

Abstract
Renal dopamine D1-like receptors (D1R and D5R) and the gastrin receptor (CCKBR) are involved in the maintenance of sodium homeostasis. The D1R has been found to interact synergistically with CCKBR in renal proximal tubule (RPT) cells to promote natriuresis and diuresis. D5R, which has a higher affinity for dopamine than D1R, has some constitutive activity. Hence, we sought to investigate the interaction between D5R and CCKBR in the regulation of renal sodium excretion. In present study, we found D5R and CCKBR increase each other’s expression in a concentration- and time-dependent manner in the HK-2 cell, the specificity of which was verified in HEK293 cells heterologously expressing both human D5R and CCKBR and in RPT cells from a male normotensive human. The specificity of D5R in the D5R and CCKBR interaction was verified further using a selective D5R antagonist, LE-PM436. Also, D5R and CCKBR colocalize and co-immunoprecipitate in BALB/c mouse RPTs and human RPT cells. CCKBR protein expression in plasma membrane-enriched fractions of renal cortex (PMFs) is greater in D5R-/- mice than D5R+/+ littermates and D5R protein expression in PMFs is also greater in CCKBR-/- mice than CCKBR+/+ littermates. High salt diet, relative to normal salt diet, increased the expression of CCKBR and D5R proteins in PMFs. Disruption of CCKBR in mice caused hypertension and decreased sodium excretion. The natriuresis in salt-loaded BALB/c mice was decreased by YF476, a CCKBR antagonist and Sch23390, a D1R/D5R antagonist. Furthermore, the natriuresis caused by gastrin was blocked by Sch23390 while the natriuresis caused by fenoldopam, a D1R/D5R agonist, was blocked by YF476. Taken together, our findings indicate that CCKBR and D5R synergistically interact in the kidney, which may contribute to the maintenance of normal sodium balance following an increase in sodium intake.
Introduction
Hypertension occurs as a consequence of a complex interplay among multiple genetic, epigenetic, and environmental determinants [1]. Salt consumption is an important non-genetic determinant, and excessive dietary salt intake can increase blood pressure in genetically susceptible individuals [2]. Recent population-based studies have revealed a nonlinear with even a J-shaped correlation between salt intake and blood pressure or cardiovascular disease mortality [3–5]. An increasing number of hormones, via their receptors, have been reported to regulate ion exchangers, transporters, channels, and pumps in renal tubules, including the renal proximal tubule (RPT), that are crucial in maintaining normal sodium balance [6,7].
Dopamine, secreted in the kidney mainly by RPT cells, via its receptors that are classified into “D1-like” (D1R and D5R) and “D2-like” (D2R, D3R and D4R) receptors, is responsible for over 50% of renal sodium excretion during conditions of mild volume and sodium excess [8–10]. The acute infusion of fenoldopam, a D1-like receptor agonist, induces natriuresis and diuresis in humans, rats, and mice [8–14]. Disruption of any of the dopamine receptor gene subtypes in mice causes hypertension which can be aggravated by salt loading that is dopamine receptor subtype dependent [10].
Gastrointestinal hormones have been reported to be involved in the regulation of renal sodium excretion and blood pressure [14,15]. An oral sodium load causes a greater natriuresis than an intravenous infusion of the same amount of sodium [16–18], suggesting that gastrointestinal hormones have a role in regulating the postprandial natriuretic response. One such hormone may be gastrin. Mice lacking the gastrin gene are hypertensive and salt-sensitive [18]. The receptor of gastrin, CCKBR, has been reported to be expressed in several nephron segments, including the RPT and collecting duct [18–21]. Gastrin, which is taken up by RPT to a greater extent than other gut hormones [22], via CCKBR, can induce natriuresis and diuresis by inhibiting the activities of renal Na+-K+-ATPase and sodium/hydrogen exchanger type 3 (NHE3) [14,18–20].
Our recent study reported a synergistic interaction between gastrin, via CCKBR, and D1R, one of the two D1-like receptors, in promoting water and sodium excretion [14]. The other D1-like receptor, D5R, has a 30% homology in the N and C termini and an 80% homology in the transmembrane domain with the D1R. D5R and D1R, via a D1R/D5R heteromer, cooperatively decrease sodium transport in RPT cells by inhibition of NHE3 and Na+-K+-ATPase activities [23]. The D5R may be more important than D1R in regulating salt balance because D5R has some constitutive activity and a higher affinity for dopamine than D1R [24,25]. Therefore, in this study, we tested the hypothesis that D5R and CCKBR synergistically regulate each other in the kidney, specifically in RPT cells, which may have important implications in the regulation of renal sodium excretion.
Materials and Methods
Materials
We used immortalized human RPT cells (HK-2) (China Center for Type Culture Collection, 3115CNCB00336, Wuhan, China), as well as well-characterized human embryonic kidney 293 (HEK293) cells, heterologously expressing human D5R [26,27], and RPT cells from a normotensive Caucasian male (NT) [28]. Adult (4-month old) male BALB/c mice were bought from Beijing HFK Bioscience Co, LTD. Sixth generation progeny D5R-/- and CCKBR-/- mice were obtained from Jackson Laboratory and bred in an AAALAC-accredited facility. The 4-month male D5R-/- and CCKBR-/- mice and their littermates were used for further study. All animal-related studies were approved by the Institutional Animal Care and Use Committee of the Institute of Laboratory Animal Science, Peking Union Medical Collage, China. The information of all the chemical drugs, antibodies, and related test kits are listed in S1 Table.
