

Abstract
The Rac1 GTPase is an essential and ubiquitous protein that signals through numerous pathways to control critical cellular processes, including cell growth, morphology, and motility. Rac1 deletion is embryonic lethal, and its dysregulation or mutation can promote cancer, arthritis, cardiovascular disease, and neurological disorders. Rac1 activity is highly regulated by modulatory proteins and posttranslational modifications. Whereas much attention has been devoted to guanine nucleotide exchange factors that act on Rac1 to promote GTP loading and Rac1 activation, cellular oxidants may also regulate Rac1 activation by promoting guanine nucleotide exchange. Herein, we show that Rac1 contains a redox-sensitive cysteine (Cys18) that can be selectively oxidized at physiological pH because of its lowered pKa. Consistent with these observations, we show that Rac1 is glutathiolated in primary chondrocytes. Oxidation of Cys18 by glutathione greatly perturbs Rac1 guanine nucleotide binding and promotes nucleotide exchange. As aspartate substitutions have been previously used to mimic cysteine oxidation, we characterized the biochemical properties of Rac1C18D. We also evaluated Rac1C18S as a redox-insensitive variant and found that it retains structural and biochemical properties similar to those of Rac1WT but is resistant to thiol oxidation. In addition, Rac1C18D, but not Rac1C18S, shows greatly enhanced nucleotide exchange, similar to that observed for Rac1 oxidation by glutathione. We employed Rac1C18D in cell-based studies to assess whether this fast-cycling variant, which mimics Rac1 oxidation by glutathione, affects Rac1 activity and function. Expression of Rac1C18D in Swiss 3T3 cells showed greatly enhanced GTP-bound Rac1 relative to Rac1WT and the redox-insensitive Rac1C18S variant. Moreover, expression of Rac1C18D in HEK-293T cells greatly promoted lamellipodia formation. Our results suggest that Rac1 oxidation at Cys18 is a novel posttranslational modification that upregulates Rac1 activity.
Keywords: Rac1 GTPases, Reactive oxygen species, Reactive nitrogen species, Glutathiolation, Cysteine oxidation, Free radicals
1. Introduction
Rac1 is a member of the Rho subclass of Ras superfamily GTPases. It functions as a molecular switch by cycling between active GTP- and inactive GDP-bound states to control the timing and specificity of cellular pathways that regulate diverse cell functions, including gene expression, cell motility, cell morphology, and cell cycling [1]. Owing to the slow intrinsic rates of nucleotide exchange and hydrolysis, temporal regulation of Rac1 activity requires modulatory factors, such as guanine nucleotide exchange factors (GEFs)2, which facilitate exchange of GDP for GTP; GTPase-activating proteins (GAPs), which catalyze GTP hydrolysis; and guanine nucleotide dissociation inhibitors (GDIs), which prevent GDP dissociation and sequester Rac1 away from cell membranes [2,3]. In addition, Rac1 is spatially and temporally regulated by various posttranslational modifications, including C-terminal lipidation [4–6], phosphorylation [7], ubiquitination [8,9], and SUMOylation [10]. Rac1 is an essential protein [11] that plays a critical role in regulating multiple cellular processes. Its dysregulation is correlated with many diseases, including cancer [12,13], osteoarthritis [14], cardiovascular disease [15], and neurological disorders [16]. Until recently, upregulation of the activity of Rho family GTPases in cancer was believed to result solely from altered Rac1 expression levels and/or aberrant expression or regulation of Rac1 GEFs, GAPs, and GDIs [17,18]. However, oncogenic mutations have been discovered in Rac1, Rac2, and Cdc42 [19]. In particular, a Rac1P29S mutant has recently been identified in 9% of primary melanomas [20], which drives oncogenic transformation in melanocytes due to increased nucleotide cycling [21–23], and Rac2P29L has been sporadically observed in human melanoma and breast cancers [24]. Another mutant, Rac1N92I, was identified in the fibrosarcoma cell line HT1080 [24]. Further, the Rac1 splice variant Rac1b also promotes fast nucleotide cycling and has been shown to promote cellular transformation [25] as well as sustaining tumor survival [26]. Whereas most oncogenic mutations identified in Ras family proteins cause chronic activation by impairing GAP-mediated GTP hydrolysis [27], oncogenic mutations in Rho family GTPases promote increased guanine nucleotide exchange in a GEF-independent manner, which results in constitutive activation [19].
In addition to GEF-mediated regulation and mutation, reactive oxygen and nitrogen species (ROS and RNS) can directly facilitate guanine nucleotide exchange and Rac1 activation [28]. Although ROS and RNS are best known for their role in oxidative stress, in which they induce DNA damage as well as oxidize lipids and proteins [29], ROS and RNS have been shown to regulate cellular signaling at physiological levels [30,31]. Cellular oxidants, such as nitrogen dioxide , superoxide , hydrogen peroxide (H2O2), and peroxynitrite (ONOO−), have been shown to specifically react with protein thiols and alter protein activity [32]. In fact, we have previously shown that oxidants, such as nitrogen dioxide, superoxide, and peroxide, can react directly with Rac1 Cys18 and regulate Rac1 activity by promoting guanine nucleotide exchange in vitro [28]. Moreover, Rac1 can be activated by exogenous addition of peroxide to HeLa cells [33].
Osteoarthritis is a degenerative joint disease that is characterized by high levels of oxidative stress [34], and increased Rac1 activity has been shown to regulate disease progression [14]. Rac1 activity has also been found to be elevated in human osteoarthritis cells [14,35]. Moreover, in primary chondrocytes, Rac1 activation increases the expression of matrix metalloproteinase 13 (MMP-13) [35], an enzyme known to play a role in cartilage matrix degradation. In both of these studies, ectopic expression of inactive Rac1 or use of a chemical inhibitor to Rac1 reduced the level of cartilage destruction and disease progression, indicating a direct role for Rac1 activity in osteoarthritis.
In addition to direct regulation of Rac1 activity by ROS and RNS, Rac1 associates with and can regulate enzymes that produce ROS and RNS. Rac1 binds to and activates NADPH oxidase (NOX) isoforms (Nox1, Nox2, and Nox3) [36,37]. Once activated, the NOX complex produces superoxide, a common cellular ROS. Superoxide has a short half-life and can be reduced by superoxide dismutase 1 (SOD1) to peroxide, a less reactive ROS [38]. Rac1 has also been shown to directly interact with SOD1 in a redox- and nucleotide-dependent manner [39]. In a study by Harraz et al. [39], dithiothreitol (DTT)-reduced GTPγS-loaded Rac1 associated with SOD1, whereas exposure of GTPγS-bound Rac1 to peroxide at concentrations as low as 50 pM reduced association with SOD1 in vitro. These data suggest that oxidation of Rac1 can regulate its activity and alter interactions with regulatory proteins and effectors. Rac1 can also associate with and directly regulate the activity of endothelial and neuronal nitric oxide synthases (eNOS and nNOS) [40]. Nitric oxide (NO•) generated from NOS is involved in numerous physiological processes [41], including vascular function [42], neurotransmission [43], and pathogen defense [44]. As Rac1 can regulate both nitric oxide production from NOS enzymes and superoxide production from NOX complexes, Rac1 likely modulates peroxynitrite generation in cells. Peroxynitrite is a potent oxidant that can easily cross membranes and directly oxidize thiols and iron–sulfur centers in proteins via direct (nonradical-mediated oxidation) or indirect (radical-mediated oxidation) mechanisms [45].
Given that Rac1 colocalizes with and modulates the activity of several redox enzymes, including Nox1/2/3, SOD1, eNOS, and nNOS, we investigated the effects of oxidative modification on Rac1 activity in vitro and in cells. We found that Rac1 has a redox-sensitive cysteine in the phosphoryl-binding loop (p-loop) that can be selectively oxidized by glutathione in vitro, and Rac1 was observed to be glutathiolated in primary chondrocytes. Further, Rac1 Cys18 glutathiolation perturbs guanine nucleotide binding and promotes guanine nucleotide cycling. We prepared a Rac1C18D variant designed to place a negative charge in the nucleotide-binding pocket, similar to sulfinic/sulfonic acid oxidation and glutathiolation, and found that the Rac1C18D variant shows greatly increased nucleotide dissociation rates in vitro, similar to glutathiolated Rac1 (Rac1S–SG). Further, the Rac1C18D variant shows increased activation in HEK-293T cells and enhances lamellipodia formation in Swiss 3T3 cells. We have also generated and char-acterized Rac1C18S, a redox-insensitive Rac1 variant that is resistant to glutathiolation. Taken together, our findings suggest that Rac1 oxidation may promote enhanced nucleotide cycling in a GEF-independent manner, which could lead to increased Rac1 cellular activation. Thus, given the role of Rac1 in regulating cellular oxidant production, dysregulation of Rac1 via oxidative posttranslational modifications may contribute to a variety of disease states in which the redox state is altered.
2. Materials and methods
2.1. Rac1 glutathiolation in primary chondrocytes
Human articular chondrocytes were isolated from normal articular cartilage obtained from tissue donors and cultured as previously described [35]. Confluent cultures were made serum-free overnight before treatment with 25 μM menadione for 0, 10, or 30 min to induce ROS production with or without 100 ng/ml insulin-like growth factor-1 (IGF-1) for 10 min. Cell lysates were prepared as previously described [35] with the addition of 20 mM iodoacetamide and 200 U/ml bovine liver catalase to the lysis buffer. Cell lysates with equal amounts of total protein were immunoprecipitated with antibodies to Rac1 (clone 23A8 from EMD Millipore; Darmstadt, Germany) using the Pierce Classic IP Kit (Thermo Scientific; San Jose, CA, USA). Immunoprecipitated proteins were separated by SDS–PAGE under nonreducing conditions and then immunoblotted with a mouse monoclonal antibody (Arbor Assays; Ann Arbor, MI, USA) to detect protein glutathiolation, which was followed by stripping the blot and reprobing for Rac1.
2.2. Expression and purification of recombinant proteins
Human Rac1WT (1–188, C178S) and the Rac1 Cys18 variants were cloned into pET15b (EMD Millipore), transformed into Escherichia coli BL21 (DE3) Rosetta2 cells (Stratagene; La Jolla, CA, USA), and grown at 37 °C to 0.6 OD600. Rac1 expression was induced upon adding 1 mM isopropyl-β-D-1-thiogalactopyrano-side. The cells were grown for 4 h at 37 °C before lysis in 50 mM KH2PO4 (pH 7.5), 150 mM NaCl, 1 mM MgCl2, 10 μM GDP, and 5 mM β-mercaptoethanol (βME). All Rac1 and Rac1 Cys18 variants were purified using a Ni–NTA column (Qiagen; Venlo, Limburg, The Netherlands) with a linear elution gradient from 0 to 300 mM imidazole. For longer term storage, purified Rac1 proteins were stored in 50% glycerol at −20 °C. The RhoGAP domain (residues 244–431) was cloned into the pQlinkH vector (Addgene), and human Tiam1 (GEF domain containing the DH/PH domain, residues 1040–1397) was cloned into pET28a. Similar to Rac1 expression and purification, these constructs were transformed into E. coli BL21 (DE3) Rosetta2 cells. The cells were lysed in 20 mM Na2HPO4 (pH 7.4), 150 mM NaCl, 20 mM imidazole, and 5 mM βME and purified using Ni–NTA agarose affinity chromatography (Qiagen).
2.3. Rac1 glutathiolation
Oxidized glutathione (GSSG) was added to Rac1 at 1000-fold excess for 15–60 min at 25 or 37 °C (time and temperature were varied to increase yield) in glutathiolation buffer (50 mM KH2PO4 (pH 6.5), 150 mM NaCl, 5 mM MgCl2, 50 μM GDP, and 0.1 mM diethylenetriaminepentaacetic acid (DTPA)). The sample was dialyzed against prechilled buffer (20 mM KH2PO4 (pH 6.5), 50 mM NaCl, 5 mM MgCl2, 10 μM GDP, and 0.1 mM DTPA) overnight.
