

Abstract
The liver plays an essential role in glucose and lipid metabolism, synthesis of plasma proteins, and detoxification of xenobiotics and other toxins. Chronic disease of this important organ is one of the leading causes of death in the United States. Following loss of tissue, liver mass can be restored by two mechanisms. Under normal conditions, or after massive loss of parenchyma by surgical resection, liver mass is maintained by division of hepatocytes. After chronic injury, or when proliferation of hepatocytes is impaired, facultative adult hepatic progenitor cells (HPCs) proliferate and differentiate into hepatocytes and cholangiocytes (biliary epithelial cells). HPCs are attractive candidates for cell transplantation because of their potential contribution to liver regeneration. However, until recently, the lack of highly specific markers has hampered efforts to better understand the origin and physiology of HPCs. Recent advances in cell isolation methods and genetic lineage tracing have enabled investigators to explore multiple aspects of HPC biology. In this review, we describe the potential origins of HPCs, the markers used to detect them, the contribution of HPCs to recovery, and the signaling pathways that regulate their biology. We end with an examination of the therapeutic potential of HPCs and their derivatives.
1. INTRODUCTION
The mammalian liver has a remarkable ability to regenerate its functional mass in response to tissue loss. In surgical models of liver resection, such as 70% partial hepatectomy (PH) in rat, the remaining uninjured hepatocytes proliferate and replace the parenchyma within 20 days (Martins, Theruvath, & Neuhaus, 2008). Under circumstances in which hepatocyte proliferation is blocked, as is the case after toxic liver injury, small cells that have scant cytoplasm and oval-shaped nuclei proliferate in the portal area and are thought to contribute to replacement of the parenchyma (Libbrecht & Roskams, 2002; Yovchev et al., 2008). These hepatic progenitor cells (HPCs), also called “oval cells” due to their morphology (Farber, 1956; Yovchev et al., 2008), can undergo bidirectional differentiation into hepatocytes and cholangiocytes (biliary epithelial cells) at least in experimental conditions (Fig. 10.1; Okabe et al., 2009; Shin et al., 2011). Hepatic progenitors are very different from tissue-resident stem cells in other epithelial tissue such as intestine and skin. In the latter tissues, stem and progenitor cells are required throughout life to replenish cells lost daily, and without continued replication of the tissue-resident stem cells, the epithelia of intestine and skin fail rapidly. In contrast, hepatic progenitors are “facultative,” meaning (1) they are not needed to replenish liver tissue under normal, healthy conditions and (2) that many markers of HPCs are only expressed in the liver after injury and when the progenitor cells are activated.
Figure 10.1.

Adult hepatic progenitor cells. A subset of cholangiocytes is activated upon injury and gives rise to HPCs that can differentiate into hepatocytes and cholangiocytes.
The terms “HPCs,” “oval cells,” “liver progenitor cells,” and “hepatic stem cells” describe the heterogeneous population of cells that have been proposed to maintain and regenerate liver during the repair process. To avoid confusion, in this review we will use the term HPCs to denote the epithelial component located within ductular reactions in the injured adult liver (Roskams et al., 2004). The “ductular reaction” is defined as the proliferation of apparent ductules that accompanies leukocyte infiltration and deposition of extracellular matrix (ECM) in response to liver injury (Roskams & Desmet, 1998; Roskams et al., 2004). The markers, origin, fate, and regenerative capability of these HPCs remain the subject of controversy in the field. In this review, we attempt to provide an overview of recent advances and to point out unanswered questions.
2. CONDITIONS THAT ACTIVATE HPC PROLIFERATION
The mechanism of ductular reaction initiation and proliferation of HPCs are poorly characterized. In humans, ductular reactions have been observed in multiple diseases such as fulminant hepatic failure, focal nodular hyperplasia, primary biliary cirrhosis, primary sclerosing cholangitis (Turanyi et al., 2010), cancer (Farber, 1956; Libbrecht & Roskams, 2002), pediatric nonalcoholic fatty liver disease (Nobili et al., 2012), genetic hemochromatosis, alcoholic liver disease, and chronic hepatitis C (Lowes, Brennan, Yeoh, & Olynyk, 1999). The number of HPCs correlates with the severity of liver diseases in human and dog (Lowes et al., 1999; Schotanus et al., 2009). In humans, a minimum of 50% hepatocyte loss is required for significant activation of the HPC compartment (Katoonizadeh, Nevens, Verslype, Pirenne, & Roskams, 2006), and there is an inverse correlation between the number of HPCs and the number of hepatocytes that express the proliferation marker Ki67 (Katoonizadeh et al., 2006). This suggests that a combination of hepatocyte loss and impaired hepatocyte proliferation is required to activate HPC proliferation. In rats, treatment with carbon tetrachloride (CCl4) preferentially induced proliferation of hepatocytes, and proliferation of nonparenchymal cells required simultaneous administration of D-galactosamine, which blocks RNA and protein synthesis in hepatocytes (Dabeva, Alpini, Hurston, & Shafritz, 1993; Dabeva & Shafritz, 1993). Furthermore, in rats that had undergone PH, the expansion of HPCs was negligible (Dusabineza et al., 2012). Significant HPC induction was only observed when 2-acetaminofluorene (2-AAF) was added to block cell cycle progression in hepatocytes (Dusabineza et al., 2012; Trautwein et al., 1999). An inverse correlation between the activation of HPCs and hepatocyte proliferation was also observed in the canine liver (Schotanus et al., 2009), providing strong evidence that suppression of hepatocyte proliferation is required for activation of the HPC compartment. In mice, however, inhibition of the hepatocyte cell cycle might not be a definitive criterion for activation of the ductular reaction. Various treatments such as CCl4 (Chu et al., 2011; Espanol-Suner et al., 2012; Malato et al., 2011; Rountree, Barsky, et al., 2007), choline-deficient, ethionine-supplemented diet (CDE) (Espanol-Suner et al., 2012; Knight & Yeoh, 2005; Tirnitz-Parker, Tonkin, Knight, Olynyk, & Yeoh, 2007), methionine choline-deficient diet supplemented with ethionine (MCDE) (Huch et al., 2013), bile duct ligation (BDL) (Chu et al., 2011; Malato et al., 2011; Sackett, Li, et al., 2009), PH (Malato et al., 2011), alpha-naphthylisothiocyanate (ANIT) (Rountree, Barsky, et al., 2007), high-fat diet combined with ethanol (Jung et al., 2008), and 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) (Carpentier et al., 2011; Malato et al., 2011; Rountree, Barsky, et al., 2007; Sackett, Li, et al., 2009; Shin et al., 2011) have been successfully used to induce proliferation of HPCs in mice in the absence of impaired hepatocyte proliferation. Rountree, Barsky, et al. (2007) tested multiple means of inducing HPC reaction in mouse models. CCl4 preferentially induced proliferation of hepatocytes, ANIT preferentially induced proliferation of cholangiocytes, and DDC-induced cycling of both hepatocytes and cholangiocytes, and the models also induced proliferation of HPCs (Rountree, Barsky, et al., 2007). Moreover, another group reported that administration of CCl4 or PH led to formation of new hepatocytes from HPCs (Malato et al., 2011). These results suggest that inhibition of hepatocyte proliferation is not necessarily a prerequisite for the activation of HPCs under all physiological conditions.
Studies using genetic mouse models have provided further insight. Endo and colleagues have demonstrated that deletion of damaged DNA-binding protein 1 (Ddb1), an adaptor for Cullin4 ubiquitin ligase (Higa & Zhang, 2007), in hepatocytes led to the activation of HPC proliferation (Endo, Zhang, Yamaji, & Cang, 2012). Ablation of Ddb1 caused only minor liver damage compared to the degree of injury induced by administration of DDC. Deletion of Ddb1 also led to a blockade of hepatocyte proliferation, which was partially due to accumulation of p21. It is also worth noting that thermoreversible gelation polymer, a synthetic material that polymerizes when exposed to temperatures above a defined threshold, induced ductular reactions when injected directly into normal rat liver (Nagaya, Kubota, Suzuki, Akashi, & Mitaka, 2006). This suggests that ECM can provide cues for activation of the HPC compartment. In Section 5, we will delve more deeply into the signaling pathways and molecular mechanism that regulate the biology of HPCs.
3. ORIGINS AND MARKERS FOR HPCs
While there is little doubt about the presence of facultative HPCs in the injured liver, the existence and identity of HPC precursors in a healthy, uninjured liver remains controversial. For example, a study using incorporation of 5-bromo-2′-deoxyuridine identified multiple niches for label-retaining cells such as the proximal biliary branches, including the canal of Hering and intralobular bile ducts (Kuwahara et al., 2008). This review summarizes the ongoing debate about the possible origins of HPCs which include: (1) biliary epithelial cells, (2) progenitor cells persisting from the fetal liver, (3) bone marrow cells, (4) hepatic stellate cells, and (5) mature adult hepatocytes.