Cell culture
All the cells (HK-2, HEK293, and NT), in DMEM/F12 with 4.5 g/L D-glucose, supplemented with 10% fetal bovine serum, 100 μg/ml penicillin and 10 μg/ml streptomycin, were cultured in a humidified cell culture incubator maintained at 37°C and supplied with 5% CO2 and 95% O2. We used cells with low passage numbers (<20 for HK-2 and NT cells and <40 for HEK293 cells) to avoid the confounding effects of cellular senescence. The cells tested negative for mycoplasma infection.
Co-transfection
HEK293 cells stably overexpressing full-length human D5R with blasticidin resistance (HEK293-D5R) were previously generated in our laboratory [26,27]. HEK293-D5R cells were transfected with pCMV6-AC vector with full-length human CCKBR cDNA, using Lipofectamine 2000 transfection reagents, according to the manufacturer’s protocol. Stably transfected single colonies, selected with 600 μg/ml neomycin for the CCKBR-positive colonies, were designated as HEK293-D5R-CCKBR cells.
Immunoblotting
Whole cell lysates were extracted in ice-cold RIPA lysis buffer, sonicated, kept on ice for 30 minutes, and centrifuged with 14000 g for 30 minutes at 4°C. PMFs were extracted using a membrane protein extraction Kit (Sango Biotech, China), in accordance with the manufacturer’s instruction. Detailed steps are shown in S1 File. Protein concentration was determined by bicinchoninic acid assay using bovine serum albumin as a standard. Equal amounts of protein (60 μg for whole cell lysates and 40 μg for PMFs) were subjected to immunoblotting. The densitometry values of whole cell lysates were normalized by the expression of GAPDH. The primary antibodies are mouse polyclonal anti-D5R (Santa Cruz, USA) and rabbit polyclonal anti-CCKBR (NOVUS, USA) whose specificities have been reported [29,30].
Quantitative Real-Time PCR
Total mRNA was purified using 1ml Trizol and quantified using a spectrophotometer. The RNA samples were reverse-transcribed using SuperScript III. Gene expression was quantified by real-time PCR, using an Applied Biosystem 7500 Real-Time PCR System. The assay used gene specific primers and One Step SYBR PrimeScript RT-PCR Kit, as described in the manufacturer’s manual. All the primers used in this study are provided in S2 Table. Data were analyzed using the △△Ct method [31].
Co-immunoprecipitation
Co-immunoprecipitation was performed using an immunoprecipitation kit. Equal amounts of whole cell lysates (500 μg protein) were mixed with mouse anti-D5R antibody (Santa Cruz, USA), non-immune mouse serum (negative control), or mouse anti-CCKBR antibody (positive control, Santa Cruz, USA) whose specificity has been reported [32]. Protein A/G agarose beads were added and incubated overnight at 4°C. The bound proteins were eluted using 30 μl of Laemmli buffer. The samples were subjected to immunoblotting and probed for CCKBR using a rabbit anti-CCKBR antibody (Novus, USA). Reverse co-immunoprecipitation was performed using the same method; cell lysates were mixed with mouse anti-CCKBR antibody (Santa Cruz, USA), non-immune mouse serum (negative control), or mouse anti-D5R (positive control, Santa Cruz, USA) and the bound proteins were subjected to immunoblotting and probed for D5R, using a rabbit anti-D5R antibody (Santa Cruz, USA) whose specificity has been reported [33,34].
Confocal microscopy of double-strained HK-2 cells and RPTs of BALB/c mouse
HK-2 cells, grown on coverslips, were fixed with ice-cold methanol for 30 minutes. Five-micron sections were cut from formalin-fixed and paraffin-embedded BALB/c mouse kidneys. CCKBR was visualized using a polyclonal rabbit anti-CCKBR antibody (NOVUS, USA), followed by Alexa Fluor 568-labeled goat anti-rabbit secondary antibody (Abcam, USA). D5R was visualized using a polyclonal mouse anti-D5R antibody (Santa Cruz, USA), followed by Alexa Fluor 488-labeled goat anti-mouse secondary antibody (Abcam, USA). For a negative control, the primary antibodies were substituted with non-immune rabbit or mouse serum at an appropriate dilution. Colocalization of the D5R and CCKBR was identified by the development of a yellow color in the merged images.
Blood pressure measurement
Blood pressure was measured from the aorta, via the left carotid artery, under pentobarbital (60 mg/kg, administered intraperitoneally) anesthesia. Subsequently, the mice were sacrificed by neck dislocation; the kidneys were harvested and samples were prepared for immunoblotting.
Sodium excretion detection
BALB/c mice, CCKBR-/- mice and CCKBR+/+ littermates were acclimatized in metabolic cages for 3 days, then divided into two groups and fed normal (0.4% NaCl) or high-salt (3% NaCl) diet for two weeks. Afterwards, BALB/c mice on high salt diet were separated into seven groups and intraperitoneally injected with vehicle (normal saline, 0.5 ml), Sch23390 (a D1-like receptor antagonist, 0.1 mg/kg) [14,27,33,35], YF476 (a CCKBR antagonist, 0.1 mg/kg) [36,37], fenoldopam (a D1-like receptor agonist, 1mg/kg) [8–14], gastrin (a CCKBR ligand, 10 μg/kg) [14,18–20], fenoldopam (1mg/kg) coupled with YF476 (0.1 mg/kg) and gastrin (10 μg/kg) coupled with Sch23390 (0.1 mg/kg), respectively, daily for one week. The BALB/c mice on normal salt diet were also injected intraperitoneally with 0.5 ml normal saline, daily for one week. At the end of drug treatment, urine was collected for 24 hours. Urine sodium concentration was measured using a Synchron EL-ISE Electrolyte system (Beckman, USA). Urine creatinine concentration was measured by an automated enzymatic method [38]. Thereafter, the mice were sacrificed by neck dislocation and the kidneys were harvested and frozen in liquid nitrogen until use.
