

Abstract
This article summarizes and reviews recent progress in the development of catalysts for the ring-opening copolymerization of carbon dioxide and epoxides. The copolymerization is an interesting method to add value to carbon dioxide, including from waste sources, and to reduce pollution associated with commodity polymer manufacture. The selection of the catalyst is of critical importance to control the composition, properties and applications of the resultant polymers. This review highlights and exemplifies some key recent findings and hypotheses, in particular using examples drawn from our own research.
Keywords: catalysis, polycarbonate, ring-opening copolymerization, CO2
1. Introduction
The ring-opening copolymerization (ROCOP) of carbon dioxide and epoxides is an interesting method to synthesize a range of aliphatic polycarbonates (figure 1) [1–5]. The reaction was discovered more than 40 years ago and has since continued to attract attention as a means to reduce pollution associated with polymer manufacture and to ‘add value’ to carbon dioxide [1]. It should be made clear that the use of CO2 in any chemical manufacturing process is unable to make a large impact on overall CO2 levels in the atmosphere. However, it remains important to pursue CO2 utilization as a means to reduce emissions, particularly those associated with existing, large-scale industrial processes and as an economic driver to support carbon capture [6,7]. The ROCOP process is strongly dependent on the selection of the catalyst, with various homogeneous and heterogeneous catalysts having been reported [5,8–15]. The focus for this review article will be to highlight and exemplify some of the key findings in this area of catalysis, in particular using examples drawn from our own research. The intention is not to provide a comprehensive review of all known catalysts; indeed, such reviews are already available [5,8–15].
Figure 1.

The ROCOP of carbon dioxide and epoxides to produce aliphatic polycarbonates. (Online version in colour.)
The ROCOP reaction is a rare example of a truly catalytic process with the potential to deliver large-scale quantities of product, which genuinely consumes carbon dioxide. The most commonly studied epoxides are cyclohexene oxide (CHO) and propylene oxide (PO). Depending on the epoxide and the selectivity of the catalyst, up to 31% (polycyclohexene carbonate) or 43% (polypropylene carbonate) of the polymer mass derives from CO2. The primary application for the polymer products is as low-molecular-weight (Mn), (poly)hydroxyl-terminated ‘polyols’, which are widely used in the manufacture of polyurethanes [16]. Polyurethanes themselves are applied as flexible/rigid foams, adhesives, coatings and elastomers, as well as in many other areas. It has been shown that the properties of CO2-derived polyols are suitable to replace polyether polyols in some applications [16,17]. Given that polyether polyols are prepared by epoxide homopolymerization, the ROCOP of CO2 and epoxides can also be viewed as a means to ‘replace’ a substantial portion of petrochemically derived resource (epoxide) with a renewable one (CO2). A recent detailed life-cycle analysis study compared these two types of polyols, showing significant reductions (approx. 20%) in both fossil resource depletion and greenhouse gas emissions for the CO2-derived polymers [18].
Furthermore, the replacement of fossil-derived epoxides is economically attractive and is stimulating a number of commercialization studies [16,19] (http://www.empowermaterials.com/; http://www.novomer.com/; http://www.covestro.com/en/Sustainability/Productions/Polyols; http://www.econic-technologies.com/ [accessed 4 September 2015]). It has also recently been demonstrated that ROCOP catalysts are compatible with carbon capture and storage (CCS) processes [20]. Studies have shown that some homogeneous magnesium catalysts can be used in polymerizations where the carbon dioxide is captured at a CCS demonstrator plant attached to a UK power station. The catalysts showed near-equivalent performances using such captured gases compared to using ‘pure’ carbon dioxide. Furthermore, the catalyst showed a high tolerance to various impurities present in captured CO2, including water, N2, CO, thiols and amines [20].
2. Polymerization pathways and mechanisms
A range of different catalysts are known but all catalysts contain metals, with Zn(II), Co(III) and Cr(III) being particularly common [5,8–14]. Prior to an examination of these catalysts, it is worth considering the series of reactions that are proposed to occur at the metal active site during polymerization, the major ones of which are illustrated in figure 2 [3,11]. The polymerization is initiated by coordination of an epoxide molecule and the subsequent ring opening by the nucleophilic attack of a carbonate group or ligand (X), so as to form a metal alkoxide intermediate. During chain propagation, carbon dioxide inserts into the metal alkoxide intermediate to form a metal carbonate species. The metal coordinates another molecule of epoxide, and nucleophilic attack by the carbonate group leads to the ring opening of the epoxide and formation of a new metal alkoxide species. Propagation therefore involves the ‘cycling’ between metal alkoxide and carbonate intermediates. The polymerization is terminated by exposure to conditions/reagents that lead to hydrolysis of the growing polymer chain and formation of a polymer chain end-capped with a hydroxyl group.
Figure 2.

Catalytic cycle of CO2/epoxide copolymerization.
There are also side reactions within this process, the proportions of which depend on the conditions, substrate and catalyst selected. The formation of ether linkages in the polymer chain can occur due to the occurrence of a metal alkoxide attack on an epoxide molecule instead of CO2 insertion. Such linkages change the polymer properties, which may be beneficial depending on the application, but nevertheless reduce the CO2 sequestered in the polymer backbone [16,17].
Five-membered ring cyclic carbonate by-products can also form; indeed, this is the thermodynamic product of the reaction between CO2 and epoxides and thus favoured under forcing conditions. The cyclic carbonates can form by depolymerization or ‘back-biting’ reactions; the extent of these depend on the catalyst and the ceiling temperature for the given polymer [21,22]. The cyclic carbonates can also form ‘off-metal’ and are particularly common, and sometimes problematic, contaminants when co-catalysts or ionic additives are applied [5,13]. For some types of catalyst, most commonly with metal salen catalysts, it is proposed that the addition of ionic co-catalysts leads to the polymer chains being in equilibrium between metal coordination and ‘free’ anionic polymer chains [5]. Such ‘off-metal’ chains have been shown to undergo cyclization to form cyclic carbonate by-products [23].
Chain transfer reactions also need to be considered. These reactions occur when the polymerizations are conducted under so-called ‘immortal’ conditions in the presence of protic compounds, such as alcohols, amines and water, among others [24–27]. When such protic reagents are present, the metal-alkoxide-terminated polymer chain is proposed to be in rapid exchange with the protic reagent, generating metal alkoxides and ‘free’ hydroxyl-terminated polymer chains. The chain transfer processes are proposed to occur more rapidly than propagation, leading to highly controlled polymerizations where the Mn of the polymer is determined by both the catalyst and chain transfer agent (protic reagent) concentrations. It is worth noting that in ROCOP the polycarbonate Mn is commonly experimentally observed to be rather lower than those expected for living polymerizations (where the Mn would depend only on the catalyst concentration) [25]. This is due to the presence or formation of chain transfer agents, such as diols, in the epoxide monomers used [28–32]. In fact, the exploitation of catalysts able to operate under immortal polymerization conditions, i.e. where such chain transfer agents are added in larger excess compared to the catalyst, is essential to selectively prepare the polyols that are required in polyurethane manufacture [18,20,24].
The catalysts for ROCOP have some general features: the metals are Lewis acids; the metals’ redox reactivity should be limited; the metal alkoxide and carbonate intermediates are labile; and initiating ligands (X) include alkoxides, carboxylates, halides and other anionic groups [5,8–14]. It is common in this area of catalysis that dinuclear or bimetallic catalysts show good performances and bimetallic pathways are proposed to accelerate epoxide ring opening [10]. Finally, the polymerization catalysts should ideally be colourless, odourless, inexpensive and have low toxicity, as they may contaminate the polymer product. In this context, there have been a number of reports of strategies to remove and recycle homogeneous catalysts [33–35].
3. Heterogeneous catalysts
The two major classes of heterogeneous catalysts are zinc glutarate (or other carboxylates) and double metal cyanides [4,36–52]. Both are well known, and in some cases industrially applied, as epoxide homopolymerization catalysts [52]. For copolymerizations using carbon dioxide, these heterogeneous catalysts require much more forcing conditions than homogeneous species. In particular, high pressures of carbon dioxide are necessary, and perfectly alternating enchainment does not occur, but rather ether-enriched polycarbonates are produced.
Zinc glutarate has been extensively studied, and two features stand out that increase activity: (i) the addition of ethylsulfinate groups and (ii) an increase in crystallinity of the catalyst [4,36–44]. The most commonly studied double metal cyanide catalyst is Zn3(CoCN6)2 (figure 3), although mixed Zn/Fe(III) species are also reported. These catalysts are typically applied as part of mixtures with so-called ‘complexation’ agents, including salts, alcohols and solvents [45–52]. They generally show very high activities but low CO2 uptakes; indeed, they commonly yield poly(ether carbonates) rather than perfectly alternating copolymers. Studying the mechanisms of such heterogeneous catalysts is very challenging; nevertheless, theoretical and experimental studies have led to the proposal that bimetallic polymerization processes are important. In particular, one study proposes that the active sites in zinc carboxylates should be separated by 4–5 Å [44].