3.1. Biliary origin of facultative HPCs
Multiple lines of evidence suggest that cholangiocytes (biliary epithelial cells) serve as facultative stem cells in the liver. Thus, these differentiated cells may acquire stem cell-like characteristics under particular conditions (Yanger & Stanger, 2011). Recent work has demonstrated the utility of protein markers in yielding insights into HSC physiology. Table 10.1 summarizes markers that are commonly used to isolate or detect HPCs in different injury models or disease contexts. Unfortunately, many of the widely used markers such as epithelial cell adhesion molecule (EPCAM) (Inada et al., 2008; Jozefczuk et al., 2010; Kamiya, Kakinuma, Yamazaki, & Nakauchi, 2009; Okabe et al., 2009; Schmelzer et al., 2007; Yovchev et al., 2008), CD133 (encoded by the Prom1 gene) (Dorrell et al., 2011; Jozefczuk et al., 2010; Kamiya et al., 2009; Rountree, Barsky, et al., 2007; Suzuki et al., 2008), SRY (sex-determining region Y)-box 9 (Sox9) (Carpentier et al., 2011; Dorrell et al., 2011), and osteopontin (OPN) (Espanol-Suner et al., 2012) are found both in HPCs of the injured liver and in cholangiocytes and/or non-parenchymal cells of the healthy liver (Dorrell et al., 2008, 2011; Espanol-Suner et al., 2012; Furuyama et al., 2011; Inada et al., 2008; Rountree, Barsky, et al., 2007). Furthermore, facultative HPCs were reported to express the ductal markers cytokeratin 19 (CK19) (Espanol-Suner et al., 2012; Furuyama et al., 2011; Grozdanov, Yovchev, & Dabeva, 2006; Inada et al., 2008; Kamiya et al., 2009; Takase et al., 2013; Theise et al., 1999; Yovchev et al., 2008) and MIC1-1C3 (Dorrell et al., 2011).
Table 10.1.
Markers for HPCs
| Marker | Diseases (models) | Species | References |
|---|---|---|---|
| A6 | DDC, CDE, viral hepatitis, deletion of Ddb1 | Mouse | Endo et al. (2012), Fan et al. (2012), and Tonkin, Knight, Curtis, Abraham, and Yeoh (2008) |
| AFP | Acute necrotizing hepatitis, HCV, primary biliary cirrhosis | Human | Spee et al. (2010) |
| 2-AAF/PH | Rat | Jensen et al. (2004) and Yovchev et al. (2008) | |
| CD44 | Acute necrotizing hepatitis, HCV, primary biliary cirrhosis | Human | Spee et al. (2010) |
| 2-AAF/PH | Rat | Yovchev et al. (2008) | |
| DDC, deletion of Ddb1 | Mouse | Endo et al. (2012) and Shin et al. (2011) | |
| CD133 | Alcoholic hepatitis, acute necrotizing hepatitis, HCV, HBV, primary biliary cirrhosis | Human | Porretti et al. (2010), Sancho-Bru et al. (2012), and Spee et al. (2010) |
| 2-AAF/PH | Rat | Yovchev et al. (2008) | |
| DDC, ANIT, CCl4 | Mouse | Dorrell et al. (2011), Rountree, Barsky, et al. (2007), and Suzuki et al. (2008) | |
| CK7 | Pediatric nonalcoholic fatty liver disease, alcoholic hepatitis, HCV, primary biliary cirrhosis | Human | Nobili et al. (2012), Sancho-Bru et al. (2012), and Spee et al. (2010) |
| 2-AAF/PH | Rat | Yovchev et al. (2008) | |
| DDC | Mouse | Suzuki et al. (2008) | |
| CK19 | Acetaminophen-induced necrosis, genetic hemochromatosis, alcoholic liver disease, HCV, HBV, acute necrotizing hepatitis, primary biliary cirrhosis | Human | Feng et al. (2012), Lowes et al. (1999), Spee et al. (2010), and Theise et al. (1999) |
| 2-AAF/PH | Rat | Dusabineza et al. (2012) and Yovchev et al. (2008) | |
| DDC, CDE, deletion of Ddb1 | Mouse | Carpentier et al. (2011), Endo et al. (2012), Fan et al. (2012), and Takase et al. (2013) | |
| CLDN4 | 2-AAF/PH | Rat | Yovchev et al. (2008) |
| Deletion of Ddb1 | Mouse | Endo et al. (2012) | |
| DLK1 | Retrorsine, 2-AAF/PH | Rat | Jensen et al. (2004) |
| EPCAM | Alcoholic hepatitis, HCV, HBV | Human | Porretti et al. (2010) and Sancho-Bru et al. (2012) |
| 2-AAF/PH | Rat | Yovchev et al. (2008) | |
| DDC, deletion of Ddb1 | Mouse | Endo et al. (2012), Fan et al. (2012), Okabe et al. (2009), Shin et al. (2011), and Takase et al. (2013) | |
| Foxl1 | BDL, DDC | Mouse | Sackett, Li, et al., (2009) and Shin et al. (2011) |
| GGT | 2-AAF/PH, GalN | Rat | Dabeva and Shafritz (1993) and Yovchev et al. (2008) |
| ITGB4 | AAF-PH | Rat | Yovchev et al. (2008) |
| Deletion of Ddb1 | Mouse | Endo et al. (2012) | |
| ITGA6 | HCV, HBV | Human | Porretti et al. (2010) |
| 2-AAF/PH | Rat | Yovchev et al. (2008) | |
| Lgr5 | CCl4, DDC, MCDE | Mouse | Huch et al. (2013) |
| M2PK | Genetic hemochromatosis, alcoholic liver disease, HCV | Human | Lowes et al. (1999) |
| CDE | Mouse | Davies, Knight, Tian, Yeoh, and Olynyk (2006) | |
| NCAM | HCV, HBV, acute necrotizing hepatitis, primary biliary cirrhosis | Human | Porretti et al. (2010) and Spee et al. (2010) |
| OPN | DDC, CDE, CCl4, PH | Mouse | Carpentier et al. (2011) and Espanol-Suner et al. (2012) |
| Sox9 | DDC, CDE | Mouse | Carpentier et al. (2011) |
| Trop2 | DDC, CDE | Mouse | Carpentier et al. (2011), Okabe et al. (2009), and Shin et al. (2011) |
2-AAF, 2-acetaminofluorene; ANIT, alpha-naphthylisothiocyanate; CCl4, carbon tetrachloride; CDE, choline-deficient, ethionine-supplemented diet; Ddb1, damaged DNA-binding protein 1; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; GalN, D-galactosamine; HBV, Hepatitis B virus; HCV, Hepatitis C virus; MCDE, methionine choline-deficient diet supplemented with ethionine; PH, partial hepatectomy.
The application of the above markers has been limited by their lack of cell-type specificity. Forkhead box L1 (Foxl1) (Shin et al., 2011), Trop2 (encoded by tumor-associated calcium signal transducer 2) (Okabe et al., 2009), and Lgr5 (leucine-rich-repeat-containing G-protein-coupled receptor 5) (Huch et al., 2013) are three newly described markers expressed exclusively by HPCs of the injured liver. Furthermore, Foxl1+ cells and Trop2+ cells express EPCAM and CK19 (Okabe et al., 2009; Sackett, Li, et al., 2009; Shin et al., 2011), and organoids expressing Lgr5 could be grown from biliary duct fragments in culture (Huch et al., 2013), further supporting the concept that at least some types of HPCs can originate from cholangiocytes. Furthermore, gene expression analyses comparing Foxl1+ HPCs, cholangiocytes, and hepatocytes demonstrate that the gene expression profile of Foxl1+ HPCs resembles that of cholangiocytes rather than hepatocytes, supporting the idea that HPCs are derived from a cholangiocytic lineage (Shin et al., 2011).
Moreover, a number of reports have demonstrated that the proliferating HPC compartment is connected to preexisting ductules, an observation that strongly supports a ductal origin for HPCs (Alpini et al., 1992; Dusabineza et al., 2012; Theise et al., 1999). Interestingly, although CD133+ cells are found in both normal and injured liver, only CD133+ cells isolated from injured liver were self-renewable (Suzuki et al., 2008). Kamiya et al. (2009) described the existence of “cholangiocytic precursor cells” that were present in the healthy liver. These cells expressed CD13 (also known as alanyl (membrane) aminopeptidase), integrin alpha 6 (ITGA6 or CD49f), CD133, and CK19 in vivo, and when these cells were cultured, they were self-renewable and began to express markers of HPCs (CD44) and hepatocytes (albumin). These results further support the idea that at least a subset of cholangiocytes can act as precursors of facultative HPCs. It has been difficult to ascertain precisely how activated cholangiocytes and HPCs differ from each other, as the two compartments share protein markers (Fig. 10.1; Kamiya et al., 2009). It is also worth mentioning that cholangiocytes comprise a heterogeneous population of cells (Roskams et al., 2004), and it would therefore be interesting to investigate whether cholangiocytic precursor cells are different from other cholangiocytes in terms of function, gene expression profile, or physiology.
