

Abstract
Bistable switches are widely used in synthetic biology to trigger cellular functions in response to environmental signals. All bistable switches developed so far, however, control the expression of target genes without access to other layers of the cellular machinery. Here, we propose a bistable switch to control the rate at which cells take up a metabolite from the environment. An uptake switch provides a new interface to command metabolic activity from the extracellular space and has great potential as a building block in more complex circuits that coordinate pathway activity across cell cultures, allocate metabolic tasks among different strains or require cell-to-cell communication with metabolic signals. Inspired by uptake systems found in nature, we propose to couple metabolite import and utilization with a genetic circuit under feedback regulation. Using mathematical models and analysis, we determined the circuit architectures that produce bistability and obtained their design space for bistability in terms of experimentally tuneable parameters. We found an activation–repression architecture to be the most robust switch because it displays bistability for the largest range of design parameters and requires little fine-tuning of the promoters' response curves. Our analytic results are based on on–off approximations of promoter activity and are in excellent qualitative agreement with simulations of more realistic models. With further analysis and simulation, we established conditions to maximize the parameter design space and to produce bimodal phenotypes via hysteresis and cell-to-cell variability. Our results highlight how mathematical analysis can drive the discovery of new circuits for synthetic biology, as the proposed circuit has all the hallmarks of a toggle switch and stands as a promising design to control metabolic phenotypes across cell cultures.
Keywords: synthetic biology, genetic circuits, bistability, cellular uptake, dynamic metabolic engineering, piecewise affine models
1. Introduction
Bistable switches are ubiquitous components in natural and engineered biological systems. They play a key role in controlling cellular decisions [1,2] and are common building blocks in synthetic gene circuits [3–6]. The aim of all synthetic switches developed so far has been to produce bistable expression of target genes. One of the major goals in synthetic biology, however, is to scale up biomolecular circuits to systems that interface gene expression with metabolic activity. These have great potential to expand the functionality of biomolecular devices, for example, to dynamically reroute flux through heterologous pathways [7] or to design self-adaptive pathways in metabolic engineering [8–10].
The ‘metabolator’, a genetic circuit designed to generate an oscillatory metabolic flux [11], showcased how complex responses could be engineered by coupling the genetic and metabolic machinery. To date, however, little progress has been made in engineering other metabolic phenotypes. A bistable uptake switch has been particularly elusive, although it is a key building block for more complex circuits that require metabolic control with extracellular metabolites. An uptake switch can be used to coordinate pathway activity in multicellular systems, for example, by allocating metabolic tasks among several strains [12] or by acting as a communication device via metabolic signals [13]. In microbial consortia, an uptake switch can control the division of labour through diversified phenotypes of slow and fast feeders. Bistable uptake can also serve as a mechanism to engineer bacterial bet-hedging that favours survival in adverse environments [14,15] or as a research tool to study cellular adaptation strategies [16], e.g. in competition assays where subpopulations of switchers and non-switchers adapt to limited carbon sources or fluctuations in nutrient abundance.
Metabolic uptake is typically carried out by transport enzymes in the cell membrane, but their kinetics do not naturally display bistability (figure 1a). Although ultrasensitive kinetics [17] can generate a switch-like response in the uptake rate, or even generate bistability through covalent modifications [18], tuneable implementations of such kinetic mechanisms require protein engineering beyond our current capabilities.
Figure 1.

Bistable switches for cellular uptake with a two-promoter circuit under feedback regulation. (a) Graded, ultrasensitive and bistable uptake. A bistable system switches between slow and fast uptake, and displays hysteresis because of different switching points. (b) Circuit architecture for the uptake systems: a transport enzyme (e1) imports a metabolite into the intracellular space. A second enzyme (e2) metabolizes the internalized metabolite (s). The circuit contains two promoters (P1 and P2) that control enzyme expression through feedback from the intracellular metabolite, including four combinations of (A)ctivation and (R)epression feedback loops; the main design parameters are the dynamic ranges μ of both promoters, defined as the ratio between maximal and baseline expression levels (see inset). (c) Schematics for the four uptake circuits. Each circuit has two interlinked positive and negative feedback loops (shown in dashed lines); a faster utilization causes a decrease in the concentration of metabolite, and thus upregulation (downregulation) of the utilization enzyme corresponds to negative (positive) feedback. (Online version in colour.)
In this paper we propose a bistable switch to control the rate at which cells take up a metabolite from the environment. We identify a genetic–metabolic system that reversibly toggles between slow and fast uptake in response to the amount of metabolite in the extracellular space. Our design relies on coupling enzyme activity with a gene regulatory circuit designed to shape the uptake response as a bistable switch. We borrowed this strategy from two well-known bistable uptake systems found in nature: the lactose operon in Escherichia coli [19] and the galactose pathway in Saccharomyces cerevisiae [14,20]. Both systems produce bistability through an underlying gene regulatory network that controls the expression of key enzymes.
We consider uptake circuits based on feedback regulation of the expression of transport and utilization enzymes (figure 1b). The circuit architecture requires two metabolite-responsive promoters and includes four regulatory motifs depending on whether the internalized metabolite activates or represses enzyme expression (figure 1c). Two of these motifs can be found in natural and engineered systems. The activation–activation circuit (labelled AA in figure 1c) has a similar architecture to the lactose operon [19], where an intracellular metabolite (allolactose) upregulates a transporter and a metabolic enzyme through binding to a transcriptional repressor. A repression–activation circuit (labelled RA in figure 1c), on the other hand, has been used to improve the production of fatty acids by balancing the supply and consumption of the intermediate malonyl-CoA [10].
From a general model for the uptake circuits, we identified those that produce bistability and determined analytic conditions for bistability in terms of the promoters' dynamic ranges and transcriptional thresholds. Our approach combines a qualitative on–off model for promoter activity together with a separation of timescales [21]. This leads to a reduced model based on piecewise affine differential equations where bistability can be studied analytically [22]. The analysis revealed that the circuits have a rich diversity of bistable regimes, i.e. qualitatively different combinations of stable steady states that lead to a bistable uptake flux, all of which can be linked to different design spaces for the promoter parameters. The multiple number of bistable regimes contrasts with, for example, the original genetic toggle switch that has only one regime for bistability [3]. Our analysis method relies on a coarse approximation of the true system dynamics, but uncovers useful relationships between experimentally tuneable parameters and the resulting metabolic phenotypes. We used the derived conditions to compare the bistable regimes in terms of the shape and size of their design spaces [23]. With the size of the design space as a proxy for robustness, we found that the AR circuit is the best candidate for an uptake switch, as it displays bistability for a large range of promoter dynamic ranges and, therefore, it is likely to be more robust in face of cell-to-cell variability and unreliable estimates for the enzyme kinetic parameters. Further analyses of the AR system revealed analytic conditions for hysteresis and design rules to maximize the promoter design space with the transcriptional thresholds. We validated our results and the performance of the AR circuit with extensive simulations of a more realistic, continuous, model for promoter activity. Population-wide simulations show that the circuit diversifies the uptake phenotypes by splitting a culture into two subpopulations with slow and fast uptake.
Our study puts forward the AR circuit as a promising design for an uptake switch in future synthetic biology applications that require metabolic control across cell cultures. A key challenge in the implementation of the uptake switch is the lack of intracellular sensors that interface metabolic signals with gene expression. This limitation is not specific to our study and pervades all current efforts to engineer genetic–metabolic circuits [24,25]. Successful implementations have relied on metabolite-responsive promoters [8,10,11], but the design and construction of such sensing mechanisms requires a substantial amount of experimental work. Our study demonstrates how mathematical design can be an effective tool to identify circuit architectures for a new biological function and to single out the key design parameters that need to be tuned, both of which can help to focus and accelerate the experimental work in the field.
2. General model for synthetic uptake circuits
We consider uptake circuits composed of two enzymes and an internalized metabolite (s) as illustrated in figure 1b. A transport enzyme (e1) imports the extracellular metabolite (s0) into the cell, which is then metabolized by different cellular processes represented by a utilization enzyme (e2). The network has two independent promoters that control the expression of enzymes in response to the internalized metabolite, thus forming two coupled feedback loops. We assume that the extracellular metabolite concentration is constant so that the circuits are thermodynamically open and sustain a non-zero flux. We model the dynamics of the metabolite and enzymes as
| 2.1 |
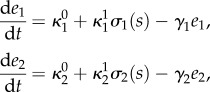 |
2.2 |
where (s, e1, e2) are the species concentrations, 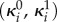 are the baseline and induced enzyme expression rates, and γi is a first-order kinetic rate that accounts for protein degradation and dilution by cell growth.
are the baseline and induced enzyme expression rates, and γi is a first-order kinetic rate that accounts for protein degradation and dilution by cell growth.