Statistics
The data are expressed as mean ± SEM. Significant difference between two groups was determined by Student’s t-test and one-way factorial ANOVA followed by Duncan’s multiple range test for groups>2. P<0.05 was considered significant.
Results
D5R and CCKBR co-regulation in HK-2 cells
In HK-2 cells, fenoldopam, a D1R and D5R agonist, increased CCKBR protein expression in a concentration- and time-dependent manner. The ability of fenoldopam (24 hours) to increase CCKBR protein was significant at ≥10−9 with a concentration for half-maximal stimulation (EC50) of 1.39 x 10−10 M (Fig 1A). The stimulatory effect of fenoldopam (10−6 M) was evident as early as 8 hours and lasted for at least 30 hours (Fig 1B). We next verified the specificity of the D1-like receptor stimulatory effect of fenoldopam (10−6 M, 24 hours) on CCKBR expression by using Sch23390, a D1-like receptor antagonist. As shown in Fig 1C, CCKBR protein expression was significantly increased with fenoldopam treatment, the effect of which was blocked by pre-incubation with Sch23390 (10−6 M, 24 hours), which by itself had no effect on CCKBR protein expression.
Fig 1. D5R and CCKBR co-regulation in HK-2 cells.

(A) CCKBR protein expression in response to varying concentrations of the D1R and D5R agonist fenoldopam (10−11–10−5 mol/L, 24 hours, n = 6, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). (B) CCKBR protein expression in response to varying durations of incubation with fenoldopam (0–30 hours, 10−6 mol/L, n = 6, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). (C) Effects of fenoldopam (10−6 mol/L, 24 hours) and D1R and D5R antagonist Sch23390 (10−6 mol/L, 24 hours) on CCKBR protein expression (n = 5, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). (D) D5R protein expression in response to varying concentrations of gastrin (10−12–10−6 mol/L, 24 hours, n = 6, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). (E) D5R protein expression in response to varying durations of incubation with gastrin (0–30 hours, 10−8 mol/L, n = 6, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). (F) Effects of gastrin (10-8mol/L, 24 hours) and CCKBR antagonist YF476 (10−8 mol/L, 24 hours) on D5R protein expression (n = 5, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). All immunoblotting results are expressed as relative density units (DU) and normalized by GAPDH expression. Immunoblots of D5R, CCKBR, and GAPDH are shown in the inset.
Gastrin, a CCKBR agonist, also increased D5R protein expression in a concentration- and time-dependent manner. The ability of gastrin to increase D5R protein was significant at ≥10−10 M with an EC50 of 1.76 x 10−11 M (Fig 1D). The stimulatory effect of gastrin (10−8 M) was evident as early as 8 hours and lasted for at least 30 hours (Fig 1E). The specificity of the stimulatory effect of gastrin (10−8 M, 24 hours) was determined by using CCKBR antagonist YF476 (10−8 M, 24 hours). The stimulatory effect of gastrin on D5R expression was abrogated by YF476, which by itself had no effect on D5R expression (Fig 1F).
D5R and CCKBR co-regulation in HEK293-D5R-CCKBR cells
Fenoldopam cannot distinguish the 2 subtypes of D1-like receptors, D1R and D5R, from each other [8–10, 12–14]. HEK293 cells express no endogenous D1R and some D5R [39]. Therefore, HEK293-D5R-CCKBR cells were generated to avoid the confounding effect of D1R. PCR and immunoblotting studies demonstrated stable HEK293-D5R-CCKBR cells over-expressing human D5R and CCKBR (S1 Fig). In these cells, gastrin increased D5R protein (1.7±0.2-fold, P<0.05) and mRNA (3.9±0.2-fold, P<0.05) expressions that were blocked by YF476, a CCKBR antagonist, which by itself had no effect (Fig 2A and 2B). Fenoldopam also significantly increased CCKBR protein (1.7±0.1 fold, P<0.05) and mRNA (5.1±0.3 fold, P<0.05) expressions, that were blocked by Sch23390, a specific D5R antagonist in the absence of D1R (Fig 2C and 2D). The results of the studies using HEK293-D5R-CCKBR cells combined with the results in HK-2 cells suggest a specific CCKBR-D5R interaction, independent of D1R, at both the transcriptional and translational levels.
Fig 2. D5R and CCKBR co-regulation in HEK293-D5R-CCKBR cells.
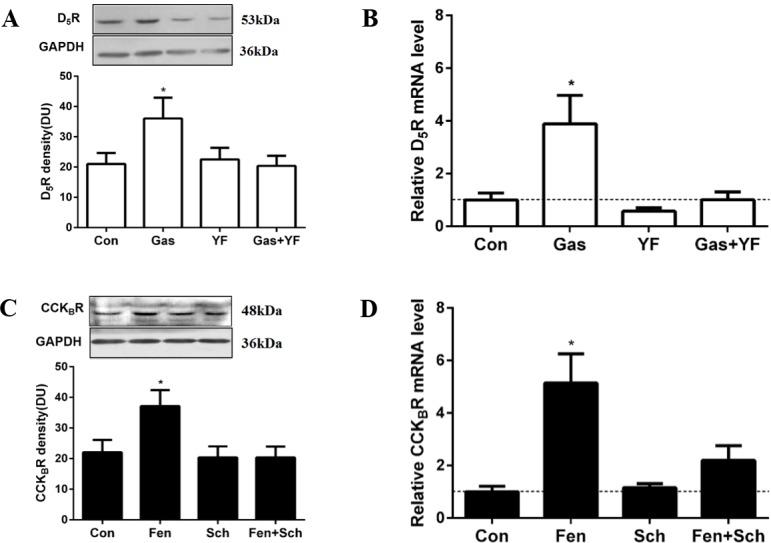
(A and B) Effects of gastrin (Gas, 10−8 mol/L, 3 hours) and CCKBR antagonist YF476 (YF, 10−8 mol/L, 3 hours) on D5R protein and mRNA expressions in HEK293-D5R-CCKBR cells (n = 6 for protein, n = 5 for mRNA, *P<0.05 versus others, one-way factorial ANOVA, Duncan’s test). (C and D) Effects of D1R and D5R agonist fenoldopam (Fen, 10−6 mol/L, 3 hours) and D1R and D5R antagonist Sch23390 (Sch, 10−6 mol/L, 3 hours) on CCKBR protein and mRNA expressions in HEK293-D5R-CCKBR cells (n = 6 for protein, n = 5 for mRNA, *P<0.05 versus others, one-way factorial ANOVA, Duncan’s test). Immunoblotting results are expressed as relative density units (DU). Immunoblots of D5R, CCKBR and GAPDH are shown in the inset. mRNA expression was determined by qRT-PCR and corrected for the expression of GAPDH mRNA.