In humans, Schmelzer, Wauthier, and Reid (2006) identified cells found in the normal liver from fetal and postnatal donors that are capable of being expanded in culture. These cells, termed human hepatic stem cells (hHpSCs), expressed EPCAM, CK19, neural cell adhesion molecule 1 (NCAM), claudin 3 (CLDN3), Kit, and aquaporin 4. The same group reported that EPCAM-expressing cells exist in the Canal of Hering in the livers of postnatal donors, and that these cells can repopulate the liver of immunodeficient mice (Schmelzer et al., 2007). Furthermore, the existence of an even more primitive stem population in the biliary tree has been reported. These stem cells are a heterogeneous population that give rise to hepatocytes, cholangiocytes, and pancreatic islets. At least a subset of these multipotent stem cells were positive for pancreatic and duodenal homeobox 1 (PDX1), SRY-box 17 (SOX17), and EPCAM (Carpino et al., 2012). At present, the relationship between the fetally derived hHpSCs and the facultative hepatic progenitors that appear in the adult liver following injury described earlier remains unresolved.
3.2. Embryonic origin of HPCs
Hepatoblasts are bipotential cells found in the fetal liver (between E9.5 and E14.5 in mice) that have the ability to differentiate into hepatocytes and cholangiocytes in vivo. Because both hepatoblasts and HPCs are proliferating and are capable of differentiating to hepatocytes and cholangiocytes, it has been suggested that fetal hepatoblasts give rise to HPCs (Schmelzer et al., 2007). The fact that hepatoblasts and HPCs share the same markers, such as delta-like 1 homolog (Jensen et al., 2004; Qiu, Hernandez, Dean, Rao, & Darlington, 2011; Yovchev et al., 2008) and alpha-fetoprotein (AFP) (Jensen et al., 2004; Yovchev et al., 2008), is cited as evidence that HPCs are derived from hepatoblasts. Fetal cells also share other characteristics of HPCs, such as clonogenicity and the ability to repopulate liver in vivo. For instance, the CD13+DLK+ fraction of cells from murine fetal liver has the ability to form colonies in culture (Kakinuma et al., 2009), and the AFP+CK19+ fraction from rat fetal liver has the ability to repopulate liver in vivo (Dabeva et al., 2000). As previously mentioned, the existence of hHpSCs that are different from hepatoblasts has also been reported in human fetal liver. These hHpSCs were observed in the ductal plate of the fetal liver and in the Canal of Hering of adult liver (Schmelzer et al., 2007). In mice, Carpentier et al. (2011) reported that the ductal plate of the fetal liver serves as the precursor for cholangiocytes, periportal hepatocytes, and HPCs in adult liver. However, one cannot rule out the possibility that the ductal plate gives rise to the fetal/neonatal adult cholangiocytes, which in turn give rise to facultative HPCs when the need arises. The main issue with this model, however, is that it requires that fetal hepatoblasts persist in the adult liver. Most studies have not observed either a CD13+DLK+ or a AFP+CK19+ fraction of cells in the healthy, uninjured liver, which argues against the continued presence of fetal hepatoblasts in the adult organ. Furthermore, the expression of AFP in the adult liver is not sufficient to prove descendancy from a persistent fetal hepatoblast, it could simply the case that the Afp promoter (or that of other fetally expressed genes) is reactivated in adult facultative HPCs. An analogous line of reasoning holds for hepatocellular carcinoma: merely because some tumors share characteristics with fetal hepatoblasts (such as rapid cycling and AFP expression) does not prove that the tumors originated from fetal hepatoblasts residing in the adult organ.
3.3. Bone marrow as the source of HPCs
In 1999, Petersen et al. (1999) demonstrated that when they transplanted bone marrow from male rats into female rats, 0.14% of hepatocytes carried the Y chromosome. They also reported that bone marrow-derived cells differentiated into hepatocytes, although the extent of their contribution to the parenchyma was less than 1% (Piscaglia, Shupe, Oh, Gasbarrini, & Petersen, 2007). The hypothesis that HPCs can originate from bone marrow is further supported by the fact that HPCs and hematopoietic stem cells express the same markers, such as Thy1 (Petersen, Goff, Greenberger, & Michalopoulos, 1998), CD34 (Omori et al., 1997), and Kit (Fujio, Evarts, Hu, Marsden, & Thorgeirsson, 1994). Subsequently, two independent groups transplanted bone marrow cells and detected donor-derived hepatic nodules in the livers of fumarylacetoacetate hydrolase (FAH)-deficient recipient mice (Vassilopoulos, Wang, & Russell, 2003; Wang et al., 2003). However, hepatocytes positive for FAH derived from transplanted bone marrow cells were shown subsequently to be the result of cell fusion rather than actual differentiation of bone marrow cells into HPCs or hepatocytes (Wang et al., 2003).
To address this discrepancy, two independent research groups transplanted GFP-expressing bone marrow cells or lacZ-transgenic bone marrow cells, respectively, and demonstrated that bone marrow cells played a negligible role in liver regeneration when recipient mice were subjected to CCl4, ANIT, DDC, or CDE diet-mediated liver injury (Rountree, Wang, et al., 2007; Tonkin et al., 2008). It has also been demonstrated that bone marrow cells do not contribute to HPCs and hepatocytes in rats (Menthena et al., 2004). Finally, recent studies demonstrated that EPCAM-positive HPCs do not express Thy1, Kit, and CD34 (Okabe et al., 2009; Yovchev et al., 2008). Similarly, FACS-sorted CD34+ and Thy1+ cells do not express genes associated with HPCs, hepatocytes, and cholangiocytes (Rountree, Barsky, et al., 2007). In sum, these results strongly suggest that the bone marrow is not a major source of HPCs.
3.4. Hepatic stellate cells
A recent report proposed hepatic stellate cells as a type of HPCs. This study was based on the use of glial fibrillary acidic protein (GFAP), a marker of stellate cells, for genetic lineage tracing with the Cre-loxP technology (Fig. 10.2). Gfap-Cre-labeled cells contributed to the HPC compartment when mice were fed an MCDE diet (Yang et al., 2008). However, the Gfap-Cre driver employed is not only active in hepatic stellate cells, but also in a subset of cholangiocytes (Ueberham, Bottger, Ueberham, Grosche, & Gebhardt, 2010). Thus, the authors could not exclude the possibility that the facultative HPCs observed in their mice arose from cholangiocytes. In fact, a subsequent study by Scholten et al. (2010) provided strong evidence that stellate cells do not contribute to the HPC compartment. Given that stellate cells are derived from a different embryonic germ layer than are hepatocytes and cholantiocytes (from the mesoderm and endoderm, respectively), the transdifferentiation of stellate cells into HPCs might require too large a shift in the gene expression program to occur naturally.
Figure 10.2.
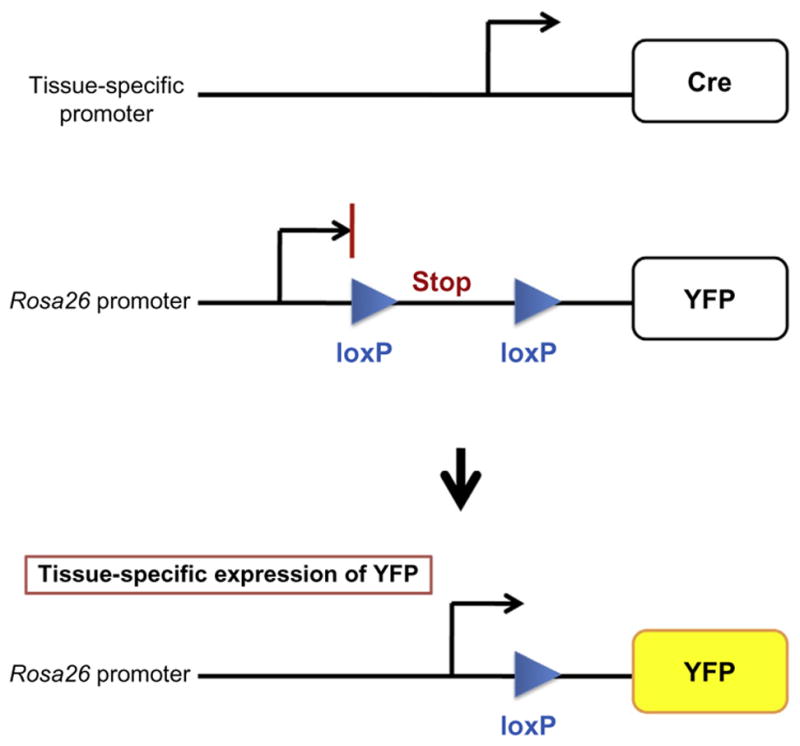
Lineage tracing using the Cre-LoxP approach. Rosa26 promoter is ubiquitously active in mice. However, transcription of YFP reporter is blocked because of the transcriptional stop sequence (Srinivas et al., 2001). The Cre recombinase excises the sequence located between two loxP sites thus allowing for the expression of YFP in a specific cell type.