2.4. Mass spectrometry of unmodified Rac1, glutathiolated Rac1, and ABD-modified Rac1
Rac1 mass measurements were performed on an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific). The mass analysis of intact Rac1 samples was achieved in full-MS, single-ion monitoring, and electron transfer dissociation–tandem mass spectrometry (ETD–MS/MS) modes with a resolution of 120,000 at m/z 400 Da. The intact MS spectra were mass deconvoluted using ProMass, and ETD–MS/MS product ion spectra were processed manually by assigning sequence ions to theoretical masses corresponding to glutathiolated Rac1 or ABD-modified Rac1.
2.5. GDP dissociation assay
Rac1 was preloaded with 2′-/3′-O-(N′-methylanthraniloyl)guanosine-5′-O-diphosphate (mant-GDP; Biolog; Bremen, Germany) in 20 mM Tris (pH 7.5), 50 mM NaCl, 200 mM (NH4)2SO4, and 0.1 mM ethylenediaminetetraacetic acid (EDTA) for 1 h at 37 °C. The reaction was incubated on ice for at least 1 h upon adding 20 mM MgCl2. The excess nucleotide was removed, and Rac1 was buffer exchanged into 20 mM Tris (pH 7.5), 150 mM NaCl, and 5 mM MgCl2. Nucleotide dissociation was initiated by addition of a 1000-fold excess of unlabeled GDP, and the nucleotide dissociation rate was determined by monitoring the fluorescence emission at 435 nm (excitation 365 nm) using an LS50B spectrophotometer (PerkinElmer; Waltham, MA, USA). Where indicated, the minimal catalytic fragment of the Rac1 GEF Tiam1 containing the DH/PH domain was used to stimulate Rac1 nucleotide dissociation [46]. All dissociation experiments were performed in triplicate. The fluorescent nucleotide dissociation curves were fit to a one-phase exponential decay equation using GraphPad Prism 5.0.
2.6. GTP hydrolysis
Rac1 single-turnover GTP hydrolysis rates were determined in the presence and absence of the RhoGAP domain of p50 rhoGAP (1:1000 GAP:Rac1) essentially as described [47]. Inorganic phosphate was removed from all buffers using the “phosphate mop,” consisting of 0.5 mM inosine in each of the following buffers and dialyzing the buffer in the presence of 1 unit of nucleoside phosphorylase [48]. Rac1 was loaded with GTP by incubating with 10-fold excess GTP at 37 °C for 1 min in 20 mM HEPES (pH 8), 20 mM (NH4)2SO4, 1 mM EDTA, and 0.1 mM DTPA. Excess GTP was removed using a PD-10 column (GE Healthcare). The reaction was performed in triplicate using a buffer containing 20 mM Tris (ph 7.4), 50 mM NaCl, 0.1 mM EDTA, and 0.5 mM inosine. GTP hydrolysis was initiated by adding 100 μM MgCl2 to a sample containing 50 μM Rac1 and FlipPi 5U (Addgene). FlipPi undergoes a conformational change upon binding inorganic phosphate, which alters its intrinsic fluorescence resonance energy transfer (FRET), as previously described [49]. Phosphate released from the hydrolysis of GTP to GDP was quantified to determine Rac1 GTP hydrolysis rates by monitoring the FlipPi FRET signal change using a Spec-tramax M5e spectrometer (excitation 415 nm and the emission ratio for 475 and 515 nm). The data were normalized and fit to a single association exponential (Prism 5.0; GraphPad; San Diego, CA, USA; n=2).
2.7. Rac1 Cys18 thiol pKa measurements
Rac1WT and Rac1C18S were dialyzed into reducing buffer (15 mM HEPES (pH 8.0), 50 mM NaCl, 5 mM MgCl2, 10 μM GDP, and 10 mM DTT), and the protein was incubated under reducing conditions for 30 min. Rac1 was then buffer exchanged using a Centricon concentrator (15-kDa molecular weight cut-off, Millipore) into nonreducing buffer (15 mM MES (pH 6.5), 30 mM NaCl, 5 mM MgCl2, 200 μM DTPA, and 10 μM GDP). Rac1 was diluted into a mixed buffer system with pH values ranging from 5.5 to 8.5; each buffer contained 30 mM MES, 30 mM MOPS, 30 mM Tricine, 5 mM MgCl2, 50 mM KCl, and 200 μM DTPA. ABD-f (4-(aminosul-fonyl)-7-fluoro-2,1,3-benzoxadiazole; Anaspec; Fremont, CA, USA) was added (1 mM) to initiate cysteine thiol modification. The reaction rate was determined by monitoring ABD-f fluorescence (excitation 389 nm, emission 513 nm) using a Spectromax M5e spectrometer (Molecular Devices; Sunnyvale, CA, USA). The initial reaction rates were fitted using linear regression analysis (Prism 5.0; GraphPad).
2.8. Rac1 circular dichroism and thermal stability
Circular dichroism data were collected on a JASCO J-815 CD spectrometer (Oklahoma City, OK, USA) with a JASCO Peltier device and water bath to control the temperature. Experiments were performed in a 1-mm cuvette at a protein concentration of 15 μM in 10 mM potassium phosphate (pH 6.5). Far-UV scans were from 200 to 280 nm. Thermal denaturation of Rac1 and Rac1 variants were monitored at 222 nm to estimate the protein melting temperature. The temperature ramp rate was 1 °C/min and data points were collected every 1 °C. All data are reported in units of mean residue ellipticity, which was calculated as follows: [θ]MRE = (θraw × MRW)/(10 × c × l), where θraw is the ellipticity in degrees, MRW is (molecular mass in kilodaltons)/(No. of residues – 1), c is the protein concentration in g/ml, and l is the pathlength of the cuvette in cm, according to [50].
2.9. NMR experiments
Rac1 was expressed and purified as described above except that the cells were grown in 15N-enriched M-9 minimal medium. Two-dimensional (2D) 1H–15N HSQC (heteronuclear single-quantum coherence spectroscopy) NMR experiments were performed using a Varian Inova 700-MHz spectrometer with a cryoprobe. The sample contained 200 μM Rac1, Rac1C18S, or Rac1C18A at 25 °C in 50 mM Tris maleate (pH 6.8), 50 mM NaCl, 5 mM MgCl2, 50 μM GDP, 0.1 mM DTPA, and 1 mM DTT. The Rac1C18D variant was collected on a Bruker 700-MHz spectrometer (Billerica, MA, USA). Two-dimensional 1H–15N HSQC NMR spectra were collected and recorded using a 2500-Hz 15N spectral width and 512 complex points. The NMR data were processed using NMR Pipe and NMRViewJ [51,52].
2.10. Cell lines, plasmids, and reagents
HEK-293T cells (from the American Type Culture Collection) and Swiss 3T3 cells (a gift from Alan Hall, Memorial Sloan Kettering Cancer Center) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Sigma; St. Louis, MO, USA) and maintained at 37 °C in 5% CO2 [53]. Keith Burridge (University of North Carolina) provided full-length human Rac1 and the full-length Rac1C18S variant, which were cloned into the pCMVJ3 vector for mammalian expression; pCMVJ3-Rac1C18D was generated from Rac1WT using PCR-based mutagenesis.
2.11. PAK pull-down assays for Rac1-GTP in HEK-293T cells
Levels of active, GTP-bound Rac1 were assessed using pull-down assays with the PAK1 p21-binding domain (GST-bound PAK-PBD, a gift from Keith Burridge) as described previously [54]. Briefly, HEK-293T cells were transiently transfected with Rac1 expression plasmids using the TransIT transfection reagent (Mirus; Madison, WI, USA) according to the manufacturer’s instructions. The next day or at 80–90% confluence, the cells were starved in serum-free DMEM for 3 h. Next, the cells were washed twice with ice-cold phosphate-buffered saline (PBS; 5.4 mM KCl, 1.7 mM KH2PO4, 13 mM NaCl, and 5.4 mM Na2HPO4 (pH 7.4)) and lysed in magnesium lysis buffer (50 mM Tris (pH 7.5), 10 mM MgCl2, 150 mM NaCl, 1% NP-40, 10% glycerol, and 0.25% Na deoxycholate) with protease inhibitors. Equal volumes were removed from each lysate for total protein analysis. Glutathione (GST) agarose beads containing 20 μg of GST–PAK-PBD were added to each lysate and incubated at 4 °C for 60 min. Agarose–GST–PAK-PBD and associated Rac1 were pelleted and washed three times with 500 μl of wash buffer (25 mM Tris (pH 7.5), 40 mM NaCl, and 30 mM MgCl2). The final pellets were suspended in 1× protein sample buffer and resolved using SDS–PAGE. Rac1 protein and variants were detected through immunoblotting using an anti-Rac antibody (Millipore). Equal protein loading was confirmed using anti-tubulin (Sigma–Aldrich), and the Rac1C18S and Rac1C18D data were normalized to the Rac1WT data.
2.12. Cytoskeleton assays in Swiss 3T3 cells
Swiss 3T3 cells were plated (5000 cells/well in 12-well plates) on coverslips previously coated with fibronectin (2.5 μg/ml) for 30 min at room temperature. The next day, Myc-tagged Rac1 constructs were transiently transfected into the cells using TransIT (Mirus) according to the manufacturer’s instructions. Twenty-four hours after transfection, the cells were fixed and stained with phalloidin and for the Myc-tag, as previously described [55]. Briefly, the cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences; Hatfield, PA, USA) overnight at 4 °C, permeabilized in 0.2% Triton X-100 (Sigma) in PBS for 5 min, and incubated with anti-Myc 911B antibody (1:500, Cell Signaling) for 1 h, followed by secondary AlexaFluor 488 anti-mouse antibody (1:500, Invitrogen) for 2 h and AlexaFluor 568 phalloidin (Invitrogen, 1:40 in PBS) for 30 min. The cells were incubated in the dark and rinsed in PBS three to five times between each step. Coverslips were mounted using 6 μl Vectashield with DAPI (Vector Laboratories; Burlingame, CA, USA). Cells were visualized and counted blindly for lamellipodia using a Nikon Eclipse TS100 microscope with a 40× objective. Representative images were recorded using a Zeiss LSM 710 confocal laser-scanning microscope with a 63× oil objective.
3. Results
We have previously shown that Rac1 activity can be directly modulated by ROS and RNS [28]. Our earlier studies focused on radical-mediated regulation of Rac1 activity, in which we found that oxidants ( and ) capable of generating a Cys18 thiyl radical caused guanine base oxidation and nucleotide dissociation in Rac1; however, we also observed that peroxide, a nonradical oxidant, increased the intrinsic rate of exchange by ~10-fold [28]. As S-glutathiolation is commonly observed after thiyl radical formation because of the high cellular glutathione concentration [56], we expanded on these studies by examining whether glutathione modification can alter Rac1 activity. Given that Rac1 Cys18 is located in the nucleotide-binding pocket and forms multiple interactions with the guanine nucleotide ligand, we postulated that Cys18 is redox-sensitive, and oxidation of this thiol could alter guanine nucleotide binding and the Rac1 activation state.
3.1. Rac1 is glutathiolated in primary chondrocytes and is specifically glutathiolated at Cys18 in vitro
Rac1 has previously been shown to contribute to the development of osteoarthritis, probably because of its ability to upregulate MMP-13 expression in articular chondrocytes [14,35]. Therefore, we used primary human chondrocytes to examine whether endogenous Rac1 is glutathiolated. The cells were treated with menadione, which induces ROS production, in the absence and presence of the growth factor IGF-1, to examine the effects of ROS on IGF signaling. As shown in Fig. 1, menadione treatment, but not IGF-1, increased the level of Rac1 glutathiolation. Detection of glutathiolated Rac1 was greatest at 10 min after menadione addition and declined at 30 min.
