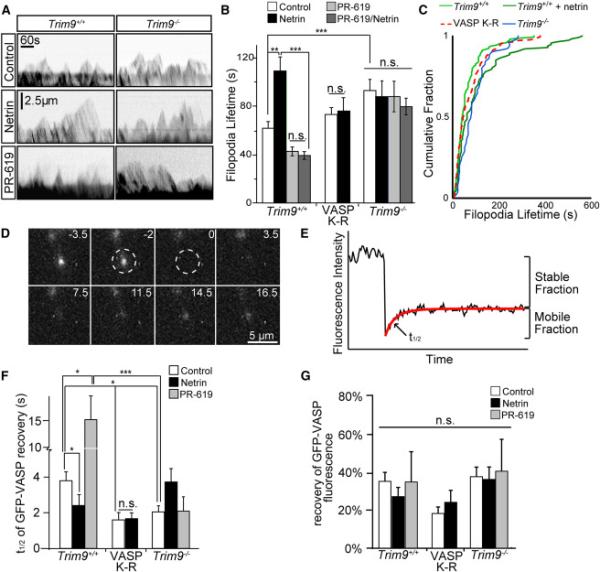
A) Example kymographs of axonal growth cone filopodia from TRIM9+/+ and TRIM9−/− neurons expressing mCherry. B) Filopodial lifetimes +/−SEM in control, netrin, and PR-619 treated TRIM9+/+ and TRIM9−/− neurons and TRIM9+/+ neurons expressing GFP-VASP K-R. C) Cumulative fraction plot of filopodial lifetime demonstrating intermediate phenotype of VASP K-R expressing neurons. D) Image montage of GFP-VASP FRAP at a filopodium tip. Bleach denoted by dashed region, time before and after bleaching in seconds. E) Example of fluorescence intensity data fit to a single exponential (red line), depicting percent fluorescence recovery and t1/2 of fluorescence recovery. F) Fluorescence recovery halftime (t1/2) +/−SEM and G) mean % fluorescence recovery +/−SEM for indicated conditions. (See also Movies S4, S5, FigS6). VASP K-R was expressed in TRIM9+/+ neurons. (See also FigS6)
TRIM9 and ubiquitination alter VASP dissociation from filopodia tips
If TRIM9-mediated ubiquitination alters VASP function and localization, VASP dynamics at filopodia tips should be affected. The dissociation of GFP-VASP from filopodia tips was measured by fluorescence recovery after photobleaching (FRAP, Fig6D, MovieS5) fit to a single exponential (Fig6E). Loss of TRIM9 accelerated GFP-VASP recovery (smaller t1/2, Fig6F, p<.05), suggesting faster VASP dissociation from tips. Following DUB inhibition, VASP recovery slowed in TRIM9+/+ filopodia (p<.05), but not in TRIM9−/− filopodia (p=.61). Netrin accelerated recovery only in TRIM9+/+ filopodia (p<.05). Fluorescence recovery of VASP K-R was faster than wildtype VASP (p<.05), and not netrin sensitive. Thus netrin stimulation, blocking VASP ubiquitination, or loss of TRIM9 accelerated VASP dissociation from filopodia tips, whereas DUB inhibition in the presence of TRIM9 slowed dissociation. This suggested that ubiquitination of VASP by TRIM9 slowed VASP dissociation from filopodia tips. Differences in recovery rate were likely not due to delayed diffusion within filopodia, as FRAP t1/2 in lamellipodia was similar to that of TRIM9+/+ untreated filopodia tips, and not significantly different between genotypes or treatments (FigS6E). The percent of GFP-VASP FRAP was unchanged (Fig6G) and similar to previous reports (Applewhite et al., 2007).
TRIM9 is required for axon turning toward netrin
We hypothesized that increased filopodia stability and density in TRIM9−/− neurons might disrupt axonal response to netrin, which we first tested in cortical explants (Fig7A). Consistent with our previous findings that deletion of TRIM9 or longer term netrin treatment did not significantly alter the length of cortical axons (Winkle et al., 2014), neurite outgrowth was unaltered by loss of TRIM9 or the presence of netrin (Fig7A). TRIM9+/+ explants exhibited biased outgrowth toward netrin, and this response was absent in TRIM9−/− explants (Fig7A, p<.05). To determine if TRIM9 was required for netrin-dependent axon turning, we utilized our recently designed micropass gradient device to create and maintain a stable netrin gradient (Fig7B, Taylor et al., 2015). Axons extended through microgrooves into a fluidically-isolated axon viewing area containing a gradient of netrin and fluorescent dextran. TRIM9+/+ axons turned toward higher netrin concentrations, but did not exhibit turning in a dextran gradient (Fig7CD, p<.01, MovieS6). Netrin-dependent turning was absent in TRIM9−/− axons (Fig7CE, p<.05). We hypothesized a netrin gradient created a gradient of deubiquitinated VASP across the growth cone. To determine if a gradient of TRIM9-mediated ubiquitination was sufficient to cause axon turning, we established a gradient of PR-619. This repulsed TRIM9+/+ axons down the PR-619 gradient (Fig7C,F, p<.005, MovieS7), but failed to induce turning of TRIM9−/− axons. Netrin-dependent axon turning was also absent in TRIM9+/+ axons overexpressing VASP K-R mutant (turning angle of −23.94 ± 23.62, n=5 axons).
