

Abstract
Vascular endothelial growth factor (VEGF) induces phosphorylation of VEGF receptor-2 (VEGFR-2) and activates the downstream signaling pathway resulting in endothelial cell migration, proliferation, and survival. Cigarette smoking is associated with abnormal vascular and endothelial function, leading to airspace enlargement. Herein, we investigated the mechanism of cigarette smoke (CS) -induced endothelial dysfunction by studying the VEGF-VEGFR-2 signaling in mouse lung and human endothelial cells. CS exposure caused oxidative stress, as shown by increased levels of 4-hydroxy-2-nonenal-adducts in mouse lung and reactive oxygen species generation in human lung microvascular endothelial cells (HMVEC-Ls). Inhibition of VEGFR-2 by a specific kinase inhibitor (NVP-AAD777) enhanced the CS-induced oxidative stress, causing augmented inflammatory cell influx and proinflammatory mediators release in mouse lung. The levels of endothelial nitric oxide synthase (eNOS) and phosphorylated (p) -eNOS in the lungs of mice exposed to CS and/or treated with VEGFR-2 inhibitor were decreased. CS down-regulated VEGFR-2 expression, eNOS levels, and VEGF-induced VEGFR-2 phosphorylation in HMVEC-Ls, resulting in impaired VEGF-induced endothelial cell migration and angiogenesis. Overall, these data show that inhibition of VEGFR-2 augmented CS-induced oxidative stress and inflammatory responses leading to endothelial dysfunction. This explains the mechanism of endothelial dysfunction in smokers and has implications in understanding the pathogenesis of pulmonary and cardiovascular diseases.
Keywords: oxidants, eNOS, angiogenesis
Vascular endothelial growth factor (VEGF) is an important regulator of vascular development. VEGF induces phosphorylation of VEGF receptor-2 (VEGFR-2), activating downstream signaling resulting in endothelial cell migration, proliferation, and survival (1). VEGF-VEGFR-2-mediated neovascularization plays an important role in maintaining the normal lung structure (2). Several lines of investigation reveal that inhibition of the VEGF-VEGFR-2 signaling pathway results in decreased lung alveolarization and arterial density (3, 4). Lung microvasculature is critical for gas exchange and alveolar integrity. Emphysema, one of the major features of chronic obstructive pulmonary disease (COPD), is defined as enlargement of the distal air spaces as a consequence of alveolar wall destruction. Cigarette smoke (CS) is a complex mixture of >4700 chemical compounds and contains a high concentration of free radicals (>1017 molecules/puff), which is known to induce oxidative stress and inflammation in the lung (5). CS-induced oxidative stress is involved in the development of emphysema, and CS-induced emphysematous alveolar septa are remarkably thin and almost avascular (6, 7). In addition, CS also plays an important role in the etiology of cardiovascular diseases and vascular dysfunction, such as atherosclerosis and pulmonary hypertension (8–12). This is due to the fact that CS induces endothelial dysfunction in the systemic and pulmonary circulation leading to the earliest morphological and pathological changes in the vessel wall (12–14). However, the mechanisms by which CS impairs endothelial function, including cell survival, proliferation, and angiogenesis, remain unknown.
Endothelial nitric oxide synthase (eNOS) plays a central role in maintaining vascular integrity (15) and is activated by VEGF and shear stress through the VEGFR-2 (16–18). It is also known that nitric oxide (NO) is an essential mediator of endothelial cell migration and VEGF-induced angiogenesis (19). VEGF acts via its receptors VEGFR-1 (Flt1) and VEGFR-2 (Flk-1/KDR) to induce vascular permeability, cell proliferation, cellular migration, and tube formation in endothelial cells (1). Both VEGF and VEGFR-2 expression levels are reduced in emphysematous lungs (20, 21). We have recently shown that both VEGF and VEGFR-2 expression are reduced in response to CS exposure in rat lungs (22). Recent studies (21) reported that chronic inhibition of global VEGF receptors using SU5416 in rats caused airway enlargement and alveolar endothelial cell death and decreased arterial density, suggesting that angiogenesis is necessary for alveolarization. However, the role of VEGFR-1 and VEGFR-2 in regulation of CS-induced alteration in alveolarization was not studied. It was postulated that oxidative stress plays a central role in alveolar septal destruction as a result of VEGFR inhibition, as shown by the preventive role of a superoxide dismutase mimetic (M40419) administrated in combination with VEGFR inhibitor (SU5416) in rat lung (23). The SU5416 compound inhibits both VEGFR-1 and VEGFR-2 kinases (21), whereas NVP-AAD777 is a specific inhibitor of VEGFR-2 kinase (22). The effect of a specific VEGFR-2 inhibitor on CS-mediated oxidative stress, inflammation, and endothelial dysfunction has not been evaluated in any in vitro or in vivo models. We hypothesized that specific inhibition of VEGFR-2 causes oxidative stress and an inflammatory response, which lead to endothelial dysfunction, and that these responses are augmented by CS.
MATERIALS AND METHODS
Unless otherwise stated, all biochemicals were of analytical grade and purchased from Sigma-Aldrich (St. Louis, MO, USA).
VEGFR-2 inhibitor treatment and CS exposure
The animal protocol was approved by the University Committee on Animal Resources (University of Rochester, Rochester, NY, USA). Male C57BL/6J mice, 8-wk-old, 25–30 g (Jackson Laboratory, Bar Harbor, ME, USA), were divided into four exposure groups (n=8): 1) air + vehicle; 2) CS + vehicle; 3) air + VEGFR-2 inhibitor [NVP-AAD777 (IC50=510 nM), Novartis Pharma AG, Basel, Switzerland]; and 4) CS+VEGFR-2 inhibitor. The VEGFR-2 inhibitor, NVP-AAD777 was dissolved in polyethylene glycol-400 (PEG 400) as a vehicle and administered at 20 mg/kg body weight subcutaneously, 3×/wk as described previously (21, 22). The details of VEGFR-2 kinase inhibition using this compound are given in our previous studies (22). Control animals were administered vehicle alone in the same manner. Mice were placed inside a ventilated chamber and exposed to whole body CS using Baumgartner-Jaeger CSM 2072i smoking machine (CH Technologies, Westwood, NJ, USA) at 300 mg/m3 total particulate matter (TPM) for 2 h/day (1 h apart 2×/day), 5 days/wk for 3 wk with 2R4F research grade cigarettes (University of Kentucky, Lexington, KY, USA; refs. 24, 25). The level of carbon monoxide in the chamber was 350 ppm. Mice were sacrificed 2 h after last exposure by intraperitoneal injection of 50 mg/kg sodium pentobarbital. The level of carboxyhemoglobin in blood was 17%. Control mice were exposed to filtered air in an identical chamber according to the same protocol described for CS exposure.
Lung fixation for morphology
Mouse lungs (4 lungs from each group) were inflated with 4% paraformaldehyde at a constant pressure of 25 cm. The inflated lungs were fixed in 4% paraformaldehyde and embedded in paraffin. Sections (5 μm) were obtained and used for morphological analyses.
Inflammatory cell counts in bronchoalveolar lavage fluid (BALF)
Lungs were lavaged via a butterfly cannula inserted into the trachea and instilled with saline. Cells were separated by centrifugation of BALF and resuspended in 1 ml of saline. Differential cell counts (minimum of 500 cells per slide) were performed on cytospin-prepared slides (Thermo Shandon, Pittsburgh, PA, USA) stained with Diff-Quick (Dade Behring, Newark, DE, USA).
Immunoblotting in lung tissue homogenate
Frozen lung tissues (0.1 g) were homogenized in 0.5 ml of ice-cold radio-immuno-precipitation assay (RIPA) lysis buffer containing complete protease inhibitor cocktail (Sigma-Al-drich). The homogenates were kept on ice for 45 min and then centrifuged at 10,000 g for 15 min at 4°C, and the supernatants were separated. Protein levels were measured using bicinchoninic acid (BCA) kit (Pierce, Rockford, IL, USA). Protein (30 μg) was electrophoresed on a 4–15% gradient PAGE gel and transblotted onto nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, USA). Membranes were blocked with 5% nonfat milk and incubated with relevant primary antibody (1:1000 dilution). Primary antibodies used were mouse anti-phosphotyrosine clone 4G10 (Upstate Cell Signaling, Temecula, CA, USA), rabbit anti-VEGFR-2, rabbit anti-eNOS (Cell Signaling Technology, Danvers, MA, USA), and mouse anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After being washed, bound antibody was detected using anti-rabbit/mouse antibody (1: 20,000 dilution) linked to horseradish peroxidase, and bound complexes were detected using enhanced chemiluminescence (Perkin Elmer, Waltham, MA, USA).
eNOS immunoprecipitation
Mouse lung homogenates were prepared in RIPA buffer as described above, and eNOS protein was immunoprecipitated using anti-eNOS antibody (1:100 dilution; rabbit anti-eNOS, Cell Signaling Technology), which was added to 250 μg of protein in a final volume of 400 μl and incubated for 1 h. Protein-A/G agarose beads (20 μl; Santa Cruz Biotechnology) were added to each sample and left overnight at 4°C on a rocker. The samples were then centrifuged at 10,000 g at 4°C for 5 min. The supernatant was discarded, and the beads were washed 3× and then resuspended in 100 μl of RIPA buffer. The samples were then mixed with 5× SDS sample buffer and boiled, and the proteins were resolved by SDS-PAGE. Immunoblotting was performed for phosphorylated (p) -eNOS [rabbit anti-p-(Ser-1177) eNOS, Cell Signaling Technology] and e-NOS (rabbit anti-eNOS, Cell Signaling Technology).