Fig. 1.
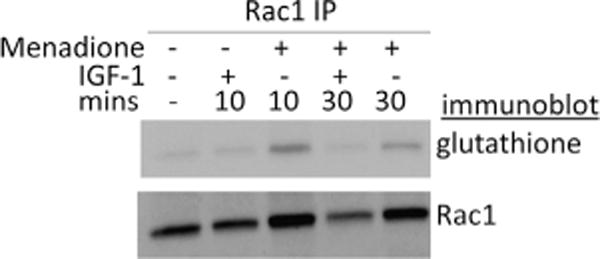
Rac1 glutathiolation in human chondrocytes. Primary human chondrocytes were treated for 10 or 30 min with 25 μM menadione to induce oxidative stress in the absence and presence of 100 ng/ml IGF-1. Cell lysates immunoprecipitated with a monoclonal antibody to Rac1 were run on a nonreducing gel and immunoblotted with a monoclonal antibody to glutathione. The blot was stripped and reprobed with the Rac1 antibody.
Given that Rac1 Cys18 is solvent accessible (Supplementary Table 1) and sensitive to ROS and RNS [28], we explored whether Cys18 in Rac1 can be specifically glutathiolated in vitro. To generate glutathiolated Rac1 (Rac1S–SG), Rac1WT was treated with 1000× GSSG. The reaction products were analyzed by MS. Fig. 2 shows the intact mass analysis of Rac1WT treated with GSSG at pH 6 and 7.5, as well as the subsequent characterization of Rac1 with a single glutathione modification by top-down mass spectrometry. We were able to semi-quantitatively assess the amount of adduct formed by comparing the normalized peak intensity of unmodified Rac1 to the intensity of the Rac1 glutathiolated peak. These analyses also show that the amount of glutathiolated Rac1 is dependent on the pH of the reaction. Fig. 2A shows the deconvoluted MS of Rac1 treated with GSSG at pH 7.5 that resulted in ~25% glutathione adduct (normalized to base peak of Rac1), and Fig. 2B shows the deconvoluted MS of Rac1 treated with GSSG at pH 6 that resulted in ~10% glutathione adduct (normalized to base peak of Rac1). The twofold drop in glutathiolation observed in the MS data of intact Rac1 suggests that a single cysteine residue in Rac1 is modified in a pH-dependent manner. These observations are consistent with data in Supplementary Fig. 1, in which ESI–MS performed on Rac1WT and Rac1C18S show glutathiolation for Rac1WT but not Rac1C18S, indicating that Cys18 is the primary site of glutathiolation in Rac1.
Fig. 2.
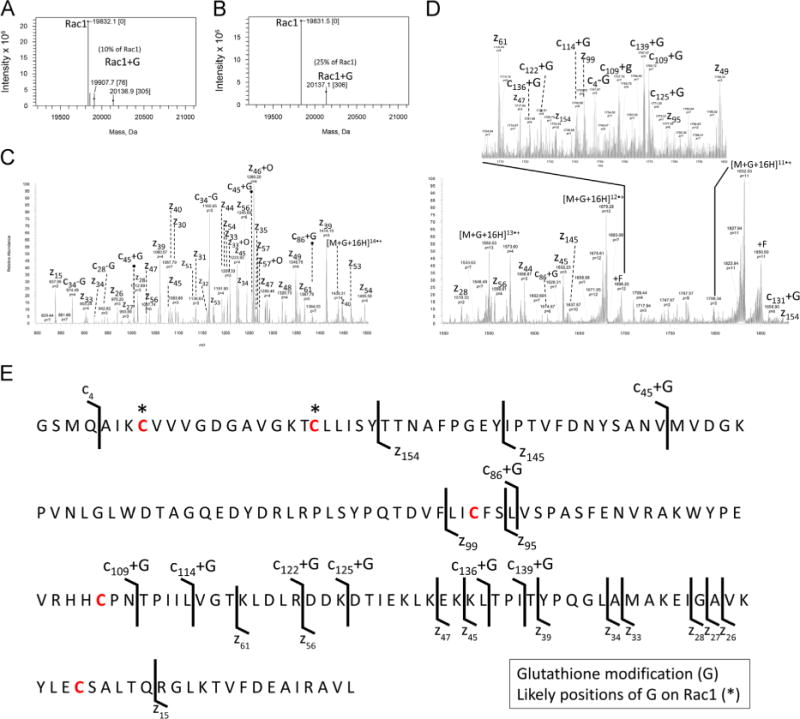
Mass spectrometry characterization of Rac1WT glutathione adducts. (A and B) Mass deconvoluted zero-charge spectra of the ESI full-MS spectra (Supplementary Fig. 1) associated with intact Rac1 treated with 1000× glutathione at pH 6 and 7.5, respectively. Note: A single glutathione adduct is seen at pH 6.0 and 7.5, with more adducts observed at higher pH. (C and D) Top-down product ion MS/MS spectra resulting from the ETD of [M+16H+G]16+ (glutathiolated Rac1, Z = 16+, m/z = 1259.6 Da) with fluoranthene anions. Note: c- and z-type ions, nondissociated precursors (electron transfer with no dissociation), and precursors with fluoranthene (m/z = 202 Da) adducts (+F) of Rac1 were observed. As glutathione is prone to cleavage at the disulfide bond during ETD, some neutral loss glutathione ions were detected. In addition, z-type ions are prone to radical-mediated oxide adduction in ETD, which were detected. (E) ETD sequence ions mapped to Rac1 with putative glutathione sites highlighted by asterisks. Confident assignment of c- and z-type ions z154, z145, z99, and c45+G leads to the localization of the glutathione adduct to either Cys6 or Cys18 of Rac1.
The site of Rac1 glutathiolation was characterized by top-down mass spectrometry by subjecting the [M+16H+G]16+ ion (Z=16+ of Rac1 containing a single glutathione) to ETD. The resulting MS/MS product ion spectrum consists of the major c- and •z-type fragment ions, shown at two different mass ranges for clarity. Data in Fig. 2C show products spanning m/z 800–1500 Da, and Fig. 2D shows all product ions spanning m/z 1500–2000 Da. The lower m/z region resulted in mostly singly charged c- and •z-type product ions from C-terminal residues, whereas the N-terminal fragment ion c45+G contained the only glutathione-containing product ion. Most of the N-terminus lacked structurally informative fragments because of the lower number of basic amino acid residues. In addition, the neutral loss of glutathione during ETD was occasionally observed. Conversely, the higher m/z region produced structurally rich c-type ions, i.e., c87+G, and a number of other larger multiply charged c-type ions that contained glutathione (Fig. 2E). Our ability to detect glutathiolated Rac1 fragments was probably due to preservation of the covalently attached disulfide bond of glutathione upon competitive fragmentation along the N–Cα backbone. These ions, along with complementary multiply charged •z-type fragment ions lacking glutathione (•z154, •z145, •z99), lead to unambiguous localization of glutathiolation to either Cys6 or Cys18. In further support of site-specific glutathiolation at Cys18, a Rac1 variant that lacks a cysteine at position 18 (Rac1C18S), but shows biochemical properties similar to Rac1WT, is not covalently modified by glutathiolation (Supplementary Fig. 1). Taken together, our data indicate that treatment of Rac1WT with GSSG results in specific glutathiolation of Rac1 Cys18.
3.2. Rac1 Cys18 glutathiolation perturbs guanine nucleotide binding and enhances the intrinsic GDP dissociation rate
As Cys18 is located in the p-loop of Rac1 and directly interacts with the bound guanine nucleotide, we hypothesized that glutathiolation at this site may interfere with nucleotide binding. To determine whether Rac1 glutathiolation alters guanine nucleotide dissociation, Rac1S–SG was preloaded with fluorescent GDP (mant-GDP), and the rate of GDP dissociation was determined. As shown in Fig. 3, oxidative modification of Rac1 Cys18 with glutathione enhances the rate of GDP dissociation by 200-fold. Adding the GEF domain (DH/PH) of Tiam1 to Rac1S–SG did not increase the GDP dissociation rate. However, as the intrinsic rate of GDP dissociation is rapid under our experimental conditions, it is unclear whether we have the dynamic range to detect GEF-mediated enhancement.
Fig.3.
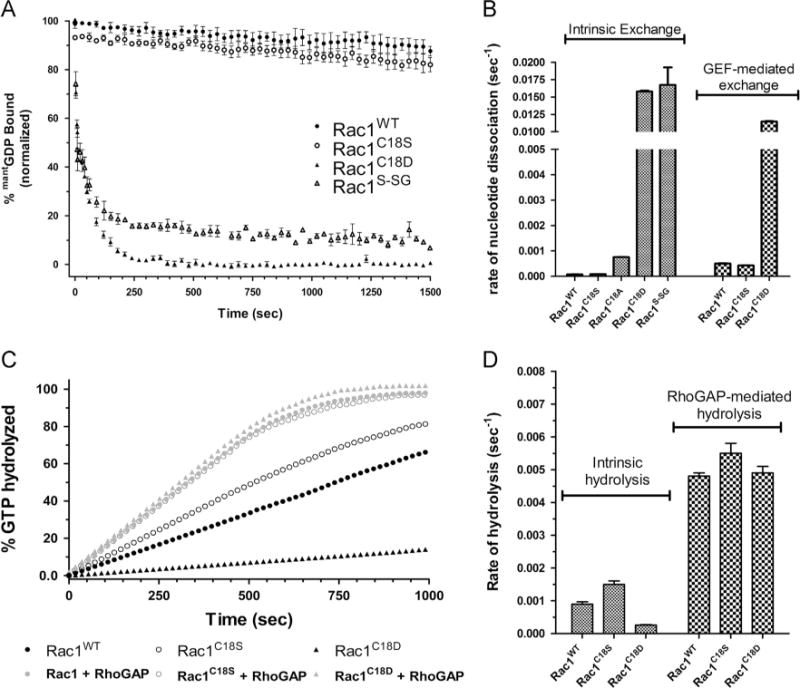
Oxidation of Rac1 Cys18 and Rac1 Cys18 variants alters guanine nucleotide exchange but not GTP hydrolysis. (A) Intrinsic mant-GDP dissociation curves for Rac1WT, Rac1C18S, Rac1C18D, and Rac1S–SG. The data were fit to a single exponential and standard errors were determined using Prism 5.0 (n = 3). (B) Bar graph of rates of intrinsic nucleotide exchange with GEF-mediated dissociation data (using the DH/PH domains of Tiam1) where applicable. (C) Graph of intrinsic and GAP-mediated single-turnover hydrolysis for Rac1WT, Rac1C18S, and Rac1C18D. The RhoGAP domain of p50 rhoGAP was used for the GTP hydrolysis measurements. The data were fit to a single exponential and standard errors were determined using Prism 5.0 (n = 2). (D) Bar graph of rates of nucleotide hydrolysis with GAP-mediated hydrolysis data (using p50 rhoGAP) where applicable. Rates of exchange and hydrolysis are presented in Table 1.
To better understand the enhanced nucleotide exchange properties associated with glutathiolated Rac1, we generated Rac1 C18D, C18S, and C18A variants. The Rac1C18D variant was generated because a Cys→Asp substitution has previously been shown to mimic a cysteine-to-sulfenic/sulfinic acid modification in recoverin [57]. As both Rac1C18D and Rac1S–SG introduce a negative charge into the phosphoryl-binding pocket, we also made more conservative Cys18 substitutions, including Rac1C18S and Rac1C18A. All variants were preloaded with fluorescent GDP (mant-GDP), and Rac1 GDP dissociation was determined by measuring the decrease in mant-GDP fluorescence as a function of time (Fig. 3A). The GDP dissociation rates of Rac1 and the Cys18 variants were also measured in the presence of the GEF domain of Tiam1. Similar to glutathiolated Rac1, Rac1C18D shows a greatly enhanced intrinsic rate of nucleotide dissociation that was approximately 200-fold faster than that of Rac1WT and was not further increased by addition of Tiam 1. In contrast, Rac1C18S shows a similar GDP dissociation rate compared to Rac1WT, and Rac1WT and Rac1C18S were similarly responsive to GEF-mediated GDP dissociation. Consistent with previous studies of Cdc42C18A [58], the Rac1C18A variant enhances the intrinsic rate of GDP dissociation by 11-fold. These results indicate that the Cys18 thiol side chain plays a role in stabilizing nucleotide binding (Fig. 3B and Table 1).