Figure 7. Deletion of TRIM9 disrupts axon guidance.
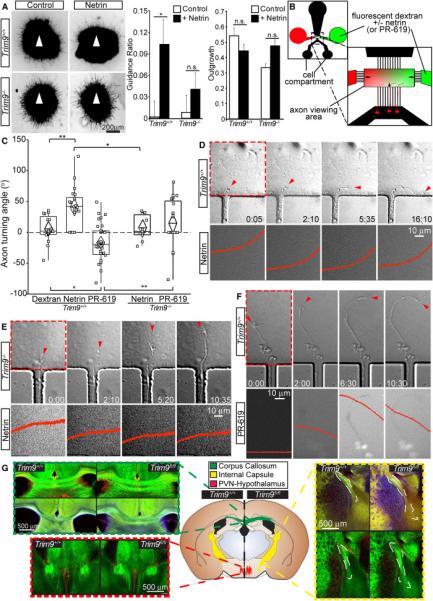
A) TRIM9+/+ but not TRIM9−/− cortical explants exhibit biased neurite outgrowth (βIII tubulin staining) towards netrin (arrowhead). B) Schematic representation of a micropass gradient device with zoomed view of the axon viewing area depicting the dextran gradient and axon growth into the viewing area, arrows indicate direction of fluid flow. C) Turning angles for TRIM9+/+ axons in a dextran gradient, dextran/netrin or dextran/PR-619 gradient and TRIM9−/− axons in a dextran/netrin gradient or dextran/PR-619 gradient. D) DIC images (top) showing a TRIM9+/+ axon turning towards a higher netrin concentration, as seen by the gradient in epifluorescence images (bottom). The red arrowhead denotes the front of the growth cone, the red box denotes the area for which epifluorescence is shown. Time in hours:minutes. Quantification of dextran fluorescence intensity across the region within each epifluorescence image (red), x and y axes are kept constant to demonstrate the stability of the gradient over time. E) TRIM9−/− axon (arrowhead) failing to turn toward the higher netrin concentrations, gradient displayed as in D. F) TRIM9+/+ axon (arrowhead) turning down a PR-619 gradient, displayed as in D. G) Regions of interest (ROI) from coronal sections of Nex-Cre/TauloxP-stop-loxPGFP/TRIM9fl/fl and TRIM9+/+ littermates reveal aberrant cortical axon projections patterns in the corpus callosum (green dashed box), the paraventricular hypothalamic nuclei (red), and the internal capsule (yellow). Green is GFP, red and blue demarcate GFAP in separate littermate pairs. The locations of ROIs and associated defects are denoted in the coronal schematic, n=3 littermate pairs. (See also Movies S6-7, FigS7)
To see if defects in cortical projections occurred in the absence of TRIM9 in vivo, we crossed TRIM9fl/fl mice to Nex-Cre/TauloxP-STOP-loxPGFP mice to delete TRIM9 and express GFP in postmitotic cortical neurons (Higginbotham et al., 2012). We compared GFP+ cortical projections in Nex-Cre/TauloxP-STOP-loxPGFP/TRIM9+/+ and Nex-Cre/TauloxP-STOP-loxPGFP/TRIM9fl/fl littermates (Fig7G). TRIM9−/− mice exhibit thickening of the corpus callosum (Winkle et al., 2014). We observed similar thickening in Nex-Cre/TRIM9fl/fl mice (green box, 14 ± 3.3%, p<0.05). A subset of cortical axons extend to the internal capsule (IC, Braisted et al., 2000; Richards et al., 1997). The length of cortical fibers projecting through the IC was increased by deletion of TRIM9 (yellow box, 32 ± 2.3%, p<0.005), suggesting these axons extended past their target. There was also an increased amount of GFP+ fibers within the paraventricular hypothalamic nuclei (PVHTN, red box, 28.5 ± 8%, p<0.05), suggesting that cortical axons (Tribollet and Dreifuss, 1981) were misguided to the PVHTN. In contrast there was no alteration in the fimbria (FigS7A, p=0.7). Thus deletion of TRIM9 induces specific cortical projection defects in vivo. Since filopodia are also dendritic spine precursors and VASP affects dendritic spines (Lin et al., 2010; Ziv and Smith, 1996), we investigated cortical spines in Thy1-GFP/TRIM9+/+ and TRIM9−/− littermates. Consistent with an early axonal role for TRIM9 in filopodia, there were no changes in cortical spine density (FigS7B).
DISCUSSION
TRIM9-mediated ubiquitination modulates VASP function within growth cone filopodia during netrin-dependent axon guidance
The filopodia tip complex contains cytoskeletal regulatory proteins that modulate filopodial growth and stability (Gupton and Gertler, 2007), but how this complex responds to extracellular cues is unknown. We propose that ubiquitination and deubiquitination of cytoskeletal proteins is a rapid, reversible means to alter the cytoskeleton. Here we demonstrate that VASP is ubiquitinated in the presence of TRIM9, which reduces the localization and dynamics of VASP at filopodia tips, as well as reduces filopodia stability, suggesting TRIM9 acts as a filopodia “off-switch”. The low stoichiometry of VASP ubiquitination indicates a small amount of modified VASP can alter filopodia stability. Tetramers of VASP regulate the polymerization and bundling of multiple actin filaments (Winkelman et al., 2014); an intriguing possibility is that ubiquitination of a single VASP protein within a tetramer is sufficient to poison the function of a Ena/VASP tetramer and inhibit the elongation of multiple filaments comprising a filopodium. Further, the colocalization of TRIM9 and VASP at filopodia tips may increase the local concentration of ubiquitinated VASP to produce filopodial-specific consequences. Consistent with this, we observed altered VASP mobility at filopodia tips that were dependent upon VASP ubiquitination status and associated with the changes in filopodial stability. In contrast, VASP mobility along the lamellipodial veil was unchanged by netrin or loss of TRIM9. This supports the hypothesis that ubiquitinated VASP may localize specifically within filopodia where it alters VASP function and filopodial stability.