3.5. Adult hepatocytes as facultative HPCs
Recently, Yanger and Stanger (2011) proposed that hepatocytes can undergo reprogramming into biliary lineage. They injected AAV8-TBG-Cre into RosaYFP mice to lineage trace fully differentiated hepatocytes in the adult liver. Upon DDC treatment, cells that expressed both biliary maker A6 and hepatocyte marker HNF4α emerged. Forty-one percent of these A6+ HNF4α+ cells were binucleated, and staining for YFP further confirmed that these cells originated from hepatocytes. They identified a substantial number of YFP+ cells that acquired a biliary morphology and expressed OPN, A6, Sox9, or CK19. They also identified binucleated HPCs in the livers of DDC-fed mice but not in the livers of control mice. These results suggest that hepatocytes can differentiate into cholangiocytes and/or HPCs in response to liver injury.
3.6. Summary
The contradictory findings regarding the origins and location of HPCs may suggest multiple origins (Kuwahara et al., 2008) or multiple maturational stages. The existence of multiple stages of HPCs has been discussed (reviewed in Duncan, Dorrell, & Grompe, 2009; Turner et al., 2011), but conclusive evidence is lacking. Future work will be required to address the exact lineage relationships between different types of precursor cells reported in the literature over the past 20 years.
4. THE CONTRIBUTION OF HPCs TO LIVER REGENERATION
Expansion of proposed HPCs has been observed in various disease contexts in human as well as in rodents (Table 10.1). However, the contribution of HPCs to restoration of parenchymal architecture and liver function has been assumed, but not proven (Takase et al., 2013). Thus far, it has been demonstrated that HPCs form both typical and atypical ducts with poorly defined lumina (Libbrecht & Roskams, 2002; Yaswen, Hayner, & Fausto, 1984), and it remains unclear whether these ducts are fully functional. While formation of tube-like structure in culture has been used as indication of HPCs’ ability to differentiate into cholangiocytes (Kamiya et al., 2009; Shin et al., 2011), this assay does not provide definitive proof that HPC-derived cholangiocytes are fully integrated into the biliary tree. Conclusive evidence to support the hypothesis that endogenous HPCs form functional ducts in vivo is still lacking.
The contribution of HPCs to hepatocytes was also the subject of debate until recently. Genetic lineage tracing using Foxl1-Cre; RosaLacZ mice demonstrated that the number of β-gal+/HNF4α+ cells increased significantly 2 weeks after BDL, suggesting that Foxl1-Cre marked HPCs can form new hepatocytes (Sackett, Li, et al., 2009). In addition, a hepatocyte fate tracing using an adeno-associated viral vector AAV8-Ttr-Cre in combination with RosaYFP mice was employed in multiple disease models such as PH, DDC, BDL, and CCl4 (Malato et al., 2011). The authors found newly formed hepatocytes derived from progenitor cells, although the contribution was limited. Similarly, another group has reported the existence of rare CK19+ or GSTP+CK19− intermediate hepatocytes in the 2-AAF/PH rat model (Dusabineza et al., 2012). In humans, intermediate hepatocytes that express CK7 or CK19 have been observed as well (Katoonizadeh et al., 2006).
Sackett, Gao, et al. (2009) demonstrated the correlation between impaired ductular reactions and impaired liver regeneration in Foxl1−/− mice after BDL. The expansion of the CK19+ ductular area was reduced 14 days after BDL in the Foxl1−/− livers, while the control livers exhibited a progressive increase in CK19+ ductular area. This effect correlated with elevated serum AST levels, delayed proliferation of hepatocytes, and increased parenchymal necrosis in Foxl1−/− mice compared to the control mice. These results strongly support the idea that HPCs contribute to liver regeneration. Similarly, Takase et al. (2013) demonstrated that forced expression of fibroblast growth factor 7 (FGF7) causes an increased HPC response in conjunction with attenuated liver damage and mortality following DDC-induced injury. FGF7, which is induced in the liver during the HPC response, was suggested to stimulate proliferation of HPCs (Takase et al., 2013). In this model, newly formed A6+CK19− hepatocytes were dramatically increased around A6+CK19+ HPCs in Fg f7-transgenic mice but not in control mice. They demonstrated that Fg f7-deficient mice exhibit decreased HPC expansion as well as higher mortality and hepatic injury upon DDC treatment.
The small number of HPC-derived hepatocytes may be explained by the finding that ECM deposition during HPC activation prevents expansion and migration of progeny to parenchyma (Espanol-Suner et al., 2012). When mice were exposed to a CDE diet for 3 weeks and then to chow diet for 2 weeks, the number of hepatocytes originating from Opn-iCreERT2+ HPCs was significantly increased compared to the mice exposed to the CDE diet alone, indicating that HPCs might play a role during the recovery phase rather than during disease progression. In sum, HPCs contribute to parenchyma of the liver following injury, and although the extent to which they do so may be minor in the experimental models tested, expanded HPCs might still be targeted for the treatment of liver diseases.
5. SIGNALING PATHWAYS AND NICHES THAT REGULATE THE BEHAVIOR OF HPCs
The signaling pathways that promote proliferation and activation of the HPC compartment, the migration of HPCs, and subsequent cell fate specification have been extensively studied. Knowledge of these pathways is essential for targeting HPCs to promote expansion and differentiation in patients with liver disease using small molecules or growth factors.
5.1. The source of signaling molecules that interact with HPCs
The ductular reaction comprises not only the expansion of HPCs, but also that of mesenchymal and inflammatory cells that surround HPCs, as well as the subsequent deposition of ECM (Roskams & Desmet, 1998; Roskams et al., 2004). Macrophages and T-cells interact with HPCs to regulate proliferation, differentiation, and migration of HPCs (Boulter et al., 2012; Lorenzini et al., 2010; Van Hul, Abarca-Quinones, Sempoux, Horsmans, & Leclercq, 2009; Viebahn et al., 2010). Hepatic stellate cells or myofibroblasts have also been observed in close proximity of HPCs both in mouse (Sackett, Li, et al., 2009) and in patients with liver diseases (Schotanus et al., 2009), suggesting that these cells may interact with HPCs. In addition, two reports demonstrated that Thy1+ mesenchymal cells can act as the niche for HPCs (Takase et al., 2013; Yovchev et al., 2008). In addition, the ECM can also be a factor that regulates HPC activity and function (Carpentier et al., 2011; Lozoya et al., 2011; Van Hul et al., 2009).
5.2. Signaling pathways that regulate activation, survival, and proliferation of HPCs
Two growth factors, FGF7 and granulocyte-colony stimulating factor (G-CSF), have been proposed to act as mitogens for HPCs (Piscaglia et al., 2007; Takase et al., 2013). FGF7 promotes the expansion of HPCs in vivo, and Thy1+ cells can act as a niche for activation of HPCs (Takase et al., 2013). G-CSF also has been shown to stimulate proliferation and migration of HPCs in rats (Piscaglia et al., 2007). Additionally, mice deficient of Bmi1, a component of the polycomb repressive complex 1 (Wu & Yang, 2011), exhibited reduced expansion of HPCs in response to DDC injury (Fan et al., 2012). Tirnitz-Parker and colleagues demonstrated that injection of “tumor necrosis factor-like weak inducer of apoptosis” to CDE-fed animals led to an increased number of HPCs and invasion of HPCs into the parenchyma. This effect correlated with activation of NF-κB (Tirnitz-Parker et al., 2010). Small molecule inhibitors of signaling pathways have also proven useful for the investigation of HPC biology. For example, SC-236, an inhibitor of cyclooxygenase 2, attenuated the CDE-induced HPC response (Davies et al., 2006). Treatment of CDE-fed mice with the PPAR gamma agonist ciglitazone resulted in reduction of HPC response as well (Knight, Yeap, Yeoh, & Olynyk, 2005).
