

Abstract
Peptide sequence analysis using a combination of gas-phase ion/ion chemistry and tandem mass spectrometry (MS/MS) is demonstrated. Singly charged anthracene anions transfer an electron to multiply protonated peptides in a radio frequency quadrupole linear ion trap (QLT) and induce fragmentation of the peptide backbone along pathways that are analogous to those observed in electron capture dissociation. Modifications to the QLT that enable this ion/ion chemistry are presented, and automated acquisition of high-quality, single-scan electron transfer dissociation MS/MS spectra of phosphopeptides separated by nanoflow HPLC is described.
Keywords: electron capture dissociation, fragmentation, ion/ion reactions, charge transfer, ion trap
Six years ago, McLafferty and coworkers (1) introduced a unique method for peptide/protein ion fragmentation: electron capture dissociation (ECD). In this method, multiply protonated peptides or proteins are confined in the Penning trap of a Fourier transform ion cyclotron resonance (FTICR) mass spectrometer and exposed to electrons with near-thermal energies. Capture of a thermal electron by a protonated peptide is exothermic by ≈6 eV (1 eV = 1.602 × 10-19 J) and causes the peptide backbone to fragment by a nonergodic process, e.g., one that does not involve intramolecular vibrational energy redistribution (2–5). One pathway for this process involves generation of an odd-electron hypervalent species (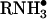 ) that dissociates to produce RNH2 and a hydrogen radical (6). As shown in Fig. 1, addition of H• to the carbonyl groups of the peptide backbone leads to a homologous series of complementary fragment ions of types c and z. Addition of H• to an amide nitrogen, a secondary pathway, leads to the formation of carbon monoxide plus a homologous series of complementary fragment ions of types a and y. Subtraction of the m/z values for the fragments within a given ion series that differ by a single amino acid affords the mass and thus the identity of the extra residue in the larger of the two fragments. The complete amino acid sequence of a peptide is deduced by extending this process to all homologous pairs of fragments within a particular ion series.
) that dissociates to produce RNH2 and a hydrogen radical (6). As shown in Fig. 1, addition of H• to the carbonyl groups of the peptide backbone leads to a homologous series of complementary fragment ions of types c and z. Addition of H• to an amide nitrogen, a secondary pathway, leads to the formation of carbon monoxide plus a homologous series of complementary fragment ions of types a and y. Subtraction of the m/z values for the fragments within a given ion series that differ by a single amino acid affords the mass and thus the identity of the extra residue in the larger of the two fragments. The complete amino acid sequence of a peptide is deduced by extending this process to all homologous pairs of fragments within a particular ion series.
Fig. 1.

Fragmentation scheme for production of c- and z-type ions after reaction of a low-energy electron with a multiply protonated peptide.
Because ECD occurs along the peptide backbone in a sequence-independent manner, preserves posttranslational modifications (PTMs) (7–14), and can be implemented on a millisecond time scale with precursor-to-product ion conversion efficiencies that approach 30% (15–21), it has become the technique of choice for the analysis of peptide and proteins with FTICR mass spectrometers (22–28). Unfortunately, ECD in its most efficient form requires that the precursor sample ions be immersed in a dense population of near-thermal electrons. Emulating these conditions in the instruments used most commonly for peptide and protein analyses, those that trap ions with radio frequency (RF) electrostatic fields rather than with static magnetic and electric fields, remains technically challenging. Thermal electrons introduced into the RF fields of RF 3D quadrupole ion trap (QIT), quadrupole time-of-flight, or RF linear 2D quadrupole ion trap (QLT) instruments maintain their thermal energy only for a fraction of a microsecond and are not trapped. Despite proposals to circumvent this difficulty (29–31), none have been implemented to date. As a result, ECD remains a technique exclusively used with FTICR, the most expensive type of MS instrumentation.
Ion fragmentation for peptide and protein sequence analysis, with QIT, quadrupole time-of-flight, and QLT instruments, is presently performed with some form of collision-activated dissociation (CAD). In this process, peptides that are protonated more or less randomly on backbone amide nitrogen atoms are kinetically excited and undergo collisions with an inert gas such as helium or argon. During each collision, imparted translational energy is converted to vibrational energy that then is rapidly distributed throughout all covalent bonds (picosecond time scale). Fragment ions are formed when the internal energy of the ion exceeds the activation barrier required for a particular bond cleavage. As shown in Fig. 2, fragmentation of protonated amide bonds affords a homologous series of complementary product ions of types b and y. Again, mass differences observed between homologous members of an ion series allow assignment of a particular amino acid to the extra residue in the larger fragment and thus facilitate peptide sequence analysis.
Fig. 2.

Fragmentation scheme for production of b- and y-type ions by CAD of a multiply protonated peptide.