Cytokines and VEGF analysis in lung tissue homogenate by the Luminex method
Whole lung tissues were homogenized in ice-cold buffer A (10 mM HEPES, pH 7.9; 10 mM KCl; 0.1 mM EDTA; 0.1 mM EGTA; 1 mM dithiothreitol; and 0.5 mM PMSF) and incubated with Nonidet P-40 for 15 min. After 15 min, samples were centrifuged, and the supernatant was used to analyze the levels of proinflammatory mediators, [neutrophil chemotactic factor (KC)/monocyte chemoattractant protein-1 (MCP-1), KC/CXCL1, MCP-1/CCL2, interleukin (IL) -6, and tumor necrosis factor (TNF) -α] and VEGF levels in lungs using a Luminex-based assay (mouse multicytokine beadmaster beadlyte kit, Upstate Cell Signaling) as instructed by the manufacturer. Data were obtained using a Luminex 100 reader (Luminex Corporation, Austin, TX, USA). Protein levels were measured with a BCA kit (Pierce).
Immunohistochemistry
Paraffin was removed from paraffin-embedded lung sections and rehydrated before immunohistochemistry. The sections were refixed, and immunostaining was performed using an antibody specific for 4-hydroxy-2-nonenal (4-HNE) adducts (anti-4-HNE antibody; OXIS International, Foster City, CA, USA) or macrophages (Mac-3 antibody, BD Pharmingen, San Jose, CA, USA). Immunodetection was performed with a biotinylated rabbit anti-mouse Ig reagent (Dako, Carpinteria, CA, USA), streptavidin-biotin complex reagent (Dako), and 3′,3-diaminobenzidine (Vector Laboratories, Burlingame, CA, USA). Counterstaining was performed with hematoxylin (22). Macrophages in each lung section (10 lung sections from different regions of the lung) were counted under the microscope (×200) in a blinded manner, and the cell numbers were averaged. The intensity of 4-HNE adducts staining was assessed by scoring method in a blinded manner (0=no staining, 1=weak staining, 2.5=moderate staining, and 5=intense staining).
In vitro studies using human lung microvascular endothelial cells (HMVEC-Ls)
HMVEC-Ls were purchased from Cambrex (Walksville, MD, USA) and were grown in endothelial growth medium-2 (EGM-2; Cambrex) media containing 10% fetal bovine albumin (FBS) at 37°C in a humidified atmosphere containing 5% CO2. Cells were grown in 75 mm flasks coated with 0.1% gelatin, and treatments were performed in 0.1% gelatin-coated 6-well plates. Cigarette smoke extract (CSE) and VEGFR-2 inhibitor treatments were performed after cells were starved for 6 h with EGM-2 media containing 0.1% FBS.
CSE preparation
Research grade cigarettes (1R3F) were obtained from the Kentucky Tobacco Research and Development Center at the University of Kentucky. The composition of 1R3F research grade cigarettes was as follows: total particulate matter, 17.1 mg/cigarette; tar, 15 mg/cigarette; and nicotine, 1.16 mg/ cigarette. CSE (10%) was prepared by bubbling smoke from one cigarette into 10 ml of EGM-2 culture media without FBS at a rate of 1 cigarette every 2 min, as described previously (26, 27), using a modification of the method described by Carp and Janoff (28). The pH of the CSE was adjusted to 7.4, and it was sterile filtered through a 0.45 μm filter (Acrodisc; Pall Corporation, Ann Arbor, MI, USA). CSE preparation was standardized by measuring the absorbance (optical density=0.86±0.05) at a wavelength of 320 nm. The pattern of absorbance (spectrogram) observed at λ320 showed very little variation between different preparations of CSE. CSE was freshly prepared for each experiment and diluted with culture media supplemented with 0.1% FBS immediately before use. Control medium was prepared by bubbling air through 10 ml of culture media without FBS, the pH was adjusted to 7.4, and the medium was sterile filtered as described above.
Effect of CSE and VEGFR-2 inhibitor on reactive oxygen species (ROS) production in HMVEC-Ls
ROS production in HMVEC-Ls in response to CSE and VEGFR-2 inhibitor was assessed using a fluorescent dye, 5-and 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCF-DA; Invitrogen, Carlsbad, CA, USA). Inside the cell, the probe is deacetylated by esterases and forms H2DCF, which is oxidized by ROS to DCF, a highly fluorescent compound. Thus, dye oxidation is an indirect measure of the presence of ROS intermediates. HMVEC-Ls were grown in 0.1% gelatin-coated 8-chamber culture slides (BD Falcon, Bedford, MA, USA) and treated with CSE (0.1–0.5%), VEGFR-2 inhibitor (0.5–1.0 μM), and CSE (0.25%) + VEGFR-2 inhibitor (0.5 μM) for 2 h and washed twice with PBS. Cells were then incubated with 10 μM of carboxy-H2DCFDA for 15 min at room temperature and rinsed with PBS. The cells were observed under a fluorescent microscope, and the green fluorescent intensity was assessed using a scoring system (1=weak staining, 2=moderate staining, and 5=intense staining).
Effect of CSE on VEGFR-2 and eNOS protein expression
HMVEC-Ls were seeded in 6-well plates containing culture media supplemented with 10% FBS in a final volume of 2 ml. The cells were grown to ~90% confluent and starved in EGM-2 media containing 0.1% FBS for 6 h before the CSE treatments. All treatments were performed in duplicate. The cells were treated with CSE (0.1, 0.25, and 0.5%) for 12 h at 37°C in a humidified atmosphere containing 5% CO2. After a 12 h treatment, cell lysates were prepared in RIPA buffer containing a protease inhibitor cocktail to investigate the level of VEGFR-2 and e-NOS protein expression. Protein concentrations were analyzed using the BCA kit (Pierce). The levels of VEGFR-2 and eNOS were assessed using immunoblotting method with a rabbit anti-VEGFR-2 antibody (Cell Signaling Technology) and rabbit anti-eNOS antibody (BD Bioscience, San Jose, CA, USA), respectively.
Determination of the effect of CS on VEGF-induced VEGFR-2 phosphorylation
HMVEC-Ls were grown (90% confluent) in 6-well culture plates and starved for 6 h in 0.1% serum and VEGF-free media. Cells were then treated with CSE (0.1–0.5%) and VEGFR-2 inhibitor (0.5 and 1.0 μM; IC50=0.51 μM) for 2 h. A stock solution of NVP-AAD777 (1 mM) was prepared in 20 μl of DMSO. Therefore, some groups were treated with DMSO (0.02 μl/ml) as controls. At the end of the 2 h period, VEGF (50 ng/ml, Cell Signaling Technology) was added and incubated for 10 min at 37°C in a humidified atmosphere containing 5% CO2. Parallel control experiments were carried out without adding VEGF. The reaction was stopped immediately by adding ice-cold PBS, and the cells were washed twice with ice-cold PBS and then lyzed using RIPA buffer containing protease inhibitor cocktail. Finally, cell lysates were sonicated for 10 s and centrifuged at 10,000 g for 15 min, and the supernatant was separated and analyzed for protein content using the BCA kit method. Phosphorylated tyrosine-1175 (rabbit anti-p-tyr1175 VEGFR-2, Cell Signaling Technology) and total VEGFR-2 (rabbit anti-VEGFR-2, Cell Signaling Technology) levels were analyzed by immunoblotting.
Assessment of the effect of CS on VEGF-induced cell migration (scratch assay)
The “scratch assay” was performed to investigate the effect of CSE on VEGF-induced endothelial cell migration. HMVEC-Ls were grown (90% confluent) in 60 mm dishes and starved for 6 h in 0.1% serum and VEGF-free media. Cells were then treated for 2 h with CSE (0.25%), N-acetyl-L-cysteine (NAC; 1 mM) + CSE (0.25%), VEGFR-2 inhibitor (1 μM), and NAC (1 mM) + VEGFR-2 inhibitor (1 μM). NAC treatment was performed 1 h before CSE treatments. At the end of the 2 h, the bottoms of the culture plates were scratched using a 200 μl pipette tip and treated with VEGF (20 ng/ml). Parallel control experiments were carried out without VEGF treatments. The number of cells that migrated beyond the scratch lines after 12 h was counted under the microscope by a single investigator in a blinded manner.