The EPCAM+ cells containing HPCs isolated from human fetal livers are enriched for components of hedgehog signaling pathway, specifically Indian hedgehog and patched 1. Cyclopamine, an inhibitor of hedgehog signaling, induced cell death of EPCAM+ cells in culture, implying that hedgehog signaling plays a role in maintaining survival of this cell population (Sicklick et al., 2006). In addition, the Rho-associated protein kinase inhibitor Y-27632 significantly improved clonogenicity of HPCs in culture (Kamiya et al., 2009; Shin et al., 2011). Thus, multiple signaling pathways act in concert to effect the activation and proliferation of HPCs, and understanding their specific contributions will require further study.
Multiple cytokines are involved in the activation of the ductular reaction and proliferation of HPCs. These cytokines include tumor necrosis factor (Knight & Yeoh, 2005; Viebahn et al., 2010), lymphotoxin alpha (Knight & Yeoh, 2005), lymphotoxin beta (Akhurst et al., 2005), interferon gamma (Akhurst et al., 2005), interleukin-22 (Feng et al., 2012), and interleukin 6 (Yeoh et al., 2007). Signal transducer and activator of transcription 3 signaling has been suggested as the central mediator of activation of the HPC response to interleukins and interferon gamma (Akhurst et al., 2005; Feng et al., 2012; Yeoh et al., 2007). These findings suggest that the inflammatory response to liver injury plays a key role in the activation of HPC response.
5.3. Signaling pathways that regulate the specification of HPCs
There is increasing evidence to suggest that the Wnt and Notch signaling pathways are key regulators of fate determination of HPCs in rodents and humans (Boulter et al., 2012; Spee et al., 2010). In humans, acute necrotizing hepatitis causes activation of the Wnt signaling pathway (Spee et al., 2010), and Notch signaling was activated in HPCs in patients with primary biliary cirrhosis. These results imply that the Wnt pathway is involved in directing HPCs toward the hepatocytic fate, while the Notch pathway promotes differentiation of HPCs toward the biliary lineage (Spee et al., 2010). Similarly, in mice, myofibroblasts were identified as a source of Jagged 1, which activates Notch signaling in HPCs and promotes specification of cholangiocytes. In contrast, Wnt3a expressed by macrophages led to specification of HPCs to hepatocytes (Boulter et al., 2012).
Interaction between HPCs and the ECM may also influence the fate of HPCs. In the liver of CDE-fed mice, deposition of ECM occurs prior to the expansion of HPCs (Van Hul et al., 2009). Inhibition of transforming growth factor beta-induced accumulation of ECM resulted in an increased number of HPC-derived hepatocytes, suggesting that the ECM is also involved in fate determination of HPCs (Espanol-Suner et al., 2012).
Interestingly, a recent report demonstrated that HPCs can also express resistin and glucagon-like peptide-1, and their expression levels correlated with the degree of fibrosis and steatosis in pediatric nonalcoholic fatty liver disease, implying existence of signaling from HPCs to the surrounding cells (Nobili et al., 2012). Further investigation of the crosstalk between HPCs and their surrounding niche will be required in order to effectively direct first proliferation and then differentiation of these cells in culture so that they can be tested as a cell therapeutic.
6. CELL THERAPY
The utilization of HPCs for cell therapy is an attractive alternative to organ transplantation for the treatment of liver disease. There are several advantages to using HPCs instead of primary hepatocytes or whole organ transplantation. First, a single HPC can be expanded in culture without losing its bidirectional differentiation potential (Shin et al., 2011; Suzuki et al., 2008). Second, the fact that HPCs are found in the liver with various types of disorders (Lowes et al., 1999; Nobili et al., 2012; Turanyi et al., 2010) suggests that HPCs can be isolated from diseased livers that cannot be used for organ transplantation. Third, it might be possible to isolate HPCs from a patient who has liver disease, expand them in culture, and transplant back to the patient. This autologous transplant protocol would obviate the need for immunosuppression after transplantation. Finally, HPCs are smaller than hepatocytes, and it has been suggested that smaller cells might have less of a tendency to cause portal hypertension following injection into the portal vein (Sandhu, Petkov, Dabeva, & Shafritz, 2001).
Multiple laboratories have demonstrated that transplanted HPCs have the ability to differentiate into hepatocytes in vivo (Dorrell et al., 2011; Endo et al., 2012; Kamiya et al., 2009; Suzuki et al., 2008; Yovchev et al., 2008). Currently, preconditioning of the recipient liver is required for engraftment and expansion of transplanted cells. For example, engraftment of hHpSCs was enhanced when the recipient mice were treated with CCl4 (Schmelzer et al., 2007). The Fah-deficient mice, a model of tyrosinemia (Grompe et al., 1993), are the most widely used recipient mice for transplantation of cells into liver in numerous studies (Dorrell et al., 2011; Huch et al., 2013; Qiu et al., 2011). FAH is a critical enzyme for tyrosine catabolism (Davern, 2001), and disruption of this enzyme results in the accumulation of toxic metabolites and cell-autonomous destruction of host hepatocytes. 2-(2-Nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC) is an inhibitor of 4-hydroxyphenylpyruvate dioxygenase (an enzyme upstream of FAH in the tyrosine catabolic pathway) (Grompe et al., 1993; Holme & Lindstedt, 1998), and it is added to the drinking water of Fah-deficient mice to maintain their liver function before transplantation. Withdrawal of NTBC favors engraftment of transplanted cells because host hepatocytes are quickly lost.
However, efficient engraftment and restoration of full liver function following transplantation of HPCs remains difficult. For reasons that are not well understood, the engraftment efficiency of HPCs is very low compared to that of primary hepatocytes (Dorrell et al., 2011; Huch et al., 2013). When HPCs were injected intrasplenically into Fah-deficient mice, engraftment was observed only in two out of 20 transplanted mice (Dorrell et al., 2011). Another report also demonstrated that injection of clonal HPCs expanded from a single cell to immunodeficient Fah-deficient mice (Fah−/−Rag2−/−Il2rg−/− mice) resulted in less than 0.1% of engraftment of the liver (Huch et al., 2013), suggesting that the low engraftment efficiency is not due to rejection of transplanted cells by the host immune system. The low engraftment efficiency may be improved by optimizing the differentiation protocol in culture (Dorrell et al., 2011; Huch et al., 2013). Current differentiation protocols fail to differentiate HPCs into fully mature hepatocytes in culture (Shin et al., 2011). Another potential problem of cell therapy is that injected cells can be dispersed to the ectopic sites throughout the body. Turner et al. (2013) demonstrated that mixing cells with specific biomaterials can improve targeting of HPCs to the liver.
The route of cell transplantation is another subject that requires investigation. While intrasplenic injection is the most widely used method for injection of hepatocytes, it has been demonstrated that small cells are more likely to become entrapped in the spleen (Cheng et al., 2009), suggesting that intrasplenic injection may not be an optimal method for the delivery of HPCs. In addition, although cell therapy possesses great potential, the injury of the host liver associated with cell transplantation should not be underestimated. Intrasplenic injection of hepatocytes or nonparenchymal epithelial cells caused disruption of gap junctions and ischemic liver injury (Gupta et al., 2000). Whether HPCs can be tumorigenic is also a concern, although it has been shown that HPCs were nontumorigenic when transplanted subcutaneously into nude mice (Tirnitz-Parker et al., 2007). In sum, although HPC-based therapies pose significant advantages compared to organ or primary hepatocytes which cannot be expanded in culture, further optimization of the technology is required before clinical application can become a reality.
7. CONCLUDING REMARKS
Recent progress in molecular biology has enabled investigation of the origin, physiology, and therapeutic potential of HPCs. However, additional studies are required to reconcile conflicting findings from different research groups based on different injury models, species, and use of markers for detecting and isolating HPCs. For example, studies revealed the heterogeneity of HPCs between rat, mouse, and human (Bisgaard et al., 2002; Jelnes et al., 2007). In addition, protocols for the differentiation and transplantation of HPCs must be optimized before this exciting cell population can be translated into clinical application.
Acknowledgments
Work in the Kaestner lab is supported by the 2012 Liver Scholar Award to S. S. from American Association for the Study of Liver Diseases/American Liver Foundation and by the P01-DK049210 grant to K. H. K. from National Institute of Diabetes and Digestive and Kidney Diseases.