Figure 3.

One of the crystal structures for zinc glutarate and a generic illustration of the structure of the repeat unit in zinc-cobalt double metal cyanide catalysts. Adapted with permission from [38]. Copyright (2004) American Chemical Society. (Online version in colour.)
4. Homogeneous catalysts
The common homogeneous catalysts can be classified into two broad types:
(a) Bicomponent catalyst systems comprising metal(III) complexes used with co-catalysts. The catalysts are usually complexes of Co(III), Cr(III), Mn(III) or Al(III) coordinated by ligands such as salens or porphyrins. The co-catalysts are typically ionic compounds, the most common of which is bis(triphenylphosphine)iminium (PPN) chloride (PPNCl), or Lewis bases, commonly 4-dimethylaminopyridine (DMAP).
(b) Dinuclear or bimetallic catalysts comprising metal(II/III) complexes. Most commonly these are complexes where two metals are coordinated by tethered ‘mononucleating’ ligands, such as Zn(II) β-diiminates (BDIs) or tethered Co(II)/Cr(III) salens. There are also examples of deliberately dinucleating ligands, such as macrocyclic ligands coordinated to Zn(II), Mg(II), Co(II/III) or Fe(III).
Most homogeneous catalysts operate under moderate/high pressures of CO2, and usually require more than 10 bar pressure, but, in contrast to heterogeneous catalysts, they do yield highly alternating copolymers [15,53–57]. In a drive to change the polymerization process conditions, catalysts have now been developed that show high activities and perfectly alternating enchainment under low pressures of carbon dioxide, including at 1 bar pressure [58–60].
(a). Bicomponent catalysts
These catalysts are used with an exogenous co-catalyst or, more recently, have the co-catalyst attached to the ancillary ligand scaffold [5,32,61–63]. The ligands are usually planar, tetradentate, dianionic compounds such as salens or porphyrins. The commonly used co-catalysts are ionic salts, such as PPNX and ammonium halides. The structures of typical catalysts in this class are illustrated in figure 4 [5,32,61–63].
Figure 4.

Bicomponent catalysts for CO2/epoxide copolymerization. (a) General porphyrin structure [32,53,63–65]. (b) General salen structure [55,57,66]. (c) Bifunctional salcy complex tethered to piperidinium moieties [67]. (d) Bifunctional salen complex tethered to quaternary ammonium groups [33].
The first well-defined homogeneous catalysts were metal(III) porphyrin complexes (e.g. figure 4a), which in combination with an ionic salt allowed the controlled ROCOP of CO2/epoxide [68]. There have subsequently been several studies to elucidate the influences of various ligand substituents and metal centres, which revealed that Co(III) centres coordinated by porphyrin ligands, substituted with electron-withdrawing groups, showed the best performances [32,53,63,64,69–75]. However, in general, metal porphyrin catalysts have lower activities and productivities compared with metal salen catalysts.
Catalysts based on salen ligands (figure 4b) are some of the most active catalysts for CO2/epoxide copolymerizations [5]. Darensbourg [5,22,62,76–79], Coates [15,31,55,80–87], Li [88], Nozaki [28,67,89–91], Lee [29,30,33,92–95], Rieger [66,69,96–98] and others have led the development of various [salenMX] complexes (where M=Cr(III), Co(III), Al(III)), many of which show very high rates and selectivities. Coates pioneered the application of chiral [Co(salen)] complexes as highly active CO2/PO ROCOP catalysts. This class of chiral cobalt salen catalysts has been extensively investigated by a number of groups and they now show outstanding levels of regio- and stereochemical control [2,31,55]. Indeed, they have been applied in the preparation of entirely new classes of stereocomplex polycarbonates, starting from racemic mixtures of epoxides [80–82,90].
The nature of the co-catalyst is also important; as mentioned PPNX salts are widely applied. Furthermore, the amount of co-catalyst is generally optimum at 1 equivalent (versus metal complex), as greater quantities reduce activity. This is proposed to be due to competitive binding (versus epoxide) at the metal centre [99,100]. Using more than 1 equivalent of co-catalyst also increases the rate of back-biting side reactions to form cyclic carbonate products [5,57,100,101]. The co-catalyst serves a number of roles in the catalytic cycle. It is proposed that the co-catalyst binds to the metal complex in order to complete an octahedral coordination geometry and, by trans-coordination, enhances the labilization of the initiating or propagating group [5]. Additionally, the co-catalyst may act as an external nucleophile which initiates polymerization. The precise mechanisms by which such catalysts operate are rather complex and not yet fully defined, but it is generally proposed that the rate-limiting step involves epoxide ring opening rather than carbon dioxide insertion [5].
Recent kinetic studies have revealed that the polymerization rates, using metal salen catalysts, are typically dependent on catalyst concentration to a fractional order, usually between 1 and 2 [89]. This is indicative of pathways involving two metal complexes and/or dimerization in the rate-limiting step. Figure 5 illustrates the two different proposed pathways for epoxide ring opening. The fractional orders in catalyst concentration could be rationalized if both pathways are feasible and occurring concurrently. Significant work has also been carried out to investigate the coordination of epoxides to such catalysts [102–104]. The gas-phase binding of epoxides in Al(III) and Cr(III) salen and porphyrin complexes was studied by Chisholm and co-workers and found to follow metal Lewis acidity trends [103]. Darensbourg also showed that the ring opening of epoxides by a nucleophile was dependent on the metal–epoxide bond length and the binding enthalpies, not solely on the rate at which epoxide binding occurred [104]. It is worth noting that the coordination of co-catalysts changes the Lewis acidity of the metal centre and so would affect epoxide binding [105].
Figure 5.
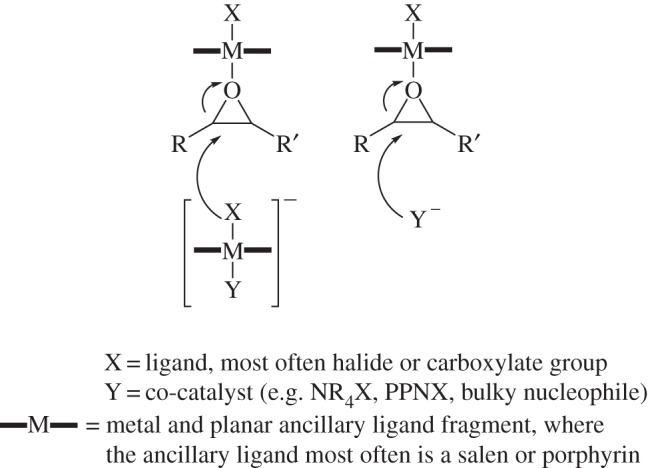
Possible pathways for epoxide ring opening by the bicomponent catalysts.
Recently, Nozaki and co-workers [89] demonstrated a theoretical model to predict the catalytic activity and selectivity in PO ROCOP using planar bicomponent catalysts. By comparing the difference between the dissociation energies for metal epoxide, carbonate and alkoxide intermediates, the preferred pathway at each stage of the catalytic cycle could be quickly estimated [89].
One challenge of exogenous co-catalysts is that, if the stoichiometry (versus catalyst) is not finely balanced, significant side reactions to produce cyclic carbonate can result. Furthermore, because the systems are bicomponent, low catalyst loadings are not generally feasible. Finally, the fractional orders in catalyst imply that dimerization may be necessary, thereby providing an entropic barrier to catalysis at low metal loading. Two strategies have been used to overcome these limitations: (i) the development of catalysts whereby the co-catalyst is attached to the ancillary ligand and (ii) the development of dinuclear salen catalysts.
Increased activities, tolerance and selectivities were observed by covalently bonding the co-catalyst moiety to the catalyst ligand, removing the need to add an exogenous co-catalyst source. Nozaki first reported this phenomenon for a Co(III) salen complex, substituted with piperidinium ‘arms’ (figure 4c) [67]. This catalyst showed very high selectivity for carbonate formation (more than 99%), a high turnover frequency (TOF) (250 h−1) and operated at room temperature, under 14 bar pressure of CO2 [67]. Lee subsequently also developed various bifunctional catalysts (figure 4d) substituted with ionic groups, which have shown some of the highest activities (TOF =26 000 h−1) ever reported and which are active under low catalyst loadings (1:25 000 catalyst : PO) [29,30,33]. Lu has also shown that a cobalt salen complex with a tethered quaternary ammonium salt is highly efficient (TOF up to 5160 h−1) for the terpolymerization of CHO with a range of aliphatic epoxides (such as PO) and CO2 [106].