Whereas ubiquitination often promotes proteasome-mediated degradation, there are increasing examples of ubiquitination altering substrate localization and function (Chau et al., 1989; Schaefer et al., 2012). The shift in VASP molecular weight indicates ~3 ubiquitins were conjugated to VASP. Since mutation of multiple lysines was required to reduce VASP ubiquitination, we hypothesize that ubiquitination of VASP can occur on a number of lysine residues. However, we found no evidence for VASP degradation, suggesting ubiquitins were removed by an unidentified DUB following netrin treatment. This DUB therefore acts as a filopodia “on-switch”. Consistent with a model that ubiquitination inhibits filopodial stability and density, netrin-dependent increases in filopodia required DUB activity. The requirement for TRIM9 in netrin-dependent axon guidance along with our findings that a gradient of DUB inhibition was sufficient to induce axon turning in the presence of TRIM9, suggests that whereas TRIM9 ubiquitinates VASP, a netrin gradient creates a gradient of VASP deubiquitination across the growth cone, which produces a spatial gradient of filopodial stability required for axon turning toward netrin. Although netrin-1 is typically described as a soluble axon guidance cue, there is evidence that netrin-1 functions as a substrate bound guidance cue (Moore et al., 2009; Moore et al., 2012). Our findings are consistent with a requirement for TRIM9 during cortical axon turning in response to netrin, whether netrin is in a soluble or bound state.
TRIM9 coordinates cytoskeletal dynamics and membrane delivery during axonal development
We recently demonstrated that TRIM9 interacts with the tSNARE SNAP25 and regulates exocytosis required for netrin-dependent axon branching (Winkle et al., 2014). Neuritogenesis, axon branching and guidance involve protrusion and expansion of the plasma membrane, which require cytoskeletal dynamics and membrane addition (Dent et al., 2007; 2004; Gupton and Gertler, 2010; Tojima et al., 2007; Winkle et al., 2014). Loss of TRIM9 is associated with elevated filopodia stability, exocytosis and axon branching. The interactions with DCC, VASP and SNAP25 poise TRIM9 to spatially and temporally coordinate robust netrin-dependent membrane protrusion and membrane delivery. This is accomplished by constraining the function of VASP and SNAP25 in the absence of netrin: for VASP by ubiquitination, and for SNAP25 by an interaction that competes with SNARE complex assembly and vesicle fusion. It is likely that TRIM9-mediated regulation of both exocytosis and filopodia is involved in both netrin-dependent axon branching and guidance.
Regulation of filopodia and axon guidance by post-translational modification
The ubiquitination of VASP mediated by TRIM9 is inhibited or reversed following netrin treatment. This would result in dis-inhibition of filopodia and localized netrin responses. TRIM9 performs autoubiquitination in vitro (Tanji et al., 2010), and may mediate its own inhibition or removal. Alternatively, since TRIM9 interacts with VASP through its dimerization domain, a change in TRIM9 conformation that promotes dimerization at the expense of interaction with VASP could occur. Consistent with this, the TRIM9 mutant compromised in Ena/VASP binding but not dimerization failed to constrain filopodia. Netrin-dependent dimerization of DCC (Stein, 2001) may promote TRIM9 dimerization. Alternatively, TRIM9 ligase activity may be masked by a competing DUB in the presence of netrin, consistent with our findings. Although the DUB is unknown a broad spectrum DUB inhibitor did not alter filopodia density and VASP mobility in TRIM9−/− neurons, supporting our hypothesis that TRIM9-mediated ubiquitination of VASP is a mechanism of filopodia regulation. Although we do not know how ubiquitination alters VASP function at the barbed end, our finding that filopodia lifetimes but not protrusion dynamics were altered by deletion of TRIM9 suggests that VASP ubiquitination may alter the anti-capping function of VASP or block the processive polymerase activity of VASP, effectively turning VASP into a capping protein.
VASP is also phosphorylated by cAMP-dependent protein kinase downstream of netrin and DCC (Lebrand et al., 2004). Phosphorylation and ubiquitination often collaborate to alter protein function (Hunter, 2007), which may be the case for VASP, or these modifications may be mutually exclusive. A relationship between ubiquitination and phosphorylation may explain our result that VASP mobility showed a trend toward decreasing in TRIM9−/− filopodia tips treated with netrin. If this response were phosphorylation-dependent, it may have been masked in TRIM9+/+ filopodia by VASP deubiquitination. The pairing of ubiquitination and phosphorylation to alter VASP localization and function may promote VASP function in a narrow spatiotemporal window constrained by the presence of netrin, and may explain why loss of TRIM9 or loss of VASP ubiquitination did not completely phenocopy netrin stimulation.