Successful peptide identification by either ECD or CAD can be achieved only when product ions from a complete or nearly complete distribution of amide backbone cleavages are observed in the corresponding tandem MS (MS/MS) spectrum. CAD often fails in this regard when the peptide contains (i) multiple Arg residues, which inhibit random protonation along the peptide backbone, or (ii) a PTM that dissociates by a lower energy pathway than that involved in cleavage of the amide linkage. Peptides containing phosphorylated Ser or Thr residues are examples of the latter case. In the gas phase, phosphate competes with the peptide backbone as a preferred site of protonation and consequently, after collision activation, undergoes nucleophilic displacement by a neighboring amide carbonyl group (Fig. 3). The resulting (M + nH)n+ - H3PO4 product ions often constitute ≥85% of the fragment ions observed under the low-energy CAD conditions. Fragment ions of types b and y that contain a phosphoserine or phosphothreonine residue also lose phosphoric acid readily.
Fig. 3.

Fragmentation scheme for loss of phosphoric acid from a multiply protonated phosphopeptide by CAD.
Unlike CAD, ECD is independent of amide bond protonation and occurs on a time scale that is short compared with internal energy distribution. Multiply protonated peptides, and those with PTMs, all fragment more or less randomly along the peptide backbone and are easily sequenced. For this reason, ECD holds great promise as a comprehensive peptide dissociation method, one that is suitable for large species and indifferent to either peptide sequence or the presence of labile PTMs.
Development of an ECD-like dissociation method for use with a low-cost, widely accessible mass spectrometer such as the QLT would have obvious utility for protein sequence analysis. Because storage of thermal electrons in an RF ion-containment field seems problematic at best, we investigated the possibility of using anions as vehicles for delivering electrons to multiply charged peptide cations. From our extensive experience with negative ion chemical ionization (CI) (32, 33), we concluded that anions with sufficiently low electron affinities could function as suitably massive, one-electron donors. Electron transfer to protonated peptides should be exothermic by 4–5.5 eV, trigger release of a hydrogen radical, and initiate fragmentation via the same nonergodic pathways accessed in ECD.
Here we describe (i) modifications of a QLT mass spectrometer to enable ion/ion experiments, (ii) a method of fragmenting multiply protonated peptides, electron transfer dissociation (ETD), and (iii) automated acquisition of high-quality, single-scan ETD tandem mass spectra from phosphopeptides separated by nanoflow HPLC (nHPLC).
Materials and Methods
Instrument Modification and Operation. All experiments were performed with a commercial QLT, the Finnigan LTQ mass spectrometer (Thermo Electron, Waltham, MA) equipped with a modified nanoflow electrospray ionization (ESI) source. The LTQ was modified to accommodate a Finnigan 4500 CI source (Thermo Electron) placed at the rear of the instrument. The anion beam was gated by on/off control of the RF voltages applied to the octopole ion guides, which transport the anions from the CI source to the QLT. Fig. 4 displays a schematic of the linear ion trap with ESI and CI sources located at either end of the three-segment device. To generate and analyze the products of ion/ion reactions, instrument control software (itcl code) was modified to incorporate the scan events diagrammed in Fig. 4 into the standard QLT MSn scan function.
Fig. 4.

Schematic of steps involved in the operation of the LTQ mass spectrometer for peptide sequence analysis by ETD. (A) Injection of multiply protonated peptide molecules (precursor ions) generated by ESI. (B) Application of a dc offset to move the precursor ions to the front section of the linear trap. (C) Injection of negatively charged reagent ions from the CI source into the center section of the linear trap. (D) Application of a supplementary dipolar broadband ac field to eject all ions except those within 3 mass-unit windows centered around the positively charged precursor ions and the negatively charged electron-donor reagent ions. (E) Removal of the dc potential well and application of a secondary RF voltage (100 V zero to peak, 600 kHz) to the end lens plates of the linear trap to allow positive and negative ion populations to mix and react. (F) Termination of ion/ion reactions by axial ejection of negatively charged reagent ions while retaining positive ions in the center section of the trap. This is followed by mass-selective, radial ejection of positively charged fragment ions to record the resulting MS/MS spectrum.
Sample Introduction. Multiply charged (protonated) peptides were generated by ESI. A 40% aqueous acetonitrile solution (with 0.1% acetic acid), containing peptides at 1 pmol/μl, was infused from a SilicaTip fused silica emitter (30-μm tip, New Objective, Woburn, MA). Samples included angiotensin I (DRVYIHPFHL, Sigma–Aldrich) and the in-house-synthesized phosphopeptides: LPISASHpSpSKTR, APVAPRPAApT-PNLSK, and DRpSPIRGpSPR. Negative CI, with methane buffer gas (MG Industries, Malvern, PA), was used to produce negative ions of anthracene (Aldrich). Anthracene was introduced to the CI source through an improvised heated-batch inlet consisting of a gas chromatograph oven and a heated transferline assembly (Thermo Electron) connected to a fused silica restrictor column.
Chromatography. An Agilent (Palo Alto, CA) 1100 series binary HPLC system was interfaced with the QLT mass spectrometer for online peptide separation and analysis by nHPLC-micro-ESI-MS (nHPLC-μESI-MS/MS).