References
- Akhurst B, Matthews V, Husk K, Smyth MJ, Abraham LJ, Yeoh GC. Differential lymphotoxin-beta and interferon gamma signaling during mouse liver regeneration induced by chronic and acute injury. Hepatology. 2005;41:327–335. doi: 10.1002/hep.20520. [DOI] [PubMed] [Google Scholar]
- Alpini G, Aragona E, Dabeva M, Salvi R, Shafritz DA, Tavoloni N. Distribution of albumin and alpha-fetoprotein mRNAs in normal, hyperplastic, and preneoplastic rat liver. The American Journal of Pathology. 1992;141:623–632. [PMC free article] [PubMed] [Google Scholar]
- Bisgaard HC, Holmskov U, Santoni-Rugiu E, Nagy P, Nielsen O, Ott P, et al. Heterogeneity of ductular reactions in adult rat and human liver revealed by novel expression of deleted in malignant brain tumor 1. The American Journal of Pathology. 2002;161:1187–1198. doi: 10.1016/S0002-9440(10)64395-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nature Medicine. 2012;18:572–579. doi: 10.1038/nm.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier R, Suner RE, van Hul N, Kopp JL, Beaudry JB, Cordi S, et al. Embryonic ductal plate cells give rise to cholangiocytes, periportal hepatocytes, and adult liver progenitor cells. Gastroenterology. 2011;141:1432–1438. 1438,e1431–1434. doi: 10.1053/j.gastro.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpino G, Cardinale V, Onori P, Franchitto A, Berloco PB, Rossi M, et al. Biliary tree stem/progenitor cells in glands of extrahepatic and intraheptic bile ducts: An anatomical in situ study yielding evidence of maturational lineages. Journal of Anatomy. 2012;220:186–199. doi: 10.1111/j.1469-7580.2011.01462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K, Benten D, Bhargava K, Inada M, Joseph B, Palestro C, et al. Hepatic targeting and biodistribution of human fetal liver stem/progenitor cells and adult hepatocytes in mice. Hepatology. 2009;50:1194–1203. doi: 10.1002/hep.23120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu AS, Diaz R, Hui JJ, Yanger K, Zong Y, Alpini G, et al. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology. 2011;53:1685–1695. doi: 10.1002/hep.24206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabeva MD, Alpini G, Hurston E, Shafritz DA. Models for hepatic progenitor cell activation. Proceedings of the Society for Experimental Biology and Medicine. 1993;204:242–252. doi: 10.3181/00379727-204-43660. [DOI] [PubMed] [Google Scholar]
- Dabeva MD, Petkov PM, Sandhu J, Oren R, Laconi E, Hurston E, et al. Proliferation and differentiation of fetal liver epithelial progenitor cells after transplantation into adult rat liver. The American Journal of Pathology. 2000;156:2017–2031. doi: 10.1016/S0002-9440(10)65074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabeva MD, Shafritz DA. Activation, proliferation, and differentiation of progenitor cells into hepatocytes in the D-galactosamine model of liver regeneration. The American Journal of Pathology. 1993;143:1606–1620. [PMC free article] [PubMed] [Google Scholar]
- Davern TJ. Molecular therapeutics of liver disease. Clinics in Liver Disease. 2001;5:381–414. vi. doi: 10.1016/s1089-3261(05)70171-9. [DOI] [PubMed] [Google Scholar]
- Davies RA, Knight B, Tian YW, Yeoh GC, Olynyk JK. Hepatic oval cell response to the choline-deficient, ethionine supplemented model of murine liver injury is attenuated by the administration of a cyclo-oxygenase 2 inhibitor. Carcinogenesis. 2006;27:1607–1616. doi: 10.1093/carcin/bgi365. [DOI] [PubMed] [Google Scholar]
- Dorrell C, Erker L, Lanxon-Cookson KM, Abraham SL, Victoroff T, Ro S, et al. Surface markers for the murine oval cell response. Hepatology. 2008;48:1282–1291. doi: 10.1002/hep.22468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell C, Erker L, Schug J, Kopp JL, Canaday PS, Fox AJ, et al. Prospective isolation of a bipotential clonogenic liver progenitor cell in adult mice. Genes & Development. 2011;25:1193–1203. doi: 10.1101/gad.2029411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Dorrell C, Grompe M. Stem cells and liver regeneration. Gastroenterology. 2009;137:466–481. doi: 10.1053/j.gastro.2009.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusabineza AC, Van Hul NK, Abarca-Quinones J, Starkel P, Najimi M, Leclercq IA. Participation of liver progenitor cells in liver regeneration: Lack of evidence in the AAF/PH rat model. Laboratory Investigation; A Journal of Technical Methods and Pathology. 2012;92:72–81. doi: 10.1038/labinvest.2011.136. [DOI] [PubMed] [Google Scholar]
- Endo Y, Zhang M, Yamaji S, Cang Y. Genetic abolishment of hepatocyte proliferation activates hepatic stem cells. PloS One. 2012;7:e31846. doi: 10.1371/journal.pone.0031846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espanol-Suner R, Carpentier R, Van Hul N, Legry V, Achouri Y, Cordi S, et al. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology. 2012;143(1564–1575):e1567. doi: 10.1053/j.gastro.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Fan L, Xu C, Wang C, Tao J, Ho C, Jiang L, et al. Bmi1 is required for hepatic progenitor cell expansion and liver tumor development. PloS One. 2012;7:e46472. doi: 10.1371/journal.pone.0046472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber E. Similarities in the sequence of early histological changes induced in the liver of the rat by ethionine, 2-acetylamino-fluorene, and 3′-methyl-4-dimethylaminoazobenzene. Cancer Research. 1956;16:142–148. [PubMed] [Google Scholar]
- Feng D, Kong X, Weng H, Park O, Wang H, Dooley S, et al. Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology. 2012;143(188–198):e187. doi: 10.1053/j.gastro.2012.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujio K, Evarts RP, Hu Z, Marsden ER, Thorgeirsson SS. Expression of stem cell factor and its receptor, c-kit, during liver regeneration from putative stem cells in adult rat. Laboratory Investigation; A Journal of Technical Methods and Pathology. 1994;70:511–516. [PubMed] [Google Scholar]
- Furuyama K, Kawaguchi Y, Akiyama H, Horiguchi M, Kodama S, Kuhara T, et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nature Genetics. 2011;43:34–41. doi: 10.1038/ng.722. [DOI] [PubMed] [Google Scholar]
- Grompe M, al-Dhalimy M, Finegold M, Ou CN, Burlingame T, Kennaway NG, et al. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes & Development. 1993;7:2298–2307. doi: 10.1101/gad.7.12a.2298. [DOI] [PubMed] [Google Scholar]
- Grozdanov PN, Yovchev MI, Dabeva MD. The oncofetal protein glypican-3 is a novel marker of hepatic progenitor/oval cells. Laboratory Investigation; A Journal of Technical Methods and Pathology. 2006;86:1272–1284. doi: 10.1038/labinvest.3700479. [DOI] [PubMed] [Google Scholar]
- Gupta S, Rajvanshi P, Malhi H, Slehria S, Sokhi RP, Vasa SR, et al. Cell transplantation causes loss of gap junctions and activates GGT expression permanently in host liver. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2000;279:G815–G826. doi: 10.1152/ajpgi.2000.279.4.G815. [DOI] [PubMed] [Google Scholar]
- Higa LA, Zhang H. Stealing the spotlight: CUL4-DDB1 ubiquitin ligase docks WD40-repeat proteins to destroy. Cell Division. 2007;2:5. doi: 10.1186/1747-1028-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holme E, Lindstedt S. Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione) Journal of Inherited Metabolic Disease. 1998;21:507–517. doi: 10.1023/a:1005410820201. [DOI] [PubMed] [Google Scholar]
- Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494:247–250. doi: 10.1038/nature11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada M, Benten D, Cheng K, Joseph B, Berishvili E, Badve S, et al. Stage-specific regulation of adhesion molecule expression segregates epithelial stem/progenitor cells in fetal and adult human livers. Hepatology International. 2008;2:50–62. doi: 10.1007/s12072-007-9023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelnes P, Santoni-Rugiu E, Rasmussen M, Friis SL, Nielsen JH, Tygstrup N, et al. Remarkable heterogeneity displayed by oval cells in rat and mouse models of stem cell-mediated liver regeneration. Hepatology. 2007;45:1462–1470. doi: 10.1002/hep.21569. [DOI] [PubMed] [Google Scholar]