In many instances, a single Ena/VASP member compensates for loss of Ena/VASP function (Dent et al., 2007; Furman et al., 2007). Although TRIM9 interacts with the EVH1 domain of all three Ena/VASP proteins in vitro, we detected ubiquitination of VASP, not Mena or EVL, in the presence of TRIM9. Since VASP exhibits unbiased tetramerization with itself, Mena and EVL (Riquelme et al., 2015), ubiquitination and inhibition of VASP may indirectly impact Mena or EVL function, however we saw no evidence of changes in Mena or EVL localization. The phenotypes caused by expression of VASP K-R support this possibility and may explain why modulation of VASP was sufficient to alter filopodia and axon guidance. In light of increasing examples of unique functions and interactions for Mena and VASP (Gupton et al., 2012; Lin et al., 2010; Worth et al., 2010), TRIM9-mediated regulation of VASP alone may be sufficient to induce the observed changes in filopodia. However, since ubiquitin ligases often have multiple substrates, TRIM9 may have additional substrates involved in modulating neuronal morphology, possibly also localized at filopodia tips.
Conservation and divergence of the netrin pathway in axon guidance
The invertebrate paralogs of netrin, DCC, VASP and TRIM9 all function in axon guidance (Gertler et al., 1995; Gitai et al., 2003; Hao et al., 2010; Kolodziej et al., 1996; Morikawa et al., 2011; Serafini et al., 1994). Given this and conserved domain organization of TRIM9 and VASP orthologs, the mechanism uncovered here may link invertebrate TRIM9 and VASP orthologs. The phenotypes associated with loss of TRIM9 however are not conserved. Loss of madd2 in C. elegans phenocopied loss of unc-6 (netrin) or unc-40 (DCC, Hao et al., 2010). Similar commissural phenotypes were observed in D. Melanogaster with loss of function mutations in dTRIM9, netrin or frazzled (DCC, Morikawa et al., 2011). In contrast, the corpus callosum agenesis caused by deletion of NTN1 or DCC (Fazeli et al., 1997; Serafini et al., 1996) or lack of the IC and corpus callosum associated with deletion of Ena/VASP genes (Kwiatkowski et al., 2007) are in contrast to the thickened corpus callosum and longer IC associated with deletion of TRIM9. Despite TRIM9 having the highest sequence homology with the single invertebrate class I TRIM (Morikawa et al., 2011), the contrasting phenotypes indicate divergence of TRIM9 function. Whether another class I TRIM is the functional mammalian ortholog remains to be seen.
EXPERIMENTAL PROCEDURES
Animals
All mouse lines were on a C57BL/6J background and bred at UNC with approval from the Institutional Animal Care and Use Committee. Timed pregnant females were obtained by placing male and female mice together overnight; the following day was designated as E0.5 if the female had a vaginal plug. TRIM9−/−, TRIM9fl/fl and Thy1-GFP mice were described (Feng et al., 2000; Winkle et al., 2014). A Nex-Cre line (from Dr. Klaus Nave) and a TauloxP-stop-loxPGFP line (from Dr. Eva Anton, Higginbotham et al., 2012) were crossed with TRIM9fl/fl mice.
Immunoblotting, Co-immunoprecipitation, Binding Assays, Ubiquitination Assays
SDS-PAGE and immunoblot analysis were performed using standard procedures with HRP 2° antibodies for His-EVH1 or far-red conjugated 2°antibodies (Licor) for other 1° antibodies. Signal was detected with ECL plus (Amersham) or Odyssey Imager (Licor). All co-immunoprecipitation assays were performed using standard procedures. For binding assays GST-tagged or His-tagged proteins were expressed in E. Coli strain BL21 (DE3) Codon Plus (Agilent). Purified proteins were incubated in lysates prepared from E15.5 mouse cortices or HEK293 lysates expressing Myc-tagged TRIM9 variants. HEK293 TRIM9−/− cells were generated by CRISPR/Cas9 gene editing. To measure ubiquitination, HEK cells transfected with mCherry or HA-DCC, FLAG-Ub, and GFP-VASP, GFP-EVL, GFP-Mena or GFP-Mena+ using Lipofectamine 2000 and cultured for 24 hrs or cortical neurons at 2DIV were treated with 10 μM MG132 for 4 hours, and 600 ng/ml netrin-1 or 600 ng/ml netrin-1 and 4 μM PR-619 50 min. Lysates were prepared as described in the supplementary section. Immunoprecipitations were performed under conditions in which only covalent interactions were preserved with anti-GFP (mouse) antibody or anti-VASP (rabbit) antibody coupled to Protein A/G agarose beads. For additional information, see Supplement.
Cortical neuron culture
E15.5 dissociated cortical neuron cultures were prepared as described (Kwiatkowski et al., 2007). Briefly, cortices were micro-dissected and neurons were dissociated with trypsin and plated on Poly-D-lysine (Sigma)-coated coverglass or tissue culture plastic in Neurobasal media supplemented with B27 (Invitrogen). To assay growth cones and filopodia, 600 ng/ml netrin-1 (+/− 4 μM PR-619 or +/− 100 nM CytoD) or 24 ng/ml recombinant FGF-2 (MBL International) was bath applied after 48 hrs in vitro for 40 min followed by fixation and immunostaining. Widefield epifluorescence images of pyramidal-shaped neurons were analyzed. Growth cone perimeter and area were measured using ImageJ. Filopodium length was measured from the filopodium tip to lamellipodial veil. Number of filopodia was counted per growth cone, and density is reported per 10 μm of growth cone perimeter. To assay filopodia dynamics, dynamic colocalization and FRAP, time-lapse imaging was performed with a stage top incubator that maintained humidity, 37°C and 5% CO2 (Tokai Hit). Cortical explants were prepared and cultured and analyzed as described (Fothergill et al., 2014). Briefly ~500 μm cortical explants from TRIM9+/+ and TRIM9−/− embryos (E13-E15) were embedded in collagen matrices. Agarose cubes soaked in netrin-1 (10 μg/ml) were asymmetrically placed ~500 μm from the explant. After 48 hrs explants were fixed, stained for βIII tubulin. Transfection procedures, microscopy and imaging parameters, and image analysis are described in the Supplement.