Direct and/or indirect interaction between D5R and CCKBR
In order to affirm the potential for a direct or indirect interaction between D5R and CCKBR, we studied the co-localization of D5R and CCKBR in HK-2 cells and RPTs of BALB/c mice. Immunofluorescent staining showed that both D5R and CCKBR were mainly expressed and colocalized at the cell surface membranes of HK-2 cells (Fig 3A) and RPTs of BALB/c mice (Fig 3B). We also performed a co-immunoprecipitation study to determine whether there is a physical interaction between D5R and CCKBR and found that D5R co-immunoprecipitated with CCKBR in both HK-2 (Fig 4A) and HEK293-D5R-CCKBR cells (Fig 4B). These data indicate that CCKBR and D5R can interact with each other via a direct and/or indirect way.
Fig 3. D5R and CCKBR colocalization in RPTs of BALB/c mice.

Colocalization of Alexa Fluor 488-labeled D5R (green) and Alexa Fluor 568-labeled CCKBR (red) in (A) HK-2 cells and (B) the RPTs of BALB/c mice. The colocalization of D5R and CCKBR is illustrated by the yellow color in the merge images. Scale bar: 50 μm for HK-2 cells and 250 μm for the RPTs of BALB/c mice. The white arrows are pointing to the RPTs.
Fig 4. D5R and CCKBR physical interaction in HK-2 and HEK293-D5R-CCKBR cells.

Co-immunoprecipitation of D5R and CCKBR in HK-2 cells (A) and HEK293-D5R-CCKBR cells (B). Whole cell lysates were subjected to immunoprecipitation (IP) with mouse anti-D5R antibody, mouse anti-CCKBR antibody, or non-immune mouse serum (negative control). Immunoprecipitated complexes were analyzed by immunoblotting (western blot, WB), using rabbit anti-D5R antibody or rabbit anti-CCKBR antibody. PC: positive control; NC: negative control. These experiments were repeated three times with similar results.
Blood pressures in CCKBR-/- mice and CCKBR+/+ littermates
Mean arterial pressures (MAPs, Fig 5) measured under anesthesia were significantly higher in CCKBR-/- mice than that in CCKBR+/+ littermates (101.0±5.3 mmHg vs 82.5±4.3 mmHg) on normal salt diet. Even though high salt diet further increased the MAPs of both CCKBR-/- mice (110.4±4.0 mmHg vs 101.0±5.3 mmHg) and the corresponding littermates (96.0±3.3 mmHg vs 82.5±4.3 mmHg) compared with normal salt diet. High salt diet increased the MAPs of CCKBR-/- mice than those of CCKBR+/+ littermates (110.4±4.0 mmHg vs 96.0±3.3 mmHg).
Fig 5. MAPs in CCKBR-/- mice and CCKBR+/+ littermates.
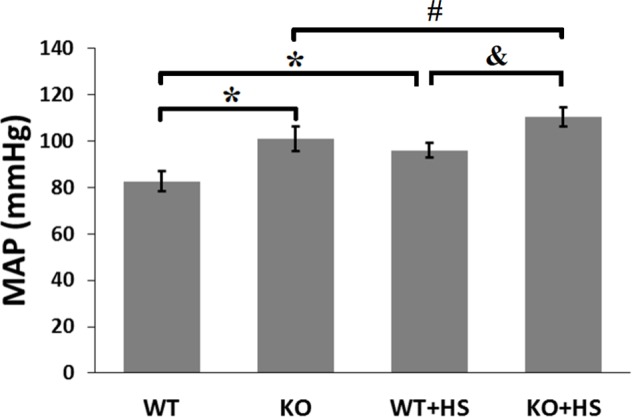
MAPs were measured from the aorta, via the left carotid artery, under pentobarbital anesthesia. WT and KO indicate CCKBR+/+ littermates (n = 19) and CCKBR-/- mice (n = 21) on normal salt diet, respectively; WT+HS and KO+HS indicate CCKBR+/+ littermates (n = 11) and CCKBR-/- mice (n = 8) on high salt diet, respectively. *P<0.05 vs WT, #P<0.05 vs KO, &P<0.05 vs WT+HS, one-way factorial ANOVA.
D5R and CCKBR interaction and their expression in mouse kidney
To test whether or not D5R and CCKBR can regulate each other’s expression in vivo, we measured, by immunoblotting, CCKBR protein expression in PMFs of D5R-/- mice and D5R protein expression in PMFs of CCKBR-/- mice. As shown in Fig 6A and 6B, CCKBR protein expression in PMFs was greater in D5R-/- mice than D5R+/+ littermates (73.3±4.3 vs 26.7±6.4; n = 3–4; P<0.05); D5R protein expression in PMFs was also greater in CCKBR-/- mice than CCKBR+/+ littermates (83.8±6.5 vs 16.2±2.2; n = 3–4; P<0.05).