The functions gi in equation (2.1) are the enzyme turnover rates, i.e. the reaction rate per unit of enzyme, and describe their kinetics for different substrate concentrations. We focus our analysis on a broad class of kinetic rate functions that includes the common Michaelis–Menten kinetics as a special case. To this end, we assume that the turnover rates are increasing functions of their substrate, so that 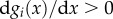 with a saturation value
with a saturation value 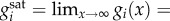
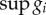 . In the case of Michaelis–Menten kinetics, the turnover rate is
. In the case of Michaelis–Menten kinetics, the turnover rate is 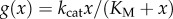 and has a saturation value
and has a saturation value 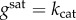 .
.
The enzyme equations in (2.2) describe the balance between protein synthesis and degradation. The functions σi(s) are lumped models for the promoter response curves and describe the activation/repression of transcriptional activity by the internalized metabolite. We assume that the promoter response curves satisfy dσi/ds > 0, σi(0) = 0 and σi(∞) = 1 in case of activation (conversely, dσi/ds < 0, σi(0) = 1 and σi(∞) = 0 in case of repression). The promoters therefore control the enzymes between a baseline (‘off’) and a maximal concentration (‘on’):
| 2.3 |
Promoter strengths are key parameters in promoter engineering [26] and one of the most easily tuneable parameters in synthetic circuits. Here, we quantify the strength of promoters via their dynamic range (μi), i.e. the ratio between their maximal and baseline activity levels
| 2.4 |
As we shall see in the next section, we found that bistability depends critically on an additional design parameter, the relative dynamic range (μ12):
| 2.5 |
which corresponds to the maximal level of the utilization enzyme relative to the baseline level of the transport enzyme. We note that because the functions σi lump transcription and translation together, our model can also account for the strength of ribosomal binding sites, another common tuneable parameter in gene circuits [27], via a linear scaling factor of enzyme expression rates in equation (2.2).
As shown in figure 1c, the general circuit architecture includes four uptake circuits, which we call AA, RR, AR and RA depending on the particular combination of gene (A)ctivation or (R)epression. Each network can be seen as combinations of two interlinked positive and negative feedback loops. In particular, we can readily rule out bistability in the RA network, because it does not contain any positive feedback loops, a well-known necessary condition for bistability [28]. In the next section, we determine conditions under which the other three circuits in figure 1c display two steady-state fluxes.
3. Bistability in the synthetic uptake circuits
The steady-state uptake flux in the circuits is
| 3.1 |
where the bars denote steady-state concentrations. The steady-state transport flux depends only on the concentration of the transport enzyme (e1). The utilization reaction, by contrast, depends on both the metabolite and enzyme, and therefore different steady-state concentrations can lead to the same utilization flux. For example, in equation (3.1) a slow utilization flux can be sustained by a lowly abundant metabolite and an overexpressed utilization enzyme, or a highly abundant metabolite and an underexpressed utilization enzyme. These two scenarios require promoters to operate at different activity levels and lead to steady states that are qualitatively different. Next we will show that a single uptake circuit can display a number of bistable regimes, i.e. qualitatively different combinations of steady-state concentrations that lead to a bistable flux. We will then provide analytic conditions that describe all combinations of promoter dynamic ranges that produce bistability, which we will refer to as the promoter design space.
3.1. Identification of all bistable regimes
Using the model in (2.1) and (2.2), we can obtain an equation for the steady-state metabolite concentration
| 3.2 |
from where both enzyme concentrations can be computed as 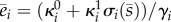 . The ideal would be to have analytic solutions of (3.2) that show how bistability depends on the promoter dynamic ranges, potentially revealing structural differences among the circuits. However, the steady-state equation is analytically intractable because of the nonlinearities in the enzyme kinetics (g2) and promoter response curves (σi). For some parameter combinations, the circuits may also lead to unbounded accumulation of the metabolite due to saturation of the utilization enzyme. This happens when the steady-state equation does not have a solution because the right-hand side of (3.2) is higher than the saturation value of g2.
. The ideal would be to have analytic solutions of (3.2) that show how bistability depends on the promoter dynamic ranges, potentially revealing structural differences among the circuits. However, the steady-state equation is analytically intractable because of the nonlinearities in the enzyme kinetics (g2) and promoter response curves (σi). For some parameter combinations, the circuits may also lead to unbounded accumulation of the metabolite due to saturation of the utilization enzyme. This happens when the steady-state equation does not have a solution because the right-hand side of (3.2) is higher than the saturation value of g2.
In general, the number of steady states and their stability depend intricately on the model parameters and the shape of the nonlinearities. A common strategy to detect bistability is to use phase plane analysis to identify the number of steady-state solutions and their behaviour with respect to model parameters. This approach becomes cumbersome in highly nonlinear models and requires case-by-case analyses for each uptake circuit. An alternative is to solve the steady-state equation numerically for many parameter combinations and use linear stability analysis in each solution, or to run long model simulations for many initial conditions and single out those that lead to two final states. It is generally difficult, however, to establish whether bistability properties found with numerical search are structural features of the model, or if instead they are a consequence of the form of the nonlinearities and the specific choice for parameter values.
We can avoid the above difficulties with an analysis technique based on piecewise affine models for gene regulation [22,29] and a separation of timescales [21,30]. This approach leads to a reduced model in which we can study bistability analytically. To obtain a tractable model, we assume that promoters switch between ‘on’ and ‘off’ activity levels depending on the amount of metabolite:
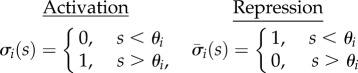 |
3.3 |
where θi is a threshold for transcriptional activation or repression. Enzymatic catalysis occurs on a much faster timescale than enzyme expression, with kinetic time constants typically in the millisecond range [31] and gene expression in the order of tens of minutes or longer. We incorporated this timescale separation to obtain a reduced model that can be extensively analysed in terms of its bistability properties.
Our analysis revealed that the uptake circuits can sustain a rich variety of bistable regimes. The results, summarized in figure 2 (details in appendix A.1 and the electronic supplementary material), indicate a total of nine qualitatively different regimes: one for the RR circuit, three for the AA circuit and five for the AR circuit.
Figure 2.

Bistable regimes in the uptake circuits. The bar plots (left panels) show all the qualitatively different regimes that sustain a bistable uptake flux. Each bistable regime corresponds to a combination of steady-state concentrations from the right panels, marked with circled numbers. The steady-state flux is proportional to the concentration of transporter enzyme, as shown in equation (3.1); therefore, low or high concentration of transporter (e1) correspond to slow or fast uptake flux, respectively. For the enzymes, the bar height represents a baseline (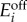 ) or maximal (
) or maximal (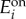 ) steady-state concentration; for the metabolite, the bar height represents the qualitative steady-state concentration of metabolite relative to the thresholds: e.g. if θ2 < θ1, the bars denote a low (
) steady-state concentration; for the metabolite, the bar height represents the qualitative steady-state concentration of metabolite relative to the thresholds: e.g. if θ2 < θ1, the bars denote a low (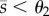 ), intermediate (
), intermediate (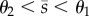 ) or high (
) or high ( ) concentration. The threshold-independent regimes (highlighted in grey) exist for any combination of regulatory thresholds. All the threshold-dependent regimes are sustained by an intermediate concentration of metabolite; these regimes vanish if both promoters have similar thresholds. The bistable regimes can be verified through numerical simulation of a continuous model with promoter responses described by steep sigmoids. Details on how to find the bistable regimes can be found in appendix A.1 and the electronic supplementary material. (Online version in colour.)
) concentration. The threshold-independent regimes (highlighted in grey) exist for any combination of regulatory thresholds. All the threshold-dependent regimes are sustained by an intermediate concentration of metabolite; these regimes vanish if both promoters have similar thresholds. The bistable regimes can be verified through numerical simulation of a continuous model with promoter responses described by steep sigmoids. Details on how to find the bistable regimes can be found in appendix A.1 and the electronic supplementary material. (Online version in colour.)