- Jensen CH, Jauho EI, Santoni-Rugiu E, Holmskov U, Teisner B, Tygstrup N, et al. Transit-amplifying ductular (oval) cells and their hepatocytic progeny are characterized by a novel and distinctive expression of delta-like protein/preadipocyte factor 1/fetal antigen 1. The American Journal of Pathology. 2004;164:1347–1359. doi: 10.1016/S0002-9440(10)63221-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozefczuk J, Stachelscheid H, Chavez L, Herwig R, Lehrach H, Zeilinger K, et al. Molecular characterization of cultured adult human liver progenitor cells. Tissue Engineering. Part C, Methods. 2010;16:821–834. doi: 10.1089/ten.TEC.2009.0578. [DOI] [PubMed] [Google Scholar]
- Jung Y, Brown KD, Witek RP, Omenetti A, Yang L, Vandongen M, et al. Accumulation of hedgehog-responsive progenitors parallels alcoholic liver disease severity in mice and humans. Gastroenterology. 2008;134:1532–1543. doi: 10.1053/j.gastro.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakinuma S, Ohta H, Kamiya A, Yamazaki Y, Oikawa T, Okada K, et al. Analyses of cell surface molecules on hepatic stem/progenitor cells in mouse fetal liver. Journal of Hepatology. 2009;51:127–138. doi: 10.1016/j.jhep.2009.02.033. [DOI] [PubMed] [Google Scholar]
- Kamiya A, Kakinuma S, Yamazaki Y, Nakauchi H. Enrichment and clonal culture of progenitor cells during mouse postnatal liver development in mice. Gastroenterology. 2009;137:1114–1126. 1126 e1111–1114. doi: 10.1053/j.gastro.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Katoonizadeh A, Nevens F, Verslype C, Pirenne J, Roskams T. Liverregeneration in acute severe liver impairment: A clinicopathological correlation study. Liver International: Official Journal of the International Association for the Study of the Liver. 2006;26:1225–1233. doi: 10.1111/j.1478-3231.2006.01377.x. [DOI] [PubMed] [Google Scholar]
- Knight B, Yeap BB, Yeoh GC, Olynyk JK. Inhibition of adult liver progenitor (oval) cell growth and viability by an agonist of the peroxisome proliferator activated receptor (PPAR) family member gamma, but not alpha or delta. Carcinogenesis. 2005;26:1782–1792. doi: 10.1093/carcin/bgi138. [DOI] [PubMed] [Google Scholar]
- Knight B, Yeoh GC. TNF/LTalpha double knockout mice display abnormal inflammatory and regenerative responses to acute and chronic liver injury. Cell and Tissue Research. 2005;319:61–70. doi: 10.1007/s00441-004-1003-6. [DOI] [PubMed] [Google Scholar]
- Kuwahara R, Kofman AV, Landis CS, Swenson ES, Barendswaard E, Theise ND. The hepatic stem cell niche: Identification by label-retaining cell assay. Hepatology. 2008;47:1994–2002. doi: 10.1002/hep.22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbrecht L, Roskams T. Hepatic progenitor cells in human liver diseases. Seminars in Cell & Developmental Biology. 2002;13:389–396. doi: 10.1016/s1084952102001258. [DOI] [PubMed] [Google Scholar]
- Lorenzini S, Bird TG, Boulter L, Bellamy C, Samuel K, Aucott R, et al. Characterisation of a stereotypical cellular and extracellular adult liver progenitor cell niche in rodents and diseased human liver. Gut. 2010;59:645–654. doi: 10.1136/gut.2009.182345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes KN, Brennan BA, Yeoh GC, Olynyk JK. Oval cell numbers in human chronic liver diseases are directly related to disease severity. The American Journal of Pathology. 1999;154:537–541. doi: 10.1016/S0002-9440(10)65299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozoya OA, Wauthier E, Turner RA, Barbier C, Prestwich GD, Guilak F, et al. Regulation of hepatic stem/progenitor phenotype by microenvironment stiffness in hydrogel models of the human liver stem cell niche. Biomaterials. 2011;32:7389–7402. doi: 10.1016/j.biomaterials.2011.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malato Y, Naqvi S, Schurmann N, Ng R, Wang B, Zape J, et al. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. The Journal of Clinical Investigation. 2011;121:4850–4860. doi: 10.1172/JCI59261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins PN, Theruvath TP, Neuhaus P. Rodent models of partial hepatectomies. Liver International: Official Journal of the International Association for the Study of the Liver. 2008;28:3–11. doi: 10.1111/j.1478-3231.2007.01628.x. [DOI] [PubMed] [Google Scholar]
- Menthena A, Deb N, Oertel M, Grozdanov PN, Sandhu J, Shah S, et al. Bone marrow progenitors are not the source of expanding oval cells in injured liver. Stem Cells. 2004;22:1049–1061. doi: 10.1634/stemcells.22-6-1049. [DOI] [PubMed] [Google Scholar]
- Nagaya M, Kubota S, Suzuki N, Akashi K, Mitaka T. Thermoreversible gelation polymer induces the emergence of hepatic stem cells in the partially injured rat liver. Hepatology. 2006;43:1053–1062. doi: 10.1002/hep.21153. [DOI] [PubMed] [Google Scholar]
- Nobili V, Carpino G, Alisi A, Franchitto A, Alpini G, De Vito R, et al. Hepatic progenitor cells activation, fibrosis, and adipokines production in pediatric non-alcoholic fatty liver disease. Hepatology. 2012;56:2142–2153. doi: 10.1002/hep.25742. [DOI] [PubMed] [Google Scholar]
- Okabe M, Tsukahara Y, Tanaka M, Suzuki K, Saito S, Kamiya Y, et al. Potential hepatic stem cells reside in EpCAM+ cells of normal and injured mouse liver. Development. 2009;136:1951–1960. doi: 10.1242/dev.031369. [DOI] [PubMed] [Google Scholar]
- Omori N, Omori M, Evarts RP, Teramoto T, Miller MJ, Hoang TN, et al. Partial cloning of rat CD34 cDNA and expression during stem cell-dependent liver regeneration in the adult rat. Hepatology. 1997;26:720–727. doi: 10.1002/hep.510260325. [DOI] [PubMed] [Google Scholar]
- Petersen BE, Bowen WC, Patrene KD, Mars WM, Sullivan AK, Murase N, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284:1168–1170. doi: 10.1126/science.284.5417.1168. [DOI] [PubMed] [Google Scholar]
- Petersen BE, Goff JP, Greenberger JS, Michalopoulos GK. Hepatic oval cells express the hematopoietic stem cell marker Thy-1 in the rat. Hepatology. 1998;27:433–445. doi: 10.1002/hep.510270218. [DOI] [PubMed] [Google Scholar]
- Piscaglia AC, Shupe TD, Oh SH, Gasbarrini A, Petersen BE. Granulocyte-colony stimulating factor promotes liver repair and induces oval cell migration and proliferation in rats. Gastroenterology. 2007;133:619–631. doi: 10.1053/j.gastro.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porretti L, Cattaneo A, Colombo F, Lopa R, Rossi G, Mazzaferro V, et al. Simultaneous characterization of progenitor cell compartments in adult human liver. Cytometry Part A: The Journal of the International Society for Analytical Cytology. 2010;77:31–40. doi: 10.1002/cyto.a.20834. [DOI] [PubMed] [Google Scholar]
- Qiu Q, Hernandez JC, Dean AM, Rao PH, Darlington GJ. CD24-positive cells from normal adult mouse liver are hepatocyte progenitor cells. Stem Cells and Development. 2011;20:2177–2188. doi: 10.1089/scd.2010.0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskams T, Desmet V. Ductular reaction and its diagnostic significance. Seminars in Diagnostic Pathology. 1998;15:259–269. [PubMed] [Google Scholar]
- Roskams TA, Theise ND, Balabaud C, Bhagat G, Bhathal PS, Bioulac-Sage P, et al. Nomenclature of the finer branches of the biliary tree: Canals, ductules, and ductular reactions in human livers. Hepatology. 2004;39:1739–1745. doi: 10.1002/hep.20130. [DOI] [PubMed] [Google Scholar]
- Rountree CB, Barsky L, Ge S, Zhu J, Senadheera S, Crooks GM. A CD133-expressing murine liver oval cell population with bilineage potential. Stem Cells. 2007;25:2419–2429. doi: 10.1634/stemcells.2007-0176. [DOI] [PubMed] [Google Scholar]
- Rountree CB, Wang X, Ge S, Barsky L, Zhu J, Gonzales I, et al. Bone marrow fails to differentiate into liver epithelium during murine development and regeneration. Hepatology. 2007;45:1250–1260. doi: 10.1002/hep.21600. [DOI] [PubMed] [Google Scholar]
- Sackett SD, Gao Y, Shin S, Esterson YB, Tsingalia A, Hurtt RS, et al. Foxl1 promotes liver repair following cholestatic injury in mice. Laboratory Investigation. 2009;89:1387–1396. doi: 10.1038/labinvest.2009.103. [DOI] [PubMed] [Google Scholar]