Fig 6. D5R and CCKBR interaction in mouse kidney.
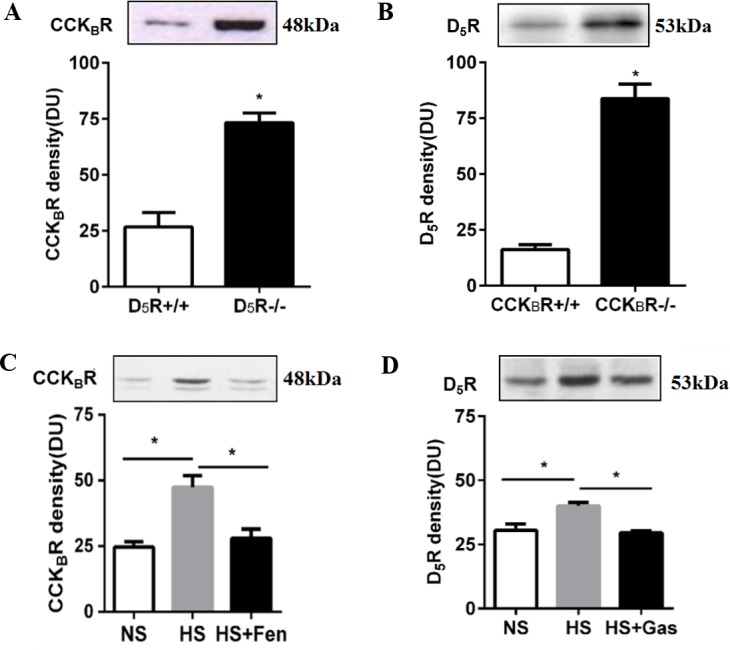
(A) CCKBR protein expression in PMFs is increased in D5R gene knockout mice (D5R-/-), relative to wild-type littermates (D5R+/+). (B) D5R protein expression in PMFs is increased in CCKBR gene knockout mice (CCKBR-/-) relative to wild-type littermates (CCKBR+/+). (C) Effect of the D1R and D5R agonist fenoldopam (Fen, 1mg/kg/day, one week) on the renal membrane protein expression of CCKBR in BALB/c mice fed high salt (HS) diet. NS = normal salt. (D) Effect of gastrin (Gas, 10g/kg/day, one week) on the renal membrane protein expression of D5R in BALB/c mice on high salt (HS) diet. NS = normal salt. *P<0.05, n = 3–5 per group, Student’s t test. All immunoblotting results are expressed as relative density units (DU). Sample loading amount was quantified by bicinchoninic acid assay. Immunoblots of D5R and CCKBR are shown in the inset.
We next assessed the interaction between renal CCKBR and D5R in BALB/c mice fed normal and high salt diets. High salt diet, relative to normal salt diet, increased both CCKBR protein expression in PMFs (47.4±4.4 vs 24.6±2.0; n = 3; P<0.05) (Fig 6C) and D5R protein expression in PMFs (40.0±1.3 vs 30.4±2.5; n = 3; P<0.05) (Fig 6D). Chronic stimulation of D1-like receptors by the intraperitoneal injection of fenoldopam, significantly decreased CCKBR protein expression in PMFs of BALB/c mice on high salt diet (27.9±3.6 vs 47.4±4.4; n = 3; P<0.05) (Fig 6C). Similarly, chronic stimulation of CCKBR, by the intraperitoneal injection of gastrin, also decreased D5R protein expression in PMFs of BALB/c mice on high salt diet (29.5±0.8 vs 40.0±1.3; n = 3; P<0.05) (Fig 6D).
Natriuretic effect of CCKBR-/- mice
D5R has been extensively reported as a natriuretic receptor [8,10,12,14,23,40]. In order to further explore the role of CCKBR in mediating renal sodium excretion, we studied the natriuretic effect of CCKBR-/- mice and their littermates (Fig 7). CCKBR-/- mice, in comparison with their littermates, had a notable decrease in natriuresis either on normal salt diet (59.1±6.6 vs 82.4±8.0) or high salt diet (135.7±17.0 vs 184.6±19.5).
Fig 7. Natriuretic effect of CCKBR-/- mice.

24-hour urine sodium excretion (UNa, mmol/L) was corrected for 24-hour creatinine excretion (UCr, mmol/L). WT = CCKBR+/+ littermates on normal salt diet (n = 8); KO = CCKBR-/- mice on normal salt diet (n = 12); WT+HS = CCKBR+/+ littermates on high salt diet (n = 8); KO+HS = CCKBR-/- mice on high salt diet (n = 8). *P<0.05 vs WT, &P<0.05 vs WT+HS, #P<0.05 vs KO, one-way factorial ANOVA.
D5R and CCKBR interaction and natriuresis
To determine if there is role of the interaction between D5R and CCKBR on their expression in modulating renal sodium transport, the natriuretic effect of the D1R and D5R agonist fenoldopam and gastrin in the presence or absence of their antagonists was investigated in BALB/c mice fed a high salt (3% NaCl) diet. As shown in Fig 8, the urine sodium excretion was significantly increased (241.4±22.4 vs 132.1±2.5; n = 7; P<0.05) after the high salt diet. Intraperitoneal administration of the D1R/D5R antagonist Sch23390 or the CCKBR antagonist YF476 evidently decreased the sodium excretion (169.4±16.8 vs 241.4±22.4 and 164.5±13.4 vs 241.4±22.4; n = 7; P<0.05), compared with the group of high salt diet. Fenoldopam significantly increased the natriuresis (377.4±38.0 vs 241.4±22.4; n = 6–7; P<0.05) in the mice fed high salt diet that was blocked by the CCKBR antagonist YF476. Similarly, gastrin evidently increased the natriuresis (443.7.4±85.3 vs 241.4±22.4; n = 6–7; P<0.05) in the mice fed high salt diet that was also blocked by the D1R/D5R antagonist, Sch23390. These results demonstrate that CCKBR and D5R interact with each other in increasing the natriuresis caused by their respective agonists in mice fed high salt diet.