As shown in figure 2, each bistable regime requires the promoters to operate at different activity levels depending on the steady-state concentration of metabolite. Moreover, if promoters respond at different regulatory thresholds, the circuits can reach steady states with intermediate metabolite concentrations (i.e. 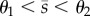 ). As a consequence, the bistable regimes depend strongly on the promoter thresholds: we observe three threshold-independent regimes (RR-0, AA-0 and AR-0 in figure 2) and six threshold-dependent regimes that emerge depending on whether θ1 < θ2 or θ1 > θ2. The threshold-dependent regimes vanish when both promoters have similar thresholds. The results in figure 2 also uncover several qualitative differences among the circuits:
). As a consequence, the bistable regimes depend strongly on the promoter thresholds: we observe three threshold-independent regimes (RR-0, AA-0 and AR-0 in figure 2) and six threshold-dependent regimes that emerge depending on whether θ1 < θ2 or θ1 > θ2. The threshold-dependent regimes vanish when both promoters have similar thresholds. The results in figure 2 also uncover several qualitative differences among the circuits:
— The RR circuit has only one bistable regime. The circuit cannot reach an intermediate concentration of metabolite because this would cause transport to be too fast to be matched by a slow utilization (in the case θ1 > θ2), or too slow to be matched by a fast utilization (in the case θ1 < θ2). Such imbalance would ultimately lead to accumulation or depletion of the internalized metabolite.
— The AA and AR circuits, by contrast, admit intermediate steady-state metabolite concentration, and consequently they can display two additional bistable regimes each (AA-1 and AA-2, AR-1 and AR-3, respectively).
— The AR circuit has two extra bistable regimes (AR-2 and AR-4) sustained by three stable steady states. We note that in these regimes, the three stable steady-state concentrations translate into two stable uptake fluxes, because the steady states have only two different concentrations for the transporter (e1), which in turn determines the uptake flux via the relation
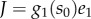 in equation (3.1).
in equation (3.1).
3.2. Shape and size of the promoter design space
To decide which circuit is the best candidate for an uptake switch, we determined the promoter design spaces and proposed a measure to assess the robustness of each bistable regime. With the simplified model in §3.1, we obtained analytic conditions for each bistable regime in terms of the promoter dynamic ranges. The conditions are summarized in figures 3 and 4, and the details on how to obtain them are in appendix A.1 and the electronic supplementary material. In particular, the design spaces for the threshold-independent regimes are
| 3.4a |
| 3.4b |
| 3.5a |
| 3.5b |
| 3.6a |
| 3.6b |
The above conditions, illustrated in figure 3, describe all combinations of dynamic ranges that lead to a bistable uptake flux. In all cases, the shape and size of the design space depend on three parameters:
| 3.7 |
Figure 3.

Promoter design space for the threshold-independent bistable regimes in figure 2. The design spaces can be visualized as three-dimensional solids in a (μ1, μ2, μ12) space. The solids correspond to the inequalities in (3.4a)–(3.6b) for fixed enzyme kinetics and transcriptional thresholds. The bar plots represent the species concentrations as in figure 2. Details on how to find the design spaces analytically are in appendix A.1 and the electronic supplementary material. The size of the design spaces provides a metric for the robustness of each regime. We quantified robustness as the volume of the design space relative to the total volume of the full parameter space for fixed enzyme kinetics and different regulatory thresholds spanning two orders of magnitude. The (θ1, θ2) values shown are relative to a nominal metabolite concentration s0 = 1 µM (the white line marks the equal threshold case). Details of the simulations and parameter values can be found in appendix A.2. (Online version in colour.)
Figure 4.

Promoter design space for the threshold-dependent bistable regimes in figure 2. The design spaces correspond to the shown inequalities for fixed enzyme kinetics and transcriptional thresholds. Details on how to find the design spaces analytically are in appendix A.1 and the electronic supplementary material. The robustness index was computed as in figure 3. (Online version in colour.)
These parameters reflect how the interplay between enzyme kinetics and gene regulation affects bistability. The βi parameters correspond to the ratio of enzyme turnover rates at a given concentration of extracellular metabolite and transcriptional threshold. They take maximal or minimal values when thresholds are far away from the Michaelis constant of the utilization enzyme (Km). The third parameter, 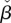 describes the saturation level of the transport enzyme relative to the maximal utilization rate.
describes the saturation level of the transport enzyme relative to the maximal utilization rate.
The conditions in (3.4a)–(3.6b) assume that the thresholds are ordered as θ1 ≤ θ2 (and therefore that β1 ≥ β2), but the converse conditions for θ1 > θ2 can be obtained by swapping β1 and β2 in the inequalities. The conditions for bistability in (3.4a)–(3.6b) have two parts: a-conditions guarantee two stable steady states for the enzyme concentrations, while b-conditions prevent the accumulation of metabolite in both steady states. The b-conditions arise due to the saturation of the enzyme kinetics: if not satisfied, then the uptake flux will be higher than the saturation rate of the utilization reaction and cause the metabolite to accumulate in the intracellular space.
As shown in figures 3 and 4, the shape and size of the design spaces varies significantly across regimes. Bistability in the RR-0 and AA-0 regimes is particularly constrained, as it requires asymmetric designs where one promoter has a much broader dynamic range than the other. The AR-0 regime, by contrast, is much more flexible as it produces bistability for more combinations of promoter dynamic ranges. We can compare the regimes using the size of their design spaces as a metric for robustness:
| 3.8 |
where Volbistable is the volume of the three-dimensional solid defined by the design space, and Voltotal is the volume of the full parameter space defined as a cube
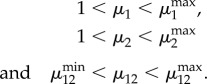 |
If we choose the same parameter cube for each circuit, the relative volume provides an effective measure to compare the design spaces. A robust circuit should ideally have a large design space to ensure bistability without a laborious fine-tuning of the promoters' response curve. The design space should also be symmetric with respect to μ1 and μ2 to allow for an independent design of both promoters. The most robust bistable circuit would therefore have a 100% relative volume (i.e. all parameter combinations lead to bistability), while fragile designs would have a much smaller volume. Because the design spaces depend strongly on the transcriptional thresholds (through the βi parameters in (3.7)), we numerically computed the relative volumes of the bistable regimes for different combinations of regulatory thresholds. The results, shown in figures 3 and 4, show that most regimes are fragile, with only two (the AA-2 and AR-0 regimes) standing out with a robustness index above 70%. As observed in figure 2, however, the AA-2 regime requires θ1 < θ2 while the AA-0 regime does not impose constraints on the transcriptional thresholds. We therefore conclude that the AR-0 regime is the most robust design for a bistable uptake switch.
The quality of the AR circuit as a bistable switch can be intuitively understood from the interaction diagrams in figure 1c. The AR circuit corresponds to two positive feedback loops, where the internalized metabolite increases its own abundance by speeding up its import and slowing down its consumption. Interlinked positive feedback loops are known to improve the bistability properties in a number of natural networks [32–34] and there is evidence that nature favours bistability through interlinked regulation [2]. In the next sections, we carry out a deeper analysis of the AR circuit.
4. Further design criteria for the activation–repression circuit
4.1. Optimization of the promoter design space
Here we use the derived conditions for bistability to learn how to maximize the circuit's design space with the transcriptional thresholds. We focus on the AR circuit designed to operate in the robust AR-0 regime, but analyses of the other regimes can be carried out analogously. For this part, we further assume that both promoters have equal baseline expression levels, i.e. 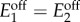 and thus μ12 = μ2. Substituting μ12 = μ2 in the conditions in (3.6a) and (3.6b), we obtain simplified conditions for bistability
and thus μ12 = μ2. Substituting μ12 = μ2 in the conditions in (3.6a) and (3.6b), we obtain simplified conditions for bistability
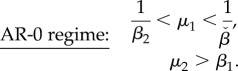 |
4.1 |
The conditions in (4.1) describe the design space as an open box in a (μ1, μ2) parameter space, illustrated in figure 5. The effect of the βi parameters on the conditions in (4.1) suggests a trade-off between the transcriptional thresholds and the size of the design space (figure 5): a low repression threshold θ2 (i.e. a larger β2 parameter) enlarges the design space for the activating promoter (μ1) and, conversely, a high activation threshold θ1 enlarges the design space for the repressing promoter (μ2). Further, we can derive criteria to maximize the design space:
- — The upper limit for μ1 grows if
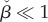 . Recalling that
. Recalling that 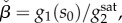 we conclude that
we conclude that 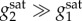 is a sufficient condition for
is a sufficient condition for 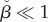 for any concentration of intermediate metabolite. In the case of Michaelis–Menten kinetics, the condition is equivalent to
for any concentration of intermediate metabolite. In the case of Michaelis–Menten kinetics, the condition is equivalent to
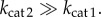
4.2 - — If β1 = β2 = 1 we minimize the lower limits for the dynamic ranges and get an optimal design space

4.3
Figure 5.

Maximization of the design space in the activation-repression circuit. The diagrams illustrate the promoter design space for different combinations of transcriptional thresholds, as described by the conditions in (4.1). The optimal design has regulatory thresholds chosen according to the criteria in (4.5) taken as equality. (Online version in colour.)