Synthetic Peptide Analysis. A mixture of 10 synthetic peptides (1–100 fmol) was loaded onto a polyimide-coated, fused-silica microcapillary “precolumn” (360 μm o.d. × 75 μm i.d.; Polymi-cro Technologies, Phoenix) that was butt-connected with polytetrafluoroethylene tubing [0.06 in o.d. × 0.012 in i.d. (1 in = 2.54 cm), Zeus Industrial Products, Orangeburg, SC] to an analytical column. This column (360 μm o.d. × 50 μm i.d.) was made with 5 cm of 5-μm C18 reverse-phase packing material (YMC, Kyoto) and equipped with an integrated, laser-pulled, electrospray emitter tip (34). Peptides were eluted at a flow rate of 60 nl/min with the following gradient: 0–100% B in 17 min, 100–0% B in 18 min [A, 100 mM aqueous acetic acid (Sigma–Aldrich); B, 100 mM acetic acid in 70:30 acetonitrile (Mallinckrodt)/water]. Spectra were recorded under data-dependent settings. The instrument cycled through acquisition of a full-scan mass spectrum (300–600 m/z) and three ETD MS/MS spectra recorded on the three most abundant ions in the full-scan mass spectrum (≈1 sec per cycle).
Complex Mixture Analysis. A 300-μg aliquot of purified nuclear proteins was digested with trypsin (Promega; 1:20, enzyme/substrate) in 100 mM NH4HCO3 (pH 8.5) overnight at 37°C. The solution was acidified with acetic acid and taken to dryness. Peptides were converted to methyl esters as described in ref. 35. Reagents were removed by lyophilization, and the sample was reconstituted in a mixture containing equal parts MeOH, MeCN, and 0.01% acetic acid. Enrichment of phosphopeptides was performed by loading half of the sample onto an Fe3+-activated immobilized metal affinity chromatography column (360 μm o.d. × 100 μm i.d.) packed with 6 cm of POROS 20 MC metal chelate affinity-chromatography packing material (Per-Septive Biosystems, Framingham, MA). Phosphopeptides were eluted onto a C18 microcapillary precolumn (described above) by using 15 μl of 250 mM ascorbic acid (Sigma). The precolumn was butt-connected to an analytical column (described above), and phosphopeptides were eluted with the following gradient: 0–60% B in 60 min; 60–100% B in 70 min. Spectra were recorded as described above except that the top five most abundant ions in the full-scan MS were selected for MS/MS (ETD; cycle time, ≈1.5 sec).
Results
Shown in Fig. 5 is a single-scan ETD mass spectrum recorded on (M + 3H)+3 ions from the doubly phosphorylated, synthetic peptide LPISASHpSpSKTR. Total acquisition time for this spectrum was 300 msec. Positive ions to be dissociated were generated at the front end of the QLT by ESI of an infused solution containing the sample at the 1 pmol/μl level. The resulting full-scan spectrum contained a mixture of (M + 3H)+3 and (M + 2H)+2 ions at m/z 482 and 722, respectively.
Fig. 5.

Single-scan ETD MS/MS spectrum resulting from a 50-msec reaction of the triply charged phosphopeptide, LPISASHpSpSKTR, at m/z 482, with anthracene anions. Predicted m/z values for fragment ions of types c and z are shown above and below the sequence, respectively. Those observed are underlined. Note that both z5 and c7 have m/z values that overlap with the ion cluster containing the product of proton abstraction, the (M + 2H)+2 ion at m/z 722. All other possible ions of types c and z appear in the spectrum. The total experiment time was ≈300 msec.
Reagent anions for the electron transfer reaction were generated in a CI source attached to the back of the QLT. Bombardment of methane gas at 1 torr (1 torr = 133 Pa) pressure with 70-eV electrons generates positively charged reagent ions,  and
and 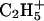 , plus a population of thermal or near-thermal electrons (32, 33). When anthracene, C14H10, is volatilized into the CI source, the major anions produced are even-electron species, m/z 177 and 179, having the formulas
, plus a population of thermal or near-thermal electrons (32, 33). When anthracene, C14H10, is volatilized into the CI source, the major anions produced are even-electron species, m/z 177 and 179, having the formulas 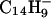 and
and 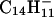 , respectively. We assume that the latter species is formed by a two-step process that involves electron capture and hydrogen atom abstraction from methane (Eqs. 1 and 2). This conclusion is supported by the finding that m/z 179 is not observed when argon is used as the CI reagent gas (data not shown).
, respectively. We assume that the latter species is formed by a two-step process that involves electron capture and hydrogen atom abstraction from methane (Eqs. 1 and 2). This conclusion is supported by the finding that m/z 179 is not observed when argon is used as the CI reagent gas (data not shown).