Table 1.
Intrinsic and GEF-mediated GDP dissociation rates for Rac1WT, Rac1C18S, Rac1C18A, Rac1C18D, and Rac1S–SG.
| Rac1 construct | koff (s−1) intrinsic | Fold increase (relative to WT) | koff (s−1) with Tiam1 |
|---|---|---|---|
| Rac1WT | 0.71 ± 0.02 × 10−4 | – | 5.02 ± 0.01 × 10−4 |
| Rac1C18S | 0.81 ± 0.00 × 10−4 | 1.1 | 4.28 ± 0.03 × 10−4 |
| Rac1C18A | 7.58 ± 0.02 × 10−4 | 10.7 | n/d |
| Rac1C18D | 157.87 ± 1.38 × 10−4 | 222.4 | 114.73 ± 0.61 × 10−4 |
| Rac1S–SG | 167.50 ± 25.13 × 10−4 | 235.9 | n/d |
In-cell regulation of Rac1 activity requires exchange of GDP for GTP to activate Rac1 and GTP hydrolysis for inactivation. To characterize the effects of Rac1 Cys18 variants on intrinsic and GAP-mediated GTP hydrolysis, we determined the rates of GTP hydrolysis of Rac1WT, Rac1C18D, and Rac1C18S in the presence and absence of the minimal catalytic domain of p50 rhoGAP. Single-turnover GTP hydrolysis rates were determined by monitoring phosphate release upon GTP hydrolysis by detecting the FRET change associated with the phosphate-binding protein FlipPi 5U. As shown in Fig. 3C and quantified in Table 2, mutating Rac1 Cys18 to either Asp or Ser did not significantly affect either the intrinsic or the GAP-mediated GTP hydrolysis rates. These data indicate that perturbation of Rac1 Cys18 by mutation, including the oxidation mimetic, does not alter GAP-mediated down regulation in vitro.
Table 2.
Intrinsic and GAP-mediated GTP hydrolysis rates for Rac1WT, Rac1C18S, and Rac1C18D.
| Rac1 construct | Intrinsic GTP hydrolysis (s−1) | p50 rhoGAP GTP hydrolysis (s−1) |
|---|---|---|
| Rac1WT | 0.90 ± 0.07 × 10−3 | 4.8 ± 0.1 × 10−3 |
| Rac1C18S | 1.50 ± 0.11 × 10−3 | 5.5 ± 0.3 × 10−3 |
| Rac1C18D | 0.26 ± 0.01 × 10−3 | 4.9 ± 0.2 × 10−3 |
3.3. The Rac1 Cys18 thiol has a depressed pKa and can be selectively modified by ABD-f at physiological pH
As Rac1 Cys18 can be selectively modified by oxidized glutathione at physiological pH, we investigated whether the pKa of the Rac1 Cys18 thiol is altered relative to a typical free cysteine thiol by measuring the cysteine reactivity of Rac1WT and Rac1C18S to ABD-f. We used the thiol-modifying reagent ABD-f, which preferentially reacts with the thiolate form of cysteine [59], to measure thiol reactivity over a wide pH range, as described previously by us for Ras [60]. ABD-f fluorescence was measured from pH 5.5 to 8.5 for Rac1WT and Rac1C18S (Fig. 4A). The rate of ABD-f reactivity for Rac1WT was normalized to produce a pH titration curve for Rac1 Cys18 (Fig. 4B) and resulted in a pKa for the Rac1 Cys18 thiol of ~7.25, which is approximately 1.5 pH units lower than a typical pKa for a free cysteine [61]. No significant reactivity was detected for Rac1C18S.
Fig. 4.
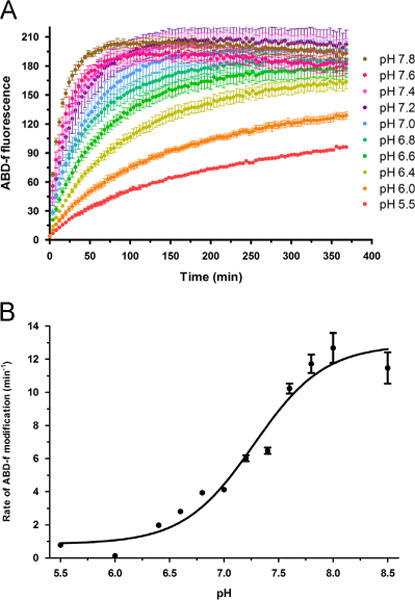
Rac1 Cys18 has an altered pKa and populates the thiolate state at physiological pH. (A) ABD-f reactivity data for Rac1WT over a pH range of 5.5–8.5. (B) Using the initial rate of modification by ABD-f to Rac1WT, the initial rate of modification was plotted vs the pH to determine the pKa of the Cys18 thiol. The resulting curve was fit to a Boltzmann sigmoidal curve to determine the inflection point, indicative of the pKa, using GraphPad Prism 5.0. Curve fitting resulted in an estimated pKa of 7.25 for Rac1 Cys18.
The Rac1 construct (Rac1 1–188, C178S) contains five cysteines, some of which are partially solvent accessible (Supplementary Table 1). Fig. 5 shows the intact mass analysis of Rac1WT and Rac1C18S that were reacted with ABD-f under the conditions used for glutathione modification. A single ABD-f modification site was identified by top-down mass spectrometry. The deconvoluted mass spectra of Rac1WT shown in Fig. 5A consists of major peaks corresponding to unmodified intact Rac1, Rac1 with a single ABD adduct mass, and Rac1 with a single deoxygenated adduct mass. Conversely, the deconvoluted mass spectra of the Rac1C18S variant, in Fig. 5B, show peaks corresponding to unmodified intact Rac1C18S, Rac1C18S with a sodium adduct, and two unidentified peaks ([M+76] and [M+99]). However, an ABD adduct is not observed for Rac1C18S, suggesting that the Rac1WT ABD adduct occurs exclusively at Cys18. This result is consistent with Rac1WT glutathiolation MS data (Fig. 2) in which a single glutathione adduct was observed at Cys18. The ESI–MS spectra for Figs. 5A and 5B are shown in Supplementary Figs. 2A and 2B, respectively. The site of the ABD adduct was further characterized by top-down mass spectrometry. Both [M+18H+ABD]18+ (Z = 18+ of Rac1 containing a single ABD) and [M + 18H]18+ (Z = 18+ of unmodified Rac1) were subjected to ETD to comprehensively assign peaks modified by ABD-f. Fig. 5C shows the ABD-modified Rac1 sequence map annotated with observed major c- and •z-type fragment ions. The c27+ABD, c54+ABD −H2O, c123+ABD, c132+ABD, and c133+ABD ions as well as the z148 ion allow localization of the ABD adduct to either Cys18 or Cys6. The absence of the ABD adduct for the Rac1C18S variant confirms that Cys18 is the most likely site of ABD-f modification.
Fig. 5.
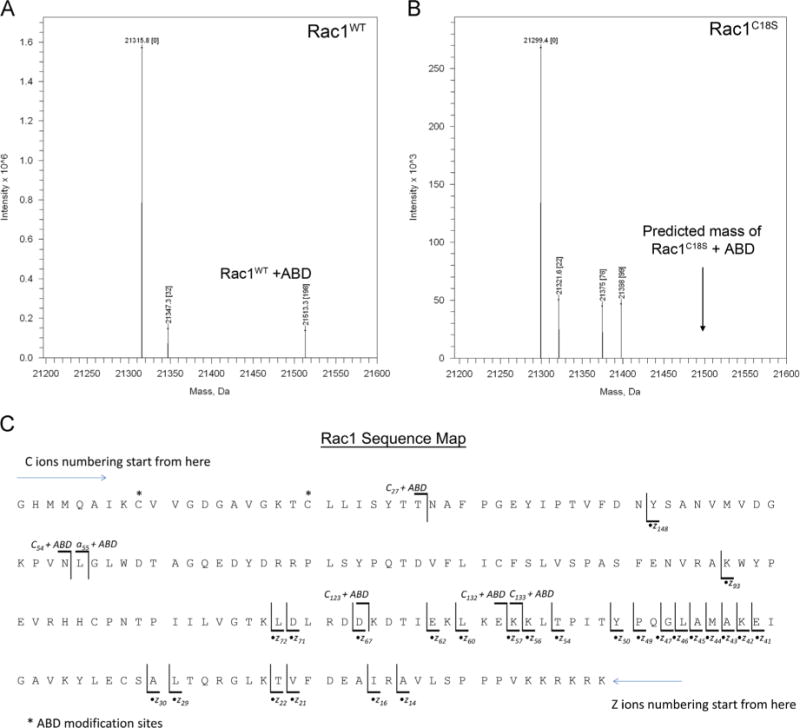
MS of Rac1WT and Rac1C18S treated with ABD-f. (A) Mass deconvoluted spectrum of ABD-f-treated Rac1WT. (B) Mass deconvoluted spectrum of ABD-f-treated Rac1C18S. Note: Rac1WT has a peak corresponding to an ABD adduct, whereas a peak at the predicted mass (shown by the arrow) for the corresponding ABD adduct of Rac1C18S is not observed. The unassigned peaks +76 and +99 Da from Rac1C18S are adducts of unknown origin. (C) ETD map of ABD-modified Rac1WT. Product ions generated by ETD of ABD-f-treated Rac1 are presented in Supplementary Fig. 3. Note that the N-terminal three residues (GHM) are not part of the native Rac1WT sequence. The c27+ABD, c54+ABD−H2O, c123+ABD, c132+ABD, and c133+ABD ions as well as the z148 ion allow for the localization of ABD to either Cys18 or Cys6. The absence of the ABD adduct in the Rac1C18S variant supports Cys18 as the site of ABD modification.
Supplementary Figure 3 shows the resulting ETD-MS/MS product ion spectra of Rac1 and Rac1+ABD as a tiled view to compare the fragmentation of unmodified and ABD-modified Rac1. Comparative analysis of peaks associated with top and bottom spectra show differences (difference spectral peaks) that assist in narrowing down the fragments containing ABD-f modification. The distinct product ion peaks along with all other observed product ions were used for the annotation of the sequence map shown in Fig. 5C. The major c- and •z-type fragment ions are shown at several different mass ranges for clarity.
Taken together, our data show that Cys18 is the only cysteine that appreciably reacts with ABD-f or GSSG at physiological pH values. These results indicate that Rac1 Cys18 has a depressed pKa, which populates the more reactive thiolate state under physiological conditions.
3.4. Thermal stability and structural analysis
As a decrease in nucleotide binding affinity is correlated with a decrease in stability for the Ras GTPase [62], we used CD to determine the total secondary structure content and thermal stability of Rac1, Rac1S–SG, and Rac1 variants. By comparing the overall secondary structure and thermal stability, we can evaluate whether Rac1S–SG and the Rac1 Cys18 variant disrupts the overall protein fold. As shown in Fig. 6, the overall secondary structure content determined by CD spectroscopy was unchanged for Rac1S–SG. In contrast, the thermal stability was decreased by 9 °C relative to Rac1WT. As the nucleotide exchange rate was significantly increased in Rac1S–SG relative to Rac1WT, the change in Tm is probably a reflection of decreased nucleotide binding affinity. We also find that the secondary structure content and thermal stability of Rac1C18D are similar to those of Rac1S–SG. In contrast to mutations and oxidative modifications that promote fast cycling, the thermal denaturation of the redox-insensitive Rac1C18S variant was slightly higher than that of Rac1WT, with a Tm of 69 °C compared to 67 °C for Rac1WT.