Assay of the effect of CS on VEGF-induced angiogenesis (capillary-like tube formation assay)
To determine the effect of CSE on VEGF-induced tube formation, cells were starved for 6 h in EGM-2 media without VEGF containing 0.1% serum and treated with CSE (0.25%) or VEGFR-2 inhibitor (NVP-AAD777; 1 μM) for 2 h. After 2 h, HMVEC-Ls were washed and plated in 24-well plate that had been precoated with 100 μl of growth factor reduced Matrigel Matrix (BD Bioscience) and cultured at 37°C in a humidified incubator containing 5% CO2 for 24 h with or without 20 ng/ml of VEGF. At the end of 24 h, capillary-like tubes were photographed under a microscope and all side branches were counted by a single investigator in a blinded manner.
Statistical analysis
The Sigma Stat 3.0 statistical program was used to analyzed the data. The results are shown as the mean ± SE. All the data were tested for the normality, and equal variance test to determine whether our sample size is appropriate to analyze the data using ANOVA. If the equal variance or normality test was failed, the data were analyzed using nonparametric analysis (Kruskal-Wallis test). All pairwise multiple comparisons were performed using the Student-Newman-Keuls method; values of P < 0.05 were considered significant.
RESULTS
Inhibition of VEGFR-2 augmented CS-induced macrophage influx into mouse lung
To determine the effect of CS and VEGFR-2 inhibitor-mediated inflammation in mouse lung, inflammatory cell counts in BALF and lung tissue sections were investigated. CS exposure significantly increased the neutrophil counts in both vehicle- and VEGFR-2 inhibitor-treated groups (P<0.001). However, macrophage counts in BALF were not significantly changed among the groups (Fig. 1A, B). The numbers of macrophages in the lung tissue sections were counted after immunostaining was performed with a Mac-3 antibody. The number of macrophages in the lung sections was significantly increased in both CS-exposed groups with and without VEGFR-2 inhibitor treatments compared with their respective air-exposed groups (P<0.05). Furthermore, the number of macrophages in the VEGFR-2 inhibitor-treated CS-exposed group was increased significantly compared with vehicle-treated CS-exposed group (P<0.05; Fig. 1C, D). The number of neutrophils was also counted in the lung sections after immunostaining was performed with an antibody specific for neutrophils. There was no significant change in the number of neutrophils in the lungs with different treatment groups (data not shown). PEG-400 was used as a vehicle control, and no effect of PEG-400 was observed on any parameters measured. Overall, these data suggested that inhibition of VEGFR-2 augmented CS-induced macrophages influx into the mouse lung.
Figure 1.

CS and/or VEGFR-2 inhibition increased inflammatory cell influx in BALF and the number of macrophages in mouse lung. Animals were treated with vehicle or VEGFR-2 inhibitor and exposed to air or CS as described in Materials and Methods. All the animals were sacrificed 2 h after the last CS exposure. Lungs were lavaged, and inflammatory cells were counted in cytospin slides after Diff-Quick staining. A) Total number of macrophages in BALF. B) Total number of neutrophils in BALF. Histograms represent mean ± SE cells/ml (n=4). ***P < 0.001 vs. both air groups. C) Representative immunohistological sections (×200) of the lungs showing macrophages (arrows). Insets on the right corner show enlarged macrophages in mouse lungs (arrows). Macrophages in the lung were immunostained using a Mac-3 antibody. Macrophages in each section were counted in a blinded manner, as described in Materials and Methods. D) Histograms represent the number of macrophages as mean ± SE counted in 4 different treatment groups (n=4). *P < 0.05 vs. both air groups; #P < 0.05 vs. vehicle-treated CS-exposed group.
CS and VEGFR-2 inhibitor increased proinflammatory mediators in mouse lung
The effects of CS and VEGFR-2 inhibitor on the levels of proinflammatory mediators (KC, MCP-1, IL-6, and TNF-α) in mouse lung homogenates were assessed using a Luminex beadlyte assay kit and were normalized with protein levels in lung homogenates. The levels of KC were significantly increased in the vehicle-treated CS group and both the VEGFR-2 inhibitor-treated air and CS groups compared with the vehicle-treated air group (P<0.001). Furthermore, KC levels were significantly increased in the VEGFR-2 inhibitor-treated CS-exposed group compared with the vehicle-treated CS-exposed groups (P<0.05), suggesting that VEGFR-2 inhibition augmented the CS-induced KC levels in mouse lung. Although no increase in MCP-1 level was observed in vehicle-treated CS-exposed group, a synergistic increase in MCP-1 level was observed in the VEGFR-2 inhibitor-treated CS-exposed group (P<0.05). The levels of IL-6 and TNF-α in the lung homogenates were significantly increased in both the vehicle and VEGFR-2 inhibitor-treated CS-exposed groups compared with their respective air groups (Fig. 2). However, the synergistic effects of CS exposure + VEGFR-2 inhibition was not observed for IL-6 and TNF-α release. Overall, these data showed that inhibition of VEGFR-2 augmented CS-induced release of key proinflammatory mediators in mouse lung.
Figure 2.

Increased levels of proinflammatory mediators in response to CS exposure and/or VEGFR-2 inhibition in mouse lung homogenate. Proinflammatory cytokines (KC/CXCL1, MCP-1/CCL2, IL-6, and TNF-α) were assessed in lung homogenates using a Luminex-based assay kit (Upstate Cell Signaling). The levels of KC and MCP-1 were significantly increased in VEGFR-2 inhibitor-treated CS-exposed group animals compared to the CS-exposed group (A, B). Furthermore, the levels of IL-6 and TNF-α were increased in response to CS exposure and in combination of CS exposure with VEGFR-2 inhibition compared with both air groups (C, D). A) #P < 0.05 vs. CS-exposed group; ***P < 0.001 vs. vehicle-treated air-exposed group. B) *P < 0.05 vs. vehicle-treated air-exposed groups; #P < 0.05 vs. vehicle-treated CS-exposed group. C) ***P < 0.001 vs. both air-exposed groups. D) *P < 0.05; **P < 0.01 vs. both air-exposed groups.
4-HNE adducts levels were increased in response to CS exposure and VEGFR-2 inhibition in mouse lung
CS exposure and VEGFR-2 inhibitor-mediated oxidative stress response were investigated by assessing 4-HNE adducts levels by immunohistochemistry in mouse lung sections. 4-HNE is a stable end product of lipid peroxidation in the cells. Higher levels of 4-HNE are found in the cells during oxidative stress due to increased lipid peroxidation, and therefore, it is used as a marker to assess oxidative stress. The 4-HNE adduct levels (staining score) were significantly increased in both airway and alveolar cells of the lung sections of mice treated with either VEGFR-2 inhibitor (P<0.001) or exposed to CS (P<0.001) or a combination of both (P<0.001) compared with vehicle-treated air group (Fig. 3A, B). Furthermore, the 4-HNE adducts level (staining score) were significantly increased in combined VEGFR-2 inhibitor-treated CS-exposed group compared with CS exposure alone group in airway epithelial cells (P<0.01). These data indicated that CS exposure or VEGFR-2 inhibitor treatment alone induces oxidative stress and in combination of VEGFR-2 inhibitor treatment and CS exposure oxidative stress was further increased particularly in airway epithelial cells.
Figure 3.

CS and VEGFR-2 inhibitor caused oxidative stress in vivo in mouse lungs and in vitro in HMVEC-Ls. 4-HNE adducts staining were increased in lung sections of mice treated with VEGFR-2 inhibitor and/or exposed to CS compared with vehicle-treated air-exposed group. A) Representative histological sections (×200) of mouse lungs showing 4-HNE adducts levels by staining (arrows) as assessed by immunohistochemistry method using mouse monoclonal antibody-specific for 4-HNE (n=4). Insets show enlarged alveolar and airway epithelium showing stained 4-HNE adducts. Alv.Epi. = alveolar epithelium; AW Epi. = airway epithelium. B) Intensity of 4-HNE adducts staining was assessed using a scoring system as described in Materials and Methods. ##P < 0.01 vs. vehicle-treated CS-exposed group; ***P < 0.001 vs. vehicle-treated air group; n = 4. C) ROS production in HMVEC-Ls in response to CSE and VEGFR-2 inhibitor was assessed using florescent dye carboxy-H2DCF-DA (Invitrogen) as described in Materials and Methods. Green fluorescent staining in HMVEC-Ls was observed under the fluorescent microscope (×200; n=3). D) Green fluorescent intensity was assessed using scoring system as described in Materials and Methods. #P < 0.05 vs. CSE or VEGFR-2 inhibitor-treated groups; **P < 0.01, ***P < 0.001 vs. control groups; n = 3.