(b). Dinuclear or bimetallic catalysts
An important development for the field was the report from Coates, in 1998, of highly active zinc BDI catalysts [107]. These catalysts have shown good activity and selectivity for CO2/CHO ROCOP, at 50°C and 7 bar pressure of CO2. A wide range of [(BDI)ZnX] complexes have since been synthesized where ligand substituents exert a strong effect on activity (figure 6a) [107,111–114]. In a landmark paper investigating the mechanism, it was shown that the most active catalysts existed as ‘loosely associated’ dimers under the polymerization conditions [56]. Rate studies revealed that the order in zinc varied from 1.0 to 1.8, implicating dimeric active sites [56].
Figure 6.

Dinuclear catalysts for CO2/epoxide copolymerization: (a) zinc BDI complex [56]; (b) zinc bis(anilido-aldimine) catalysts [108]; (c) tethered zinc BDI complex (TMS = trimethylsilyl) [109]; (d) tethered cobalt salen complex [91]; (e) dinuclear macrocyclic catalyst [110].
This study has inspired many others to develop dinuclear catalysts so as to increase activity and selectivity. Lee pioneered this with a series of bis(anilido-aldimine) Zn(II) complexes which showed high activities (TOF =2860 h−1) in copolymerization, at 1 : 50 000 [Zn] : [epoxide] loadings (figure 6b) [108]. The activity is sensitive to the N-aryl ortho-substituents and the fluorination of the aromatic rings significantly increased the TOF values [108]. Many researchers have investigated methods to ‘tether’ Zn(II) BDI catalysts [109,115–117]. The geometry, site and flexibility of the tether exert a significant influence over the activity, the best system for CO2/CHO being a dinuclear Zn(II) catalyst (figure 6c), which showed TOF values of 155 000 h−1, at 100°C and 30 bar [109,116,117].
Nozaki and Rieger have investigated the tethering together of two salen ligands (figure 6d) so as to target dinuclear catalysts [91,97]. When no co-catalyst was used, these catalysts operated via a bimetallic mechanism and showed improved performances (6–11 times better than the monometallic counterparts). Additionally, these catalysts were able to operate at low catalyst loadings (up to 20 000, epoxide/catalyst), supporting the notion that two metal sites may be involved in the pathway [91,97].
Our research group has focused on dinuclear catalysts coordinated by macrocyclic diphenolate ancillary ligands for CO2/CHO ROCOP [20,26,60,110,118–129]. The di-zinc catalyst (figure 6e) was the first catalyst to show promising activity under 1 bar pressure of CO2 [60]. Subsequently, we have explored a range of ligands, metals and co-ligands, including the first reports of active Fe(III) and Mg(II) catalysts for ROCOP [20,26,60,110,118–128]. Changing the metal centre significantly affects the polymerization rates, with the order of activity being Co(III) > Mg(II) > Fe(III) > Zn(II) using the same symmetrical ancillary ligand [20,26,60,110,118–128]. Kinetic studies of CO2/epoxide ROCOP, using the di-zinc acetate complex, showed a first-order dependence on CHO and catalyst concentrations and zero-order dependence on CO2 (1–40 bar) [124].
Furthermore, the detailed analysis of the temperature dependence of the rate coefficients for both polymerization and cyclic carbonate formation enables a comparison of the relative barriers for polymerization (polycyclohexene carbonate, PCHC) versus cyclic carbonate formation (cyclohexene carbonate, CHC), the results of which are qualitatively illustrated in figure 7 [124]. It is apparent that the di-zinc catalyst shows a high selectivity for polymer formation due to the barrier to polymerization being approximately half the value for cyclic carbonate formation (Ea(PCHC) of 96.8 kJ mol−1 and Ea(CHC) of 137.5 kJ mol−1).
Figure 7.

The different reaction energy barriers to the formation of PCHC versus cyclic carbonate. As part of the full kinetic analysis of the polymerization, the relative barriers were determined to be Ea(PCHC) of 96.8 kJ mol−1 and Ea(CHC) of 137.5 kJ mol−1. Adapted with permission from [124]. Copyright (2011) American Chemical Society.
The rate law shows a zero-order dependence of carbon dioxide pressure (1–40 bar), which suggests that, under these conditions, the ring opening of the epoxide is rate-determining. The first-order catalyst dependence also supports the proposal of dinuclear propagation pathways. Although the catalyst is dinuclear, it also has two acetate co-ligands and therefore could potentially initiate two polymer chains per catalyst [123]. The polymerization kinetics show first-order dependences on both catalyst and epoxide concentrations, which implies that only one acetate co-ligand initiates polymerization, although more complex pathways cannot be ruled out on the basis of the rate law. This led to further investigations into the possible roles for the co-ligands.
A series of di-cobalt halide catalysts were prepared, with various neutral co-ligands being coordinated to them, including pyridine, methyl imidazole and DMAP (figures 8 and 9) [122]. The complexes all showed closely related solid-state structures, with the ancillary ligand adopting a ‘bowl’ conformation, whereby the NH substituents occupy the same ‘face’ of the molecule. In the concave face of the molecule, a halide ligand bridges the two cobalt centres. On the opposite convex face, a halide and Lewis base donor molecule coordinate to the cobalt centres (figure 8). It was discovered that the complexes showed rates strongly dependent on the nature of the donor ligand, with DMAP complexes having low activities and those coordinated by the weaker pyridine donor being equivalently active to catalysts without any co-ligand coordination. The finding that coordination of a single DMAP molecule could significantly slow the catalysis supported the notion that polymerization dominates from the convex face of the molecule and that two metals are involved.
Figure 8.

The structure, as determined using X-ray crystallography, of the di-cobalt catalyst [LCo2Cl2(methyl imidazole)]. The ‘bowl’ shape of the ligand is notable in the structure, as are the twofaces. The convex face refers to the outside of the ‘bowl’ (i.e. where Cl and MeIm are coordinated) and the concave face refers to the inside of the ‘bowl’ (i.e. where the bridging Cl ligand is coordinated). Adapted from [122].
Figure 9.

The structures of the di-Co(II) complexes and the corresponding activities in the copolymerization of CO2/CHO. Polymerization conditions: CHO : catalyst = 1000 : 1, 80°C, 1 bar CO2 [122].
A detailed spectroscopic and density functional theory (DFT) study also shed light on the polymerization pathways and key intermediates [123]. Using in situ attenuated total reflectance–infrared (ATR-IR) spectroscopy to characterize the reaction between di-zinc acetate complex and CHO showed that there were different environments for the acetate co-ligand. One of the acetate groups shows resonances consistent with attack at the CHO group, while the other has a resonance consistent with it maintaining a ‘bridging’ coordination mode between the two zinc centres.
The DFT study, carried out in solution, using appropriate functionals to model the polar environment, and using the complete catalyst structure, also substantiated the proposal that, although both metals are involved in propagation, only one of the two co-ligands initiates and propagates polymerization. The remaining co-ligand remains coordinated to the metal centres and mediates the polymerization process. Indeed, it was revealed by DFT that the polymer chain ‘switches’ coordination site between the two metal centres with each monomer insertion (epoxide or carbon dioxide). The chain shuttling is counter-balanced by an equal but opposite change in coordination site for the acetate co-ligand. It was shown that, by changing the co-ligand, it was possible to alter the rate of polymerization and DFT could be used to predict activity [123].
The chain shuttling mechanism implies that there are distinct roles for the two metal centres, as sketched in figure 10. Thus, one of the metal centres (illustrated in blue) coordinates the epoxides, while the other centre (illustrated in red) inserts carbon dioxide. The requirements for these processes are distinct, with epoxide coordination being accelerated by Lewis acidic/electrophilic metal centres, while the carbonate formation and attack step is favoured by metals showing labile carbonate groups. Such a mechanism would be expected to be improved by having distinct metals serving the two roles and thus provided an impetus to study heterodinuclear catalysts.
Figure 10.

Comparison of the experimentally determined free energy barrier (a) to polymerizationwith the value determined by DFT (b) [123]. (Online version in colour.)
In 2014, our group reported the first example of such a system [120]. The catalyst is a mixture of compounds, including both homodinuclear complexes and the heterodinuclear complex. Figure 11 compares the activity and productivity of this catalyst mixture, the homodinuclear complexes alone and in a 50 : 50 combination. It is clear that the mixture containing the heterodinculear catalyst shows an activity that is greater than either of the homodinuclear complexes or combinations of them. This finding provides the first evidence, albeit of a mixture of components, that heterodinuclear complexes are worth investigation in this field of catalysis [120].
Figure 11.

The structures of di-zinc (1), di-magnesium (2) and the heterodinuclear mixed catalystsystem (3) which were compared for the ROCOP of CO2/CHO [120]. (Online version in colour.)
5. Conclusion
The ROCOP of carbon dioxide and epoxides provides a useful means to reduce pollution, to consume carbon dioxide and to produce aliphatic polycarbonates. Low-Mn, hydroxyl-end-capped, polycarbonate polyols are emerging as useful materials for the production of higher polymers, notably polyurethanes. There remain significant opportunities and challenges to understand and optimize the material properties of the polymers depending on the catalyst and raw materials available.