Fig.6.
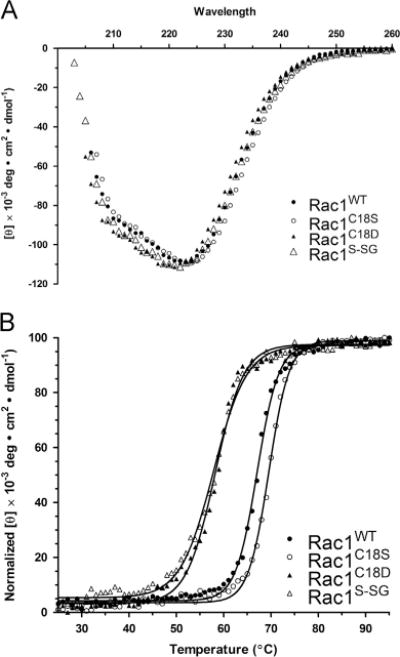
Oxidation and mutation of Rac1 Cys18 does not perturb protein secondary structure or stability. (A) CD spectra (scan from 200 to 280 nm) measuring the secondary structure elements of Rac1WT, Rac1C18S, Rac1C18D, and Rac1S–SG. (B) Thermal denaturation of Rac1WT, Rac1C18S, Rac1C18D, and Rac1S–SG at 220 nm at temperatures ranging from 25 to 95 °C. Tm values were calculated by fitting the data to a Boltzmann sigmoidal curve. Data are representative of three thermal melts and were fit using GraphPad Prism 5.0.
To determine how mutations at Cys18 perturb Rac1 in a site-specific manner, we employed NMR on 15N-enriched Rac1-GDP. We performed a 2D 1H–15N HSQC on Rac1WT, Rac1C18D, and Rac1C18S and compared the variants to the HSQC spectrum of Rac1WT, which has been previously assigned [63]. As we are able to detect one peak for every backbone amide with the exception of proline, this technique allows us to probe whether mutation at Cys18 causes localized or more global structural perturbations in Rac1. The 2D 1H–15N HSQC overlay of Rac1WT and Rac1C18D is shown in Fig. 7A, with residues showing chemical shift changes mapped onto the Rac1 structure (pdb 1MH1) (Fig. 7B). Comparison of the NMR spectra of Rac1C18D and Rac1WT showed chemical shift differences (22) in a small percentage (14%) of the total amides (chemical shifts greater than one line width). In addition, two amide peaks showed changes in linewidth. As shown in Fig. 7B, residues that undergo chemical shift changes upon mutation of Cys18 to Asp are highlighted in purple and are primarily localized to the p-loop, switch I, and SAx motif of Rac1 (switch II was not detected in the Rac1WT NMR spectrum).
Fig. 7.
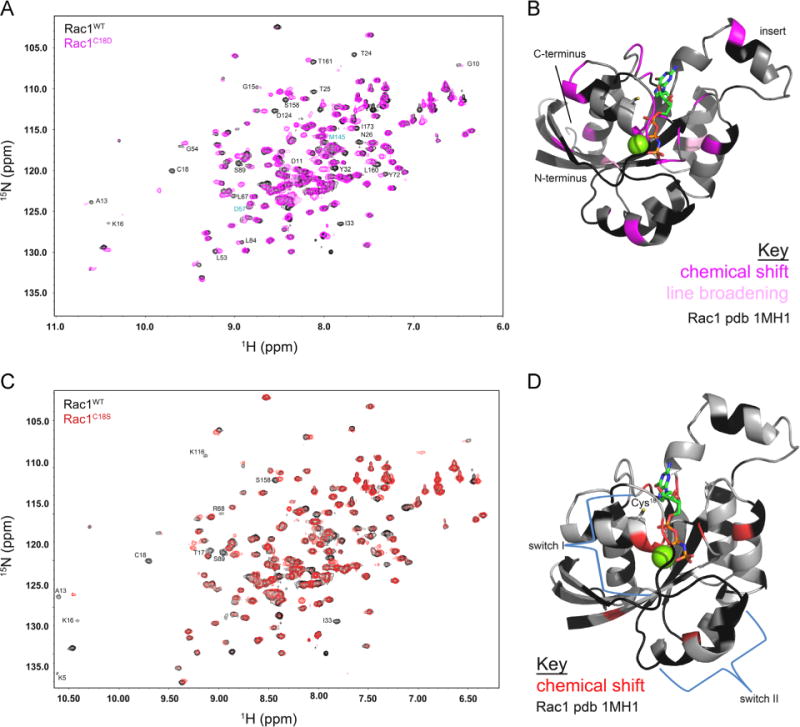
2D NMR spectra of Rac1 and Rac1 variants. (A) 2D 1H–15N HSQC spectra overlay of Rac1WT (black) and Rac1C18D (magenta). Peaks that showed line broadening are shown in cyan and peaks with a chemical shift greater than one linewidth are in black. (B) The major chemical shift perturbations (broadening, where the Rac1C18D resonance linewidth is less than 50% of the corresponding Rac1WT resonance (shown in light pink), or shifted, where there was no peak detected in Rac1C18D within one linewidth of a peak in Rac1WT (shown in magenta) are mapped onto a ribbon diagram of GTP-bound Rac1 (pdb 1MH1). (C) 2D 1H–15N HSQC spectra overlay of Rac1WT (black) and Rac1C18S (red). Peaks that showed line broadening are shown in cyan and peaks with a chemical shift greater than one linewidth are in black. (D) The major chemical shift perturbations (broadening, where the Rac1C18S resonance linewidth is less than 50% of the corresponding Rac1WT resonance (shown in light red), or shifted, where there was no peak detected in Rac1C18S within one linewidth of a peak in Rac1WT (shown in red) are mapped onto a ribbon diagram of GTP-bound Rac1 (pdb 1MH1). In (B) and (D), residues with chemical shifts or line broadening are as indicated. The unassigned and undetected residues are indicated in black, and the remaining residues are gray. GTP is depicted as a multicolored stick structure, and Mg2+ is shown as a green sphere.
In contrast, only nine residues show chemical shift changes for Rac1C18S compared to Rac1WT. Moreover, the residues displaying chemical shift changes correspond to residues proximal to the site of mutation (Fig. 7C). In Fig. 7D, a ribbon diagram of the structure of Rac1 (pdb 1MH1) is presented with residues showing chemical shift perturbations highlighted. Most of the residues that show peak changes are near the site of mutation, indicating that mutation of Cys18 to Ser does not significantly perturb the structure of Rac1, consistent with our findings that the biochemical properties and secondary structure of Rac1C18S are not significantly altered compared to Rac1WT. Further, these data support the use of Rac1C18S as a redox-insensitive variant of Rac1.
3.5. Rac1C18D is hyperactivated in HEK-293T cells
Our data indicate that both the Rac1C18D variant and glutathiolated Rac1 show greatly enhanced rates of GDP dissociation, similar to the Rac1 fast-cycling mutant (P29S) that promotes Rac1-mediated oncogenesis. The Rac1P29S mutant has a significantly faster nucleotide exchange rate and induces an activated phenotype in COS-7 cells [21]. As Rac1C18D shows greatly enhanced nucleotide dissociation (>200-fold), we postulated that this oxidative mimetic will populate Rac1 in the active, GTP-bound state in cells. Therefore, we examined the activation state of Rac1WT, Rac1C18D, and Rac1C18S in HEK-293T cells using a PAK1 pull-down assay. As shown in Fig. 8, Rac1WT and Rac1C18S show similar levels of association with PAK1-PBD. In contrast, Rac1C18D showed a 5-fold higher level of activation compared to Rac1WT and Rac1C18S, which is consistent with our observation that Rac1C18D has a greatly enhanced in vitro nucleotide dissociation rate compared with Rac1WT. As enhanced Rac1 activation has been previously observed in HeLa cells upon H2O2 addition [33], our data further support the hypothesis that oxidation at Rac1 Cys18 can modulate the Rac1 activation state.
Fig. 8.
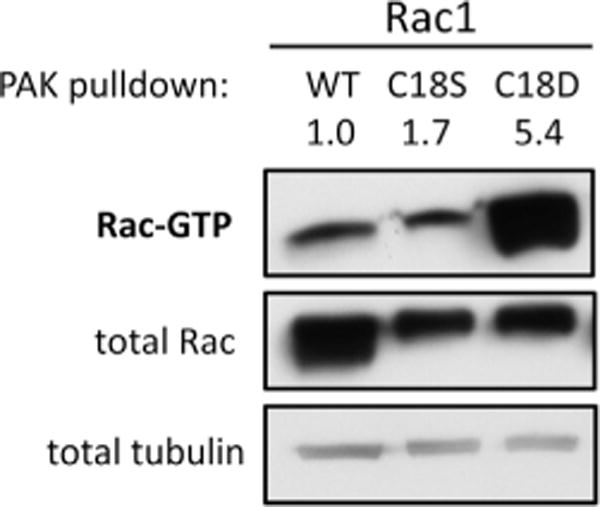
The Rac1C18D variant is hyperactivated in HEK-293T cells. Rac1WT, Rac1C18S, and Rac1C18D were transiently expressed in HEK-293T cells and PAK pull-down assays were used to assess the levels of active, GTP-bound Rac1. Active Rac1 was pulled down from cell lysates with PAK-PBD (p21-binding domain) coupled to agarose beads and was detected by immunoblotting for Rac1. A representative pull-down assay (n = 6) is shown.
3.6. Rac1C18D enhances Rac1-mediated lamellipodia formation in Swiss 3T3 cells
As we determined through PAK1 pull-down assays that Rac1C18D is more populated in its GTP-bound state compared with Rac1WT or Rac1C18S (Fig. 8), we sought to determine if the increased activity of Rac1C18D could induce a biological phenotype that indicates enhanced Rac1 signaling. A canonical function of active Rac1 is actin cytoskeleton changes that induce lamellipodia formation [64,65]. Therefore, we visualized the actin cytoskeleton through phalloidin staining in Swiss 3T3 cells expressing Rac1WT, Rac1C18D, or Rac1C18S. Cells expressing the Myc-tagged Rac1 constructs were identified by immunofluorescence, and lamellipodia formation in the Rac1-expressing cells was quantified. We determined that Rac1C18D expression increased lamellipodia formation 5- to 10-fold compared with Rac1WT and Rac1C18S (Fig. 9). Therefore, Rac1C18D can function similar to activated Rac1WT and promote lamellipodia formation.
Fig. 9.
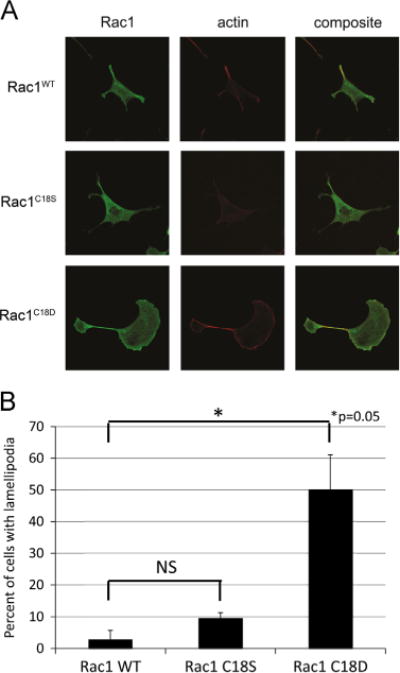
Rac1C18D enhances lamellipodia formation in Swiss 3T3 cells. To examine the effect of Rac1 oxidation on its ability to regulate the actin cytoskeleton, Swiss 3T3 cells were transfected with Rac1WT, Rac1C18S, or Rac1C18D. The actin cytoskeleton was visualized using phalloidin staining. (A and B) Rac1C18D induces lamellipodia formation, whereas Rac1C18S does not. Twenty-four hours after transfection with Myc-tagged Rac1 constructs, cells were fixed and stained for Myc-tag and phalloidin. Using the Myc-tag antibody to identify cells that express the Rac1 constructs, cells were counted (blindly with respect to the specific Rac1 construct expressed) for the presence or absence of lamellipodia (n = 2, Rac1WT, 88 cells total; Rac1C18S, 78 cells; and Rac1C18D, 92 cells). Representative images were collected using a Zeiss 710 microscope with a 63× oil objective and are shown in (A). The percentage of cells with lamellipodia was quantified (B). Statistical significance was determined using Student’s t test (*p = 0.05); error bars represent SEM.