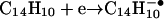 |
[1] |
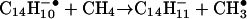 |
[2] |
When 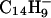 and
and 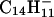 are reacted (≈50 msec) with (M + 3H)+3 ions from LPISASHpSpSKTR, they function both as bases and as one-electron reducing agents. Proton abstraction generates the (M + 2H)+2 and (M+H)+ products observed in the ion clusters centered at m/z 722 and 1443, respectively. Also present in these clusters is a population of ions having compositions corresponding to (M + 3H)+2• and (M + 3H)+••, respectively. Isolation and collision activation of these ion clusters yields a mixture of c- and z-type fragment ions from the odd-electron ion components and b- and y-type fragment ions from the even-electron ion components (data not shown). From the isotopic distribution we estimate that 30–50% of the charge-transfer product ions are noncovalently bound yet dissociated precursor ions. These are easily dissociated by CAD.
are reacted (≈50 msec) with (M + 3H)+3 ions from LPISASHpSpSKTR, they function both as bases and as one-electron reducing agents. Proton abstraction generates the (M + 2H)+2 and (M+H)+ products observed in the ion clusters centered at m/z 722 and 1443, respectively. Also present in these clusters is a population of ions having compositions corresponding to (M + 3H)+2• and (M + 3H)+••, respectively. Isolation and collision activation of these ion clusters yields a mixture of c- and z-type fragment ions from the odd-electron ion components and b- and y-type fragment ions from the even-electron ion components (data not shown). From the isotopic distribution we estimate that 30–50% of the charge-transfer product ions are noncovalently bound yet dissociated precursor ions. These are easily dissociated by CAD.
Electron transfer also leads to the direct generation of c- and z-type fragment ions. In Fig. 5, m/z values for predicted c- and z-type product ions derived from this sample are shown above and below the peptide sequence. Those observed are underlined and account for 31% of the total product-ion current. Note that only four members of c- and z-type ion series are missing. Two of these occur at m/z values that overlap with the ion cluster corresponding to the abundant (M + 2H)+2 species. The other two c- and z-type fragment ions are not produced because their formation involves cleavage of the N–CH bond in the ring system of Pro. When this bond is broken, the new fragments remain attached through the other atoms in the ring. Accordingly, formation of c- and z-type product ions containing the N and C termini of Pro, respectively, are not observed in either ECD or ETD spectra.
To demonstrate the feasibility of generating ETD spectra on a chromatographic time scale, we analyzed a mixture containing 10 synthetic peptides at the 1- to 100-fmol level by nHPLC-μESI-MS/MS. Shown in Fig. 6A is an ion chromatogram containing signals corresponding to (M + 3H)+3 ions at m/z 433, 524, and 434 for DRVYIHPFHL (≈100 fmol), APVAPR-PAApTPNLSK (≈10 fmol), and DRpSPIRGpSPR (≈1 fmol), respectively. Peak widths are in the range of 10–14 sec.
Fig. 6.

Data-dependent analysis of a peptide mixture by using a combination of nHPLC-μESI and ETD-MS/MS. (A) Total ion chromatogram (peaks are ≈10 sec wide). (B) Single-scan, 500- to 600-msec, ETD spectrum recorded on 100 fmol of the triply protonated peptide, DRVYIHPFHL. (C) Single-scan, 500- to 600-msec, ETD spectrum recorded on 1 fmol of the triply protonated peptide, DRpSPIRGpSPR.
With the instrument operating in the data-dependent mode, multiple single-scan ETD spectra (in this case 500–600 msec per spectrum) were recorded for each sample. Single-scan ETD spectra for two of the peptides, DRVYIHPFHL and DRpSPIR-GpSPR, are shown in Fig. 6 B and C, respectively. For angiotensin, DRVYIHPFHL, 14 of 18 possible c- and z-type product ions are present in the ETD spectrum (Fig. 6B). Those that are absent either occur at very low m/z values or cannot be observed because they are formed by cleavage of the Pro ring system. The single-scan ETD spectrum recorded on 1 fmol of the doubly phosphorylated peptide, DRpSPIRGpSPR, is displayed in Fig. 6C. Even at this sample level the spectrum contains 12 of 18 possible c- and z-type fragment ions. Four of the six missing are involved in the cleavage of the five-membered ring systems of the two Pro residues. Both of the above peptides are easily sequenced from the observed fragment ions.
Displayed in Fig. 7 are the conventional CAD and ETD MS/MS spectra generated from (M + 3H)+3 ions (m/z 412) of doubly phosphorylated peptide ERpSLpSRER. Both spectra were acquired during nHPLC analyses of phosphopeptides generated in a tryptic digest of human nuclear proteins. All peptides were converted to methyl esters and subjected to immobilized metal affinity chromatography before analysis by MS. Fragmentation observed in the low-energy CAD spectrum (Fig. 7A) is dominated by ions corresponding to the loss of one and two molecules of phosphoric acid from the side chains of Ser residues. Abundant ions, formed by the additional loss of water or methanol, are also observed. Fragment ions derived from cleavage of the peptide backbone are either absent or present at <0.5% relative ion abundance. Sequence analysis, therefore, is impossible.
Fig. 7.