ROS production was increased in response to CSE and VEGFR-2 inhibition in vitro in HMVEC-Ls
CSE- and VEGFR-2 inhibitor-induced ROS production were investigated using the fluorescent dye carboxy-H2DCF-DA in vitro utilizing HMVEC-Ls and confirmed the data of oxidative stress effect of VEGFR-2 inhibitor in mouse lung. ROS production was significantly increased by CSE treatments in a concentration-dependent manner in HMVEC-Ls (data not shown). VEGFR-2 inhibitor alone caused significantly increased ROS production compared with control experiments in HMVEC-Ls (P<0.01). Furthermore, ROS production was significantly increased when the cells were treated with CSE (0.25%) in combination with the VEGFR-2 inhibitor (0.5 μM) compared with CSE (0.25%) or VEGFR-2 inhibitor treatments alone in HMVEC-Ls (P<0.05; Fig. 3C, D). These data showed that both CSE and VEGFR-2 inhibition caused increased ROS production in HMVEC-Ls.
VEGFR-2 levels and its phosphorylation were decreased in response to CS and/or VEGFR-2 inhibition in mouse lung
The effects of CS and VEGFR-2 inhibitor on tyrosine-phosphorylated VEGFR-2 levels in mouse lungs were assessed by immunoblotting. The levels of phosphorylated VEGFR-2 were normalized with total VEGFR-2 found on the same blot. Significantly decreased levels of phosphorylated VEGFR-2 were observed in mouse lung tissues of the VEGFR-2 inhibitor-treated groups compared with vehicle groups (P<0.05), suggesting that subcutaneous injection of VEGFR-2 inhibitor (NVP-AAD777) impaired VEGFR-2 phosphorylation in the mouse lung (Fig. 4A, B). These data also confirmed the effectiveness of the dose of VEGFR-2 inhibitor administered (20 mg/kg body weight). The levels of VEGFR-2 expression were significantly reduced in both VEGFR-2 inhibitor-treated air- and CS-exposed groups and the vehicle-treated CS-exposed group compared with vehicle-treated air group (P<0.05; Fig. 4A, C). These data revealed that both CS and VEGFR-2 inhibition caused down-regulation of VEGFR-2 receptor protein expression in mouse lung.
Figure 4.

VEGFR-2 levels and its phosphorylation were decreased in response to CS and/or VEGFR-2 inhibition in mouse lung. Soluble proteins from lung homogenates were electrophoresed in a 4 –15% gradient PAGE gel and electroblotted into nitro-cellulose membranes. A) p-VEGFR-2 and VEGFR-2 protein were measured using immunoblotting with rabbit anti-tyr(p)-VEGFR-2 (Clone 4G10-universal p-tyr antibody, Upstate Cell Signaling) and anti-VEGFR-2 primary antibodies (Cell Signaling), respectively. B) Histograms represent mean ± SE relative levels of p-VEGFR-2 (n=4). *P < 0.05 vs. both vehicle-treated air- and CS-exposed groups. C) Histograms represent mean ± SE relative VEGFR-2 expression in mouse lungs (n=4). *P < 0.05 vs. vehicle-treated air-exposed group.
CSE down-regulated VEGFR-2 expression and impaired VEGF-induced VEGFR-2 phosphorylation in HMVECs
Because VEGFR-2 is an essential mediator of VEGF-induced endothelial function, the effect of CSE on VEGFR-2 expression level and VEGF-induced VEGFR-2 phosphorylation was assessed in vitro in HMVEC-Ls. We investigated the effect of CSE on basal VEGFR-2 protein expression level in a time- and concentration-dependent manner. Our data showed that at 2 h of CSE treatments, VEGFR-2 protein levels were neither down-regulated nor phosphorylated, but at 12 h of CSE treatment, VEGFR-2 levels were down-regulated. Therefore, to determine whether CSE had any effect on VEGF-induced VEGFR-2 phosphorylation, the 2 h time point was selected so as to rule out the direct effect CSE on VEGFR-2 down-regulation. The levels of total and phosphorylated (tyrosine 1175) VEGFR-2 were measured by immunoblotting after the cells were treated with VEGF. VEGFR-2 phosphorylation was significantly decreased in CSE-treated cells in concentration-dependent manner (0.1– 0.5%) compared with the control experiment (P<0.05; P<0.01; Fig. 5A, B). These data revealed that the CSE concentration dependently down-regulated VEGF-induced VEGFR-2 phosphorylation in vitro in HMVEC-Ls. Furthermore, it is confirmed that VEGF-induced VEGFR-2 phosphorylation was completely inhibited by the VEGFR-2 inhibitor NVP-AAD777 at the concentration of 1.0 μM in vitro in HMVEC-Ls. DMSO vehicle treatment had no effects on VEGF-induced VEGFR-2 phosphorylation. To determine the effect of CSE on VEGFR-2 expression levels, HMVEC-Ls were starved and treated for 12 h, and VEGFR-2 levels were measured by immunoblotting. CSE concentration dependently (0.1– 0.5%) down-regulated the VEGFR-2 expression levels (P<0.05; Fig. 5C, D). These new finding showed that CSE concentration dependently down-regulated the levels of VEGFR-2 in vitro in HMVEC-Ls.
Figure 5.

CSE concentration dependently decreased VEGFR-2 and VEGF-induced phosphorylation of VEGFR-2 (tyr 1175) in HMVEC-Ls. A) CSE- or VEGFR-2 inhibitor-treated cells (2 h) were incubated with VEGF as mentioned in Materials and Methods. Phosphorylated and total VEGFR-2 levels were measured using immunoblotting with rabbit monoclonal anti-p-VEGFR (tyr-1175, Cell Signaling Technology) and rabbit monoclonal anti-VEGFR-2 antibodies (Cell Signaling Technology), respectively. The VEGFR-2 blot showed two bands: 230 kDa, fully glycosylated functional receptor; and 200 kDa, semiglycosylated nonfunctional receptor. Phosphorylation was seen only in fully glycosylated functional receptor. CSE down-regulated VEGF-induced VEGFR-2 phosphorylation in a concentration-dependent manner. Pretreatment of cells with VEGFR-2 inhibitor (NVP-AAD777, 1 μM) abolished the VEGF-induced VEGFR-2 phosphorylation. B) Histograms represent mean ± SE relative VEGFR-2 phosphorylation (n=3). *P < 0.05, **P < 0.01 vs. control groups. C) VEGFR-2 levels were measured after cells were treated with CSE (0.1–0.5%) for 12 h using immunoblotting. VEGFR-2 blot showed two bands: 230 kDa, fully glycosylated functional receptor; and 200 kDa, semiglycosylated nonfunctional receptor. VEGFR-2 levels (230 kDa band) were significantly decreased in response to CSE in a concentration-dependent manner. D) Histograms represent mean ± SE relative expression levels of VEGFR-2 (n=3). *P < 0.05, **P < 0.01 vs. control groups.
VEGF levels were decreased in response to CS exposure and/or VEGFR-2 inhibition in mouse lung
To investigate the effect of CS and VEGFR-2 inhibitor on VEGF expression, VEGF protein levels were assessed by a Luminex beadlyte kit and normalized with protein levels in mouse lung homogenates. The VEGF levels in mouse lung were significantly decreased in the vehicle-treated CS-exposed group and VEGFR-2 inhibitor-treated air and CS-exposed groups when compared with the vehicle-treated air group (P<0.05; P<0.01). Further decreases in VEGF levels were observed in combination of the VEGFR-2 inhibitor- and CS-exposed group compared with vehicle-treated CS-exposed group in mouse lungs, suggesting that VEGFR-2 inhibition caused further decreased of CS-induced down-regulation of VEGF (P<0.05). These data suggested that both CS and VEGFR-2 inhibition led to down-regulation of VEGF protein expression in mouse lungs (Fig. 6).
Figure 6.