Using the definition of the βi parameters 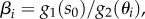 we can impose the condition βi = 1 to obtain an optimal threshold
we can impose the condition βi = 1 to obtain an optimal threshold
| 4.4 |
where 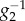 is the inverse function of g2. We therefore conclude that if the transcriptional thresholds are designed as
is the inverse function of g2. We therefore conclude that if the transcriptional thresholds are designed as
| 4.5 |
then the AR circuit has a maximal design space for bistability. Note that we state the conditions in (4.5) as inequalities because, by definition, the dynamic ranges are μi > 1 and consequently any combination of thresholds that satisfies (4.5) leads to the same maximal design space.
The conditions in (4.2) and (4.5) provide quantitative criteria to design an AR circuit with maximal design space for bistability. Condition (4.2) relaxes the upper limit for the first promoter (dynamic range μ1), but it is generally difficult to satisfy because catalytic enzymes in the same pathway tend to have similar kcat values. On the other hand, condition (4.5), illustrated in figure 5, loosens the lower limit for the promoter dynamic ranges, and therefore may prove useful in implementations with weak promoters and tuneable regulatory thresholds.
4.2. Conditions for hysteresis
So far we have focused on a bistable uptake flux under a fixed amount of extracellular metabolite. A hallmark feature of bistable switches, however, is that they display hysteresis to changes in the input stimulus. As shown in the bifurcation diagram in figure 1a, hysteresis causes cells to switch between slow and fast uptake at different metabolite concentrations. This mechanism filters out spurious switching from extracellular fluctuations, and implements a form of memory where the response of a cell to intermediate metabolite concentrations depends on its previous exposure to it. Because equal transcriptional thresholds enlarge the design space (figure 3), we assumed a nominal threshold for both promoters, θ1 = θ2 = θ and obtained conditions for the AR circuit to display hysteresis:
| 4.6a |
 |
4.6b |
| 4.6c |
| 4.6d |
The above conditions can be obtained by examining the effect of the extracellular metabolite (s0) on the circuit's steady states through changes in the βi parameters (details in appendix A.1 and the electronic supplementary material). The conditions depend on two extra parameters
| 4.7 |
which correspond to the original βi and 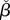 parameters in (3.7) under saturation of the transport enzyme. The conditions in (4.6a) and (4.6b) are the same as the design space in (3.6), while the conditions in (4.6c) and (4.6d) add further constraints to the design space. Condition (4.6c) guarantees that uptake can be bidirectionally switched, i.e. from slow to fast and vice versa (if not satisfied, the circuit can only be switched off). Condition (4.6d) ensures that the metabolite steady-state
parameters in (3.7) under saturation of the transport enzyme. The conditions in (4.6a) and (4.6b) are the same as the design space in (3.6), while the conditions in (4.6c) and (4.6d) add further constraints to the design space. Condition (4.6c) guarantees that uptake can be bidirectionally switched, i.e. from slow to fast and vice versa (if not satisfied, the circuit can only be switched off). Condition (4.6d) ensures that the metabolite steady-state 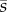 exists for all concentrations of extracellular metabolite. Note that condition (4.6d) becomes less tight under the kinetic condition in (4.2).
exists for all concentrations of extracellular metabolite. Note that condition (4.6d) becomes less tight under the kinetic condition in (4.2).
5. Activation–repression circuit with graded promoters
5.1. Validation of the design criteria
In the previous sections, we obtained design criteria for the AR circuit based on a coarse approximation for promoter activity. The approximation assumes that promoters behave in an on–off fashion, i.e. having either a maximal or baseline activity without intermediate levels of expression. In practical implementations, promoter sensitivities are severely constrained and therefore it is unclear whether the derived design criteria are useful when using realistic promoters with graded, low-sensitivity, response curves.
To test the utility of the AR circuit in a more realistic model, we ran extensive simulations of the circuit with sigmoidal models for promoter response curves [35]. We modelled the promoter response curves as Hill functions
| 5.1 |
where θ is the regulatory threshold and h is the promoter sensitivity (Hill coefficient). We computed the parameter regions for bistability in the continuous model in (2.1) and (2.2) with low, intermediate and high promoter sensitivities. The results in figure 6a suggest that although the design spaces for the continuous model are smaller than those predicted by our approximation, they preserve the predicted qualitative properties (cf. figures 5 and 6a). We can distinguish among designs that are monostable, bistable, or that do not have a steady state. Optimization of the regulatory threshold, i.e. according to the criterion in (4.5), effectively enlarges the design space in the continuous model, even in the case of low-sensitivity promoters (h = 2). These results thus suggest that the derived design criteria can guide the design in more realistic models for promoter activities.
Figure 6.

Bistability in the activation-repression circuit. (a) Regions for bistability in non-optimized and optimized designs for a continuous model of the activation-repression circuit (cf. figure 5). In the non-optimized designs, the regulatory threshold was chosen as θ = s0 for an extracellular metabolite concentration s0 = 4.7 µM; in the optimized designs, the threshold was chosen to maximize the region for bistability according to the criterion in (4.5) taken as equality; promoter sensitivities are h = {2, 4, 8}. (b) Domains of attraction of two specific designs with low-sensitivity promoters (h = 2) and promoter dynamic ranges (μ1, μ2) = (19, 2) and (μ1, μ2) = (10, 10), marked as and in panel (a). (c) Bifurcation diagrams of the enzyme concentrations and internalized metabolite as a function of the extracellular concentration; solid (dashed) lines indicate the stable (unstable) steady states. The design is optimized for a nominal concentration s0 = 1 µM according to (4.5) taken as equality, and promoter dynamic ranges (μ1, μ2) = (6, 5). The baseline enzyme expression levels were fixed to 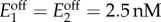 in panels (a,b), and
in panels (a,b), and 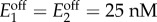 in panel (c). Details of the simulations and parameter values can be found in appendix A.2. (Online version in colour.)
in panel (c). Details of the simulations and parameter values can be found in appendix A.2. (Online version in colour.)
In figure 6b, we plot the domains of attraction for each steady state in particular instances of AR circuits with low-sensitivity promoters. The results suggest that domains of attraction can depend strongly on the transcriptional thresholds and, in particular, threshold optimization can also help to equalize the domains of attraction and prevent a bias towards one uptake flux more than the other. The bifurcation diagrams in figure 6c indicate that the AR circuit effectively functions as a bidirectional switch with hysteresis, toggling between low/high states for enzyme expression.
5.2. Emergence of bimodal phenotypes across cell populations
Our results describe conditions under which single cells display a bistable uptake when exposed to the extracellular metabolite. At a population level, however, each individual cell will switch to slow or fast uptake depending on its intracellular state before exposure to the metabolite. Owing to numerous factors that affect the cellular composition, cell populations can exhibit a large cell-to-cell variability. In the case of synthetic gene circuits, variability can arise from, e.g. fluctuations in plasmid copy numbers, variability in transcriptional and translational resources (RNA polymerases, sigma factors and ribosomes), mutations in the promoter sequences and stochastic fluctuations inherent to gene expression [36].
We ran population-wide simulations of the AR circuit to test how it would perform in bacterial cultures with significant cell-to-cell variability. The domains of attraction in figure 6b suggest that single cells switch to a slow or fast uptake depending only on the abundance of enzymes and not the intracellular metabolite. We therefore focused on how variability in enzyme levels propagates to the flux phenotypes produced by the uptake switch [37,38]. We modelled variability in enzyme expression through deterministic simulations for many cells in a culture with randomized promoter dynamic ranges. The results, shown in figure 7, indicate that the proposed AR circuit can effectively toggle the uptake flux in a population. The resulting population-wide histograms show the hysteretic response of the uptake switch, with individual cells switching to a slow or fast uptake depending on their previous exposure to extracellular metabolite. As predicted by the bifurcation diagrams in figure 6c, the range for hysteresis grows with more sensitive promoters and further, we found that the high-flux state has a narrow distribution that is largely insensitive to the Hill coefficient. This indicates that the AR circuit tightly controls the uptake flux across a population even for low-sensitivity promoters. As a consequence of cell-to-cell variability, individual cells switch at different extracellular concentrations of the metabolite, leading to the observed bimodal phenotypes when the metabolite is close to the switching threshold.
Figure 7.