Axon Turning Assay
Micropass gradient devices were used to measure axon turning. Device preparation and experimental protocol is as described (Taylor et al., under revision). Briefly, TRIM9+/+ and TRIM9−/− E15.5 cortical neurons were plated in devices; after axons entered the axon viewing area (2-4 days), a control gradient of dextran (starting at 1 μM), a gradient of netrin+dextran (600 ng/ml), or PR-619+dextran (1 μM) was established. DIC (axons) and epifluorescence (dextran) images were acquired every 5 min for 8-18 hrs at 20× magnification. The angle of axon turning relative to the initial trajectory of the axon before the gradient is reported, with positive angles indicating turning up gradient. Angles for turning were measured for axons in attractive netrin concentrations as described (Taylor et al., 2015).
Neuroanatomical Measurements
Maximal projections of multi-area Z stacks of serial coronal sections of 5-week-old Nex-Cre/TauloxP-stop-loxPGFP/TRIM9fl/fl and Nex-Cre/TauloxP-stop-loxPGFP/TRIM9+/+ littermates were collected. Position-matched sections between littermates were analyzed for axon projection comparisons. Dendritic spine density was measured from Z-stacks of 60 μm coronal sections of 3-week-old Thy1-GFP/TRIM9+/+ and Thy1-GFP/TRIM9−/− littermates. See Supplement for additional information.
Statistics
At least 3 independent experiments were performed for each assay, except where noted. Data distribution normality was determined using the Shapiro-Wilk test. Normally distributed data was compared by unpaired t-test for two independent samples, or ANOVA with Tukey post-hoc correction, for >2 comparisons. For non-normal data, the Mann-Whitney test was used or Kruskal-Wallis nonparametric ANOVA with Bonferonni posthoc correction for >2 comparisons. All data are presented as means +/− standard error of the mean (SEM), unless where 95% confidence interval (CI) was reported. Statistical significance is represented as such: *p<.05, **p<.01, ***p<.005).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Charles Park, Haejin Song, Maite Ghazelah for technical assistance and Saskia Neher for molecular expertise. This work was supported by the National Institutes of Health: GM108970 (SLG), R41MH097377 (AMT), K12HD073945 (AMT), F31 NS087837-01A1 (CW), T32 NS007431-16 (NB), a pilot grant from UNC SOM (SLG), American Heart Association fellowships 14POST20450085 (SM) and 0615692T (LM) and DFG grants Ro2525/51 and Schm2670/1-1 (SR). The work performed at MIT was supported by NIH grant GM68678 to Frank Gertler, whom we thank for generous support and invaluable discussions. AMT is an Alfred P. Sloan Fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
SM performed and analyzed ubiquitination and binding assays, filopodial density assays, and axon guidance assays and wrote the paper. NB performed and analyzed live cell filopodial imaging assays, filopodial density assays and binding assays and wrote the paper; C.W. performed in vivo analysis of axon projection defects, LMperformed yeast two hybrid screens and binding assays. CH performed competitive binding assays and analyzed imaging data. DP and SR generated TRIM9−/− 293 cells and wrote associated methodological text; AMT assisted in design and analysis of axon guidance assays using the micropass gradient device; SLG conceptualized, designed, performed and supervised experiments, analyzed data, and wrote the paper. All authors reviewed and made comments on the manuscript.