Catalysis plays a key role in the success and scope of this reaction. The selection of the catalyst is central to being able to control features such as the rate, productivity, selectivity, regio-/stereo-chemistry, polymer molecular weight, polymer end-groups and polymer composition and to produce block copolymers. So far, several successful heterogeneous and homogeneous catalyst types have been studied. A common theme is that the pathways are often proposed to occur via dinuclear or bimetallic routes, whereby one metal activates the epoxide, while the other metal provides the nucleophile (carbonate) to attack and ring-open the epoxide. Thus, recently, a number of improvements to catalyst rate and selectivity have been achieved by targeting structures that optimize the coordination chemistry of dinuclear catalysts. The discovery and further development of dinuclear and bimetallic homo- and heterogeneous catalysts is an important area for future research.
Authors' contributions
All authors have contributed to the review article.
Competing interests
C.K.W. is a director and CSO of Econic Technologies.
Funding
The Engineering and Physical Sciences Research Council (EPSRC) are acknowledged for funding (EP/K035274/1; EP/K014070/1; EP/K014668; EP/L017393/1).
References
- 1.Inoue S, Koinuma H, Tsuruta T. 1969. Copolymerization of carbon dioxide and epoxide. J. Poly. Sci. B Polym. Lett. 7, 287–292. ( 10.1002/pol.1969.110070408) [DOI] [Google Scholar]
- 2.Childers MI, Longo JM, Van Zee NJ, LaPointe AM, Coates GW. 2014. Stereoselective epoxide polymerization and copolymerization. Chem. Rev. 114, 8129–8152. ( 10.1021/cr400725x) [DOI] [PubMed] [Google Scholar]
- 3.Paul S, Zhu YQ, Romain C, Saini PK, Brooks R, Williams CK. 2015. Ring-opening copolymerization (ROCOP): synthesis and properties of polyesters and polycarbonates. Chem. Commun. 51, 6459–6479. ( 10.1039/C4CC10113H) [DOI] [PubMed] [Google Scholar]
- 4.Luinstra GA. 2008. Poly(propylene carbonate), old copolymers of propylene oxide and carbon dioxide with new interests: catalysis and material properties. Polym. Rev. 48, 192–219. ( 10.1080/15583720701834240) [DOI] [Google Scholar]
- 5.Darensbourg DJ. 2007. Making plastics from carbon dioxide: salen metal complexes as catalysts for the production of polycarbonates from epoxides and CO2. Chem. Rev. 107, 2388–2410. ( 10.1021/cr068363q) [DOI] [PubMed] [Google Scholar]
- 6.Markewitz P, Kuckshinrichs W, Leitner W, Linssen J, Zapp P, Bongartz R, Schreiber A, Müller TE. 2012. Worldwide innovations in the development of carbon capture technologies and the utilization of CO2. Energy Environ. Sci. 5, 7281–7305. ( 10.1039/c2ee03403d) [DOI] [Google Scholar]
- 7.MacDowell N. et al 2010. An overview of CO2 capture technologies. Energy Environ. Sci. 3, 1645–1669. ( 10.1039/c004106h) [DOI] [Google Scholar]
- 8.Darensbourg DJ, Yeung AD. 2014. A concise review of computational studies of the carbon dioxide–epoxide copolymerization reactions. Polym. Chem. 5, 3949–3962. ( 10.1039/c4py00299g) [DOI] [Google Scholar]
- 9.Darensbourg DJ, Wilson SJ. 2012. What’s new with CO2? Recent advances in its copolymerization with oxiranes. Green Chem. 14, 2665–2671. ( 10.1039/c2gc35928f) [DOI] [Google Scholar]
- 10.Romain C, Thevenon A, Saini PK, Williams CK. 2015. Dinuclear metal complex-mediated formation of CO2-based polycarbonates. In Carbon dioxide and organometallics (ed.Lu X-B.), Topics in Organometallic Chemistry, no. 53, pp. 101–141. Cham, Switzerland: Springer International; ( 10.1007/3418_2015_95) [DOI] [Google Scholar]
- 11.Kember MR, Buchard A, Williams CK. 2011. Catalysts for CO2/epoxide copolymerisation. Chem. Commun. 47, 141–163. ( 10.1039/C0CC02207A) [DOI] [PubMed] [Google Scholar]
- 12.Nozaki K. 2004. Asymmetric catalytic synthesis of polyketones and polycarbonates. J. Macromol. Sci. Pure Appl. Chem. 76, 541–546. ( 10.1351/pac200476030541) [DOI] [Google Scholar]
- 13.Lu X-B, Ren W-M, Wu G-P. 2012. CO2 copolymers from epoxides: catalyst activity, product selectivity, and stereochemistry control. Acc. Chem. Res. 45, 1721–1735. ( 10.1021/ar300035z) [DOI] [PubMed] [Google Scholar]
- 14.Klaus S, Lehenmeier MW, Anderson CE, Rieger B. 2011. Recent advances in CO2/epoxide copolymerization—new strategies and cooperative mechanisms. Coord. Chem. Rev. 255, 1460–1479. ( 10.1016/j.ccr.2010.12.002) [DOI] [Google Scholar]
- 15.Coates GW, Moore DR. 2004. Discrete metal-based catalysts for the copolymerization of CO2 and epoxides: discovery, reactivity, optimization, and mechanism. Angew. Chem. Int. Ed. 43, 6618–6639. ( 10.1002/anie.200460442) [DOI] [PubMed] [Google Scholar]
- 16.Langanke J, Wolf A, Hofmann J, Bohm K, Subhani MA, Muller TE, Leitner W, Gürtler C. 2014. Carbon dioxide (CO2) as sustainable feedstock for polyurethane production. Green Chem. 16, 1865–1870. ( 10.1039/C3GC41788C) [DOI] [Google Scholar]
- 17.Lee SH, Cyriac A, Jeon JY, Lee BY. 2012. Preparation of thermoplastic polyurethanes using in situ generated poly(propylene carbonate)-diols. Polym. Chem. 3, 1215–1220. ( 10.1039/c2py00010e) [DOI] [Google Scholar]
- 18.von der Assen N, Bardow A. 2014. Life cycle assessment of polyols for polyurethane production using CO2 as feedstock: insights from an industrial case study. Green Chem. 16, 3272–3280. ( 10.1039/c4gc00513a) [DOI] [Google Scholar]
- 19.Korashvili R, Noernberg B, Bornholdt N, Borchardt E, Luinstra GA. 2013. Poly(propylene carbonate) from carbon dioxide: challenges for large-scale application. Chem. Ing. Tech. 85, 437–446. ( 10.1002/cite.201200215) [DOI] [Google Scholar]
- 20.Chapman AC, Keyworth C, Kember MR, Lennox AJJ, Williams CK. 2015. Adding value to power station captured CO2: tolerant Zn and Mg homogeneous catalysts for polycarbonate polyol production. ACS Catal. 5, 1581–1588. ( 10.1021/cs501798s) [DOI] [Google Scholar]
- 21.Darensbourg DJ, Wei S-H. 2012. Depolymerization of polycarbonates derived from carbon dioxide and epoxides to provide cyclic carbonates. A kinetic study. Macromolecules 45, 5916–5922. ( 10.1021/ma301110c) [DOI] [Google Scholar]
- 22.Darensbourg DJ, Moncada AI, Wei S-H. 2011. Aliphatic polycarbonates produced from the coupling of carbon dioxide and oxetanes and their depolymerization via cyclic carbonate formation. Macromolecules 44, 2568–2576. ( 10.1021/ma2002323) [DOI] [Google Scholar]
- 23.Wu G-P, Wei S-H, Ren W-M, Lu X-B, Li B, Zu Y-P, Darensbourg DJ. 2011. Alternating copolymerization of CO2 and styrene oxide with Co(III)-based catalyst systems: differences between styrene oxide and propylene oxide. Energy Environ. Sci. 4, 5084–5092. ( 10.1039/c1ee02566j) [DOI] [Google Scholar]
- 24.Cyriac A, Lee SH, Varghese JK, Park ES, Park JH, Lee BY. 2010. Immortal CO2/propylene oxide copolymerization: precise control of molecular weight and architecture of various block copolymers. Macromolecules 43, 7398–7401. ( 10.1021/ma101259k) [DOI] [Google Scholar]
- 25.Inoue S. 2000. Immortal polymerization: the outset, development, and application. J. Polym. Sci. A Polym. Chem. 38, 2861–2871. () [DOI] [Google Scholar]
- 26.Kember MR, Copley J, Buchard A, Williams CK. 2012. Triblock copolymers from lactide and telechelic poly(cyclohexene carbonate). Polym. Chem. 3, 1196–1201. ( 10.1039/c2py00543c) [DOI] [Google Scholar]