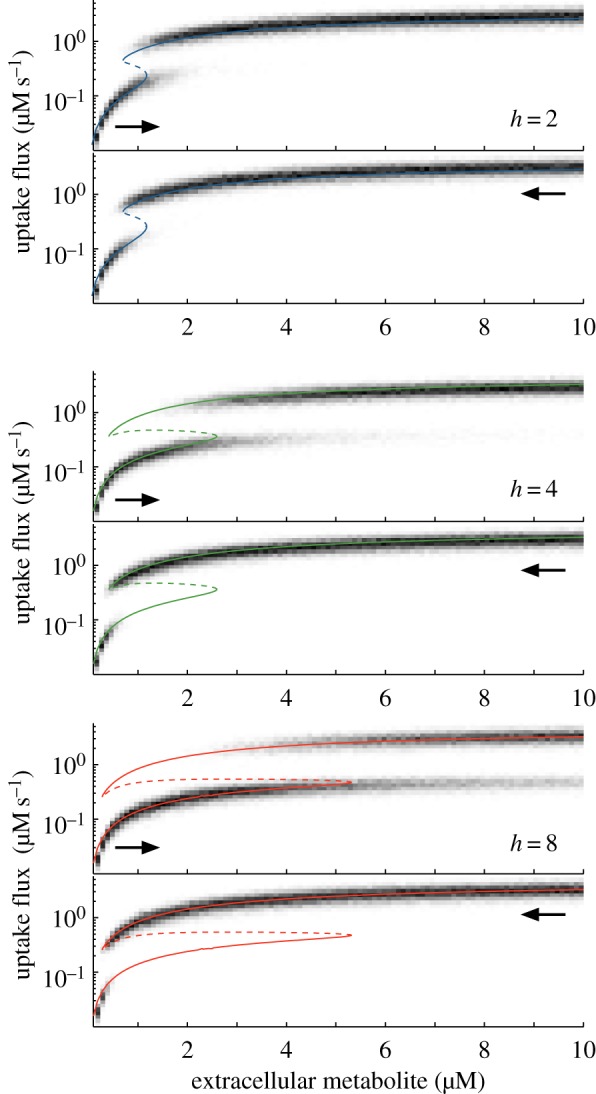
Hysteresis and bimodal phenotypes in the activation-repression circuit. Response of a cell population with variability in the enzyme expression levels. The heat maps are histograms of the population-wide distributions of the uptake flux for increasing or decreasing levels of extracellular metabolite. Cell populations were initialized at low (top panels) or high (bottom panels) uptake fluxes. The greyscale represents the number of cells with a given uptake flux. The histograms were obtained from a population with 500 cells with enzyme expression levels (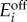 and
and 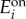 ) sampled from Gamma distributions [39] with means corresponding to the designs in figure 6c and a coefficient of variation of 20%, representative of measured genome-wide fluctuations in protein abundance [40]. The circuit is optimized for a nominal metabolite concentration s0 = 1 µM according to the criterion in (4.5) taken as equality. The histograms are overlaid with the flux bifurcation diagrams computed from figure 6c. Details of the simulations and parameter values can be found in appendix A.2. (Online version in colour.)
) sampled from Gamma distributions [39] with means corresponding to the designs in figure 6c and a coefficient of variation of 20%, representative of measured genome-wide fluctuations in protein abundance [40]. The circuit is optimized for a nominal metabolite concentration s0 = 1 µM according to the criterion in (4.5) taken as equality. The histograms are overlaid with the flux bifurcation diagrams computed from figure 6c. Details of the simulations and parameter values can be found in appendix A.2. (Online version in colour.)
6. Discussion
In this paper we proposed a bistable switch to control the rate at which cells take up a metabolite from the environment. The switch couples enzyme activity with a two-promoter gene network under feedback regulation. We examined mathematical models for four candidate networks and obtained the design spaces for promoter dynamic ranges that produce a bistable uptake system. Using the size of the promoter design space as a proxy for robustness, we singled out one network, an AR circuit (AR in figure 1c), that is significantly more robust than the others and is the best candidate for an uptake switch.
The proposed AR circuit effectively toggles between slow and fast uptake depending on the abundance of the extracellular metabolite. The shape of its design space suggests that both promoters can be tuned independently and we found criteria to maximize the design space by tuning the transcriptional thresholds. The large design space also indicates that the switch is robust to variability in promoter strengths and thus requires little fine-tuning of promoter response curves. Population-wide simulations show the emergence of bimodal phenotypes due to cell-to-cell variability and hysteresis. The circuit thus works as a memory device where individual cells lock into slow or fast uptake depending on their previous exposure to the extracellular metabolite, while protecting them from spurious switching caused by stochastic environmental fluctuations.
A key element in the proposed switch is the use of feedback regulation of enzyme expression levels. This strategy was inspired by the regulation of the lactose operon in E. coli [19] and the galactose pathway in S. cerevisiae [14], two well-known uptake systems where bistability emerges from the interplay between metabolism and gene regulation. In the lactose operon, bistability emerges from a regulatory architecture similar to the AA circuit studied here, but our results indicate it is not as robust as the AR circuit because it requires more careful fine-tuning of the design parameters. Natural systems may achieve such fine-tuning through evolution, but this is extremely laborious in engineered systems. In the galactose pathway, on the other hand, bistability emerges from a more complex gene regulatory network with multiple components and interactions that are difficult to tease apart. Other bacterial systems that display switching metabolic phenotypes, e.g. the carbon catabolite repression system [41], the glycolytic–gluconeogenic switch [42] and the central carbon metabolism [43], rely on even more intricate regulation and are too complex to be used as templates for design. Although other strategies to produce bistability may exist, either using different genetic circuits or regulatory mechanisms, the proposed AR circuit is a simple architecture for a robust uptake switch, and thus a promising backbone for future implementations.
The uptake switch provides an interface to control metabolic activity from the extracellular space. This could be useful, for example, in metabolic engineering applications that need to regulate production with extracellular inducers or to trigger pathways only when substrates reach an activation threshold. The hysteretic response of the switch can help to control production in face of substrate variability or heterogeneous bioreactor conditions. Another promising application for a bistable uptake switch is the control and coordination of metabolism in microbial consortia. Although most synthetic cell-to-cell communication systems rely on mechanisms drawn from quorum sensing or hormone signalling, recent studies have also explored the use of metabolic signals to coordinate pathways distributed among different strains [44]. The field is in its early days, but it is becoming increasingly clear that metabolites may not only provide a new channel for synthetic communication between cells [12], but also that consortia can outperform single-strain cultures [13]. A plausible scenario for this is, for example, to split a large synthetic pathway among different strains and thus alleviate the genetic burden caused by expression of multiple heterologous proteins in a single strain [45]. The general principle is to have a ‘sender’ strain that secretes a metabolite that is then taken up by a ‘receiver’ strain. If the exchanged metabolite is a precursor for a target product in the receiver strain, an uptake switch can serve as a mechanism to lock receivers in a high uptake flux and, through hysteresis, insulate them from extracellular fluctuations in the transmitted signal. Another possibility is to use the uptake switch in receiver cells to diversify their phenotypes. Upon command from sender cells, receivers can split into slow and fast feeders, opening up the possibility to use bet-hedging to control metabolic activity upon changes in growth conditions, a well-known survival strategy used by microbes [15]. Such synthetic systems could also be used to study the evolution of social interactions in microbes. A number of studies have successfully used synthetic gene circuits to uncover how strains evolve their phenotypes in different conditions, e.g. under competition for shared carbon sources or cooperation through exchange of nutrients and signalling molecules [16,46,47]. These diverse applications suggest that bistable uptake switches will become increasingly relevant as efforts to engineer synthetic consortia intensify in the future.
Asserting whether a biochemical network is bistable is a challenging mathematical and computational problem. For specific classes of models, a number of approaches have addressed bistability by, e.g. exploiting the structure of the model's Jacobian [28,48] or using notions from chemical reaction network theory [49] (see table 3 in [50]) for a list of existing approaches). Finding parameter regions for bistability is even harder and, although promising approaches exist for specific model types [51], for more general models, the problem remains largely unsolved and we do not have effective methods other than numerical exploration of the parameter space.
We overcame the above limitations with an analysis technique that combines a piecewise affine model for gene expression, a kinetic model for metabolic reactions and a separation of timescales between both [21]. This strategy proved useful to single out networks that display bistability and to identify the parameter design spaces analytically. A salient conclusion of our analysis is that the uptake systems display a diverse range of bistable regimes. The AR circuit, in particular, displays five qualitatively different bistable regimes depending on the promoter dynamic ranges and their transcriptional thresholds. Our approach also offers a number of other advantages: it requires minimal assumptions on the enzyme kinetics, it accounts for the four regulatory circuits simultaneously without separate ad hoc analyses, and it reveals the underlying geometry of the design space for bistability in terms of experimentally accessible parameters. The latter uncovers how the interplay between promoter design and enzyme kinetics affects the shape and size of the design space, giving a first idea of which design parameters are most relevant to achieve a prescribed phenotype.
We point out that because our analysis relies on a coarse on–off approximation of promoters, its predictions are not guaranteed to hold in more realistic models for promoter activity. Our simulation results show, however, that the derived design criteria can effectively guide circuit design in models with standard sigmoidal descriptions of promoter response curves, even in the case of low Hill numbers, and that the derived design spaces provide an excellent starting point to search for bistability.