- Sackett SD, Li Z, Hurtt R, Gao Y, Wells RG, Brondell K, et al. Foxl1 is a marker of bipotential hepatic progenitor cells in mice. Hepatology. 2009;49:920–929. doi: 10.1002/hep.22705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Bru P, Altamirano J, Rodrigo-Torres D, Coll M, Millan C, Jose Lozano J, et al. Liver progenitor cell markers correlate with liver damage and predict short-term mortality in patients with alcoholic hepatitis. Hepatology. 2012;55:1931–1941. doi: 10.1002/hep.25614. [DOI] [PubMed] [Google Scholar]
- Sandhu JS, Petkov PM, Dabeva MD, Shafritz DA. Stem cell properties and repopulation of the rat liver by fetal liver epithelial progenitor cells. The American Journal of Pathology. 2001;159:1323–1334. doi: 10.1016/S0002-9440(10)62519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzer E, Wauthier E, Reid LM. The phenotypes of pluripotent human hepatic progenitors. Stem Cells. 2006;24:1852–1858. doi: 10.1634/stemcells.2006-0036. [DOI] [PubMed] [Google Scholar]
- Schmelzer E, Zhang L, Bruce A, Wauthier E, Ludlow J, Yao HL, et al. Human hepatic stem cells from fetal and postnatal donors. The Journal of Experimental Medicine. 2007;204:1973–1987. doi: 10.1084/jem.20061603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholten D, Osterreicher CH, Scholten A, Iwaisako K, Gu G, Brenner DA, et al. Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology. 2010;139:987–998. doi: 10.1053/j.gastro.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotanus BA, van den Ingh TS, Penning LC, Rothuizen J, Roskams TA, Spee B. Cross-species immunohistochemical investigation of the activation of the liver progenitor cell niche in different types of liver disease. Liver International: Official Journal of the International Association for the Study of the Liver. 2009;29:1241–1252. doi: 10.1111/j.1478-3231.2009.02024.x. [DOI] [PubMed] [Google Scholar]
- Shin S, Walton G, Aoki R, Brondell K, Schug J, Fox A, et al. Foxl1-Cre-marked adult hepatic progenitors have clonogenic and bilineage differentiation potential. Genes & Development. 2011;25:1185–1192. doi: 10.1101/gad.2027811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicklick JK, Li YX, Melhem A, Schmelzer E, Zdanowicz M, Huang J, et al. Hedgehog signaling maintains resident hepatic progenitors throughout life. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2006;290:G859–G870. doi: 10.1152/ajpgi.00456.2005. [DOI] [PubMed] [Google Scholar]
- Spee B, Carpino G, Schotanus BA, Katoonizadeh A, Vander Borght S, Gaudio E, et al. Characterisation of the liver progenitor cell niche in liver diseases: Potential involvement of Wnt and Notch signalling. Gut. 2010;59:247–257. doi: 10.1136/gut.2009.188367. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Developmental Biology. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Sekiya S, Onishi M, Oshima N, Kiyonari H, Nakauchi H, et al. Flow cytometric isolation and clonal identification of self-renewing bipotent hepatic progenitor cells in adult mouse liver. Hepatology. 2008;48:1964–1978. doi: 10.1002/hep.22558. [DOI] [PubMed] [Google Scholar]
- Takase HM, Itoh T, Ino S, Wang T, Koji T, Akira S, et al. FGF7 is a functional niche signal required for stimulation of adult liver progenitor cells that support liver regeneration. Genes & Development. 2013;27:169–181. doi: 10.1101/gad.204776.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theise ND, Saxena R, Portmann BC, Thung SN, Yee H, Chiriboga L, et al. The canals of Hering and hepatic stem cells in humans. Hepatology. 1999;30:1425–1433. doi: 10.1002/hep.510300614. [DOI] [PubMed] [Google Scholar]
- Tirnitz-Parker JE, Tonkin JN, Knight B, Olynyk JK, Yeoh GC. Isolation, culture and immortalisation of hepatic oval cells from adult mice fed a choline-deficient, ethionine-supplemented diet. The International Journal of Biochemistry & Cell Biology. 2007;39:2226–2239. doi: 10.1016/j.biocel.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Tirnitz-Parker JE, Viebahn CS, Jakubowski A, Klopcic BR, Olynyk JK, Yeoh GC, et al. Tumor necrosis factor-like weak inducer of apoptosis is a mitogen for liver progenitor cells. Hepatology. 2010;52:291–302. doi: 10.1002/hep.23663. [DOI] [PubMed] [Google Scholar]
- Tonkin JN, Knight B, Curtis D, Abraham LJ, Yeoh GC. Bone marrow cells play only a very minor role in chronic liver regeneration induced by a choline-deficient, ethionine-supplemented diet. Stem Cell Research. 2008;1:195–204. doi: 10.1016/j.scr.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Trautwein C, Will M, Kubicka S, Rakemann T, Flemming P, Manns MP. 2-acetaminofluorene blocks cell cycle progression after hepatectomy by p21 induction and lack of cyclin E expression. Oncogene. 1999;18:6443–6453. doi: 10.1038/sj.onc.1203045. [DOI] [PubMed] [Google Scholar]
- Turanyi E, Dezso K, Csomor J, Schaff Z, Paku S, Nagy P. Immunohistochemical classification of ductular reactions in human liver. Histopathology. 2010;57:607–614. doi: 10.1111/j.1365-2559.2010.03668.x. [DOI] [PubMed] [Google Scholar]
- Turner R, Lozoya O, Wang Y, Cardinale V, Gaudio E, Alpini G, et al. Human hepatic stem cell and maturational liver lineage biology. Hepatology. 2011;53:1035–1045. doi: 10.1002/hep.24157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RA, Wauthier E, Lozoya O, McClelland R, Bowsher JE, Barbier C, et al. Successful transplantation of human hepatic stem cells with restricted localization to liver using hyaluronan graftsdagger. Hepatology. 2013;57:775–784. doi: 10.1002/hep.26065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueberham E, Bottger J, Ueberham U, Grosche J, Gebhardt R. Response of sinusoidal mouse liver cells to choline-deficient ethionine-supplemented diet. Comparative Hepatology. 2010;9:8. doi: 10.1186/1476-5926-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hul NK, Abarca-Quinones J, Sempoux C, Horsmans Y, Leclercq IA. Relation between liver progenitor cell expansion and extracellular matrix deposition in a CDE-induced murine model of chronic liver injury. Hepatology. 2009;49:1625–1635. doi: 10.1002/hep.22820. [DOI] [PubMed] [Google Scholar]
- Vassilopoulos G, Wang PR, Russell DW. Transplanted bone marrow regenerates liver by cell fusion. Nature. 2003;422:901–904. doi: 10.1038/nature01539. [DOI] [PubMed] [Google Scholar]
- Viebahn CS, Benseler V, Holz LE, Elsegood CL, Vo M, Bertolino P, et al. Invading macrophages play a major role in the liver progenitor cell response to chronic liver injury. Journal of Hepatology. 2010;53:500–507. doi: 10.1016/j.jhep.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Wang X, Willenbring H, Akkari Y, Torimaru Y, Foster M, Al-Dhalimy M, et al. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature. 2003;422:897–901. doi: 10.1038/nature01531. [DOI] [PubMed] [Google Scholar]
- Wu KJ, Yang MH. Epithelial-mesenchymal transition and cancer stemness: The Twist1-Bmi1 connection. Bioscience Reports. 2011;31:449–455. doi: 10.1042/BSR20100114. [DOI] [PubMed] [Google Scholar]
- Yang L, Jung Y, Omenetti A, Witek RP, Choi S, Vandongen HM, et al. Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells. 2008;26:2104–2113. doi: 10.1634/stemcells.2008-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanger K, Stanger BZ. Facultative stem cells in liver and pancreas: Fact and fancy. Developmental Dynamics: An Official Publication of the American Association of Anatomists. 2011;240:521–529. doi: 10.1002/dvdy.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen P, Hayner NT, Fausto N. Isolation of oval cells by centrifugal elutriation and comparison with other cell types purified from normal and preneoplastic livers. Cancer Research. 1984;44:324–331. [PubMed] [Google Scholar]
- Yeoh GC, Ernst M, Rose-John S, Akhurst B, Payne C, Long S, et al. Opposing roles of gp130-mediated STAT-3 and ERK-1/2 signaling in liver progenitor cell migration and proliferation. Hepatology. 2007;45:486–494. doi: 10.1002/hep.21535. [DOI] [PubMed] [Google Scholar]
- Yovchev MI, Grozdanov PN, Zhou H, Racherla H, Guha C, Dabeva MD. Identification of adult hepatic progenitor cells capable of repopulating injured rat liver. Hepatology. 2008;47:636–647. doi: 10.1002/hep.22047. [DOI] [PubMed] [Google Scholar]