Comparison of single-scan (500- to 600-msec) CAD and ETD mass spectra recorded during data-dependent analyses (nHPLC-μESI-MS/MS) of phosphopeptides generated in a tryptic digest of human nuclear proteins. All peptides were converted to methyl esters and subjected to immobilized metal affinity chromatography before analysis by MS. (A) CAD spectrum dominated by fragment ions corresponding to the loss of phosphoric acid and either methanol or water. (B) ETD spectrum containing 13 of 14 possible c- and z-type product ions. Note that the spectrum is devoid of fragment ions corresponding to the loss of phosphoric acid.
The information content of the ETD spectrum (Fig. 7B) is dramatically different from that observed in the low-energy CAD spectrum. Ions produced by loss of phosphoric acid are absent, and fragmentation occurs predominately along the peptide backbone. Of 14 possible c- and z-type product ions, 13 are found in the spectrum. These are more than sufficient to define the sequence ERpSLpSRER.
Discussion
ETD. Ion/ion reactions of multiply protonated peptides with singly charged anions in the QLT yields products associated with both proton and electron transfer. Some anions function as strong bases and react exclusively by proton abstraction (data not shown). Still others participate in both proton transfer and electron transfer. Observed product ratios depend on the anion structure (data not shown). Appropriately, we expect additional investigation to reveal anions that react primarily via electron transfer. The latter pathway initiates a fragmentation cascade to produce c- and z-type product ions. In some instances fragmentation occurs, but the product ions remain noncovalently bound. Collisional activation of these product ions generates c- and z-type fragments. This same phenomena has been observed in ECD experiments (17–19, 36).
Both the quality and extent of fragmentation shown in Figs. 5, 6, 7 and the sample level detected (Fig. 6C) are typical of that observed for spectra recorded to date on hundreds of peptides, including those with PTMs (data not shown). These spectra can be interpreted manually (de novo) or used to search databases, with an algorithm such as SEQUEST, to generate peptide sequences (37). Typically, CAD tandem mass spectra of non-phosphorylated tryptic peptides generate crosscorrelation scores of ≈2.0–4.0 (38). With ETD, tandem mass spectra of tryptic peptides with or without PTMs produce crosscorrelation scores ranging from 3.0 to 6.5. Further enhancement of these scores is likely once SEQUEST is adapted to consider features that are uniquely characteristic of ETD fragmentation (e.g., absence of c- and z-type fragments adjacent to Pro).
Instrumentation. A by-product of this investigation is the development of an ion/ion instrument based on a radial ejection QLT mass spectrometer. Conventional QLT devices use dc potentials to provide axial ion containment. Consequently, only ions of a single polarity can be confined within any given region or segment of the trap, which precludes use of the commercial QLT for ion/ion experiments. In our modified QLT, co-trapping of cations and anions is accomplished entirely by RF confinement fields. Secondary RF fields, imposed by superposition of RF voltages to the end lenses of the QLT, provided the required charge-sign-independent axial trapping.
The QLT instrument has several unique advantages over QIT instruments for performing ion/ion experiments, including greater ion capacity (≈30-fold) and higher ion-injection efficiency (≈10- to 30-fold) (39). As illustrated in Fig. 4, manipulation of the dc bias potentials, applied to the QLT's segments and end lenses, permits axial segregation of precursor cations and reagent anions during anion injection and isolation. Initiation and termination of the ion/ion reaction is controlled by adjustment of these dc bias potentials. The physical geometry of the apparatus is also advantageous, because different types of ions may be injected from either end of the device by using two different ion sources. Furthermore, because anions are injected along the null axis of the RF quadrupole field, they suffer a minimal kinetic excitation. Anions injected in this manner are less likely to undergo electron detachment during stabilizing collisions with the helium buffer gas. “Soft” injection of anions was a key consideration in choosing a QLT for this work, because it was anticipated that the best anion electron donors might also be susceptible to premature electron detachment.
Future Directions. The commercial QLT instrument, modified for this research, was not engineered for experiments involving ion/ion reactions. Future linear trap instruments designed for this purpose will undoubtedly contain additional segments plus controls for superposition of multiple RF, ac, and dc fields. These features will allow implementation of even more extensive ion/ion manipulations, which almost certainly will include fully independent isolation of precursor and reagent ion clusters plus the experiments pioneered by McLuckey et al. (40–46) for charge-state reduction and gas-phase (charge-state) concentration on the QIT instrument. Additionally, we envision implementing techniques that prevent ETD products from undergoing subsequent ion/ion reactions that lead to either neutralization of charge or formation of additional fragments.
Because the time scales for performing CAD, ETD, and proton transfer (charge reduction) in the QLT are short (tens of milliseconds), multiple ion-reaction steps can be incorporated into individual MS/MS or MSn experiments. In the context of “bottom-up” proteomics-type experiments (data-dependent HPLC MS/MS analyses of peptides generated by enzymatic digestion of protein mixtures), we expect ETD to promote the use of proteolytic enzymes such as Lys-C or Asp-N, which generate peptides having an average length of 20–25 residues. With ESI, such peptides are converted to precursor ions having three to six charges and thus are ideal candidates for ETD. Following ETD, we foresee use of an ion/ion, proton-abstraction reaction (charge reduction) to ensure that c- and z-type fragment ions are predominately in the +1 charge state before mass analysis.