In this work we focused on the promoter dynamic ranges as the main tuneable parameters of the circuits. Although new technologies in DNA engineering are ever expanding the number of tuneable ‘knobs’ in synthetic circuits [52,53], promoter dynamic ranges are particularly flexible in that they can be altered with many techniques, e.g. by random mutagenesis [26], by manipulation of polymerase binding sites [54] or by the addition of sequence repeats [55]. In its current form, our model analysis can also be used to study the effect of tuneable protein half-lives [56], the strength of ribosomal binding sites [27], and in general, other genetic modifications that can be modelled as a linear scaling of protein expression rates. Other tuning strategies, e.g. affinity of transcription factors or post-translational modifications, however, cannot be directly included in our analysis and require a more mechanistic model for gene expression beyond the lumped model used here.
Our main goal in this paper has been to investigate the mathematical design of an uptake switch. We sought to draw analytic links between bistability and design parameters, for which we studied a tractable model that retains the typical nonlinearities encountered in enzyme kinetics and gene regulation. The costs of this analytic treatment were a number of model simplifications that should be addressed in future molecular implementations of the switch. First, the model should include the mechanistic details for gene regulation. By including the detailed interactions between the internalized metabolite and enzyme expression, the model will predict the effect of the particular strategy used to tune the circuit function. Second, the model should be tailored to the specific metabolite and enzymes employed, including features such as reversible transport or regulatory mechanisms of kinetic activity. Third, the model should account for the interactions between the uptake switch and its host. These can significantly degrade the function of genetic circuits [57] and recent progress in models for bacterial growth allows systematic incorporation of host–circuit interactions into the circuit design [58]. This will be particularly relevant for switches designed to take up carbon sources or other essential nutrients, as these will likely interfere with central metabolic functions of the host and trigger some of its native regulatory mechanisms [59].
The molecular implementation of the proposed switch remains a challenge because of the lack of mechanisms to sense intracellular metabolites and control gene expression. Natural systems have evolved a number of mechanisms to sense intracellular metabolites, see e.g. the comprehensive discussions in [59,60], but in general it is not easy to make them respond to metabolites they have not evolved to sense [25]. The lack of metabolite sensors is the most important bottleneck in dynamic metabolic engineering [24] and limits all current efforts to engineer synthetic gene circuits for metabolism. In our study, we have assumed that the intracellular metabolite controls enzyme expression by direct activation or repression of the promoters, but in implementations the regulation will be mediated by a specific molecular mechanism, e.g. natural metabolite-responsive transcription factors [8] or hybrid promoter–regulator systems [61]. Although currently there are no modular mechanisms to sense intracellular metabolites, recent progress in the field has led to novel sensors [25] and the implementation of an RA circuit in E. coli [10], bringing us increasingly close to building complex genetic–metabolic circuitry. This makes the role of mathematical design evermore important, as it is a powerful tool to discover useful circuit architectures that could be built once metabolite sensors are available.
Supplementary Material
Acknowledgements
We thank Jordan Ang, John Heap, Andrea Weiße and Fuzhong Zhang for helpful suggestions.
Appendix A
A.1. Analytic results
A.1.1. Identification of bistable regimes and parameter spaces for bistability
The details on how to identify each bistable regime in figure 2, together with their conditions for bistability (the design spaces in figures 3 and 4), can be found in the electronic supplementary material. The general idea is to first approximate the promoter response curves in model (2.1) and (2.2) by step functions:
| A 1 |
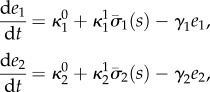 |
A 2 |
where 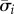 are the step functions in (3.3). Using the separation of timescales, we reduce the model assuming the metabolite to be in a quasi-steady state with respect to the evolution of enzyme concentrations. Details on the technical conditions for the separation of timescales can be found in [62]. We take ds/dt ≈ 0 in equation (A 1) to get an algebraic equation for the metabolite concentration
are the step functions in (3.3). Using the separation of timescales, we reduce the model assuming the metabolite to be in a quasi-steady state with respect to the evolution of enzyme concentrations. Details on the technical conditions for the separation of timescales can be found in [62]. We take ds/dt ≈ 0 in equation (A 1) to get an algebraic equation for the metabolite concentration
| A 3 |
The key observation is that, since g2(s) is an increasing function of s, the condition s < θi implies that g2(s) < g2(θi), which after substituting in (A 3) leads to the equivalences:
| A 4 |
where 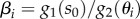 are the parameters that appear in all the conditions for bistability (figures 3 and 4). We can then get a reduced model in the form of a two-dimensional discontinuous differential equation
are the parameters that appear in all the conditions for bistability (figures 3 and 4). We can then get a reduced model in the form of a two-dimensional discontinuous differential equation
| A 5 |
where e = [e1, e2] is the vector of enzyme concentrations and the matrix Γ = diag{γ1, γ2} contains the degradation rates. The vector ϕ is piecewise constant and is formed by different combinations of baseline (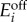 ) and maximal (
) and maximal (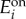 ) enzyme expression levels, depending on whether e1/e2 > βi or e1/e2 < βi. The model in (A 5) is a piecewise affine differential equation defined in conic domains (because conditions such as e1/e2 > βi describe a cone in an (e1, e2) plane). We can then identify its bistable regimes and the conditions for bistability by examining the geometry of the partitioned state space. The conditions for bistability arise naturally in terms of
) enzyme expression levels, depending on whether e1/e2 > βi or e1/e2 < βi. The model in (A 5) is a piecewise affine differential equation defined in conic domains (because conditions such as e1/e2 > βi describe a cone in an (e1, e2) plane). We can then identify its bistable regimes and the conditions for bistability by examining the geometry of the partitioned state space. The conditions for bistability arise naturally in terms of 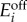 and
and 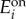 concentrations, but we can convert them to conditions on the promoter dynamic ranges with the following equivalences (recall the definitions in (2.3)–(2.5)):
concentrations, but we can convert them to conditions on the promoter dynamic ranges with the following equivalences (recall the definitions in (2.3)–(2.5)):
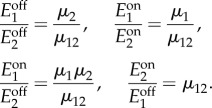 |
A 6 |
A.1.2. Conditions for hysteresis in the activation–repression circuit
We can obtain the conditions for hysteresis in the AR circuit (the inequalities in (4.6a)–(4.6d)) by examining the model's bistability with the parameters 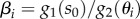 regarded as functions of the extracellular metabolite (s0). The key idea is to ensure that: for low s0 concentrations the model is monostable with a slow uptake flux, for intermediate s0 concentrations the model is bistable, and for high s0 concentrations the model is monostable with a fast uptake flux. These three conditions guarantee that the piecewise model has two saddle-node-like bifurcations and thus displays hysteresis. Further details can be found in the electronic supplementary material.
regarded as functions of the extracellular metabolite (s0). The key idea is to ensure that: for low s0 concentrations the model is monostable with a slow uptake flux, for intermediate s0 concentrations the model is bistable, and for high s0 concentrations the model is monostable with a fast uptake flux. These three conditions guarantee that the piecewise model has two saddle-node-like bifurcations and thus displays hysteresis. Further details can be found in the electronic supplementary material.
A.2. Model simulations
Simulations were done in Matlab with enzyme kinetic parameters kcat1 = 32 s−1, kcat2 = 320 s−1, KM1 = KM2 = 4.7 µM, and enzyme degradation rate constants γ1 = γ2 = 2 × 10−4 s, corresponding to a half-life of approximately 1 h.
A.2.1. Size of promoter design spaces
To compute the volumes of the solids in figures 3 and 4, we computed the convex hull of points satisfying the inequalities that define each design space. The μ1 and μ2 axes contains 50 linearly spaced points each, with 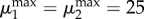 . The μ12 axis contains 50 log-spaced points with
. The μ12 axis contains 50 log-spaced points with 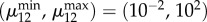 .
.
A.2.2. Simulations of the continuous model
We determined the parameter regions for bistability (figure 6a) from long simulations of the model in (2.1) and (2.2) for 104 pairs of promoter dynamic ranges (μ1,
μ2) and μ12 = μ2 sampled from a regular grid with increasing promoter sensitivities (hi = {2, 4, 8} for i = 1, 2). We ran two simulations for each (μ1, μ2) pair, initialized at the two stable steady states predicted by the piecewise affine model. We discriminated between monostability and bistability using the Euclidean distance between the final time points of each simulation. We determined the regions for metabolite accumulation by checking the condition 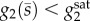 at the final time points (in which case the steady-state equation in (2.7) does not have a solution).
at the final time points (in which case the steady-state equation in (2.7) does not have a solution).