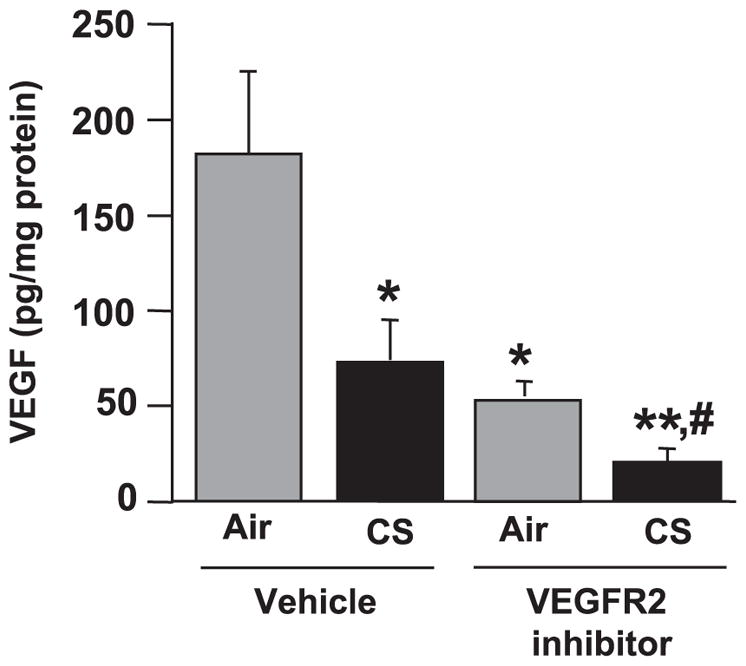
VEGF levels were decreased in response to CS exposure and/or VEGFR-2 inhibition in mouse lung. VEGF protein levels, as assessed by a Luminex-based beadlyte kit method (mouse multicytokine beadmaster kit, Upstate Cell Signaling), were significantly decreased after CS exposure, VEGFR-2 inhibition and in combination of both treatments compared with vehicle-treated air-exposed animals. Histograms represent mean ± SE VEGF levels (n=4). *P < 0.05, **P < 0.01 vs. vehicle-treated air-exposed group; #P < 0.05 vs. vehicle-treated CS-exposed group.
Phosphorylated and total eNOS protein levels were decreased in mouse lungs in response to CS and/or VEGFR-2 inhibition
VEGF-induced VEGFR-2 phosphorylation and downstream signaling induce activation of eNOS, a key enzyme linked to endothelial function. Therefore, the effects of CS exposure and VEGFR-2 inhibition on p-eNOS and total eNOS protein levels were assessed in mouse lung. Our data showed that CS exposure or VEGFR-2 inhibition significantly reduced the level of p-eNOS and total eNOS protein in mouse lung as compared with that of the vehicle-treated air-exposed group (Fig. 7). Furthermore, inhibition of VEGFR-2 augmented CS-induced reduction of p-eNOS levels in mouse lung (P<0.05). These data suggested that eNOS is more susceptible to the effect of CS when its signaling is impaired by VEGFR-2 inhibition in mouse lung.
Figure 7.

Phosphorylated and total eNOS protein levels were decreased in mouse lungs in response to CS and/or VEGFR-2 inhibition. A) Soluble eNOS proteins from mouse lung were immunoprecipitated (IP) as described in Materials and Methods and immunoblotted (IB) for p-eNOS (Ser-1177) and eNOS (Cell Signaling Technology). B) Histograms represent mean ± SE relative p-eNOS levels (n=4). *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle-treated air-exposed group; #P < 0.05 vs. vehicle-treated smoke-exposed group. C) Soluble proteins from mouse lung homogenates were electrophoresed in SDS-PAGE gel. eNOS protein levels were measured using immunoblotting with rabbit monoclonal anti-eNOS primary antibody (Cell Signaling technology). D) Histograms represent mean ± SE relative eNOS expression (n=4). *P < 0.05, **P < 0.01 vs. vehicle-treated air-exposed group; #P < 0.05 vs. VEGFR-2 inhibitor-treated air-exposed group.
CS down-regulated the levels of eNOS in vitro in HMVEC-Ls
Since VEGFR-2 and eNOS are essential mediators of VEGF-induced endothelial function, the effect of CSE on eNOS protein levels in vitro in HMVEC-Ls was determined. Cells were starved and treated for 12 h with CSE, and eNOS protein levels were measured by immunoblotting method. CSE concentration dependently (0.25 and 0.5%) down-regulated the eNOS protein levels (P<0.05; Fig. 8A, B). However, the levels of eNOS were unaffected after 12 h at a relatively low level of CSE (0.1%). Nevertheless, these data suggested that CSE down-regulated the eNOS protein levels in HMVECs.
Figure 8.
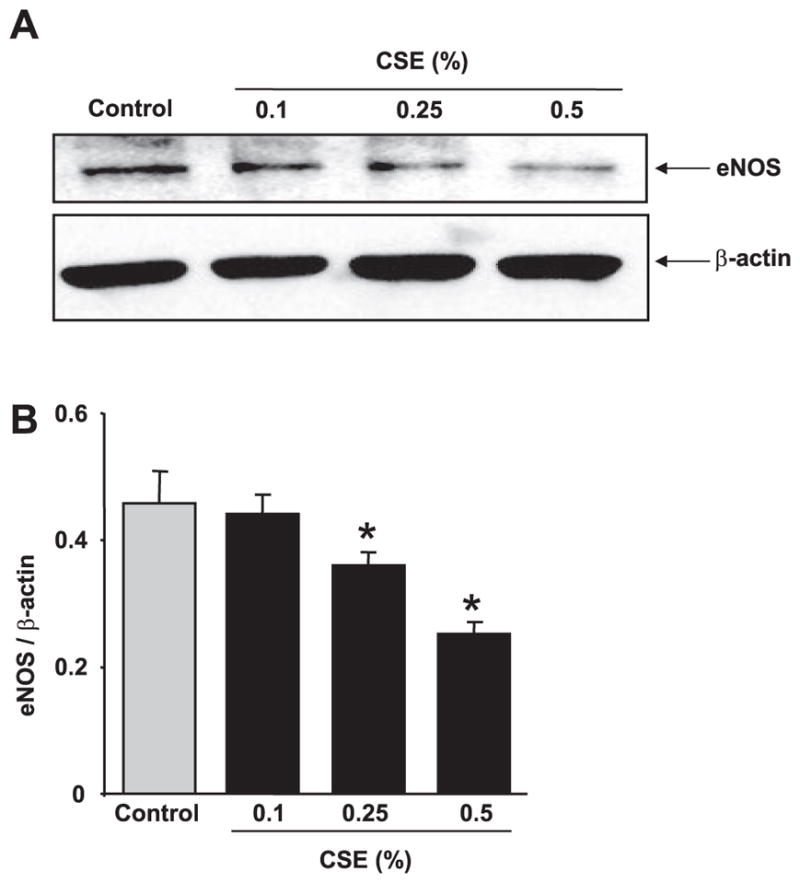
CSE concentration dependently down-regulated the level of eNOS in HMVEC-Ls. A) HMVEC-Ls were treated with CSE (0.1–0.5%) for 12 h in 0.1% FBS containing EGM-2 media, and eNOS protein levels were measured using immunoblotting. eNOS protein levels were down-regulated in a concentration-dependent manner (0.25 and 0.5%. B) Histograms represent mean ± SE relative expression levels of VEGFR-2 and eNOS (n=3). *P < 0.05 vs. control groups.
CS impaired VEGF-induced cell migration and angiogenesis in vitro in HMVEC-Ls
We further investigated whether the CSE-mediated decrease in the level of VEGFR-2 and VEGF-induced VEGFR-2 phosphorylation caused impaired cell migration and angiogenesis using the scratch and capillary-like tube formation assays, respectively. Specific VEGFR-2 inhibitor, NVP-AAD777 was used to compare the effect of CSE-mediated VEGF-induced cell migration and angiogenesis. VEGF-induced cell migration (P<0.01; Fig. 9A, B) and capillary-like tube formation (P<0.05; Fig. 9C, D) were significantly reduced in CSE-treated HMVEC-Ls compared with control (VEGF-treated) experiments. Furthermore, pretreatment of HMVEC-Ls with NAC, a thiol antioxidant, significantly attenuated the CSE-induced impaired cell migration (P<0.05). However, VEGFR-2 inhibitor-mediated VEGF-induced impaired cell migration was not reversed by NAC pretreatment. These data suggested that CSE-induced impaired cell migration is due to ROS generated from CSE or the presence of electrophilic compounds in CSE. Significantly inhibited cell migration and capillary-like tube formations were observed in HMVEC-Ls treated with VEGFR-2 inhibitor, demonstrating the involvement of VEGF-induced VEGFR-2 phosphorylation and its downstream signaling pathway in endothelial cell migration and angiogenesis (P<0.001). In both scratch and tube formation assays, we observed that VEGFR-2 inhibition (2 h) prevents attachment of cells to the culture plates and some morphological changes. However, cell viability was not significantly affected with VEGFR-2 inhibition as assessed by trypan blue exclusion test.
Figure 9.