Fig 8. D5R and CCKBR interaction and natriuresis.
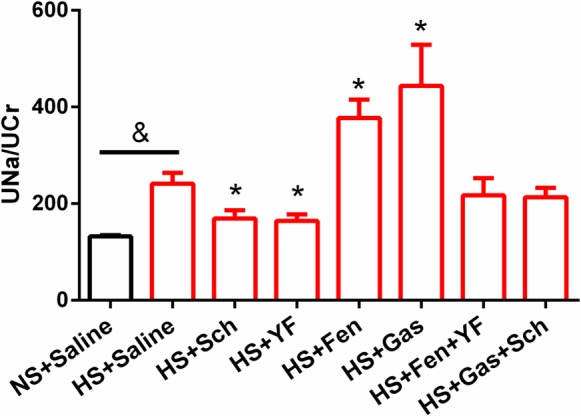
24-hour urinary sodium to creatinine ratio (UNa/UCr) was used to evaluate natriuresis. Red bars represent the groups fed high salt (HS) diet. Black bar represents the normal salt (NS) diet group. Sch = Sch23390 (D1R and D5R antagonist), Fen = Fenoldopam (D1R and D5R agonist), Gas = Gastrin, and YF = YF476 (CCKBR antagonist). n = 5–7, &P<0.05 vs NS, Student’s t test; *P<0.05 vs HS, one-way factorial ANOVA.
Discussion
Cross-transplantation studies between hypertensive and normotensive strains of rats and mice have provided convincing evidence for the role of the kidney in the regulation of blood pressure [40–42]. The kidney is the paramount organ in the regulation of sodium balance, an impairment of which causes a shift of the pressure-natriuresis curve “to the right”; a higher blood pressure is needed to excrete the same amount of sodium [43]. Relative to the other nephron segments, the RPT is responsible for reabsorption of >65% of filtered salt and water [42,44]. The renal regulation of sodium balance involves a cross-talk between natriuretic or anti-natriuretic factors acting on RPT, exemplified by the dopaminergic and renin-angiotensin (AT1R and AT2R) systems [6,8–14,23,33,35,40,42,44–48].
Our previous study, in agreement with other studies, verified that another natriuretic hormone, gastrin, from the gastrointestinal tract, may act in the kidney, including the RPT, to inhibit sodium transport [14,18–20]. Our results demonstrate that knockout of gastrin receptor (CCKBR) gene in mice results in high blood pressure that can be aggravated in response to an oral salt load, which is in accordance with an early study [18]. Gastrin can inhibit Na+-K+-ATPase activity in intestinal mucosa [49] and RPT cells [14]. Gastrin may increase the level of cAMP and the activities of some signal transducers, for example, protein kinase A and C (PKA and PKC), to cause a decrease in Na+-K+-ATPase activity, directly or indirectly [19,50–53]. Gastrin has been reported to increase NHE activity in pancreatic acini [54,55] but this is due mainly to NHE1 [56], although NHE3 is expressed in pancreatic duct cells [57]. By contrast, we have reported that gastrin inhibits NHE3 activity in human RPT cells by a phosphoinositide 3-kinase-/PKC dependent pathway [19] and Na+-K+-ATPase activity in rat RPT cells [14]. To test the possibility that the high blood pressure of CCKBR-/- mice is related to a decreased ability to excrete a sodium load that elicited by the inhibition of sodium handling, sodium excretion studies in CCKBR-/- mice and their littermates were performed. Our results show that CCKBR-/- mice excrete less sodium than CCKBR+/+ littermates either on normal salt diet or high salt diet.
Our previous study demonstrated a synergistic interaction between gastrin and renal dopamine, presumably acting at the D1R, in increasing renal sodium excretion [14]. Dopamine produced by the RPT, independent of renal nerves and not converted to norepinephrine, is important in facilitating the excretion of sodium after a moderate sodium load [8–14,40,42,44–48,58]. Prevention of the RPT production of dopamine [59] or deletion of any of the dopamine receptor subtypes [10] results in hypertension that is dopamine receptor subtype specific. Dopamine, via all its receptors, decreases renal sodium transport by inhibiting the activity of sodium exchangers, channels, and pump [8–14,23,42,44,46–48,58–60]. In the present study, we found a concentration- and time-dependent synergistic interaction between CCKBR and the other dopamine D1-like receptor, D5R. Because the over expression of proteins can result in promiscuous associations, we first used HK-2 and human RPT cells that endogenously express D5R and CCKBR. Although HK-2 cells retain many functional characteristics of RPTCs, a study discovered that HK-2 cells are uncoupled from D1R adenylyl cyclase stimulation [61]. Therefore, we verified this effect in NT cells obtained from a normotensive white male (S2 Fig); the D1-like receptor agonist fenoldopam-stimulated cAMP accumulation was similar in HK-2 and NT cells (S2 File and S3 Fig). These results demonstrated that our HK-2 cells have normal D1-like receptor adenylyl cyclase coupling, in agreement with other reports [60,62]. Although there is no agonist that is selective to D1R or D5R [9,10,12–14], we used a specific D5R antagonist, LE-PM436 [23,33] to verify the involvement of the D5R in our studies (S4 Fig). In order to rule out any confounding effect of the D1R, we co-expressed human D5R and CCKBR in HEK-293 cells (HEK293-D5R-CCKBR). In HEK293-D5R-CCKBR cells, we found that D5R and CCKBR regulated each other’s total cellular expression at both the protein and mRNA levels, suggesting that the regulation may occur at the transcriptional level.