We computed the domains of attraction in figure 6b from long simulations of (2.1) and (2.2) for 8 × 103 initial conditions sampled from a uniform grid. The bifurcation diagrams in figure 6c were computed with the MatCont package for Matlab [63].
A.2.3. Population-wide simulations
We computed the histograms in figure 7 from simulations of the deterministic model (2.1) and (2.2) with randomized parameters. The top/bottom panels in figure 7 are simulations of cells initialized at a low/high uptake fluxes in steady state, respectively, and each simulation was run for 100 increasing/decreasing concentrations of extracellular metabolite in the range [0.1, 10] µM. For each metabolite concentration s0, we sampled the baseline and maximal enzyme concentrations (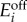 and
and 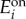 for i = 1,2) from Gamma distributions with means
for i = 1,2) from Gamma distributions with means 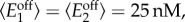
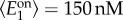 and
and 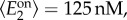 which correspond to dynamic ranges (μ1, μ2) = (6,5); these are the same parameters as in the bifurcation diagrams in figure 6c. We used a coefficient of variation of 20%, representative of measured fluctuations in protein abundance reported in the literature [40]. The gene expression parameters were then computed as
which correspond to dynamic ranges (μ1, μ2) = (6,5); these are the same parameters as in the bifurcation diagrams in figure 6c. We used a coefficient of variation of 20%, representative of measured fluctuations in protein abundance reported in the literature [40]. The gene expression parameters were then computed as 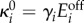 and
and 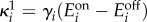 . The histograms were obtained from simulations of 500 cells for each concentration s0; we discarded and resampled all samples that led to metabolite accumulation by checking the condition
. The histograms were obtained from simulations of 500 cells for each concentration s0; we discarded and resampled all samples that led to metabolite accumulation by checking the condition 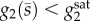 at the final time points of the simulation.
at the final time points of the simulation.
Competing interests
We have no competing interests.
Funding
D.A.O. was supported in part by Imperial College London through a Junior Research Fellowship and the Human Frontier Science Program through a Young Investigator Grant (RGY0076/2015). M.C. was supported in part by projects GeMCo (ANR-2010-BLAN-0201-01) and Labex Signalife (ANR-11-LABX-0028-01).
References
- 1.Tyson JJ, Chen KC, Novak B. 2003. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 15, 221–231. ( 10.1016/S0955-0674(03)00017-6) [DOI] [PubMed] [Google Scholar]
- 2.Brandman O, Ferrell JE, Li R, Meyer T. 2005. Interlinked fast and slow positive feedback loops drive reliable cell decisions. Science 310, 496–498. ( 10.1126/science.1113834) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardner TS, Cantor CR, Collins JJ. 2000. Construction of a genetic toggle switch in Escherichia coli. Nature 403, 339–342. ( 10.1038/35002131) [DOI] [PubMed] [Google Scholar]
- 4.Atkinson MR, Savageau MA, Myers JT, Ninfa AJ. 2003. Development of genetic circuitry exhibiting toggle switch or oscillatory behavior in Escherichia coli. Cell 113, 597–607. ( 10.1016/S0092-8674(03)00346-5) [DOI] [PubMed] [Google Scholar]
- 5.Kramer BP, Fussenegger M. 2005. Hysteresis in a synthetic mammalian gene network. Proc. Natl Acad. Sci. USA 102, 9517–9522. ( 10.1073/pnas.0500345102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen D, Arkin AP. 2012. Sequestration-based bistability enables tuning of the switching boundaries and design of a latch. Mol. Syst. Biol. 8, 620 ( 10.1038/msb.2012.52) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farmer WR, Liao JC. 2000. Improving lycopene production in Escherichia coli by engineering metabolic control. Nat. Biotechnol. 18, 533–537. ( 10.1038/75398) [DOI] [PubMed] [Google Scholar]
- 8.Zhang F, Carothers JM, Keasling JD. 2012. Design of a dynamic sensor–regulator system for production of chemicals and fuels derived from fatty acids. Nat. Biotechnol. 30, 354–359. ( 10.1038/nbt.2149) [DOI] [PubMed] [Google Scholar]
- 9.Oyarzún DA, Stan G-BV. 2013. Synthetic gene circuits for metabolic control: design trade-offs and constraints. J. R. Soc. Interface 10, 20120671 ( 10.1098/rsif.2012.0671) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu P, Li L, Zhang F, Stephanopoulos G, Koffas M. 2014. Improving fatty acids production by engineering dynamic pathway regulation and metabolic control. Proc. Natl Acad. Sci. USA 111, 11 299–11 304. ( 10.1073/pnas.1406401111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fung E, Wong WW, Suen JK, Bulter T, Lee S-G, Liao JC. 2005. A synthetic gene-metabolic oscillator. Nature 435, 118–122. ( 10.1038/nature03508) [DOI] [PubMed] [Google Scholar]
- 12.Silva-Rocha R, de Lorenzo V. 2014. Engineering multicellular logic in bacteria with metabolic wires. ACS Synth. Biol. 3, 204–209. ( 10.1021/sb400064y) [DOI] [PubMed] [Google Scholar]
- 13.Brenner K, You L, Arnold FH. 2008. Engineering microbial consortia: a new frontier in synthetic biology. Trends Biotechnol. 26, 483–489. ( 10.1016/j.tibtech.2008.05.004) [DOI] [PubMed] [Google Scholar]
- 14.Acar M, Becskei A, van Oudenaarden A. 2005. Enhancement of cellular memory by reducing stochastic transitions. Nature 435, 228–232. ( 10.1038/nature03524) [DOI] [PubMed] [Google Scholar]
- 15.Solopova A, van Gestel J, Weissing FJ, Bachmann H, Teusink B, Kok J, Kuipers OP. 2014. Bet-hedging during bacterial diauxic shift. Proc. Natl Acad. Sci. USA 111, 7427–7432. ( 10.1073/pnas.1320063111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kashiwagi A, Urabe I, Kaneko K, Yomo T. 2006. Adaptive response of a gene network to environmental changes by fitness-induced attractor selection. PLoS ONE 1, e49 ( 10.1371/journal.pone.0000049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldbeter A, Koshland DE. 1981. An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl Acad. Sci. USA 78, 6840–6844. ( 10.1073/pnas.78.11.6840) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ciliberto A, Capuani F, Tyson JJ. 2007. Modeling networks of coupled enzymatic reactions using the total quasi-steady state approximation. PLoS Comput. Biol. 3, e45 ( 10.1371/journal.pcbi.0030045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ozbudak E, Thattai M, Lim H, Shraiman B, van Oudenaarden A. 2004. Multistability in the lactose utilization network of Escherichia coli. Nature 427, 737–740. ( 10.1038/nature02298) [DOI] [PubMed] [Google Scholar]
- 20.Cosentino C, Salerno L, Passanti A, Merola A, Bates DG, Amato F. 2012. Structural bistability of the GAL regulatory network and characterization of its domains of attraction. J. Comput. Biol. 19, 148–162. ( 10.1089/cmb.2011.0251) [DOI] [PubMed] [Google Scholar]
- 21.Oyarzún DA, Chaves M, Hoff-Hoffmeyer-Zlotnik M. 2012. Multistability and oscillations in genetic control of metabolism. J. Theoret. Biol. 295, 139–153. ( 10.1016/j.jtbi.2011.11.017) [DOI] [PubMed] [Google Scholar]
- 22.Casey R, de Jong H, Gouzé J-L. 2006. Piecewise-linear models of genetic regulatory networks: equilibria and their stability. J. Math. Biol. 52, 27–56. ( 10.1007/s00285-005-0338-2) [DOI] [PubMed] [Google Scholar]
- 23.Dayarian A, Chaves M, Sontag ED, Sengupta AM. 2009. Shape, size, and robustness: feasible regions in the parameter space of biochemical networks. PLoS Comput. Biol. 5, e1000256 ( 10.1371/journal.pcbi.1000256) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brockman IM, Prather KLJ. 2015. Dynamic metabolic engineering: new strategies for developing responsive cell factories. Biotechnol. J. 10, 1360–1369. ( 10.1002/biot.201400422) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu D, Evans T, Zhang F. 2015. Applications and advances of metabolite biosensors for metabolic engineering. Metab. Eng. 31, 35–43. ( 10.1016/j.ymben.2015.06.008) [DOI] [PubMed] [Google Scholar]