- 27.Yi N, Unruangsri J, Shaw J, Williams CK. 2015. Carbon dioxide capture and utilization: using dinuclear catalysts to prepare polycarbonates. Faraday Discuss. 183, 67–82. ( 10.1039/c5fd00073d) [DOI] [PubMed] [Google Scholar]
- 28.Nakano K, Nakamura M, Nozaki K. 2009. Alternating copolymerization of cyclohexene oxide with carbon dioxide catalyzed by (salalen)CrCl complexes. Macromolecules 42, 6972–6980. ( 10.1021/ma9012626) [DOI] [Google Scholar]
- 29.Na SJ, Sujith S, Cyriac A, Kim BE, Yoo J, Kang YK, Han SJ, Lee C, Lee BY. 2009. Elucidation of the structure of a highly active catalytic system for CO2/epoxide copolymerization: a salen–cobaltate complex of an unusual binding mode. Inorg. Chem. 48, 10 455–10 465. ( 10.1021/ic901584u) [DOI] [PubMed] [Google Scholar]
- 30.Noh EK, Na SJ, Sujith S, Kim SW, Lee BY. 2007. Two components in a molecule: highly efficient and thermally robust catalytic system for CO2/epoxide copolymerization. J. Am. Chem. Soc. 129, 8082–8083. ( 10.1021/ja071290n) [DOI] [PubMed] [Google Scholar]
- 31.Cohen CT, Chu T, Coates GW. 2005. Cobalt catalysts for the alternating copolymerization of propylene oxide and carbon dioxide: combining high activity and selectivity. J. Am. Chem. Soc. 127, 10 869–10 878. ( 10.1021/ja051744l) [DOI] [PubMed] [Google Scholar]
- 32.Chatterjee C, Chisholm MH. 2011. The influence of the metal (Al, Cr, and Co) and the substituents of the porphyrin in controlling the reactions involved in the copolymerization of propylene oxide and carbon dioxide by porphyrin metal(III) complexes. 1. Aluminum chemistry. Inorg. Chem. 50, 4481–4492. ( 10.1021/ic200142f) [DOI] [PubMed] [Google Scholar]
- 33.Sujith S, Min JK, Seong JE, Na SJ, Lee BY. 2008. A highly active and recyclable catalytic system for CO2 propylene oxide copolymerization. Angew. Chem. Int. Ed. 47, 7306–7309. ( 10.1002/anie.200801852) [DOI] [PubMed] [Google Scholar]
- 34.Nakano K, Fujie R, Shintani R, Nozaki K. 2013. Efficient catalyst removal and recycling in copolymerization of epoxides with carbon dioxide via simple liquid–liquid phase separation. Chem. Commun. 49, 9332–9334. ( 10.1039/c3cc45622f) [DOI] [PubMed] [Google Scholar]
- 35.Yu K, Jones CW. 2003. Silica-immobilized zinc β-diiminate catalysts for the copolymerization of epoxides and carbon dioxide. Organometallics 22, 2571–2580. ( 10.1021/om030209w) [DOI] [Google Scholar]
- 36.Ree M, Hwang Y, Kim JS, Kim H, Kim G. 2006. New findings in the catalytic activity of zinc glutarate and its application in the chemical fixation of CO2 into polycarbonates and their derivatives. Catal. Today 115, 134–145. ( 10.1016/j.cattod.2006.02.068) [DOI] [Google Scholar]
- 37.Hwang Y, Kim H, Ree M. 2005. Zinc glutarate catalyzed synthesis and biodegradability of poly(carbonate-co-ester)s from CO2, propylene oxide, and ε-caprolactone. Macromol. Symp. 224, 227–237. ( 10.1002/masy.200550620) [DOI] [Google Scholar]
- 38.Kim JS, Kim H, Ree M. 2004. Hydrothermal synthesis of single-crystalline zinc glutarate and its structural determination. Chem. Mater. 16, 2981–2983. ( 10.1021/cm035358j) [DOI] [Google Scholar]
- 39.Kim JS. et al 2003. X-ray absorption and NMR spectroscopic investigations of zinc glutarates prepared from various zinc sources and their catalytic activities in the copolymerization of carbon dioxide and propylene oxide. J. Catal. 218, 209–219. ( 10.1016/S0021-9517(03)00082-4) [DOI] [Google Scholar]
- 40.Kim JS. et al 2003. NEXAFS spectroscopy study of the surface properties of zinc glutarate and its reactivity with carbon dioxide and propylene oxide. J. Catal. 218, 386–395. ( 10.1016/S0021-9517(03)00122-2) [DOI] [Google Scholar]
- 41.Ree M, Bae JY, Jung JH, Shin TJ. 1999. A new copolymerization process leading to poly(propylene carbonate) with a highly enhanced yield from carbon dioxide and propylene oxide. J. Polym. Sci. A Polym. Chem. 37, 1863–1876. () [DOI] [Google Scholar]
- 42.Eberhardt R, Allmendinger M, Zintl M, Troll C, Luinstra GA, Rieger B. 2004. New zinc dicarboxylate catalysts for the CO2/propylene oxide copolymerization reaction: activity enhancement through Zn(II)-ethylsulfinate initiating groups. Macromol. Chem. Phys. 205, 42–47. ( 10.1002/macp.200350081) [DOI] [Google Scholar]
- 43.Wu X, Zhao H, Nörnberg B, Theato P, Luinstra GA. 2014. Synthesis and characterization of hydroxyl-functionalized poly(propylene carbonate). Macromolecules 47, 492–497. ( 10.1021/ma401899h) [DOI] [Google Scholar]
- 44.Klaus S, Lehenmeier MW, Herdtweck E, Deglmann P, Ott AK, Rieger B. 2011. Mechanistic insights into heterogeneous zinc dicarboxylates and theoretical considerations for CO2–epoxide copolymerization. J. Am. Chem. Soc. 133, 13 151–13 161. ( 10.1021/ja204481w) [DOI] [PubMed] [Google Scholar]
- 45.Varghese JK, Park DS, Jeon JY, Lee BY. 2013. Double metal cyanide catalyst prepared using H3Co(CN)6 for high carbonate fraction and molecular weight control in carbon dioxide/propylene oxide copolymerization. J. Polym. Sci. A Polym. Chem. 51, 4811–4818. ( 10.1002/pola.26905) [DOI] [Google Scholar]
- 46.Sebastian J, Srinivas D. 2013. Influence of method of preparation of solid, double-metal cyanide complexes on their catalytic activity for synthesis of hyperbranched polymers. Appl. Catal. A 464, 51–60. ( 10.1016/j.apcata.2013.05.024) [DOI] [Google Scholar]
- 47.Zhang X-H, Wei R-J, Sun X-K, Zhang J-F, Du B-Y, Fan Z-Q, Qi G-R. 2011. Selective copolymerization of carbon dioxide with propylene oxide catalyzed by a nanolamellar double metal cyanide complex catalyst at low polymerization temperatures. Polym. J. 52, 5494–5502. ( 10.1016/j.polymer.2011.09.040) [DOI] [Google Scholar]
- 48.Sun XK, Zhang XH, Liu F, Chen S, Du BY, Wang Q, Fan Z-Q, Qi G-R. 2008. Alternating copolymerization of carbon dioxide and cyclohexene oxide catalyzed by silicon dioxide/Zn–CoIII double metal cyanide complex hybrid catalysts with a nanolamellar structure. J. Polym. Sci. A Polym. Chem. 46, 3128–3139. ( 10.1002/pola.22666) [DOI] [Google Scholar]
- 49.Robertson NJ, Qin ZQ, Dallinger GC, Lobkovsky EB, Lee S, Coates GW. 2006. Two-dimensional double metal cyanide complexes: highly active catalysts for the homopolymerization of propylene oxide and copolymerization of propylene oxide and carbon dioxide. Dalton Trans. 2006, 5390–5395. ( 10.1039/b607963f) [DOI] [PubMed] [Google Scholar]
- 50.Kim I, Yi MJ, Lee KJ, Park D-W, Kim BU, Ha C-S. 2006. Aliphatic polycarbonate synthesis by copolymerization of carbon dioxide with epoxides over double metal cyanide catalysts prepared by using ZnX2 (X = F, Cl, Br, I). Catal. Today 111, 292–296. ( 10.1016/j.cattod.2005.10.039) [DOI] [Google Scholar]
- 51.Chen S, Qi G-R, Hua Z-J, Yan H-Q. 2004. Double metal cyanide complex based on Zn3[Co(CN)6]2 as highly active catalyst for copolymerization of carbon dioxide and cyclohexene oxide. J. Polym. Sci. A Polym. Chem. 42, 5284–5291. ( 10.1002/pola.20334) [DOI] [Google Scholar]
- 52.Kruper WJ Jr, Swart DJ. 1983. Dow Chemical Company. Carbon dioxide oxirane copolymers prepared using double metal cyanide complexes. Patent US 4,500,704, 15 August 1983.