REFERENCES
- Altun M, Kramer HB, Willems LI, McDermott JL, Leach CA, Goldenberg SJ, Kumar KGS, Konietzny R, Fischer R, Kogan E, et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 2011;18:1401–1412. doi: 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Applewhite DA, Barzik M, Kojima S-I, Svitkina TM, Gertler FB, Borisy GG. Ena/VASP proteins have an anti-capping independent function in filopodia formation. Mol Biol Cell. 2007;18:2579–2591. doi: 10.1091/mbc.E06-11-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzik M, Kotova TI, Higgs HN, Hazelwood L, Hanein D, Gertler FB, Schafer DA. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem. 2005;280:28653–28662. doi: 10.1074/jbc.M503957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, et al. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Boëda B, Knowles PP, Briggs DC, Murray-Rust J, Soriano E, Garvalov BK, McDonald NQ, Way M. Molecular recognition of the Tes LIM2-3 domains by the actin-related protein Arp7A. J Biol Chem. 2011;286:11543–11554. doi: 10.1074/jbc.M110.171264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braisted JE, Catalano SM, Stimac R, Kennedy TE, Tessier-Lavigne M, Shatz CJ, O'Leary DD. Netrin-1 promotes thalamic axon growth and is required for proper development of the thalamocortical projection. J Neurosci. 2000;20:5792–5801. doi: 10.1523/JNEUROSCI.20-15-05792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitsprecher D, Kiesewetter AK, Linkner J, Urbanke C, Resch GP, Small JV, Faix J. Clustering of VASP actively drives processive, WH2 domain-mediated actin filament elongation. Embo J. 2008;27:2943–2954. doi: 10.1038/emboj.2008.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau V, Tobias J, Bachmair A, Marriott D, Ecker D, Gonda D, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- Chen XJ, Squarr AJ, Stephan R, Chen B, Higgins TE, Barry DJ, Martin MC, Rosen MK, Bogdan S, Way M. Ena/VASP Proteins Cooperate with the WAVE Complex to Regulate the Actin Cytoskeleton. Dev Cell. 2014;30:569–584. doi: 10.1016/j.devcel.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsen JMR, Sylvestersen KB, Bekker-Jensen S, Szklarczyk D, Poulsen JW, Horn H, Jensen LJ, Mailand N, Nielsen ML. Mass Spectrometric Analysis of Lysine Ubiquitylation Reveals Promiscuity at Site Level. Molecular & Cellular Proteomics. 2011;10:M110.003590. doi: 10.1074/mcp.M110.003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Kalil K. Axon branching requires interactions between dynamic microtubules and actin filaments. Journal of Neuroscience. 2001;21:9757–9769. doi: 10.1523/JNEUROSCI.21-24-09757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Barnes AM, Tang F, Kalil K. Netrin-1 and semaphorin 3A promote or inhibit cortical axon branching, respectively, by reorganization of the cytoskeleton. J Neurosci. 2004;24:3002–3012. doi: 10.1523/JNEUROSCI.4963-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Kwiatkowski AV, Mebane LM, Philippar U, Barzik M, Rubinson DA, Gupton S, Van Veen JE, Furman C, Zhang J, et al. Filopodia are required for cortical neurite initiation. Nat Cell Biol. 2007;9:1347–1359. doi: 10.1038/ncb1654. [DOI] [PubMed] [Google Scholar]
- Didier C, Broday L, Bhoumik A, Israeli S, Takahashi S, Nakayama K, Thomas SM, Turner CE, Henderson S, Sabe H, et al. RNF5, a RING Finger Protein That Regulates Cell Motility by Targeting Paxillin Ubiquitination and Altered Localization. Mol Cell Biol. 2003;23:5331–5345. doi: 10.1128/MCB.23.15.5331-5345.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazeli A, Dickinson SL, Hermiston ML, Tighe RV, Steen RG, Small CG, Stoeckli ET, Keino-Masu K, Masu M, Rayburn H, et al. Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature. 1997;386:796–804. doi: 10.1038/386796a0. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fothergill T, Donahoo A-LS, Douglass A, Zalucki O, Yuan J, Shu T, Goodhill GJ, Richards LJ. Netrin-DCC signaling regulates corpus callosum formation through attraction of pioneering axons and by modulating Slit2-mediated repulsion. Cereb Cortex. 2014;24:1138–1151. doi: 10.1093/cercor/bhs395. [DOI] [PubMed] [Google Scholar]
- Furman C, Sieminski AL, Kwiatkowski AV, Rubinson DA, Vasile E, Bronson RT, Fässler R, Gertler FB. Ena/VASP is required for endothelial barrier function in vivo. J Cell Biol. 2007;179:761–775. doi: 10.1083/jcb.200705002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler FB, Comer AR, Juang JL, Ahern SM, Clark MJ, Liebl EC, Hoffmann FM. enabled, a dosage-sensitive suppressor of mutations in the Drosophila Abl tyrosine kinase, encodes an Abl substrate with SH3 domain-binding properties. Genes Dev. 1995;9:521–533. doi: 10.1101/gad.9.5.521. [DOI] [PubMed] [Google Scholar]
- Gitai Z, Yu TW, Lundquist EA, Tessier-Lavigne M, Bargmann CI. The netrin receptor UNC-40/DCC stimulates axon attraction and outgrowth through enabled and, in parallel, Rac and UNC-115/AbLIM. Neuron. 2003;37:53–65. doi: 10.1016/s0896-6273(02)01149-2. [DOI] [PubMed] [Google Scholar]
- Gupton SL, Gertler FB. Filopodia: the fingers that do the walking. Sci STKE 2007. 2007:re5. doi: 10.1126/stke.4002007re5. [DOI] [PubMed] [Google Scholar]