- 26.Alper H, Fischer C, Nevoigt E, Stephanopoulos G. 2005. Tuning genetic control through promoter engineering. Proc. Natl Acad. Sci. USA 102, 12 678–12 683. ( 10.1073/pnas.0504604102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salis HM, Mirsky EA, Voigt CA. 2009. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 27, 946–950. ( 10.1038/nbt.1568) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaufman M, Soulé C, Thomas R. 2007. A new necessary condition on interaction graphs for multistationarity. J. Theoret. Biol. 248, 675–685. ( 10.1016/j.jtbi.2007.06.016) [DOI] [PubMed] [Google Scholar]
- 29.de Jong H, Gouzé J-L, Hernandez C, Page M, Sari T, Geiselmann J. 2004. Qualitative simulation of genetic regulatory networks using piecewise-linear models. Bull. Math. Biol. 2, 301–340. ( 10.1016/j.bulm.2003.08.010) [DOI] [PubMed] [Google Scholar]
- 30.Ropers D, Baldazzi V, de Jong H. 2011. Model reduction using piecewise-linear approximations preserves dynamic properties of the carbon starvation response in Escherichia coli. IEEE/ACM Trans. Comput. Biol. Bioinform. 8, 166–181. ( 10.1109/TCBB.2009.49) [DOI] [PubMed] [Google Scholar]
- 31.Alon U. 2006. An introduction to systems biology: design principles of biological circuits. London, UK: Chapman & Hall. [Google Scholar]
- 32.Sriram K, Soliman S, Fages F. 2009. Dynamics of the interlocked positive feedback loops explaining the robust epigenetic switching in Candida albicans. J. Theoret. Biol. 258, 71–88. ( 10.1016/j.jtbi.2009.01.008) [DOI] [PubMed] [Google Scholar]
- 33.Domingo-Sananes MR, Novak B. 2010. Different effects of redundant feedback loops on a bistable switch. Chaos 20, 045120 ( 10.1063/1.3526967) [DOI] [PubMed] [Google Scholar]
- 34.Tiwari A, Igoshin OA. 2012. Coupling between feedback loops in autoregulatory networks affects bistability range, open-loop gain and switching times. Phys. Biol. 9, 055003 ( 10.1088/1478-3975/9/5/055003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yagil G, Yagil E. 1971. On the relation between effector concentration and the rate of induced enzyme synthesis. Biophys. J. 11, 11–27. ( 10.1016/S0006-3495(71)86192-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raj A, van Oudenaarden A. 2009. Single-molecule approaches to stochastic gene expression. Annu. Rev. Biophys. 38, 255–270. ( 10.1146/annurev.biophys.37.032807.125928) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labhsetwar P, Cole JA, Roberts E, Price ND, Luthey-Schulten ZA. 2013. Heterogeneity in protein expression induces metabolic variability in a modeled Escherichia coli population. Proc. Natl Acad. Sci. USA 110, 14 006–14 011. ( 10.1073/pnas.1222569110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oyarzún DA, Lugagne J-B, Stan G-B. 2015. Noise propagation in synthetic gene circuits for metabolic control. ACS Synth. Biol. 4, 116–125. ( 10.1021/sb400126a) [DOI] [PubMed] [Google Scholar]
- 39.Shahrezaei V, Swain PS. 2008. Analytical distributions for stochastic gene expression. Proc. Natl Acad. Sci. USA 105, 17 256–17 261. ( 10.1073/pnas.0803850105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taniguchi Y, Choi PJ, Li G-W, Chen H, Babu M, Hearn J, Emili A, Xie XS. 2010. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329, 533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kremling A, Geiselmann J, Ropers D, de Jong H. 2015. Understanding carbon catabolite repression in Escherichia coli using quantitative models. Trends Microbiol. 23, 99–109. ( 10.1016/j.tim.2014.11.002) [DOI] [PubMed] [Google Scholar]
- 42.Kotte O, Volkmer B, Radzikowski JL, Heinemann M. 2014. Phenotypic bistability in Escherichia coli’s central carbon metabolism. Mol. Syst. Biol. 10, 736 ( 10.15252/msb.20135022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Heerden JH et al. . 2014. Lost in transition: start-up of glycolysis yields subpopulations of nongrowing cells. Science 343, 1245114 ( 10.1126/science.1245114) [DOI] [PubMed] [Google Scholar]
- 44.Ford TJ, Silver PA. 2015. Synthetic biology expands chemical control of microorganisms. Curr. Opin. Chem. Biol. 28, 20–28. ( 10.1016/j.cbpa.2015.05.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jagmann N, Philipp B. 2014. Reprint of design of synthetic microbial communities for biotechnological production processes. J. Biotechnol. 192, 293–301. ( 10.1016/j.jbiotec.2014.11.005) [DOI] [PubMed] [Google Scholar]
- 46.Shou W, Ram S, Vilar JMG. 2007. Synthetic cooperation in engineered yeast populations. Proc. Natl Acad. Sci. USA 104, 1877–1882. ( 10.1073/pnas.0610575104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Youk H, Lim WA. 2014. Secreting and sensing the same molecule allows cells to achieve versatile social behaviors. Science 343, 1242782 ( 10.1126/science.1242782) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conradi C, Flockerzi D. 2012. Switching in mass action networks based on linear inequalities. SIAM J. Appl. Dyn. Syst. 11, 110–134. ( 10.1137/10081722X) [DOI] [Google Scholar]
- 49.Siegal-Gaskins D, Mejia-Guerra MK, Smith GD, Grotewold E. 2011. Emergence of switch-like behavior in a large family of simple biochemical networks. PLoS Comput. Biol. 7, e1002039 ( 10.1371/journal.pcbi.1002039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feliu E, Wiuf C. 2013. A computational method to preclude multistationarity in networks of interacting species. Bioinformatics 29, 2327–2334. ( 10.1093/bioinformatics/btt400) [DOI] [PubMed] [Google Scholar]
- 51.Otero-Muras I, Banga JR, Alonso AA. 2012. Characterizing multistationarity regimes in biochemical reaction networks. PLoS ONE 7, e39194 ( 10.1371/journal.pone.0039194) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ang J, Harris E, Hussey BJ, Kil R, McMillen DR. 2013. Tuning response curves for synthetic biology. ACS Synth. Biol. 2, 547–567. ( 10.1021/sb4000564) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arpino JAJ, Hancock EJ, Anderson J, Barahona M, Stan G-BV, Papachristodoulou A, Polizzi K. 2013. Tuning the dials of synthetic biology. Microbiology 159(Pt 7), 1236–1253. ( 10.1099/mic.0.067975-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brewster RC, Jones DL, Phillips R. 2012. Tuning promoter strength through RNA polymerase binding site design in Escherichia coli. PLoS Comput. Biol. 8, e1002811 ( 10.1371/journal.pcbi.1002811) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Egbert RG, Klavins E. 2012. Fine-tuning gene networks using simple sequence repeats. Proc. Natl Acad. Sci. USA 109, 16 817–16 822. ( 10.1073/pnas.1205693109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McGinness KE, Baker TA, Sauer RT. 2006. Engineering controllable protein degradation. Mol. Cell 22, 701–707. ( 10.1016/j.molcel.2006.04.027) [DOI] [PubMed] [Google Scholar]
- 57.Cardinale S, Arkin AP. 2012. Contextualizing context for synthetic biology—identifying causes of failure of synthetic biological systems. Biotechnol. J. 7, 856–866. ( 10.1002/biot.201200085) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weiße AY, Oyarzún DA, Danos V, Swain PS. 2015. Mechanistic links between cellular trade-offs, gene expression, growth. Proc. Natl Acad. Sci. USA 112, E1038–E1047. ( 10.1073/pnas.1416533112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chubukov V, Gerosa L, Kochanowski K. 2014. Coordination of microbial metabolism. Nat. Rev. Microbiol. 12U, 327–340. ( 10.1038/nrmicro3238) [DOI] [PubMed] [Google Scholar]
- 60.Kotte O, Zaugg JB, Heinemann M. 2010. Bacterial adaptation through distributed sensing of metabolic fluxes. Mol. Syst. Biol. 6, 355 ( 10.1038/msb.2010.10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu P, Wang W, Li L, Bhan N, Zhang F, Koffas MAG. 2014. Design and kinetic analysis of a hybrid promoter–regulator system for malonyl-CoA sensing in Escherichia coli. ACS Chem. Biol. 9, 451–458. ( 10.1021/cb400623m) [DOI] [PubMed] [Google Scholar]
- 62.Kuntz J, Oyarzún D, Stan G-B. 2014. Model reduction of genetic–metabolic networks via time scale separation. In A systems theoretic approach to systems and synthetic biology I: models and system characterizations (eds VV Kulkarni, G-B Stan, K Raman), pp. 181–210. Berlin, Germany: Springer. [Google Scholar]
- 63.Dhooge A, Govaerts W, Kuznetsov YA. 2003. MATCONT: A MATLAB package for numerical bifurcation analysis of ODEs. ACM Trans. Math. Softw. 29, 141–164. ( 10.1145/779359.779362) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.