- 53.Sugimoto H, Ohshima H, Inoue S. 2003. Alternating copolymerization of carbon dioxide and epoxide by manganese porphyrin: the first example of polycarbonate synthesis from 1-atm carbon dioxide. J. Polym. Sci. A Polym. Chem. 41, 3549–3555. ( 10.1002/pola.10835) [DOI] [Google Scholar]
- 54.Darensbourg DJ, Wildeson JR, Yarbrough JC, Reibenspies JH. 2000. Bis 2,6-difluorophenoxide dimeric complexes of zinc and cadmium and their phosphine adducts: lessons learned relative to carbon dioxide/cyclohexene oxide alternating copolymerization processes catalyzed by zinc phenoxides. J. Am. Chem. Soc. 122, 12 487–12 496. ( 10.1021/ja002855h) [DOI] [Google Scholar]
- 55.Qin Z, Thomas CM, Lee S, Coates GW. 2003. Cobalt-based complexes for the copolymerization of propylene oxide and CO2: active and selective catalysts for polycarbonate synthesis. Angew. Chem. Int. Ed. 42, 5484–5487. ( 10.1002/anie.200352605) [DOI] [PubMed] [Google Scholar]
- 56.Moore DR, Cheng M, Lobkovsky EB, Coates GW. 2003. Mechanism of the alternating copolymerization of epoxides and CO2 using β-diiminate zinc catalysts: evidence for a bimetallic epoxide enchainment. J. Am. Chem. Soc. 125, 11 911–11 924. ( 10.1021/ja030085e) [DOI] [PubMed] [Google Scholar]
- 57.Darensbourg DJ, Yarbrough JC. 2002. Mechanistic aspects of the copolymerization reaction of carbon dioxide and epoxides, using a chiral salen chromium chloride catalyst. J. Am. Chem. Soc. 124, 6335–6342. ( 10.1021/ja012714v) [DOI] [PubMed] [Google Scholar]
- 58.Xiao Y, Wang Z, Ding K. 2005. Copolymerization of cyclohexene oxide with CO2 by using intramolecular dinuclear zinc catalysts. Chem. Eur. J. 11, 3668–3678. ( 10.1002/chem.200401159) [DOI] [PubMed] [Google Scholar]
- 59.Xiao Y, Wang Z, Ding K. 2006. Intramolecularly dinuclear magnesium complex catalyzed copolymerization of cyclohexene oxide with CO2 under ambient CO2 pressure: kinetics and mechanism. Macromolecules 39, 128–137. ( 10.1021/ma051859+) [DOI] [Google Scholar]
- 60.Kember MR, Knight PD, Reung PTR, Williams CK. 2009. Highly active dizinc catalyst for the copolymerization of carbon dioxide and cyclohexene oxide at one atmosphere pressure. Angew. Chem. Int. Ed. 48, 931–933. ( 10.1002/anie.200803896) [DOI] [PubMed] [Google Scholar]
- 61.Lu X-B, Darensbourg DJ. 2012. Cobalt catalysts for the coupling of CO2 and epoxides to provide polycarbonates and cyclic carbonates. Chem. Soc. Rev. 41, 1462–1484. ( 10.1039/C1CS15142H) [DOI] [PubMed] [Google Scholar]
- 62.Darensbourg DJ. 2012. Salen metal complexes as catalysts for the synthesis of polycarbonates from cyclic ethers and carbon dioxide. In Synthetic biodegradable polymers (eds B Rieger, A Kunkel, GW Coates, R Reichardt, E Dinjus, TA Zevaco), Advances in Polymer Science, vol. 245, pp. 1–27. Berlin, Germany: Springer; ( 10.1007/12_2011_135) [DOI] [Google Scholar]
- 63.Chatterjee C, Chisholm MH. 2012. Influence of the metal (Al, Cr, and Co) and the substituents of the porphyrin in controlling the reactions involved in the copolymerization of propylene oxide and carbon dioxide by porphyrin metal(III) complexes. 2. Chromium chemistry. Inorg. Chem. 51, 12 041–12 052. ( 10.1021/ic302137w) [DOI] [PubMed] [Google Scholar]
- 64.Chatterjee C, Chisholm MH, El-Khaldy A, McIntosh RD, Miller JT, Wu T. 2013. Influence of the metal (Al, Cr, and Co) and substituents of the porphyrin in controlling reactions involved in copolymerization of propylene oxide and carbon dioxide by porphyrin metal(III) complexes. 3. Cobalt chemistry. Inorg. Chem. 52, 4547–4553. ( 10.1021/ic400068y) [DOI] [PubMed] [Google Scholar]
- 65.Aida T, Inoue S. 1983. Activation of carbon dioxide with aluminum porphyrin and reaction with epoxide. Studies on (tetraphenylporphinato)aluminum alkoxide having a long oxyalkylene chain as the alkoxide group. J. Am. Chem. Soc. 105, 1304–1309. ( 10.1021/ja00343a038) [DOI] [Google Scholar]
- 66.Klaus S, Vagin SI, Lehenmeier MW, Deglmann P, Brym AK, Rieger B. 2011. Kinetic and mechanistic investigation of mononuclear and flexibly linked dinuclear complexes for copolymerization of CO2 and epoxides. Macromolecules 44, 9508–9516. ( 10.1021/ma201387f) [DOI] [Google Scholar]
- 67.Nakano K, Kamada T, Nozaki K. 2006. Selective formation of polycarbonate over cyclic carbonate: copolymerization of epoxides with carbon dioxide catalyzed by a cobalt(III) complex with a piperidinium end-capping arm. Angew. Chem. Int. Ed. 45, 7274–7277. ( 10.1002/anie.200603132) [DOI] [PubMed] [Google Scholar]
- 68.Takeda N, Inoue S. 1978. Polymerization of 1,2-epoxypropane: a copolymerization with carbon dioxide catalyzed by metalloporphyrins. Macromol. Chem. Phys. 179, 1377–1381. ( 10.1002/macp.1978.021790529) [DOI] [Google Scholar]
- 69.Xia W, Salmeia KA, Vagin SI, Rieger B. 2015. Concerning the deactivation of cobalt(III)-based porphyrin and salen catalysts in epoxide/ CO2 copolymerization. Chem. Eur. J. 21, 4384–4390. ( 10.1002/chem.201406258) [DOI] [PubMed] [Google Scholar]
- 70.Xia W, Vagin SI, Rieger B. 2014. Regarding initial ring opening of propylene oxide in its copolymerization with CO2 catalyzed by a cobalt(III) porphyrin complex. Chem. Eur. J. 20, 15 499–15 504. ( 10.1002/chem.201404147) [DOI] [PubMed] [Google Scholar]
- 71.Harrold ND, Li Y, Chisholm MH. 2013. Studies of ring-opening reactions of styrene oxide by chromium tetraphenylporphyrin initiators. Mechanistic and stereochemical considerations. Macromolecules 46, 692–698. ( 10.1021/ma302492p) [DOI] [Google Scholar]
- 72.Anderson CE, Vagin SI, Hammann M, Zimmermann L, Rieger B. 2013. Copolymerisation of propylene oxide and carbon dioxide by dinuclear cobalt porphyrins. ChemCatChem 5, 3269–3280. ( 10.1002/cctc.201300307) [DOI] [Google Scholar]
- 73.Anderson CE, Vagin SI, Xia W, Jin H, Rieger B. 2012. Cobaltoporphyrin-catalyzed CO2/epoxide copolymerization: selectivity control by molecular design. Macromolecules 45, 6840–6849. ( 10.1021/ma301205g) [DOI] [Google Scholar]
- 74.Sugimoto H, Kuroda K. 2008. The cobalt porphyrin–Lewis base system: a highly selective catalyst for alternating copolymerization of CO2 and epoxide under mild conditions. Macromolecules 41, 312–317. ( 10.1021/ma702354s) [DOI] [Google Scholar]
- 75.Mang S, Cooper AI, Colclough ME, Chauhan N, Holmes AB. 2000. Copolymerization of CO2 and 1,2-cyclohexene oxide using a CO2-soluble chromium porphyrin catalyst. Macromolecules 33, 303–308. ( 10.1021/ma991162m) [DOI] [Google Scholar]
- 76.Darensbourg DJ, Yeung AD. 2015. Kinetics of the (salen)Cr(III)- and (salen)Co(III)-catalyzed copolymerization of epoxides with CO2, and of the accompanying degradation reactions. Polym. Chem. 6, 1103–1117. ( 10.1039/C4PY01322K) [DOI] [Google Scholar]
- 77.Darensbourg DJ, Wilson SJ. 2013. Synthesis of CO2-derived poly(indene carbonate) from indene oxide utilizing bifunctional cobalt(III) catalysts. Macromolecules 46, 5929–5934. ( 10.1021/ma4013779) [DOI] [Google Scholar]