- Gupton SL, Gertler FB. Integrin signaling switches the cytoskeletal and exocytic machinery that drives neuritogenesis. Dev Cell. 2010;18:725–736. doi: 10.1016/j.devcel.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton SL, Riquelme D, Hughes-Alford SK, Tadros J, Rudina SS, Hynes RO, Lauffenburger D, Gertler FB. Mena binds α5 integrin directly and modulates α5β1 function. J Cell Biol. 2012;198:657–676. doi: 10.1083/jcb.201202079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SD, Mullins RD. VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J Cell Biol. 2010;191:571–584. doi: 10.1083/jcb.201003014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao JC, Adler CE, Mebane L, Gertler FB, Bargmann CI, Tessier-Lavigne M. The tripartite motif protein MADD-2 functions with the receptor UNC-40 (DCC) in Netrin-mediated axon attraction and branching. Dev Cell. 2010;18:950–960. doi: 10.1016/j.devcel.2010.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham H, Eom T-Y, Mariani LE, Bachleda A, Hirt J, Gukassyan V, Cusack CL, Lai C, Caspary T, Anton ES. Arl13b in primary cilia regulates the migration and placement of interneurons in the developing cerebral cortex. Dev Cell. 2012;23:925–938. doi: 10.1016/j.devcel.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell. 2007;28:730–738. doi: 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Kim TY, Siesser PF, Rossman KL, Goldfarb D, Mackinnon K, Yan F, Yi X, MacCoss MJ, Moon RT, Der CJ, et al. Substrate trapping proteomics reveals targets of the βTrCP2/FBXW11 ubiquitin ligase. Mol Cell Biol. 2015;35:167–181. doi: 10.1128/MCB.00857-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin AL, Tessier-Lavigne M. Mechanisms and Molecules of Neuronal Wiring: A Primer. Cold Spring Harb Perspect Biol. 2011;3:a001727–a001727. doi: 10.1101/cshperspect.a001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziej PA, Timpe LC, Mitchell KJ, Fried SR, Goodman CS, Jan LY, Jan YN. frazzled encodes a Drosophila member of the DCC immunoglobulin subfamily and is required for CNS and motor axon guidance. Cell. 1996;87:197–204. doi: 10.1016/s0092-8674(00)81338-0. [DOI] [PubMed] [Google Scholar]
- Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. doi: 10.1146/annurev.cellbio.19.050103.103356. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski AV, Rubinson DA, Dent EW, Edward van Veen J, Leslie JD, Zhang J, Mebane LM, Philippar U, Pinheiro EM, Burds AA, et al. Ena/VASP Is Required for Neuritogenesis in the Developing Cortex. Neuron. 2007;56:441–455. doi: 10.1016/j.neuron.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Lanier LM, Gates MA, Witke W, Menzies AS, Wehman AM, Macklis JD, Kwiatkowski D, Soriano P, Gertler FB. Mena is required for neurulation and commissure formation. Neuron. 1999;22:313–325. doi: 10.1016/s0896-6273(00)81092-2. [DOI] [PubMed] [Google Scholar]
- Lebrand C, Dent EW, Strasser GA, Lanier LM, Krause M, Svitkina TM, Borisy GG, Gertler FB. Critical role of Ena/VASP proteins for filopodia formation in neurons and in function downstream of netrin-1. Neuron. 2004;42:37–49. doi: 10.1016/s0896-6273(04)00108-4. [DOI] [PubMed] [Google Scholar]
- Li Y, Chin LS, Weigel C, Li L. Spring, a novel RING finger protein that regulates synaptic vesicle exocytosis. J Biol Chem. 2001;276:40824–40833. doi: 10.1074/jbc.M106141200. [DOI] [PubMed] [Google Scholar]
- Lin W-H, Nebhan CA, Anderson BR, Webb DJ. Vasodilator-stimulated phosphoprotein (VASP) induces actin assembly in dendritic spines to promote their development and potentiate synaptic strength. J Biol Chem. 2010;285:36010–36020. doi: 10.1074/jbc.M110.129841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SW, Biais N, Sheetz MP. Traction on immobilized netrin-1 is sufficient to reorient axons. Science. 2009;325:166. doi: 10.1126/science.1173851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SW, Zhang X, Lynch CD, Sheetz MP. Netrin-1 Attracts Axons through FAK-Dependent Mechanotransduction. Journal of Neuroscience. 2012;32:11574–11585. doi: 10.1523/JNEUROSCI.0999-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa RK, Kanamori T, Yasunaga K-I, Emoto K. Different levels of the Tripartite motif protein, Anomalies in sensory axon patterning (Asap), regulate distinct axonal projections of Drosophila sensory neurons. Proceedings of the National Academy of Sciences. 2011;108:19389–19394. doi: 10.1073/pnas.1109843108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer D, Feldner J, Vaughan T, Vetter I, Pujic Z, Rosoff WJ, Burrage K, Dayan P, Richards LJ, Goodhill GJ. A Bayesian model predicts the response of axons to molecular gradients. Proceedings of the National Academy of Sciences. 2009;106:10296–10301. doi: 10.1073/pnas.0900715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and Cellular Roles of Ubiquitin-Specific Deubiquitinating Enzymes. Annu Rev Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards LJ, Koester SE, Tuttle R, O'Leary DD. Directed growth of early cortical axons is influenced by a chemoattractant released from an intermediate target. J Neurosci. 1997;17:2445–2458. doi: 10.1523/JNEUROSCI.17-07-02445.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme DN, Meyer AS, Barzik M, Keating A, Gertler FB. Selectivity in subunit composition of Ena/VASP tetramers. Bioscience Reports. 2015;35:e00246–e00246. doi: 10.1042/BSR20150149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MS, Desterro JMP, Lain S, Lane DP, Hay RT. Multiple C-Terminal Lysine Residues Target p53 for Ubiquitin-Proteasome-Mediated Degradation. Mol Cell Biol. 2000;20:8458–8467. doi: 10.1128/mcb.20.22.8458-8467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosoff WJ, Urbach JS, Esrick MA, McAllister RG, Richards LJ, Goodhill GJ. A new chemotaxis assay shows the extreme sensitivity of axons to molecular gradients. Nat Neurosci. 2004;7:678–682. doi: 10.1038/nn1259. [DOI] [PubMed] [Google Scholar]