For “top-down”-type analyses (direct characterization of intact proteins or large peptides by MS/MS experiments), we envision merging the ECD MS/MS technology developed for FTICR instruments by McLafferty and coworkers (47–49) with the ion/ion CAD MS/MS technology developed for the QIT instrument by McLuckey and coworkers (50–54). Based on preliminary results (data not shown), we believe that a protocol using MS3 experiments could be ideal for generating sequence information from small proteins or large peptides. A typical MS3 experiment might include the following steps: (i) gas-phase concentration (charge reduction by proton transfer with “ion parking”) to convert the initial heterogeneous mixture of charge states observed for proteins ionized by ESI to a single charge state (precursor ion m/z); (ii) m/z isolation and CAD of the precursor ions to create a limited set of large product ions [either b-type ions produced by cleavage C-terminal to Asp residues or y-type ions formed by cleavage N-terminal to Pro residues (55)]; (iii) m/z isolation and ETD of a single CAD product ion; and (iv) m/z analysis of the second-generation product ions. The resultant ETD MS3 spectrum would yield sequence information derived from a single b- or y-type intermediate product ion (MS2 product ion). A series of such MS3 experiments would generate sequence information for all the major intermediate product ions.
In the protocol described above, the first dissociation step (CAD) is analogous to the enzymatic digestion step in the conventional bottom-up proteomics analyses. The MS3 spectra correspond to the MS2 spectra obtained from tryptic peptides. However, in the proposed MS3 protocol, each MS3 spectrum of a b- or y-type product ion provides sequence information for a portion of a protein for which the molecular mass is known.
Summary
We have developed a methodology (ETD) that facilitates peptide sequence analysis by a combination of ion/ion chemistry and MS/MS. We demonstrate that anthracene anions transfer an electron to multiply protonated peptides in a QLT instrument and induce fragmentation of the peptide backbone along pathways that are analogous to those observed in ECD. We describe modifications to the QLT instrument that enable this ion/ion chemistry and present data that document the utility of the ETD process for sequence analysis peptides in complex mixtures by nHPLC and MS/MS. ETD is particularly well suited for characterization of peptides containing PTMs. We also outline experiments that use a combination of ion/ion chemistry (proton-transfer reactions), CAD, and ETD to characterize the primary structure of intact proteins. Finally, we suggest that the gas-phase ion/ion chemistry will become an indispensable tool for peptide and protein sequence analysis in the near future and is likely to drive the development of new MS instrumentation and software.
Acknowledgments
We thank Neil Kelleher for a helpful discussion, Jarrod Marto for input during early discussions of this research, Jae Schwartz for assistance in design of the dual isolation waveforms, and Scott Quarmby for donation of the batch inlet. This work was supported by funds from National Institutes of Health Grants GM37537 (to D.F.H.), AI33993 (to D.F.H.), and RR018688 (to J.J.C.), National Science Foundation Grant MCB-0209793 (to D.F.H.), and Thermo Electron.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ECD, electron capture dissociation; FTICR, Fourier transform ion cyclotron resonance; PTM, posttranslational modification; RF, radio frequency; QIT, RF 3D quadrupole ion trap; QLT, RF linear 2D quadrupole ion trap; CAD, collision-activated dissociation; MS/MS, tandem MS; CI, chemical ionization; ETD, electron transfer dissociation; nHPLC, nanoflow HPLC; ESI, electrospray ionization; μESI, micro-ESI.