CSE impaired VEGF-induced cell migration and capillary-like tube formation in endothelial cells. A) HMVEC-Ls were starved (6 h) and pretreated with CSE (0.25%), NAC (1 mM), VEGFR-2 inhibitor (1 μM), CSE (0.25%) + NAC (1 mM), or VEGFR-2 inhibitor (1 μM) + NAC (1 mM) for 2 h in EGM-2 media containing 0.1% FBS without VEGF. The treatment with NAC (1 mM) was performed 1 h before CSE or VEGFR-2 inhibitor treatments. After 2 h, cells were washed twice with PBS, and the bottom of the culture plates were scratched using a standard 200 μl pipette tip and treated with VEGF (20 ng/ml) in EGM-2 media containing 0.1% FBS. Representative photographs of cells (×200) were taken at the beginning and after 12 h. Number of cells migrated beyond the scratch lines after 12 h was counted under the microscope by a single investigator in a blinded manner. B) Histograms represent mean ± SE cell migration beyond the scratch lines (n=3). *P < 0.05 vs. CSE + NAC-treated group; **P < 0.01 vs. CSE-treated group as indicated; ***P < 0.001 vs. VEGF-treated control group. C) HMVEC-Ls were starved (6 h) and pretreated with CSE (0.25%) or VEGFR-2 inhibitor (1 μM) for 2 h in EGM-2 media containing 0.1% FBS without VEGF. Cells were detached and allowed to grow in the plates coated with Matrigel matrix (BD Bioscience) with or without VEGF (20 ng/ml). Representative photographs (×200) were taken after 24 h, and the number of capillary-like tube branches was counted in a blinded manner (n=3). D) Histogram represents mean ± SE capillary-like tube branches (n=3). *P < 0.05, ***P < 0.001 vs. VEGF-treated control group.
DISCUSSION
The oxidative stress imposed by CS causes an inflammatory response due to the release of proinflammatory mediators and recruitment of inflammatory cells, such as macrophages and neutrophils into the lung (24, 25, 29). Our results confirmed that CS induced an influx of inflammatory cells and increased proinflammatory mediators (KC and MCP-1) in mouse lungs. Surprisingly, inhibition of VEGFR-2 augmented CSE-induced inflammatory response in the lung. However, synergistic effects of CS exposure and VEGFR-2 inhibition were not seen for IL-6 and TNF-α release. It is known that IL-6 and TNF-α act as autocrine growth factors for tumor angiogenesis and that these cytokines can be prometastatic or proangiogenic (30, 31). It is also known that inhibition of angiogenesis down-regulated the levels of IL-6 and TNF-α in mouse serum (31). Since the VEGFR-2 inhibitor (NVP-AAD777) is antiangiogenic, it is possible that inhibition of VEGFR-2 may have a confounding effect on these cytokine release in mouse lung. The other reason may be that the there was already a threshold level of induction of IL-6 and TNF-α in response to CS exposure, which was not augmented by VEGFR-2 inhibition. Nevertheless, the number of macrophages in lung interstitium and the levels of MCP-1 and KC in lung tissue was significantly increased in the group treated with VEGFR-2 inhibitor and subsequent exposure to CS, suggesting that VEGFR-2 inhibition augmented the CS-induced inflammation in mouse lungs.
Oxidative stress was increased with VEGFR-2 inhibition and CS exposure, and this was augmented in combination of VEGFR-2 inhibition with CS exposure as assessed by lipid peroxidation products (4-HNE adducts) stained in airway epithelial cells in vivo in mouse lung and carboxy-H2DCF-DA fluorescent staining (ROS release) in vitro in HMVEC-Ls. However, the mechanism for VEGFR-2 inhibitor induced ROS generation and inflammation in the lung is not clear. Increased levels of oxidative stress, as measured by 4-HNE adducts staining, occur in the lungs of smokers and in patients with COPD (32). It was also shown that chronic inhibition of global VEGFR by SU5416 resulted in oxidative stress in rat lungs (23). It was speculated that the interruption of VEGF survival signals is interpreted by lung cells as a “danger signal” resulting in oxidative stress as a downstream response (23). Hence, we propose that the mechanism of VEGFR-2 inhibitor-induced oxidative stress in vivo in mouse lung and in vitro in HMVEC-Ls may either be due to activation of NADPH oxidase or dual oxidases in endothelial cells; however, this concept requires further investigation.
Both VEGF and VEGFR-2 expression are decreased in emphysematous lungs (20, 21). VEGF levels in the BALF were also decreased in smokers (33) but not in the lungs of patients with mild COPD (34, 35). It is also interesting to note that chronic inhibition of VEGFR leads to enlargement of airspaces and decreased capillary density, which is characteristic of emphysema in rat lungs (21, 36). Moreover, patients with COPD also display reduced vascularization of the lung, including decreased capillary density (37). In the present study, we showed the decreased levels of VEGF and VEGFR-2 levels in both the VEGFR-2 inhibitor and CS-exposed groups in vivo in mouse lung and the decreased level of CSE-mediated VEGFR-2 expression in vitro in HMVEC-Ls. These results corroborate previous findings in smokers and in patients with COPD showing a decreased lung level of VEGF and VEGFR-2 (22) and implicate CS-derived free radicals as being directly involved in down-regulation of VEGF and VEGFR-2 levels in endothelial cells. However, the mechanism of CS-mediated down-regulation of VEGF and VEGFR-2 is not known. It is possible that aldehyde-mediated modifications of cellular signaling may impair VEGF release from epithelial cells and macrophages as shown by 4-HNE adducts formation in these cells in response to NVP-AAD777 treatment and CSE exposure. CS exposure may also alter kinase signaling mechanism or induce post-translational modifications of VEGFR-2 subsequently leading to its degradation.
VEGF phosphorylates VEGFR-2, and its downstream signaling activates eNOS, which is a key enzyme in the regulation of endothelial cell migration, proliferation, and survival (1). Cigarette smoking is associated with endothelial dysfunction (2, 8, 10–12). Clinical data also showed impaired vascularization in smokers and those with COPD (38). However, the exact mechanisms involved in CS-induced endothelial dysfunction and decreased vasculature in the lungs are not known. Hence, we propose that CS causes down-regulation of VEGFR-2 expression and impairs VEGF-induced VEGFR-2 phosphorylation and eNOS activation resulting in endothelial dysfunction. To support these observations, we carried out in vitro experiments using HMVEC-Ls to determine the CSE-mediated down-regulation of VEGFR-2 levels. It is known that phosphorylation of VEGFR-2 tyrosine residue at 1175 (in human) is essential for vasculogenesis and inhibition of phosphorylation may be associated with reduced VEGFR-2 down-stream signaling (39). Consistent with in vivo findings, CSE down-regulated VEGFR-2 and eNOS protein expression and VEGF-induced VEGFR-2 tyrosine (tyr-1175) phosphorylation in endothelial cells. Similarly, decreased levels of phosphorylated VEGFR-2 were shown in the lungs of smokers and COPD patients (22). The reason for CS-mediated down-regulation of VEGFR-2 phosphorylation is not known. However, our data indicated that tyrosine residues are nitrated (unpublished results) in response to CSE treatment rendering VEGFR-2 inactive for downstream kinase signaling or affecting its activity. The other likely reason would be that CS-derived lipid peroxidation products, such as 4-HNE and acrolein and/or reactive nitrogen species, react with VEGFR-2 rendering VEGFR-2 inactive for downstream kinase signaling or affecting its activity (40, 41).
VEGF-mediated VEGFR-2 phosphorylation and downstream signaling induce eNOS activation in endothelium (1). Since eNOS is an essential mediator of VEGF-VEGFR-2-mediated endothelial function, we determined whether CS exposure had any effect on phosphorylation of eNOS and its levels in mouse lung in vivo and in HMVEC-Ls in vitro. VEGFR-2 inhibition led to CS-induced down-regulation of phosphorylated eNOS and total eNOS both in HMVEC-Ls and mouse lung. However, at low levels of CSE the eNOS protein level was not affected in vitro in HMVEC-Ls. It has been shown that CSE causes phosphorylation of eNOS via activation of redox-sensitive phosphoinositide 3-kinase (PI3K) and Akt-signaling pathways (42). Furthermore, phosphorylation of eNOS released NO and up-regulated the expression of eNOS protein (43). Our data showed no effect for low concentration of CSE on eNOS levels, suggesting a VEGFR-2-independent (bypassing VEGFR-2) signaling mechanism possibly by direct activation PI3K and Akt is responsible for nullifying the effect of CSE-induced eNOS down-regulation in endothelial cells in vitro. However, at high concentrations of CSE, the PI3-kinase/Akt-induced eNOS activation is impaired by CSE-mediated aldehyde/oxidative stress-induced effects, which would then impair VEGFR-2-mediated downstream signaling for eNOS phosphorylation. Indeed, Su et al. (44, 45) have shown CSE (high concentrations) mediated irreversible inhibition of eNOS activity in endothelial cells, possibly by kinase signaling mechanisms. Nevertheless, our in vivo data clearly show down-regulation of phophorylated eNOS, suggesting CS-mediated endothelial dysfunction in the lung.