We next tested if the D5R and CCKBR interaction in vitro has significance in vivo. We found that a high salt diet increased both D5R and CCKBR proteins expression in PMFs. However, in contrast to the ability of either receptor to increase each other’s total expression in cells in vitro, we found that the stimulation of one receptor actually decreased the other receptor protein expression in PMFs of sodium-loaded BALB/c mice. We did not study the mechanism of this finding. However, the stimulation of membrane bound receptors should result in their internalization [63], thus the decrease in D5R and CCKBR expressions in PMFs with gastrin and fenoldopam treatment, respectively. This may also explain why the disruption of one receptor (e.g. D5R-/-, CCKBR-/-) increased the other receptor expression in PMFs, i.e., there is no physical interaction and therefore, no internalization. Because D5R and CCKBR are both natriuretic receptors, disruption of either receptor may cause a compensatory increase in the protein expression of the other. In other words, short-term stimulation of one receptor may result in transient activation or increased expression of the other but continuous stimulation should result in desensitization. Because D5R and CCKBR physically interact, the desensitization of one may desensitize the other by their internalization or even decreased expression, if they are routed to proteasomes or lysosomes. We have reported that D5R-/- mice have increased sympathetic tone [64], which can result in vagal inhibition, releasing the slow inhibitory effect on gastrin release [65]. Therefore, we suggest that in vivo, intricate neural and hormonal mechanisms may participate in the mutual D5R and CCKBR regulation. However, the specific mechanism remains to be further explored.
Acute renal perfusion experiments have showed the synergistic effect of D1-like receptors, presumably D1R, and CCKBR on natriuresis [14]. In this study, we used the 24-hour urinary sodium to creatinine ratio [66] to evaluate the long-term effect of salt intake, gastrin, fenoldopam, and their respective receptor antagonists on urinary sodium excretion. High salt diet significantly increased water and food intake in BALB/c mice (S3 Table), that agrees with a commonly held belief that salt intake arouses thirst and increases food consumption [67,68]. High salt diet induced an augment in urinary sodium concentration. We think this, in part, may be an indirect consequence of activation of natriuretic receptors, including D5R and CCKBR. Two weeks of high salt diet had no effect on the body weights and blood pressures (S3 Table) of BALB/c mice that are salt resistant. But, to be in balance, high salt groups may excrete a larger urine volume than normal salt diet. Intraperitoneal administration of the D1R/D5R agonist fenoldopam promoted a natriuresis in salt-loaded BALB/c mice, which was not observed in D5R-/- mice which are hypertensive [27,40,64,69]. The presence of intact D1R in D5R-/- mice does not make any difference probably because the D1R and D5R physically interact in the inhibition of renal sodium transport [23,33]. D5R-/- mice, on normal and high sodium diet have increased renal expression of sodium co-transporters, NKCC2 and NCC, and γ subunits of ENaC; on high salt diet renal NHE3 expression was also increased [69]. The increased renal expression of sodium co-transporters and channels may be responsible for the impaired ability of D5R-/- mice to maintain a normal sodium balance, shifting the pressure-natriuresis plot to the right [26,27,69]. In our results, fenoldopam enhanced the natriuresis in sodium loaded BALB/c mice, which was abrogated by YF476, a potent and selective CCKBR antagonist while the increase in natriuresis caused by gastrin was abolished by Sch23390, a D1R and D5R antagonist. The pharmacological assays have no significant effect on water intake, food intake and blood pressure (S3 Table), indicating that the variation of urinary sodium to creatinine ratio is not the results of different sodium consumption. Both fenoldopam [70] and gastrin [71] have been previously confirmed to have no effect on food intake, which can further support our results.
In summary, we have demonstrated that CCKBR and D5R synergistically interact in the kidney. The cooperative effect of D5R and CCKBR may be involved in the maintenance of normal sodium and water balance when salt intake is increased.
Supporting Information
D5R and CCKBR mRNA (A and B) and protein (C) expressions in co-transfected HEK293-D5R-CCKBR cells. HDC: HEK293-D5R-CCKBR cell total mRNA; NC: negative control, HEK293 cell total mRNA; Blank: H2O; PC: positive control, plasmid including the human D5R or CCKBR gene. GAPDH (36kDa) is used for the correction of protein loading.
(TIF)
(A) Effects of fenoldopam (10−6 mol/L, 24 hours) and D1R/D5R antagonist Sch23390 (10-6mol/L, 24 hours) on CCKBR protein expression (n = 6, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). (B) Effects of gastrin (10-8mol/L, 24 hours) and CCKBR antagonist YF476 (10-8mol/L, 24 hours) on D5R protein expression (n = 6, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). All immunoblotting results are expressed as relative density units (DU) and normalized by GAPDH expression. Immunoblots of D5R, CCKBR, and GAPDH are shown in the inset.
(TIF)
White bar represents HK-2 cells; black bar represents NT cells. cAMP production is expressed as nanogram (ng) per liter of solution (n = 6, *P<0.05 vs others, one-way factorial ANOVA, Duncan’s test).
(TIF)
(A) HK-2 cells, (B) NT cells. Effects of fenoldopam (10−6 mol/L, 24 hours) and D5R antagonist LE-PM436 (10-6mol/L, 24 hours) on CCKBR protein expression (n = 5, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). All immunoblotting results are expressed as relative density units (DU) and normalized by GAPDH expression. Immunoblots of CCKBR and GAPDH are shown in the inset.