4. Discussion
Among its many vital functions, the small GTPase Rac1 plays a key role in regulating redox enzymes, such as NOS and NOX, which generate nitric oxide and superoxide, respectively [66]. Moreover, Rac1 has been shown to interact directly with the antioxidant enzyme SOD1 in a redox- and nucleotide-dependent manner [39]. Rac1 has also been shown to be directly regulated by ROS and RNS oxidants in vitro [28] and by exposure to peroxide in REF52 and HeLa cells [33]. These findings suggest that ROS and RNS regulate both Rac1-mediated oxidant production and Rac1 signaling.
Redox regulation by modification of cysteine residues has been shown to regulate the function of diverse types of signaling proteins. For example, the transcription factor NF-κB, which is a key mediator of Rac1 function, is one prominent example of a cell signaling protein that is regulated in part by cysteine oxidation [67,68]. NF-κB can be regulated directly by S-glutathiolation of its own p50 and/or p65 subunit or indirectly by S-glutathiolation of its regulators IKK and IκBα [69]. In addition, p38 MAPK can be activated by reversible cysteine oxidation [70], whereas a number of protein tyrosine phosphatases become inactivated by oxidation of a catalytic cysteine [71]. In addition to these signaling proteins, we have previously reported that thiyl radical formation at Cys118 of the small GTPase Ras can regulate its activity [72,73]. This mechanism of activation appears to play a key role in Ras-mediated tumorigenesis [74]. However, we have previously shown that non-radical-mediated cysteine oxidation (i.e., nitrosation and glutathiolation) of this solvent-accessible cysteine does not alter Ras activity [75,76]. In contrast to Ras, the redox-sensitive cysteine in Rac1 (Cys18) has direct interactions with the bound nucleotide [77], and oxidation of this cysteine by both radical- and nonradical cysteine oxidation can impair guanine nucleotide binding, resulting in accelerated nucleotide exchange [28] and upregulation of Rac1 activity [33]. Given that cellular Rac1 colocalizes with enzymes that generate both ROS and RNS, Rac1 probably reacts with a variety of oxidants in the cell. Here, we show that cellular Rac1 is S-glutathiolated in chondrocytes. This oxidative modification leads to enhanced nucleotide exchange in vitro, which is a key step to promote Rac1 activation in cells. Consistent with these observations, we find that a Rac1 oxidative mimetic shows enhanced GTP-dependent effector binding and downstream sig-naling to the cytoskeleton.
The 305-Da S-glutathione modification is a common thiol oxidation product found under both basal conditions and oxidative stress [56,78] and has been observed in many proteins with redox-sensitive cysteine residues [69,79,80]. For example, the Ras GTPase undergoes S-glutathiolation at Cys118 upon exposure to peroxynitrite and DEA NONOate (an NO• -donating agent) in bovine aortic endothelial cells [80,81]. We now show that Rac1 is S-glutathiolated in primary chondrocytes upon menadione-induced ROS formation. Osteoarthritis is a degenerative joint disease that is characterized by high levels of oxidative stress in chondrocytes [34], and Rac1 activity is enhanced in osteoarthritic chondrocytes compared to normal cells [35]. Moreover, inhibition of Rac1 blocked production of the degra-dative matrix metalloprotease MMP-13 in chondrocytes [35] and reduced the severity of arthritis in a mouse model [14]. As we show here that S-glutathiolation of in vitro-purified Rac1 Cys18 greatly accelerates nucleotide dissociation, a key mechanism of activating Rac1, we postulate that Rac1 oxidation may promote its activation in degenerative joint disease and may play a role in driving arthritis progression.
In addition to S-glutathiolation, oxidation of cellular thiols by ROS can also promote the formation of sulfenic, sulfinic, and sulfonic acids. However, generation of a single species of oxidized thiol in vitro can prove difficult if not impossible. Previously, Permyakov et al. [57] used a Cys→Asp variant to mimic thiol oxidation in recoverin because aspartic acid shows similarity in shape and charge to sulfinic acid (Supplementary Fig. 4). As oxidation by ROS can yield several different oxidation products [32], use of the oxidation mimetic Rac1C18D allows for the study of a singly oxidized species. This variant shows greatly enhanced guanine nucleotide exchange, similar to glutathiolated Rac1, supporting the use of this variant as an oxidation mimetic. We also generated redox-insensitive Rac1C18S and Rac1C18A variants. Interestingly, we observe a trend toward increased nucleotide dissociation for Rac1C18S<Rac1C18A<Rac1C18D. Whereas the Rac1C18D variant has a dramatically enhanced (200-fold) GDP dissociation rate, similar to that observed for Rac1 glutathiolation, the Rac1C18A variant possesses an approximately 11-fold increased rate of nucleotide exchange relative to Rac1WT. In contrast, the Rac1C18S variant has an intrinsic dissociation rate that is similar to that of Rac1WT. Therefore, the effect of mutation at Cys18 on guanine nucleotide binding is dependent on the mutation type, with the less conservative negatively charged substitution (Rac1C18D) producing the largest perturbation in guanine nucleotide binding. Given these observations, we postulate that different oxidation states alter Rac1 activity in distinct ways.
Rac1 is well known as an inducer of cytoskeletal reorganization, such as membrane ruffling and lamellipodia formation [64,82]. Consistent with our in vitro data demonstrating increased nucleotide exchange, we observed that Rac1C18D is hyperactivated in HEK-293T cells and promotes lamellipodia formation in Swiss 3T3 cells, whereas Rac1C18S showed levels of cellular activation similar to those of Rac1WT. To the extent that the Rac1C18D variant mimics oxidized Rac1 (e.g., Rac1S–SG), these results suggest that oxidation of Rac1 at Cys18 can promote Rac1 activation through increased nucleotide exchange. Furthermore, our results indicate that Rac1C18D retains the ability to bind to at least a subset of effector proteins that promote Rac1-mediated cytoskeletal changes. Thus, unlike other dynamic posttranslational modifications of Rac1, such as phosphorylation [83,84] and palmitoylation of the C-terminal membrane-targeting domain [6], which decrease Rac1 activity and/or alter Rac1 effector binding by altering its localization [85], oxidation of the N-terminal Cys18 primarily enhances the population of activated Rac1.
Rac1 is also well known as an inducer of ROS and RNS [86]. However, in contrast to the enhancing effects of oxidation on Rac1-mediated cytoskeletal organization, cysteine oxidation of Rac1 has been reported to decrease its association with SOD1 [39]. Therefore, it will be interesting to determine whether its interactions with other redox enzymes, such as NOX [36,37] and NOS [40], are also negatively affected by Rac1 cysteine oxidation. If so, this could indicate the presence of a negative feedback loop that would further add to the complexity of Rac1-mediated redox signaling. Conversely, a positive feedback loop could exist in which oxidation of Rac1 may facilitate interactions with other effectors. Future studies will investigate whether Rac1 oxidation alters effector interactions.
Finally, it is intriguing to note that Rac1 and Ras are differentially regulated by thiol oxidants. We have previously reported that thiyl radical formation at Ras Cys118 can promote guanine nucleotide exchange and Ras activation [72,73], whereas non-radical-mediated nitrosation and glutathiolation does not alter Ras nucleotide binding [75,76]. In contrast, we show herein that Rac1 can be activated by nonradical oxidation at Cys18. We propose that the distinct locations of the redox-sensitive cysteines within the nucleotide binding motifs of Rac1 and Ras explain this differential regulation. We therefore performed sequence and structural analyses of the redox-sensitive motifs in the context of the redox-sensitive thiols. Fig. 10A and 10B depict the redox-sensitive motifs contained within the protein structures ([77] and pdb 3GFT) for Rac1 and Ras, respectively. The redox-sensitive thiol in Rac1, Cys18, is within the GxxxxGK [S/T] motif, otherwise known as the p-loop, which places the thiol within hydrogen-bonding distance from the α-phosphate. However, in Ras, the redox-sensitive thiol lies within the [N/T] KxD motif and does not have direct interactions with other residues in Ras or with the bound nucleotide. Further, the thiol is located 6.6 Å away from the bound nucleotide and is solvent exposed. This difference in location can explain their distinct modes of regulation. For both Rac1 and Ras, thiyl radical formation at their respective cysteines can lead to oxidation of the bound nucleotide [28,72,73], which promotes nucleotide dissociation and can result in activation. We find by several complementary methods that thiol oxidation by nonradical oxidative mechanisms perturbs nucleotide binding only in Rac1 and not Ras ([75,76] and this study). Further, the altered pKa of Rac1 Cys18 (this study) relative to Ras [61] renders Rac1 susceptible to thiol oxidation at physiological pH. Hence, Rac1 Cys18 is susceptible to a wider array of oxidative modifications, including both radical and nonradical oxidation. This differential oxidant-mediated regulation is depicted in Fig. 10C and 10D.
Fig. 10.
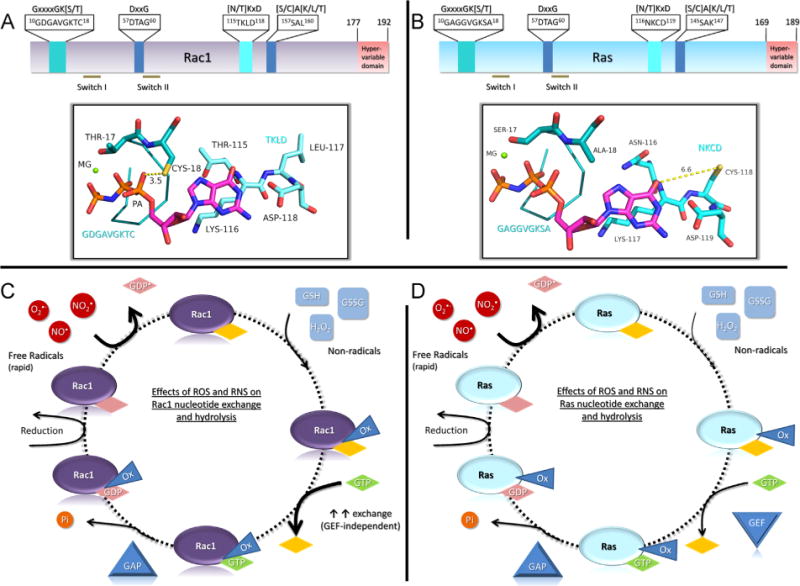
Differential modes of Rac1 and Ras activity regulation by thiol-mediated oxidants. (A) A linear depiction of Rac1 and residues critical for nucleotide binding. The top bar shows the consensus sequence for the nucleotide binding motifs with the GTPase-specific sequence highlighted within the box. Below, the structural depiction illustrates where the GxxxxGK[S/T] and [N/T]KxD motifs are located within the Rac1 structure (pdb 1MH1) and their proximity to the nucleotide. Cysteine 18 is approximately 3.5 Å from the α-phosphate (labeled PA) in Rac1 when bound to GDP or GTP. The coloring is as follows: blue, nitrogen; orange, phosphate; yellow, cysteine; red, oxygen; teal, carbon associated with Rac1; and the bound nucleotide (GMPPNP) carbons are colored magenta. (B) A linear depiction of Ras (pdb 3GFT) as in (A) with the structural depiction of the GxxxxGK[S/T] and [N/T]KxD motifs shown below. The redox-sensitive Cys118 is approximately 6.6 Å from the guanine ring of the bound GMPPNP nucleotide and does not make direct interactions with either the nucleotide or residues within Ras. In addition, the orientation of the Cys118 thiol is directed away from the nucleotide-binding pocket. (C) The redox-cycle diagram for Rac1 illustrates the effects of reactive oxygen and nitrogen species on Rac1 nucleotide binding. Rac1 is sensitive to thiol oxidation by radical oxidants through reaction with Cys18, resulting in enhanced nucleotide dissociation. In addition, reaction of Rac1 with nonradical oxidants impairs nucleotide binding by covalent modification of Cys18. Owing to the proximity of Cys18 to the bound nucleotide, oxidation at this site interferes with nucleotide binding, which is indicated by the oxidative modification triangle (Ox) that overlaps with the nucleotide, and results in increased nucleotide exchange. As oxidation does not affect GAP function, oxidized Rac1 can be inactivated by GAPs. Known nucleotide-dependent steps are labeled with diamonds with a nucleotide label and independent steps are shown as diamonds with no label. NO•, nitric oxide; , nitrogen dioxide; , superoxide; GSH, glutathione; GSSG, oxidized glutathione; H2O2, peroxide; GAP, GTPase-activating protein; GEF, guanine nucleotide exchange factor. (D) A redox-cycle diagram for Ras. The redox-sensitive thiol in Ras at Cys118 does not directly interact with either Ras residues or the bound nucleotide; consequently, the major difference between Ras and Rac1 is that covalent modification of Ras Cys118 does not alter nucleotide binding. As such, the oxidative modification does not overlap with the bound nucleotide, as depicted. Labeling is the same as in (C).