- Sanchez JG, Okreglicka K, Chandrasekaran V, Welker JM, Sundquist WI, Pornillos O. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proceedings of the National Academy of Sciences. 2014;111:2494–2499. doi: 10.1073/pnas.1318962111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A, Nethe M, Hordijk PL. Ubiquitin links to cytoskeletal dynamics, cell adhesion and migration. Biochem J. 2012;442:13–25. doi: 10.1042/BJ20111815. [DOI] [PubMed] [Google Scholar]
- Seiberlich V, Goldbaum O, Zhukareva V, Richter-Landsberg C. The small molecule inhibitor PR-619 of deubiquitinating enzymes affects the microtubule network and causes protein aggregate formation in neural cells: Implications for neurodegenerative diseases. Biochimica Et Biophysica Acta (BBA) - Molecular Cell Research. 2012;1823:2057–2068. doi: 10.1016/j.bbamcr.2012.04.011. [DOI] [PubMed] [Google Scholar]
- Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, Tessier-Lavigne M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 1996;87:1001–1014. doi: 10.1016/s0092-8674(00)81795-x. [DOI] [PubMed] [Google Scholar]
- Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M. The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell. 1994;78:409–424. doi: 10.1016/0092-8674(94)90420-0. [DOI] [PubMed] [Google Scholar]
- Shekarabi M, Kennedy TE. The netrin-1 receptor DCC promotes filopodia formation and cell spreading by activating Cdc42 and Rac1. Mol. Cell. Neurosci. 2002;19:1–17. doi: 10.1006/mcne.2001.1075. [DOI] [PubMed] [Google Scholar]
- Short KM, Cox TC. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. Journal of Biological Chemistry. 2006;281:8970–8980. doi: 10.1074/jbc.M512755200. [DOI] [PubMed] [Google Scholar]
- Short KM, Hopwood B, Yi Z, Cox TC. MID1 and MID2 homo- and heterodimerise to tether the rapamycin-sensitive PP2A regulatory subunit, alpha 4, to microtubules: implications for the clinical variability of X-linked Opitz GBBB syndrome and other developmental disorders. BMC Cell Biol. 2002;3:1. doi: 10.1186/1471-2121-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein E. Binding of DCC by Netrin-1 to Mediate Axon Guidance Independent of Adenosine A2B Receptor Activation. Science. 2001;291:1976–1982. doi: 10.1126/science.1059391. [DOI] [PubMed] [Google Scholar]
- Tanji K, Kamitani T, Mori F, Kakita A, Takahashi H, Wakabayashi K. TRIM9, a novel brain-specific E3 ubiquitin ligase, is repressed in the brain of Parkinson's disease and dementia with Lewy bodies. Neurobiology of Disease. 2010;38:210–218. doi: 10.1016/j.nbd.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Menon S, Gupton SL. Passive microfluidic chamber for long-term imaging of axon guidance in response to soluble gradients. Lab on a Chip. 2015;15:2781–2789. doi: 10.1039/c5lc00503e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojima T, Akiyama H, Itofusa R, Li Y, Katayama H, Miyawaki A, Kamiguchi H. Attractive axon guidance involves asymmetric membrane transport and exocytosis in the growth cone. Nat Neurosci. 2007;10:58–66. doi: 10.1038/nn1814. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Dreifuss JJ. Localization of neurones projecting to the hypothalamic paraventricular nucleus area of the rat: A horseradish peroxidase study. Neuroscience. 1981;6:1315–1328. doi: 10.1016/0306-4522(81)90190-1. [DOI] [PubMed] [Google Scholar]
- Webber CA, Chen YY, Hehr CL, Johnston J, McFarlane S. Multiple signaling pathways regulate FGF-2-induced retinal ganglion cell neurite extension and growth cone guidance. Molecular and Cellular Neuroscience. 2005;30:37–47. doi: 10.1016/j.mcn.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Williamson LC, Halpern JL, Montecucco C, Brown JE, Neale EA. Clostridial neurotoxins and substrate proteolysis in intact neurons: botulinum neurotoxin C acts on synaptosomal-associated protein of 25 kDa. J Biol Chem. 1996;271:7694–7699. doi: 10.1074/jbc.271.13.7694. [DOI] [PubMed] [Google Scholar]
- Winkle CC, McClain LM, Valtschanoff JG, Park CS, Maglione C, Gupton SL. A novel Netrin-1-sensitive mechanism promotes local SNARE-mediated exocytosis during axon branching. J Cell Biol. 2014;205:217–232. doi: 10.1083/jcb.201311003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelman JD, Bilancia CG, Peifer M, Kovar DR. Ena/VASP Enabled is a highly processive actin polymerase tailored to self-assemble parallel-bundled F-actin networks with Fascin. Proceedings of the National Academy of Sciences. 2014;111:4121–4126. doi: 10.1073/pnas.1322093111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth DC, Hodivala-Dilke K, Robinson SD, King SJ, Morton PE, Gertler FB, Humphries MJ, Parsons M. Alpha v beta3 integrin spatially regulates VASP and RIAM to control adhesion dynamics and migration. J Cell Biol. 2010;189:369–383. doi: 10.1083/jcb.200912014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechel S, Werner S, Unsicker K, Bohlen und Halbach, von, O Expression and functions of fibroblast growth factor 2 (FGF-2) in hippocampal formation. The Neuroscientist. 2010;16:357–373. doi: 10.1177/1073858410371513. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.