- 78.Darensbourg DJ, Chung W-C. 2013. Relative basicities of cyclic ethers and esters. Chemistry of importance to ring-opening co- and terpolymerization reactions. Polyhedron 58, 139–143. ( 10.1016/j.poly.2012.08.076) [DOI] [Google Scholar]
- 79.Darensbourg DJ, Moncada AI, Choi W, Reibenspies JH. 2008. Mechanistic studies of the copolymerization reaction of oxetane and carbon dioxide to provide aliphatic polycarbonates catalyzed by (salen)CrX complexes. J. Am. Chem. Soc. 130, 6523–6533. ( 10.1021/ja800302c) [DOI] [PubMed] [Google Scholar]
- 80.Auriemma F, De Rosa C, Di Caprio MR, Di Girolamo R, Ellis WC, Coates GW. 2015. Stereocomplexed poly(limonene carbonate): a unique example of the cocrystallization of amorphous enantiomeric polymers. Angew. Chem. Int. Ed. 54, 1215–1218. ( 10.1002/anie.201410211) [DOI] [PubMed] [Google Scholar]
- 81.Auriemma F, De Rosa C, Di Caprio MR, Di Girolamo R, Coates GW. 2015. Crystallization of alternating limonene oxide/carbon dioxide copolymers: determination of the crystal structure of stereocomplex poly(limonene carbonate). Macromolecules 48, 2534–2550. ( 10.1021/acs.macromol.5b00157) [DOI] [Google Scholar]
- 82.Liu Y, Ren WM, Wang M, Liu C, Lu XB. 2015. Crystalline stereocomplexed polycarbonates: hydrogen-bond-driven interlocked orderly assembly of the opposite enantiomers. Angew. Chem. Int. Ed. 54, 2241–2244. ( 10.1002/anie.201410692) [DOI] [PubMed] [Google Scholar]
- 83.Liu Y, Ren WM, He KK, Lu XB. 2014. Crystalline-gradient polycarbonates prepared from enantioselective terpolymerization of meso-epoxides with CO2. Nat. Commun. 5, 5687 ( 10.1038/ncomms6687) [DOI] [PubMed] [Google Scholar]
- 84.Wu G-P, Zu Y-P, Xu P-X, Ren W-M, Lu X-B. 2013. Microstructure analysis of a CO2 copolymer from styrene oxide at the diad level. Asian J. Chem. 8, 1854–1862. ( 10.1002/asia.201300115) [DOI] [PubMed] [Google Scholar]
- 85.Wu G-P, Xu P-X, Zu Y-P, Ren W-M, Lu X-B. 2013. Cobalt(III)-complex-mediated terpolymerization of CO2, styrene oxide, and epoxides with an electron-donating group. J. Polym. Sci. A Polym. Chem. 51, 874–879. ( 10.1002/pola.26444) [DOI] [Google Scholar]
- 86.Wu G-P, Xu P-X, Lu X-B, Zu Y-P, Wei S-H, Ren W-M, Darensbourg DJ. 2013. Crystalline CO2 copolymer from epichlorohydrin via Co(III)-complex-mediated stereospecific polymerization. Macromolecules 46, 2128–2133. ( 10.1021/ma400252h) [DOI] [Google Scholar]
- 87.Liu Y, Ren W-M, Liu J, Lu X-B. 2013. Asymmetric copolymerization of CO2 with meso-epoxides mediated by dinuclear cobalt(III) complexes: unprecedented enantioselectivity and activity. Angew. Chem. Int. Ed. 52, 11 594–11 598. ( 10.1002/anie.201305154) [DOI] [PubMed] [Google Scholar]
- 88.Niu Y, Li H. 2013. Alternating copolymerization of carbon dioxide and cyclohexene oxide catalyzed by salen CoIII(acetate) complexes. Colloid. Polym. Sci. 291, 2181–2189. ( 10.1007/s00396-013-2957-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ohkawara T, Suzuki K, Nakano K, Mori S, Nozaki K. 2014. Facile estimation of catalytic activity and selectivities in copolymerization of propylene oxide with carbon dioxide mediated by metal complexes with planar tetradentate ligand. J. Am. Chem. Soc. 136, 10 728–10 735. ( 10.1021/ja5046814) [DOI] [PubMed] [Google Scholar]
- 90.Nakano K, Hashimoto S, Nakamura M, Kamada T, Nozaki K. 2011. Stereocomplex of poly(propylene carbonate): synthesis of stereogradient poly(propylene carbonate) by regio- and enantioselective copolymerization of propylene oxide with carbon dioxide. Angew. Chem. Int. Ed. 50, 4868–4871. ( 10.1002/anie.201007958) [DOI] [PubMed] [Google Scholar]
- 91.Nakano K, Hashimoto S, Nozaki K. 2010. Bimetallic mechanism operating in the copolymerization of propylene oxide with carbon dioxide catalyzed by cobalt–salen complexes. Chem. Sci. 1, 369–373. ( 10.1039/c0sc00220h) [DOI] [Google Scholar]
- 92.Jeon JY, Lee JJ, Varghese JK, Na SJ, Sujith S, Go MJ, Lee J, Ok M-A, Lee BY. 2013. CO2/ethylene oxide copolymerization and ligand variation for a highly active salen–cobalt(III) complex tethering 4 quaternary ammonium salts. Dalton Trans. 42, 9245–9254. ( 10.1039/C2DT31854G) [DOI] [PubMed] [Google Scholar]
- 93.Yoo J, Na SJ, Park HC, Cyriac A, Lee BY. 2010. Anion variation on a cobalt(III) complex of salen-type ligand tethered by four quaternary ammonium salts for CO2/epoxide copolymerization. Dalton Trans. 39, 2622–2630. ( 10.1039/b920992a) [DOI] [PubMed] [Google Scholar]
- 94.Seong JE, Na SJ, Cyriac A, Kim B-W, Lee BY. 2010. Terpolymerizations of CO2, propylene oxide, and various epoxides using a cobalt(III) complex of salen-type ligand tethered by four quaternary ammonium salts. Macromolecules 43, 903–908. ( 10.1021/ma902162n) [DOI] [Google Scholar]
- 95.Kim BE, Varghese JK, Han YG, Lee BY. 2010. Cobalt(III) complexes of various salen-type ligand bearing four quaternary ammonium salts and their reactivity for CO2/epoxide copolymerization. Bull. Korean Chem. Soc. 31, 829–834. ( 10.5012/bkcs.2010.31.04.829) [DOI] [Google Scholar]
- 96.Luinstra GA, Haas GR, Molnar F, Bernhart V, Eberhardt R, Rieger B. 2005. On the formation of aliphatic polycarbonates from epoxides with chromium(III) and aluminum(III) metal–salen complexes. Chem. Eur. J. 11, 6298–6314. ( 10.1002/chem.200500356) [DOI] [PubMed] [Google Scholar]
- 97.Vagin SI, Reichardt R, Klaus S, Rieger B. 2010. Conformationally flexible dimeric salphen complexes for bifunctional catalysis. J. Am. Chem. Soc. 132, 14 367–14 369. ( 10.1021/ja106484t) [DOI] [PubMed] [Google Scholar]
- 98.Eberhardt R, Allmendinger M, Rieger B. 2003. DMAP/Cr(III) catalyst ratio: the decisive factor for poly(propylene carbonate) formation in the coupling of CO2 and propylene oxide. Macromol. Rapid Commun. 24, 194–196. ( 10.1002/marc.200390022) [DOI] [Google Scholar]
- 99.Paddock RL, Nguyen ST. 2001. Chemical CO2 fixation: Cr(III) salen complexes as highly efficient catalysts for the coupling of CO2 and epoxides. J. Am. Chem. Soc. 123, 11 498–11 499. ( 10.1021/ja0164677) [DOI] [PubMed] [Google Scholar]
- 100.Darensbourg DJ, Bottarelli P, Andreatta JR. 2007. Inquiry into the formation of cyclic carbonates during the (salen)CrX catalyzed CO2/cyclohexene oxide copolymerization process in the presence of ionic initiators. Macromolecules 40, 7727–7729. ( 10.1021/ma071206o) [DOI] [Google Scholar]
- 101.Darensbourg DJ, Mackiewicz RM, Rodgers JL, Phelps AL. 2004. (Salen)CrIIIX catalysts for the copolymerization of carbon dioxide and epoxides: role of the initiator and cocatalyst. Inorg. Chem. 43, 1831–1833. ( 10.1021/ic0352856) [DOI] [PubMed] [Google Scholar]
- 102.Jacobsen EN. 2000. Asymmetric catalysis of epoxide ring-opening reactions. Acc. Chem. Res. 33, 421–431. ( 10.1021/ar960061v) [DOI] [PubMed] [Google Scholar]