It has previously been shown that NO donors can rescue cellular migration and tube formation in endothelial cells exposed to CSE, suggesting that impaired NO biosynthesis is related to dysfunctional endothelium-dependent vasodilation in smokers (19). We further investigated whether the CSE-mediated decrease in the level of VEGFR-2 and VEGF-induced VEGFR-2 phosphorylation caused impaired cell migration and angiogenesis. Both angiogenesis and cell migration are important in neovascularization in the lung. Angiogenesis is driven by microvascular endothelial cells, which on activation by angiogenic growth factors such as VEGF migrate into the interstitial matrix, proliferate, and form new capillary-like structures (46). Our data showed that CSE impaired endothelial cell migration and angiogenesis as assessed by wound healing scratch and capillary-like tube formation assays, respectively. Hence, the endothelial dysfunction seen in smokers could possibly be related to an impaired response to VEGF. The CSE-mediated inhibition of endothelial cell migration may be associated with a reduced expression of several integrins that are involved in NO-dependent endothelial migration (1). The signaling mechanisms for CS-induced alteration in cell migration and angiogenesis are not known. We hypothesize that CS-induced ROS and free radicals (micromolar to millimolar levels) cause alterations in VEGF-VEGFR-2 downstream signaling for cell migration and angiogenesis. This may be the mechanism for CS-induced endothelial dysfunction. However, it has been shown that VEGF signaling is mediated via NADPH oxidase-derived ROS production (nanomolar to picomolar levels) in particular by the gp91phox subunit of NADPH oxidase leading to downstream signaling of angiogenesis (47, 48). Hence, a fine tuning exists between low levels of ROS, which is proangiogenic, and high levels of ROS and free radicals and aldehydes, which may be antiangiogenic.
Our data showed that pretreatment of HMVEC-Ls with NAC, a thiol nucleophilic antioxidant, significantly attenuated the CSE-mediated VEGF-induced impaired cell migration. These findings suggested that CSE-mediated impaired cell migration is associated with oxidative stress imposed by CSE. We further observed that CS-inhibited VEGF-induced phosphorylation of VEGFR-2 was reversed by NAC, suggesting that NAC is in fact conjugating free radicals and aldehydes present in CS (unpublished results). Similarly, Michaud et al. (19) have recently shown that CSE-impaired VEGF-induced endothelial cell migration and tube formation was free radical dependent. However, VEGFR-2-inhibited cell migration was not reversed by NAC pretreatment, although VEGFR-2 inhibition was associated with ROS generation. The differential effect of NAC on CSE- and VEGFR-2 inhibitor-mediated attenuation of cell migration is not known. It is possible that ROS production in response to VEGFR-2 inhibition is secondary to its ability to affect cell migration. Nevertheless, these data suggest that CSE-induced (but not VEGFR-2-induced) impaired cell migration is due to either ROS generated from CSE (via NADPH oxidase activation) or to the electrophilic compounds present in CSE (49).
Taken together, our data of mouse lung in vivo and in human endothelial cells in vitro support the concept that inhibition of VEGFR-2 led to increased oxidative stress and inflammatory response and CS exposure further augmented oxidative stress and inflammatory response. Overall, CS-induced oxidative stress impaired both VEGF-induced VEGFR-2 phosphorylation and VEGFR-2 expression in HMVEC-Ls in vitro and in mouse lungs in vivo leading to endothelial dysfunction as assessed by decreased levels of eNOS, cell migration, and angiogenesis. This, in turn, could lead to endothelial dysfunction associated with the development of atherosclerotic lesions and the triggering of acute ischemic events in patients exposed to CS. Our data also demonstrate that the CS-induced impaired endothelial function was attenuated by pretreatment with the thiol antioxidant NAC in HMVEC-Ls in vitro. These new findings not only define the basic understanding of oxidant-induced endothelial dysfunction via the VEGF-VEGFR-2 specific pathway but also have broad implications in the pathogenesis of various CS-mediated pulmonary and cardiovascular diseases where endothelial dysfunction occurs. Further studies are in progress to determine the mechanisms by which CS mediates VEGF-VEGFR-2-induced endothelial dysfunction in the lung.
Acknowledgments
This study was supported by the National Institute of Environmental Health Sciences (NIEHS) Environmental Health Sciences Center grant ES01247, NIEHS Toxicology Training grant ES07026, and Philip Morris USA Inc. and Philip Morris International. We thank J. Wood (Novartis Pharma AG, Basel, Switzerland) for providing the VEGF/ KDR tyrosine kinase inhibitor NVP-AAD777.
References
- 1.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signaling–in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 2.Voelkel NF, Vandivier RW, Tuder RM. Vascular endothelial growth factor in the lung. Am J Physiol Lung Cell Mol Physiol. 2006;290:L209–L221. doi: 10.1152/ajplung.00185.2005. [DOI] [PubMed] [Google Scholar]
- 3.Tuder RM, Flook BE, Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia: modulation of gene expression by nitric oxide. J Clin Invest. 1995;95:1798–1807. doi: 10.1172/JCI117858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, Ferrara N. VEGF is required for growth and survival in neonatal mice. Development. 1999;126:1149–1159. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
- 5.Pryor WA, Stone K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann N Y Acad Sci. 1993;686:12–27. doi: 10.1111/j.1749-6632.1993.tb39148.x. [DOI] [PubMed] [Google Scholar]
- 6.Liebow AA. Pulmonary emphysema with special emphasis to vascular changes. Am Rev Respir Dis. 1959;80:67–93. doi: 10.1164/arrd.1959.80.1P2.67. [DOI] [PubMed] [Google Scholar]
- 7.Yamato H, Sun JP, Churg A, Wright JL. Cigarette smoke-induced emphysema in guinea pigs is associated with diffusely decreased capillary density and capillary narrowing. Lab Invest. 1996;75:211–219. [PubMed] [Google Scholar]
- 8.Puranik R, Celermajer DS. Smoking and endothelial function. Prog Cardiovasc Dis. 2003;45:443–458. doi: 10.1053/pcad.2003.YPCAD13. [DOI] [PubMed] [Google Scholar]
- 9.Mathew R, Huang J, Gewitz MH. Pulmonary artery hypertension: caveolin-1 and eNOS interrelationship: a new perspective. Cardiol Rev. 2007;15:143–149. doi: 10.1097/01.crd.0000249381.49138.b9. [DOI] [PubMed] [Google Scholar]
- 10.Zeiher AM, Schächinger V, Minners J. Long-term cigarette smoking impairs endothelium-dependent coronary arterial vasodilator function. Circulation. 1995;92:94–100. doi: 10.1161/01.cir.92.5.1094. [DOI] [PubMed] [Google Scholar]
- 11.Tanriverdi H, Evrengul H, Kuru O, Tanriverdi S, Seleci D, Enli Y, Kaftan HA, Kilic M. Cigarette smoking induced oxidative stress may impair endothelial function and coronary blood flow in angiographically normal coronary arteries. Circ J. 2006;70:593–599. doi: 10.1253/circj.70.593. [DOI] [PubMed] [Google Scholar]
- 12.Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731–1737. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 13.Wright JL, Tai H, Churg A. Vasoactive mediators and pulmonary hypertension after cigarette smoke exposure in the guinea pig. J Appl Physiol. 2006;100:672–678. doi: 10.1152/japplphysiol.00274.2005. [DOI] [PubMed] [Google Scholar]
- 14.Lee JH, Lee DS, Kim EK, Choe KH, Oh YM, Shim TS, Kim SE, Lee YS, Lee SD. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med. 2005;172:987–993. doi: 10.1164/rccm.200501-041OC. [DOI] [PubMed] [Google Scholar]
- 15.Arnal JF, Dinh-Xuan AT, Pueyo M, Darblade B, Rami J. Endothelium-derived nitric oxide and vascular physiology and pathology. Cell Mol Life Sci. 1999;5:1078–1087. doi: 10.1007/s000180050358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res. 2003;93:354–363. doi: 10.1161/01.RES.0000089257.94002.96. [DOI] [PubMed] [Google Scholar]