References
- 1.Zubarev, R. A., Kelleher, N. L. & McLafferty, F. W. (1998) J. Am. Chem. Soc. 120, 3265-3266. [Google Scholar]
- 2.Zubarev, R. A., Kruger, N. A., Fridriksson, E. K., Lewis, M. A., Horn, D. M., Carpenter, B. K. & McLafferty, F. W. (1999) J. Am. Chem. Soc. 121, 2857-2862. [Google Scholar]
- 3.Zubarev, R. A. (2003) Mass Spectrom. Rev. 22, 57-77. [DOI] [PubMed] [Google Scholar]
- 4.Cerda, B. A., Horn, D. M., Breuker, K., Carpenter, B. K. & McLafferty, F. W. (1999) Eur. J. Mass Spectrom. (Chichester, England) 5, 335-338. [Google Scholar]
- 5.Zubarev, R. A., Haselmann, K. F., Budnik, B., Kjeldsen, F. & Jensen, F. (2002) Eur. J. Mass Spectrom. (Chichester, England) 8, 337-349. [Google Scholar]
- 6.McLafferty, F. W., Horn, D. M., Breuker, K., Ge, Y., Lewis, M. A., Cerda, B., Zubarev, R. A. & Carpenter, B. K. (2001) J. Am. Soc. Mass Spectrom. 12, 245-249. [DOI] [PubMed] [Google Scholar]
- 7.Mirgorodskaya, E., Roepstorff, P. & Zubarev, R. A. (1999) Anal. Chem. 71, 4431-4436. [DOI] [PubMed] [Google Scholar]
- 8.Kelleher, N. L., Zubarev, R. A., Bush, K., Furie, B., Furie, B. C., McLafferty, F. W. & Walsh, C. T. (1999) Anal. Chem. 71, 4250-4253. [DOI] [PubMed] [Google Scholar]
- 9.Stensballe, A., Andersen, S. & Jensen, O. N. (2001) Proteomics 1, 207-222. [DOI] [PubMed] [Google Scholar]
- 10.Mirgorodskaya, E., Hassan, H., Clausen, H. & Roepstorff, P. (2001) Anal. Chem. 73, 1263-1269. [DOI] [PubMed] [Google Scholar]
- 11.Shi, S. D. H., Hemling, M. E., Carr, S. A., Horn, D. M., Lindh, I. & McLafferty, F. W. (2001) Anal. Chem. 73, 19-22. [DOI] [PubMed] [Google Scholar]
- 12.Budnik, B. A., Haselmann, K. F., Elkin, Y. N., Gorbach, V. I. & Zubarev, R. A. (2003) Anal. Chem. 75, 5994-6001. [DOI] [PubMed] [Google Scholar]
- 13.Emmett, M. R. (2003) J. Chromatogr. A 1013, 203-213. [DOI] [PubMed] [Google Scholar]
- 14.Mann, M. & Jensen, O. N. (2003) Nat. Biotechnol. 21, 255-261. [DOI] [PubMed] [Google Scholar]
- 15.Hakansson, K., Cooper, H. J., Emmett, M. R., Costello, C. E., Marshall, A. G. & Nilsson, C. L. (2001) Anal. Chem. 73, 4530-4536. [DOI] [PubMed] [Google Scholar]
- 16.Hakansson, K., Emmett, M. R., Hendrickson, C. L. & Marshall, A. G. (2001) Anal. Chem. 73, 3605-3610. [DOI] [PubMed] [Google Scholar]
- 17.Tsybin, Y. O., Witt, M., Baykut, G., Kjeldsen, F. & Hakansson, P. (2003) Rapid Commun. Mass Spectrom. 17, 1759-1768. [DOI] [PubMed] [Google Scholar]
- 18.Hakansson, K., Chalmers, M. J., Quinn, J. P., McFarland, M. A., Hendrickson, C. L. & Marshall, A. G. (2003) Anal. Chem. 75, 3256-3262. [DOI] [PubMed] [Google Scholar]
- 19.Mormann, M. & Peter-Katalinic, J. (2003) Rapid Commun. Mass Spectrom. 17, 2208-2214. [DOI] [PubMed] [Google Scholar]
- 20.Tsybin, Y. O., Hakansson, P., Budnik, B. A., Haselmann, K. F., Kjeldsen, F., Gorshkov, M. & Zubarev, R. A. (2001) Rapid Comun. Mass Spectrom. 15, 1849-1854. [DOI] [PubMed] [Google Scholar]
- 21.Haselmann, K. F., Budnik, B. A., Olsen, J. V., Nielsen, M. L., Reis, C. A., Clausen, H., Johnsen, A. H. & Zubarev, R. A. (2001) Anal. Chem. 73, 2998-3005. [DOI] [PubMed] [Google Scholar]
- 22.Charlebois, J. P., Patrie, S. M. & Kelleher, N. L. (2003) Anal. Chem. 75, 3263-3266. [DOI] [PubMed] [Google Scholar]
- 23.Fagerquist, C. K., Hudgins, R. R., Emmett, M. R., Hakansson, K. & Marshall, A. G. (2003) J. Am. Soc. Mass Spectrom. 14, 682-682. [DOI] [PubMed] [Google Scholar]
- 24.Hakansson, K., Cooper, H. J., Hudgins, R. R. & Nilsson, C. L. (2003) Curr. Org. Chem. 7, 1503-1525. [Google Scholar]
- 25.Cooper, H. J., Case, M. A., McLendon, G. L. & Marshall, A. G. (2003) J. Am. Chem. Soc. 125, 5331-5339. [DOI] [PubMed] [Google Scholar]
- 26.Zabrouskov, V., Giacomelli, L., van Wijk, K. J. & McLafferty, F. W. (2003) Mol. Cell. Proteomics 2, 1253-1260. [DOI] [PubMed] [Google Scholar]
- 27.Ge, Y., Lawhorn, B. G., Elnaggar, M., Sze, S. K., Begley, T. P. & McLafferty, F. W. (2003) Protein Sci. 12, 2320-2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ge, Y., ElNaggar, M., Sze, S. K., Bin Oh, H., Begley, T. P., McLafferty, F. W., Boshoff, H. & Barry, C. E. (2003) J. Am. Soc. Mass Spectrom. 14, 253-261. [DOI] [PubMed] [Google Scholar]
- 29.Franzen, J. (2002) U.S. Patent Appl. 20,020,175,280.