5. Conclusions
In summary, our in vitro and cell-based experiments suggest that oxidative modification of Rac1 can increase Rac1 activity and downstream signaling by enhancing nucleotide exchange. In addition, our findings indicate that Rac1C18D may prove useful as an oxidation mimetic, whereas Rac1C18S is a redox-insensitive variant that displays biochemical properties, structure, and interactions with Rac1 regulators similar to Rac1WT under nonstressed conditions. Thus, the Rac1C18S variant should serve as a valuable tool to determine whether cellular oxidants act directly or indirectly on Rac1 and will aid in elucidating the role of Rac1 oxidation in normal and disease states. Our data support a potential new layer of regulation for Rac1 activation through oxidative modification. Redox regulation of Rac1 may not only directly modulate Rac1 activity but also provide a feedback mechanism for regulating major ROS- and RNS-modulating enzymes, including NOS, NOX, and SOD1, that are, in turn, regulated by Rac1. Our observations are particularly interesting given that Rac1 plays a critical role in cellular redox regulation and that both Rac1 and ROS/RNS are involved in multiple common pathophysiological conditions. For example, our finding that Rac1 is S-glutathiolated in primary chondrocytes suggests a possible mechanism for regulating Rac1 activity levels in osteoarthritis. It will be interesting to determine the role of Rac1 oxidation in this and other diseases, such as cancer, in which high levels of oxidative stress are present.
Supplementary Material
Acknowledgments
The research efforts described herein were supported by the National Institutes of Health through Grants CA089614 to S.L.C., CA161494 and CA042978 to A.D.C., AR049003 to R.F.L., and NIH 1U24CA160035 from the National Cancer Institute Clinical Proteomic Tumor Analysis Consortium to X.C. G.A.H., L.E.M., and M.E.A. were partially funded by the Program in Molecular and Cellular Biophysics (NIH T32GM008570). We also thank Min Qi Lu for his help in purifying the proteins.
Abbreviations
- ABD-f
4-(aminosulfonyl)-7-fluoro-2,1,3-benzoxadiazole
- βME
β-mercaptoethanol
- CD
circular dichroism
- DMEM
Dulbecco’s modified Eagle’s medium
- DTPA
diethylenetriaminepentaacetic acid
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- eNOS
endothelial nitric oxide synthase
- ETD–MS/MS
electron transfer dissociation–tandem mass spectrometry
- FRET
fluorescence resonance energy transfer
- GAP
GTPase-activating protein
- GDI
guanine nucleotide dissociation inhibitor
- GEF
guanine nucleotide exchange factor
- GSSG
oxidized glutathione
- HSQC
heteronuclear single-quantum coherence spectroscopy
- IκBα
nuclear factor of κ light polypeptide gene enhancer in B cells inhibitor, α
- IKK
IκB kinase
- IGF-1
insulin-like growth factor-1
- MAPK
mitogen-activated kinase
- mant-GDP
2′-/3′-O-(N′-(methylanthraniloyl)guanosine-5′-O-diphosphate
- MMP
matrix metalloproteinase
- NF-κB
nuclear factor-κB
- nNOS
neuronal nitric oxide synthase
- NOX
NADPH oxidase
- PBD
p21-binding domain
- PBS
phosphate-buffered saline
- p-loop
phosphoryl-binding loop
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SOD1
superoxide dismutase 1
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed.2014.09.027.
References
- 1.Bosco EE, Mulloy JC, Zheng Y. Rac1 GTPase: a Rac of all trades. Cell Mol Life Sci. 2009;66:370–374. doi: 10.1007/s00018-008-8552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12:493–504. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 4.Adamson P, Paterson HF, Hall A. Intracellular localization of the P21rho proteins. J Cell Biol. 1992;119:617–627. doi: 10.1083/jcb.119.3.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post-translational modifications of p21rho proteins. J Biol Chem. 1992;267:20033–20038. [PubMed] [Google Scholar]
- 6.Navarro-Lerida I, Sanchez-Perales S, Calvo M, Rentero C, Zheng Y, Enrich C, Del Pozo MA. A palmitoylation switch mechanism regulates Rac1 function and membrane organization. EMBO J. 2012;31:534–551. doi: 10.1038/emboj.2011.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwarz J, Proff J, Havemeier A, Ladwein M, Rottner K, Barlag B, Pich A, Tatge H, Just I, Gerhard R. Serine-71 phosphorylation of Rac1 modulates downstream signaling. PLoS One. 2012;7:e44358. doi: 10.1371/journal.pone.0044358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Visvikis O, Lores P, Boyer L, Chardin P, Lemichez E, Gacon G. Activated Rac1, but not the tumorigenic variant Rac1b, is ubiquitinated on Lys 147 through a JNK-regulated process. FEBS J. 2008;275:386–396. doi: 10.1111/j.1742-4658.2007.06209.x. [DOI] [PubMed] [Google Scholar]
- 9.Nethe M, Anthony EC, Fernandez-Borja M, Dee R, Geerts D, Hensbergen PJ, Deelder AM, Schmidt G, Hordijk PL. Focal-adhesion targeting links caveolin-1 to a Rac1-degradation pathway. J Cell Sci. 2010;123:1948–1958. doi: 10.1242/jcs.062919. [DOI] [PubMed] [Google Scholar]
- 10.Castillo-Lluva S, Tatham MH, Jones RC, Jaffray EG, Edmondson RD, Hay RT, Malliri A. SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nat Cell Biol. 2010;12:1078–1085. doi: 10.1038/ncb2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugihara K, Nakatsuji N, Nakamura K, Nakao K, Hashimoto R, Otani H, Sakagami H, Kondo H, Nozawa S, Aiba A, Katsuki M. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene. 1998;17:3427–3433. doi: 10.1038/sj.onc.1202595. [DOI] [PubMed] [Google Scholar]
- 12.Debidda M, Williams DA, Zheng Y. Rac1 GTPase regulates cell genomic stability and senescence. J Biol Chem. 2006;281:38519–38528. doi: 10.1074/jbc.M604607200. [DOI] [PubMed] [Google Scholar]
- 13.Mack NA, Whalley HJ, Castillo-Lluva S, Malliri A. The diverse roles of Rac signaling in tumorigenesis. Cell Cycle. 2011;10:1571–1581. doi: 10.4161/cc.10.10.15612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu S, Lu P, Liu H, Chen P, Wu Y, Wang Y, Sun H, Zhang X, Xia Q, Heng BC, Zhou Y, Ouyang HW. Inhibition of Rac1 activity by controlled release of NSC23766 from chitosan microspheres effectively ameliorates osteoarthritis development in vivo. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2013-203901. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci USA. 2006;103:7432–7437. doi: 10.1073/pnas.0510444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tudor EL, Perkinton MS, Schmidt A, Ackerley S, Brownlees J, Jacobsen NJ, Byers HL, Ward M, Hall A, Leigh PN, Shaw CE, McLoughlin DM, Miller CC. ALS2/Alsin regulates Rac–PAK signaling and neurite outgrowth. J Biol Chem. 2005;280:34735–34740. doi: 10.1074/jbc.M506216200. [DOI] [PubMed] [Google Scholar]
- 17.Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582:2093–2101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 18.Wertheimer E, Gutierrez-Uzquiza A, Rosemblit C, Lopez-Haber C, Sosa MS, Kazanietz MG. Rac signaling in breast cancer: a tale of GEFs and GAPs. Cell Signalling. 2012;24:353–362. doi: 10.1016/j.cellsig.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alan JK, Lundquist EA. Mutationally activated Rho GTPases in cancer. Small GTPases. 2013;4:159–163. doi: 10.4161/sgtp.26530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, Capatana A, Holman EC, Bosenberg M, Sznol M, Kluger HM, Brash DE, Stern DF, Materin MA, Lo RS, Mane S, Ma S, Kidd KK, Hayward NK, Lifton RP, Schlessinger J, Boggon TJ, Halaban R. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis MJ, Ha BH, Holman EC, Halaban R, Schlessinger J, Boggon TJ. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc Natl Acad Sci USA. 2013;110:912–917. doi: 10.1073/pnas.1220895110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li A, Machesky LM. Rac1 cycling fast in melanoma with P29S. Pigment Cell Melanoma Res. 2013;26:289–290. doi: 10.1111/pcmr.12074. [DOI] [PubMed] [Google Scholar]
- 23.Machesky LM, Sansom OJ. Rac1 in the driver’s seat for melanoma. Pigment Cell Melanoma Res. 2012;25:762–764. doi: 10.1111/pcmr.12004. [DOI] [PubMed] [Google Scholar]
- 24.Kawazu M, Ueno T, Kontani K, Ogita Y, Ando M, Fukumura K, Yamato A, Soda M, Takeuchi K, Miki Y, Yamaguchi H, Yasuda T, Naoe T, Yamashita Y, Katada T, Choi YL, Mano H. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc Natl Acad Sci USA. 2013;110:3029–3034. doi: 10.1073/pnas.1216141110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh A, Karnoub AE, Palmby TR, Lengyel E, Sondek J, Der CJ. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene. 2004;23:9369–9380. doi: 10.1038/sj.onc.1208182. [DOI] [PubMed] [Google Scholar]
- 26.Matos P, Jordan P. Increased Rac1b expression sustains colorectal tumor cell survival. Mol Cancer Res. 2008;6:1178–1184. doi: 10.1158/1541-7786.MCR-08-0008. [DOI] [PubMed] [Google Scholar]
- 27.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heo J, Campbell SL. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J Biol Chem. 2005;280:31003–31010. doi: 10.1074/jbc.M504768200. [DOI] [PubMed] [Google Scholar]
- 29.Pryor WA, Houk KN, Foote CS, Fukuto JM, Ignarro LJ, Squadrito GL, Davies KJ. Free radical biology and medicine: it’s a gas, man! Am J Physiol Regul Integr Comp Physiol. 2006;291:R491–511. doi: 10.1152/ajpregu.00614.2005. [DOI] [PubMed] [Google Scholar]
- 30.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signalling. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287:C246–C256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 32.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 33.Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One. 2009;4:e8045. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henrotin YE, Bruckner P, Pujol JP. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage. 2003;11:747–755. doi: 10.1016/s1063-4584(03)00150-x. [DOI] [PubMed] [Google Scholar]