- 103.Chen P, Chisholm MH, Gallucci JC, Zhang X, Zhou Z. 2005. Binding of propylene oxide to porphyrin- and salen-M(III) cations, where M = Al, Ga, Cr, and Co. Inorg. Chem. 44, 2588–2595. ( 10.1021/ic048597x) [DOI] [PubMed] [Google Scholar]
- 104.Darensbourg DJ, Billodeaux DR, Perez LM. 2004. 113Cd NMR determination of the binding parameters of alicyclic epoxides to [hydrotris(3-phenylpyrazol-1-yl)borate]Cd(II) acetate. Organometallics 23, 5286–5290. ( 10.1021/om049761r) [DOI] [Google Scholar]
- 105.Aida T, Inoue S. 1996. Metalloporphyrins as initiators for living and immortal polymerizations. Acc. Chem. Res. 29, 39–48. ( 10.1021/ar950029l) [DOI] [Google Scholar]
- 106.Ren W-M, Zhang X, Liu Y, Li J-F, Wang H, Lu X-B. 2010. Highly active, bifunctional Co(III)-salen catalyst for alternating copolymerization of CO2 with cyclohexene oxide and terpolymerization with aliphatic epoxides. Macromolecules 43, 1396–1402. ( 10.1021/ma902321g) [DOI] [Google Scholar]
- 107.Cheng M, Lobkovsky EB, Coates GW. 1998. Catalytic reactions involving C1 feedstocks: new high-activity Zn(II)-based catalysts for the alternating copolymerization of carbon dioxide and epoxides. J. Am. Chem. Soc. 120, 11 018–11 019. ( 10.1021/ja982601k) [DOI] [Google Scholar]
- 108.Lee BY, Kwon HY, Lee SY, Na SJ, Han S-I, Yun H, Lee H, Park Y-W. 2005. Bimetallic anilido-aldimine zinc complexes for epoxide/CO2 copolymerization. J. Am. Chem. Soc. 127, 3031–3037. ( 10.1021/ja0435135) [DOI] [PubMed] [Google Scholar]
- 109.Kissling S, Lehenmeier MW, Altenbuchner PT, Kronast A, Reiter M, Deglmann P, Seemann UB, Rieger B. 2015. Dinuclear zinc catalysts with unprecedented activities for the copolymerization of cyclohexene oxide and CO2. Chem. Commun. 51, 4579–4582. ( 10.1039/C5CC00784D) [DOI] [PubMed] [Google Scholar]
- 110.Kember MR, White AJP, Williams CK. 2010. Highly active di- and trimetallic cobalt catalysts for the copolymerization of CHO and CO2 at atmospheric pressure. Macromolecules 43, 2291–2298. ( 10.1021/ma902582m) [DOI] [Google Scholar]
- 111.Ellis WC, Jung Y, Mulzer M, Di Girolamo R, Lobkovsky EB, Coates GW. 2014. Copolymerization of CO2 and meso epoxides using enantioselective β-diiminate catalysts: a route to highly isotactic polycarbonates. Chem. Sci. 5, 4004–4011. ( 10.1039/C4SC01686F) [DOI] [Google Scholar]
- 112.Byrne CM, Allen SD, Lobkovsky EB, Coates GW. 2004. Alternating copolymerization of limonene oxide and carbon dioxide. J. Am. Chem. Soc. 126, 11 404–11 405. ( 10.1021/ja0472580) [DOI] [PubMed] [Google Scholar]
- 113.Allen SD, Moore DR, Lobkovsky EB, Coates GW. 2002. High-activity, single-site catalysts for the alternating copolymerization of CO2 and propylene oxide. J. Am. Chem. Soc. 124, 14 284–14 285. ( 10.1021/ja028071g) [DOI] [PubMed] [Google Scholar]
- 114.Jeske RC, Rowley JM, Coates GW. 2008. Pre-rate-determining selectivity in the terpolymerization of epoxides, cyclic anhydrides, and CO2: a one-step route to diblock copolymers. Angew. Chem. Int. Ed. 47, 6041–6044. ( 10.1002/anie.200801415) [DOI] [PubMed] [Google Scholar]
- 115.Piesik DFJ, Range S, Harder S. 2008. Bimetallic calcium and zinc complexes with bridged β-diketiminate ligands: investigations on epoxide/ CO2 copolymerization. Organometallics 27, 6178–6187. ( 10.1021/om800597f) [DOI] [Google Scholar]
- 116.Kissling S, Altenbuchner PT, Lehenmeier MW, Herdtweck E, Deglmann P, Seemann UB, Rieger B. 2015. Mechanistic aspects of a highly active dinuclear zinc catalyst for the co-polymerization of epoxides and CO2. Chem. Eur. J. 21, 8148–8157. ( 10.1002/chem.201406055) [DOI] [PubMed] [Google Scholar]
- 117.Lehenmeier MW, Kissling S, Altenbuchner PT, Bruckmeier C, Deglmann P, Brym A-K, Rieger B. 2013. Flexibly tethered dinuclear zinc complexes: a solution to the entropy problem in CO2/epoxide copolymerization catalysis? Angew. Chem. Int. Ed. 52, 9821–9826. ( 10.1002/anie.201302157) [DOI] [PubMed] [Google Scholar]
- 118.Winkler M, Romain C, Meier MAR, Williams CK. 2015. Renewable polycarbonates and polyesters from 1,4-cyclohexadiene. Green Chem. 17, 300–306. ( 10.1039/C4GC01353K) [DOI] [Google Scholar]
- 119.Saini PK, Romain C, Zhu Y, Williams CK. 2014. Di-magnesium and zinc catalysts for the copolymerization of phthalic anhydride and cyclohexene oxide. Polym. Chem. 5, 6068–6075. ( 10.1039/C4PY00748D) [DOI] [Google Scholar]
- 120.Saini PK, Romain C, Williams CK. 2014. Dinuclear metal catalysts: improved performance of heterodinuclear mixed catalysts for CO2–epoxide copolymerization. Chem. Commun. 50, 4164–4167. ( 10.1039/c3cc49158g) [DOI] [PubMed] [Google Scholar]
- 121.Kember MR, Williams CK. 2012. Efficient magnesium catalysts for the copolymerization of epoxides and CO2; using water to synthesize polycarbonate polyols. J. Am. Chem. Soc. 134, 15 676–15 679. ( 10.1021/ja307096m) [DOI] [PubMed] [Google Scholar]
- 122.Kember MR, Jutz F, Buchard A, White AJP, Williams CK. 2012. Di-cobalt(II) catalysts for the copolymerisation of CO2 and cyclohexene oxide: support for a dinuclear mechanism? Chem. Sci. 3, 1245–1255. ( 10.1039/c2sc00802e) [DOI] [Google Scholar]
- 123.Buchard A, Jutz F, Kember MR, White AJP, Rzepa HS, Williams CK. 2012. Experimental and computational investigation of the mechanism of carbon dioxide/cyclohexene oxide copolymerization using a dizinc catalyst. Macromolecules 45, 6781–6795. ( 10.1021/ma300803b) [DOI] [Google Scholar]
- 124.Jutz F, Buchard A, Kember MR, Fredrickson SB, Williams CK. 2011. Mechanistic investigation and reaction kinetics of the low-pressure copolymerization of cyclohexene oxide and carbon dioxide catalyzed by a dizinc complex. J. Am. Chem. Soc. 133, 17 395–17 405. ( 10.1021/ja206352x) [DOI] [PubMed] [Google Scholar]
- 125.Buchard A, Kember MR, Sandeman KG, Williams CK. 2011. A bimetallic iron(III) catalyst for CO2/epoxide coupling. Chem. Commun. 47, 212–214. ( 10.1039/C0CC02205E) [DOI] [PubMed] [Google Scholar]
- 126.Kember MR, White AJP, Williams CK. 2009. Di- and tri-zinc catalysts for the low-pressure copolymerization of CO2 and cyclohexene oxide. Inorg. Chem. 48, 9535–9542. ( 10.1021/ic901109e) [DOI] [PubMed] [Google Scholar]
- 127.Zhu Y, Romain C, Poirier V, Williams CK. 2015. Influences of a dizinc catalyst and bifunctional chain transfer agents on the polymer architecture in the ring-opening polymerization of ε-caprolactone. Macromolecules 48, 2407–2416. ( 10.1021/acs.macromol.5b00225) [DOI] [Google Scholar]
- 128.Romain C, Williams CK. 2014. Chemoselective polymerization control: from mixed-monomer feedstock to copolymers. Angew. Chem. Int. Ed. 53, 1607–1610. ( 10.1002/anie.201309575) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhu Y, Romain C, Williams CK. 2015. Selective polymerization catalysis: controlling the metal chain end group to prepare block copolyesters. J. Am. Chem. Soc. 137, 12 179–12 182. ( 10.1021/jacs.5b04541) [DOI] [PubMed] [Google Scholar]