- 30.Whitehouse, C. M., Welkie, D. G., Gholamreza, J., Cousins, L. & Rakov, S. (2003) International Patent Appl. PCT/US03/17436.
- 31.Park, M. A. (2002) U.S. Patent Appl. 20,020,092,980.
- 32.Hunt, D. F., Stafford, G. C., Crow, F. A. & Russell, J. W. (1976) Anal. Chem. 48, 2098-2105. [Google Scholar]
- 33.Hunt, D. F. & Crow, F. A. (1978) Anal. Chem. 50, 1781-1784. [Google Scholar]
- 34.Martin, S. E., Shabanowitz, J., Hunt, D. F. & Marto, J. A. (2000) Anal. Chem. 72, 4266-4274. [DOI] [PubMed] [Google Scholar]
- 35.Ficarro, S. B., McCleland, M. L., Stukenberg, P. T., Burke, D. J., Ross, M. M., Shabanowitz, J., Hunt, D. F. & White, F. M. (2002) Nat. Biotechnol. 20, 301-305. [DOI] [PubMed] [Google Scholar]
- 36.Hakansson, K., Hudgins, R. R. & Marshall, A. G. (2003) J. Am. Soc. Mass Spectrom. 14, 23-41. [DOI] [PubMed] [Google Scholar]
- 37.Eng, J. K., McCormack, A. L. & Yates, J. R., III (1994) J. Am. Soc. Mass Spectrom. 5, 976-989. [DOI] [PubMed] [Google Scholar]
- 38.MacCoss, M. J., Wu, C. C. & Yates, J. R., III (2002) Anal. Chem. 74, 5593-5599. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz, J. C., Senko, M. W. & Syka, J. E. P. (2002) J. Am. Soc. Mass Spectrom. 13, 659-669. [DOI] [PubMed] [Google Scholar]
- 40.Stephenson, J. L. & McLuckey, S. A. (1997) Int. J. Mass Spectrom. 162, 89-106. [Google Scholar]
- 41.McLuckey, S. A. & Stephenson, J. L. (1998) Mass Spectrom. Rev. 17, 369-407. [DOI] [PubMed] [Google Scholar]
- 42.Stephenson, J. L. & McLuckey, S. A. (1998) Anal. Chem. 70, 3533-3544. [DOI] [PubMed] [Google Scholar]
- 43.McLuckey, S. A., Stephenson, J. L. & Asano, K. G. (1998) Anal. Chem. 70, 1198-1202. [DOI] [PubMed] [Google Scholar]
- 44.Reid, G. E., Shang, H., Hogan, J. M., Lee, G. U. & McLuckey, S. A. (2002) J. Am. Chem. Soc. 124, 7353-7362. [DOI] [PubMed] [Google Scholar]
- 45.McLuckey, S. A., Reid, G. E. & Wells, J. M. (2002) Anal. Chem. 74, 336-346. [DOI] [PubMed] [Google Scholar]
- 46.Engel, B. J., Pan, P., Reid, G. E., Wells, J. M. & McLuckey, S. A. (2002) Int. J. Mass Spectrom. 219, 171-187. [Google Scholar]
- 47.Zubarev, R. A., Horn, D. M., Fridriksson, E. K., Kelleher, N. L., Kruger, N. A., Lewis, M. A., Carpenter, B. K. & McLafferty, F. W. (2000) Anal. Chem. 72, 563-573. [DOI] [PubMed] [Google Scholar]
- 48.Sze, S. K., Ge, Y., Oh, H. & McLafferty, F. W. (2002) Proc. Natl. Acad. Sci. USA. 99, 1774-1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ge, Y., Lawhorn, B. G., ElNaggar, M., Strauss, E., Park, J. H., Begley, T. P. & McLafferty, F. W. (2002) J. Am. Chem. Soc. 124, 672-678. [DOI] [PubMed] [Google Scholar]
- 50.Chrisman, P. A. & McLuckey, S. A. (2002) J. Proteome Res. 1, 549-557. [DOI] [PubMed] [Google Scholar]
- 51.Reid, G. E. & McLuckey, S. A. (2002) J. Mass Spectrom. 37, 663-675. [DOI] [PubMed] [Google Scholar]
- 52.Hogan, J. M. & McLuckey, S. A. (2003) J. Mass Spectrom. 38, 245-256. [DOI] [PubMed] [Google Scholar]
- 53.Hogan, J. M., Pitteri, S. J. & McLuckey, S. A. (2003) Anal. Chem. 75, 6509-6516. [DOI] [PubMed] [Google Scholar]
- 54.Amunugama, R., Hogan, J. M., Newton, K. A. & McLuckey, S. A. (2004) Anal. Chem. 76, 720-727. [DOI] [PubMed] [Google Scholar]
- 55.Pitteri, S. J., Reid, G. E. & McLuckey, S. A. (2004) J. Proteome Res. 3, 46-54. [DOI] [PubMed] [Google Scholar]