- 35.Long DL, Willey JS, Loeser RF. Rac1 is required for matrix metalloproteinase 13 production by chondrocytes in response to fibronectin fragments. Arthritis Rheum. 2013;65:1561–1568. doi: 10.1002/art.37922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leto TL, Morand S, Hurt D, Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signaling. 2009;11:2607–2619. doi: 10.1089/ars.2009.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ueyama T, Geiszt M, Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol Cell Biol. 2006;26:2160–2174. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liochev SI, Fridovich I. The effects of superoxide dismutase on H2O2 formation. Free Radic Biol Med. 2007;42:1465–1469. doi: 10.1016/j.freeradbiomed.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 39.Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schoneich C, Engelhardt JF. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Selvakumar B, Hess DT, Goldschmidt-Clermont PJ, Stamler JS. Co-regulation of constitutive nitric oxide synthases and NADPH oxidase by the small GTPase Rac. FEBS Lett. 2008;582:2195–2202. doi: 10.1016/j.febslet.2008.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lundberg JO, Weitzberg E. Nitrite reduction to nitric oxide in the vasculature. Am J Physiol Heart Circ Physiol. 2008;295:H477–478. doi: 10.1152/ajpheart.00611.2008. [DOI] [PubMed] [Google Scholar]
- 42.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bult H, Boeckxstaens GE, Pelckmans PA, Jordaens FH, Van Maercke YM, Herman AG. Nitric oxide as an inhibitory non-adrenergic non-cholinergic neurotransmitter. Nature. 1990;345:346–347. doi: 10.1038/345346a0. [DOI] [PubMed] [Google Scholar]
- 44.Shay AH, Choi R, Whittaker K, Salehi K, Kitchen CM, Tashkin DP, Roth MD, Baldwin GC. Impairment of antimicrobial activity and nitric oxide production in alveolar macrophages from smokers of marijuana and cocaine. J Infect Dis. 2003;187:700–704. doi: 10.1086/368370. [DOI] [PubMed] [Google Scholar]
- 45.Liaudet L, Vassalli G, Pacher P. Role of peroxynitrite in the redox regulation of cell signal transduction pathways. Front Biosci (Landmark Ed) 2009;14:4809–4814. doi: 10.2741/3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crompton AM, Foley LH, Wood A, Roscoe W, Stokoe D, McCormick F, Symons M, Bollag G. Regulation of Tiam1 nucleotide exchange activity by pleckstrin domain binding ligands. J Biol Chem. 2000;275:25751–25759. doi: 10.1074/jbc.M002050200. [DOI] [PubMed] [Google Scholar]
- 47.Shutes A, Der CJ. Real-time in vitro measurement of GTP hydrolysis. Methods. 2005;37:183–189. doi: 10.1016/j.ymeth.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 48.Brune M, Hunter JL, Corrie JET, Webb MR. Direct, real-time measurement of rapid inorganic-phosphate release using a novel fluorescent-probe and its application to actomyosin subfragment-1 ATPase. Biochemistry. 1994:8262–8271. doi: 10.1021/bi00193a013. 8262–8271; 199433. [DOI] [PubMed] [Google Scholar]
- 49.Gu H, Lalonde S, Okumoto S, Looger LL, Scharff-Poulsen AM, Grossman AR, Kossmann J, Jakobsen I, Frommer WB. A novel analytical method for in vivo phosphate tracking. FEBS Lett. 2006;580:5885–5893. doi: 10.1016/j.febslet.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751:119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 51.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 52.Johnson BA, Blevins RA. NMR View: a computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 53.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 54.Alan JK, Berzat AC, Dewar BJ, Graves LM, Cox AD. Regulation of the Rho family small GTPase Wrch-1/RhoU by C-terminal tyrosine phosphorylation requires Src. Mol Cell Biol. 2010;30:4324–4338. doi: 10.1128/MCB.01646-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Madigan JP, Bodemann BO, Brady DC, Dewar BJ, Keller PJ, Leitges M, Philips MR, Ridley AJ, Der CJ, Cox AD. Regulation of Rnd3 localization and function by protein kinase C alpha-mediated phosphorylation. Biochem J. 2009;424:153–161. doi: 10.1042/BJ20082377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hill BG, Bhatnagar A. Protein S-glutathiolation: redox-sensitive regulation of protein function. J Mol Cell Cardiol. 2012;52:559–567. doi: 10.1016/j.yjmcc.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Permyakov SE, Zernii EY, Knyazeva EL, Denesyuk AI, Nazipova AA, Kolpakova TV, Zinchenko DV, Philippov PP, Permyakov EA, Senin II. Oxidation mimicking substitution of conservative cysteine in recoverin suppresses its membrane association. Amino Acids. 2012;42:1435–1442. doi: 10.1007/s00726-011-0843-0. [DOI] [PubMed] [Google Scholar]
- 58.Rossman KL, Worthylake DK, Snyder JT, Cheng L, Whitehead IP, Sondek J. Functional analysis of Cdc42 residues required for guanine nucleotide exchange. J Biol Chem. 2002;277:50893–50898. doi: 10.1074/jbc.M208580200. [DOI] [PubMed] [Google Scholar]
- 59.Isom DG, Marguet PR, Oas TG, Hellinga HW. A miniaturized technique for assessing protein thermodynamics and function using fast determination of quantitative cysteine reactivity. Proteins. 2011;79:1034–1047. doi: 10.1002/prot.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hobbs GA, Gunawardena HP, Campbell SL. Biophysical and proteomic characterization strategies for cysteine modifications in ras GTPases. Methods Mol Biol. 2014;1120:75–96. doi: 10.1007/978-1-62703-791-4_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roos G, Foloppe N, Messens J. Understanding the pK(a) of redox cysteines: the key role of hydrogen bonding. Antioxid Redox Signaling. 2013;18:94–127. doi: 10.1089/ars.2012.4521. [DOI] [PubMed] [Google Scholar]
- 62.Zhang J, Matthews CR. Ligand binding is the principal determinant of stability for the p21(H)-ras protein. Biochemistry. 1998;37:14881–14890. doi: 10.1021/bi9811157. [DOI] [PubMed] [Google Scholar]
- 63.Thapar R, Moore CD, Campbell SL. Backbone 1H, 13C, and 15N resonance assignments for the 21 kDa GTPase Rac1 complexed to GDP and Mg2+ J Biomol NMR. 2003;27:87–88. doi: 10.1023/a:1024774230562. [DOI] [PubMed] [Google Scholar]
- 64.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 65.Guo F, Debidda M, Yang L, Williams DA, Zheng Y. Genetic deletion of Rac1 GTPase reveals its critical role in actin stress fiber formation and focal adhesion complex assembly. J Biol Chem. 2006;281:18652–18659. doi: 10.1074/jbc.M603508200. [DOI] [PubMed] [Google Scholar]
- 66.Mitchell L, Hobbs GA, Aghajanian A, Campbell SL. Redox regulation of Ras and Rho GTPases: mechanism and function. Antioxid Redox Signaling1. 2013;8:250–258. doi: 10.1089/ars.2012.4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakajima S, Kitamura M. Bidirectional regulation of NF-κB by reactive oxygen species: a role of unfolded protein response. Free Radic Biol Med. 2013;65:162–174. doi: 10.1016/j.freeradbiomed.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 68.Tobar N, Caceres M, Santibanez JF, Smith PC, Martinez J. RAC1 activity and intracellular ROS modulate the migratory potential of MCF-7 cells through a NADPH oxidase and NF kappa B-dependent mechanism. Cancer Lett. 2008;267:125–132. doi: 10.1016/j.canlet.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 69.Shelton MD, Mieyal JJ. Regulation by reversible S-glutathionylation: molecular targets implicated in inflammatory diseases. Mol Cells. 2008;25:332–346. [PMC free article] [PubMed] [Google Scholar]
- 70.Templeton DJ, Aye MS, Rady J, Xu F, Cross JV. Purification of reversibly oxidized proteins (PROP) reveals a redox switch controlling p38 MAP kinase activity. PLoS One. 2010:5. doi: 10.1371/journal.pone.0015012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ostman A, Frijhoff J, Sandin A, Bohmer FD. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem. 2011;150:345–356. doi: 10.1093/jb/mvr104. [DOI] [PubMed] [Google Scholar]
- 72.Davis MF, Zhou L, Ehrenshaft M, Ranguelova K, Gunawardena HP, Chen X, Bonini MG, Mason RP, Campbell SL. Detection of Ras GTPase protein radicals through immunospin trapping. Free Radic Biol Med. 2012;53:1339–1345. doi: 10.1016/j.freeradbiomed.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heo J, Prutzman KC, Mocanu V, Campbell SL. Mechanism of free radical nitric oxide-mediated Ras guanine nucleotide dissociation. J Mol Biol. 2005;346:1423–1440. doi: 10.1016/j.jmb.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 74.Lim KH, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646–649. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Williams JG, Pappu K, Campbell SL. Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc Natl Acad Sci USA. 2003;100:6376–6381. doi: 10.1073/pnas.1037299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hobbs GA, Bonini MG, Gunawardena HP, Chen X, Campbell SL. Glutathiolated Ras: characterization and implications for Ras activation. Free Radic Biol Med. 2013;57:221–229. doi: 10.1016/j.freeradbiomed.2012.10.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hirshberg M, Stockley RW, Dodson G, Webb MR. The crystal structure of human rac1, a member of the rho-family complexed with a GTP analogue. Nat Struct Biol. 1997;4:147–152. doi: 10.1038/nsb0297-147. [DOI] [PubMed] [Google Scholar]
- 78.Held JM, Gibson BW. Regulatory control or oxidative damage? Proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol Cell Proteomics. 2012;11(R111):013037. doi: 10.1074/mcp.R111.013037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cardaci S, Filomeni G, Ciriolo MR. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J Cell Sci. 2012;125:2115–2125. doi: 10.1242/jcs.095216. [DOI] [PubMed] [Google Scholar]
- 80.Clavreul N, Bachschmid MM, Hou X, Shi C, Idrizovic A, Ido Y, Pimentel D, Cohen RA. S-glutathiolation of p21ras by peroxynitrite mediates endothelial insulin resistance caused by oxidized low-density lipoprotein. Arterioscler Thromb Vasc Biol. 2006;26:2454–2461. doi: 10.1161/01.ATV.0000242791.28953.4c. [DOI] [PubMed] [Google Scholar]
- 81.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 82.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 83.Kwon T, Kwon DY, Chun J, Kim JH, Kang SS. Akt protein kinase inhibits Rac1–GTP binding through phosphorylation at serine 71 of Rac1. J Biol Chem. 2000;275:423–428. doi: 10.1074/jbc.275.1.423. [DOI] [PubMed] [Google Scholar]
- 84.Chang FM, Lemmon C, Lietha D, Eck M, Romer L. Tyrosine phosphorylation of Rac1: a role in regulation of cell spreading. PLoS One. 2011:6. doi: 10.1371/journal.pone.0028587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eisenberg S, Laude AJ, Beckett AJ, Mageean CJ, Aran V, Hernandez-Valladares M, Henis YI, Prior IA. The role of palmitoylation in regulating Ras localization and function. Biochem Soc Trans. 2013;41:79–83. doi: 10.1042/BST20120268. [DOI] [PubMed] [Google Scholar]
- 86.Hobbs GA, Zhou B, Cox AD, Campbell SL. Rho GTPases, oxidation, and cell redox control. Small GTPases. 2014;5:e28579. doi: 10.4161/sgtp.28579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.