- 17.Shen BQ, Lee DY, Zioncheck TF. Vascular endothelial growth factor governs endothelial nitric-oxide synthase expression via a KDR/Flk-1 receptor and a protein kinase C signaling pathway. J Biol Chem. 1999;274:33057–33063. doi: 10.1074/jbc.274.46.33057. [DOI] [PubMed] [Google Scholar]
- 18.Tanimoto T, Jin ZG, Berk BC. Transactivation of vascular endothelial growth factor (VEGF) receptor Flk-1/ KDR is involved in sphingosine 1-phosphate-stimulated phosphorylation of Akt and endothelial nitric-oxide synthase (eNOS) J Biol Chem. 2002;277:42997–43001. doi: 10.1074/jbc.M204764200. [DOI] [PubMed] [Google Scholar]
- 19.Michaud SE, Dussault S, Groleau J, Haddad P, Rivard A. Cigarette smoke exposure impairs VEGF-induced endothelial cell migration: role of NO and reactive oxygen species. J Mol Cell Cardiol. 2006;41:275–284. doi: 10.1016/j.yjmcc.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Kasahara Y, Tuder RM, Cool CD, Lynch DA, Flores SC, Voelkel NF. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med. 2001;163:737–744. doi: 10.1164/ajrccm.163.3.2002117. [DOI] [PubMed] [Google Scholar]
- 21.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106:1311–1319. doi: 10.1172/JCI10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marwick JA, Stevenson CS, Giddings J, MacNee W, Butler K, Rahman I, Kirkham PA. Cigarette smoke disrupts VEGF165-VEGFR-2 receptor signaling complex in rat lungs and patients with COPD: morphological impact of VEGFR-2 inhibition. Am J Physiol Lung Cell Mol Physiol. 2006;290:L897–L908. doi: 10.1152/ajplung.00116.2005. [DOI] [PubMed] [Google Scholar]
- 23.Tuder RM, Zhen L, Cho CY, Taraseviciene-Stewart L, Kasahara Y, Salvemini D, Voelkel NF, Flores SC. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol. 2003;29:88–97. doi: 10.1165/rcmb.2002-0228OC. [DOI] [PubMed] [Google Scholar]
- 24.Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol. 2007;292:L567–576. doi: 10.1152/ajplung.00308.2006. [DOI] [PubMed] [Google Scholar]
- 25.Thatcher TH, McHugh NA, Egan RW, Chapman RW, Hey JA, Turner CK, Redonnet MR, Seweryniak KE, Sime PJ, Phipps RP. Role of CXCR2 in cigarette smoke-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2005;289:L322–L328. doi: 10.1152/ajplung.00039.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moodie FM, Marwick JA, Anderson CS, Szulakowski P, Biswas SK, Bauter MR, Kilty I, Rahman I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-κB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J. 2004;18:1897–1899. doi: 10.1096/fj.04-1506fje. [DOI] [PubMed] [Google Scholar]
- 27.Yang SR, Chida AS, Bauter M, Shafiq N, Seweryniak K, Maggirwar SB, Kilty I, Rahman I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-κB and post-translational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006;291:L46–L57. doi: 10.1152/ajplung.00241.2005. [DOI] [PubMed] [Google Scholar]
- 28.Carp H, Janoff A. Possible mechanisms of emphysema in smokers. In vitro suppression of serum elastase-inhibitory capacity by fresh cigarette smoke and its prevention by antioxidants. Am Rev Respir Dis. 1978;118:617–621. doi: 10.1164/arrd.1978.118.3.617. [DOI] [PubMed] [Google Scholar]
- 29.Thatcher TH, Maggirwar SB, Baglole CJ, Lakatos HF, Gasiewicz TA, Phipps RP, Sime PJ. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am J Pathol. 2007;170:855–864. doi: 10.2353/ajpath.2007.060391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thejass P, Kuttan G. Antiangiogenic activity of diallyl sulfide (DAS) Int Immunopharmacol. 2007;7:295–305. doi: 10.1016/j.intimp.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 31.Thejass P, Kuttan G. Inhibition of endothelial cell differentiation and proinflammatory cytokine production during angiogenesis by allyl isothiocyanate and phenyl isothiocyanate. Integr Cancer Ther. 2007;6:389–399. doi: 10.1177/1534735407309084. [DOI] [PubMed] [Google Scholar]
- 32.Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, Stolk J, MacNee W, De Boer WI. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166:490–495. doi: 10.1164/rccm.2110101. [DOI] [PubMed] [Google Scholar]
- 33.Koyama S, Sato E, Haniuda M, Numanami H, Nagai S, Izumi T. Decreased level of vascular endothelial growth factor in bronchoalveolar lavage fluid of normal smokers and patients with pulmonary fibrosis. Am J Respir Crit Care Med. 2002;166:382–385. doi: 10.1164/rccm.2103112. [DOI] [PubMed] [Google Scholar]
- 34.Kranenburg AR, de Boer WI, Alagappan VK, Sterk PJ, Sharma HS. Enhanced bronchial expression of vascular endothelial growth factor and receptors (Flk-1 and Flt-1) in patients with chronic obstructive pulmonary disease. Thorax. 2005;60:106–113. doi: 10.1136/thx.2004.023986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santos S, Peinado VI, Ramirez J, Morales-Blanhir J, Bastos R, Roca J, Rodriguez-Roisin R, Barbera JA. Enhanced expression of vascular endothelial growth factor in pulmonary arteries of smokers and patients with moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167:1250–1256. doi: 10.1164/rccm.200210-1233OC. [DOI] [PubMed] [Google Scholar]
- 36.Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF, Abman SH. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol. 2000;279:L600–L607. doi: 10.1152/ajplung.2000.279.3.L600. [DOI] [PubMed] [Google Scholar]
- 37.Voelkel NF, Cool C. Pulmonary vascular involvement in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2003;46:28s–32s. doi: 10.1183/09031936.03.00000503. [DOI] [PubMed] [Google Scholar]
- 38.Papaioannou AI, Kostikas K, Kollia P, Gourgoulianis KI. Clinical implications for vascular endothelial growth factor in the lung: friend or foe? Respir Res. 2006;177:128. doi: 10.1186/1465-9921-7-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M. Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci U S A. 2005;102:1076–1081. doi: 10.1073/pnas.0404984102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rahman I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. J Biochem Mol Biol. 2003;36:95–109. doi: 10.5483/bmbrep.2003.36.1.095. [DOI] [PubMed] [Google Scholar]
- 41.Yamaguchi Y, Nasu F, Harada A, Kunitomo M. Oxidants in the gas phase of cigarette smoke pass through the lung alveolar wall and raise systemic oxidative stress. J Pharmacol Sci. 2007;103:275–282. doi: 10.1254/jphs.fp0061055. [DOI] [PubMed] [Google Scholar]
- 42.Wagner L, Laczy B, Tamaskó M, Mazák I, Markó L, Molnár GA, Wagner Z, Mohás M, Cseh J, Fekete A, Wittmann I. Cigarette smoke-induced alterations in endothelial nitric oxide synthase phosphorylation: role of protein kinase C. Endothelium. 2007;14:245–255. doi: 10.1080/10623320701606707. [DOI] [PubMed] [Google Scholar]
- 43.Jayachandran M, Hayashi T, Sumi D, Thakur NK, Kano H, Ignarro LJ, Iguchi A. Up-regulation of endothelial nitric oxide synthase through beta(2)-adrenergic receptor–the role of a beta-blocker with NO-releasing action. Biochem Biophys Res Commun. 2001;280:589–594. doi: 10.1006/bbrc.2000.4177. [DOI] [PubMed] [Google Scholar]
- 44.Su Y, Han W, Giraldo C, De Li Y, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. 1998;19:819–825. doi: 10.1165/ajrcmb.19.5.3091. [DOI] [PubMed] [Google Scholar]
- 45.Su Y, Cao W, Han Z, Block ER. Cigarette smoke extract inhibits angiogenesis of pulmonary artery endothelial cells: the role of calpain. Am J Physiol Lung Cell Mol Physiol. 2004;287:L794–L800. doi: 10.1152/ajplung.00079.2004. [DOI] [PubMed] [Google Scholar]
- 46.Ucuzian AA, Greisler HP. In vitro models of angiogenesis. World J Surg. 2007;31:654–663. doi: 10.1007/s00268-006-0763-4. [DOI] [PubMed] [Google Scholar]
- 47.Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C, Alexander RW. Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res. 2002;91:1160–1167. doi: 10.1161/01.res.0000046227.65158.f8. [DOI] [PubMed] [Google Scholar]
- 48.Ushio-Fukai M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2007;9:731–739. doi: 10.1089/ars.2007.1556. [DOI] [PubMed] [Google Scholar]
- 49.Orosz Z, Csiszar A, Labinskyy N, Smith K, Kaminski PM, Ferdinandy P, Wolin MS, Rivera A, Ungvari Z. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: role of NAD(P)H oxidase activation. Am J Physiol Heart Circ Physiol. 2007;292:H130–H139. doi: 10.1152/ajpheart.00599.2006. [DOI] [PubMed] [Google Scholar]