(TIF)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
These studies were supported, in part, by grants from National Natural Science Foundation (China) (81370358) and National Institutes of Health (USA) (R01DK39308, PAJ). LE-PM436 was generously provided by Dr. Christoph Enzensperger (Institut für Pharmazie, Lehrstuhl für Pharmazeutische/Medizinische Chemie, FriedrichSchiller-Universität Jena, Jena, Germany).
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
These studies were supported by grants from National Natural Science Foundation (China) (81370358, ZWY) and National Institutes of Health (USA) (R01DK39308, PAJ).
References
- 1.Liang M, Cowley AW Jr, Mattson DL, Kotchen TA, Liu Y. (2013) Epigenomics of hypertension. Semin Nephrol 33:392–399. 10.1016/j.semnephrol.2013.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forman JP, Scheven L, de Jong PE, Bakker SJ, Curhan GC, Gansevoort RT. (2012) Association between sodium intake and change in uric acid, urine albumin excretion, and the risk of developing hypertension. Circulation 125:3108–3116. 10.1161/CIRCULATIONAHA.112.096115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mente A, O'Donnell MJ, Rangarajan S, McQueen MJ, Poirier P, Wielgosz A, et al. (2014) Association of urinary sodium and potassium excretion with blood pressure. N Engl J Med 371:601–611. 10.1056/NEJMoa1311989 [DOI] [PubMed] [Google Scholar]
- 4.Stolarz-Skrzypek K, Kuznetsova T, Thijs L, Tikhonoff V, Seidlerova J, Richart T, et al. (2011) Fatal and nonfatal outcomes, incidence of hypertension, and blood pressure changes in relation to urinary sodium excretion. JAMA 305:1777–1785. 10.1001/jama.2011.574 [DOI] [PubMed] [Google Scholar]
- 5.O'Donnell M, Mente A, Rangarajan S, McQueen MJ, Wang X, Liu L, et al. (2014) Urinary sodium and potassium excretion, mortality, and cardiovascular events. N Engl J Med 371:612–623. 10.1056/NEJMoa1311889 [DOI] [PubMed] [Google Scholar]
- 6.Herrera M, Coffman TM. (2012) The kidney and hypertension: Novel insights from transgenic models. Curr Opin Nephrol Hypertens 21:171–178. 10.1097/MNH.0b013e3283503068 [DOI] [PubMed] [Google Scholar]
- 7.Hamlyn JM. (2014) Natriuretic hormones, endogenous ouabain, and related sodium transport inhibitors. Front Endocrinol (Lausanne) 5:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hussain T, Lokhandwala MF. (2003) Renal dopamine receptors and hypertension. Exp Biol Med (Maywood) 228:134–142. [DOI] [PubMed] [Google Scholar]
- 9.Zhang MZ, Harris RC. (2015) Antihypertensive mechanisms of intra-renal dopamine. Curr Opin Nephrol Hypertens 24:117–122. 10.1097/MNH.0000000000000104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanada H, Jones JE, Jose PA. (2011) Genetics of salt-sensitive hypertension. Curr Hypertens Rep 13:55–66. 10.1007/s11906-010-0167-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weder AB, Gleiberman L, Sachdeva A. (2009) Urinary dopamine excretion and renal responses to fenoldopam infusion in blacks and whites. J Clin Hypertens (Greenwich) 11:707–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salomone LJ, Howell NL, McGrath HE, Kemp BA, Keller SR, Gildea JJ, et al. (2007) Intrarenal dopamine D1-like receptor stimulation induces natriuresis via an angiotensin type-2 receptor mechanism. Hypertension 49:155–161. [DOI] [PubMed] [Google Scholar]
- 13.Trivedi M, Marwaha A, Lokhandwala M. (2004) Rosiglitazone restores g-protein coupling, recruitment, and function of renal dopamine D1A receptor in obese Zucker rats. Hypertension 43:376–382. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Asico LD, Zheng S, Villar VA, He D, Zhou L, et al. (2013) Gastrin and D1 dopamine receptor interact to induce natriuresis and diuresis. Hypertension 62:927–933. 10.1161/HYPERTENSIONAHA.113.01094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang X, Wang W, Ning B, Liu X, Gong J, Gan F, et al. (2013) Basal and postprandial serum levels of gastrin in normotensive and hypertensive adults. Clin Exp Hypertens 35:74–78. 10.3109/10641963.2012.690474 [DOI] [PubMed] [Google Scholar]
- 16.Carey RM. (1978) Evidence for a splanchnic sodium input monitor regulating renal sodium excretion in man. Lack of dependence upon aldosterone. Circ Res 43:19–23. [DOI] [PubMed] [Google Scholar]
- 17.Michell AR, Debnam ES, Unwin RJ. (2008) Regulation of renal function by the gastrointestinal tract: Potential role of gut-derived peptides and hormones. Annu Rev Physiol 70:379–403. [DOI] [PubMed] [Google Scholar]
- 18.Tarasova NI, Kopp JA, Jose P, Farnsworth DW, Michejda CJ, Wank SA. (1996) Postprandial changes in renal function are mediated by elevated serum gastrin acting at cholecystokinin type B receptors (CCKBR) in the kidney. Gastroenterology 110:A1106–A1106. [Google Scholar]
- 19.Liu T, Jose PA. (2013) Gastrin induces sodium-hydrogen exchanger 3 phosphorylation and mTOR activation via a phosphoinositide 3-kinase-/protein kinase C dependent but AKT-independent pathway in renal proximal tubule cells derived from a normotensive male human. Endocrinology 154:865–875. 10.1210/en.2012-1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Schrenck T, Ahrens M, de Weerth A, Bobrowski C, Wolf G, Jonas L, et al. (2000) CCKB/gastrin receptors mediate changes in sodium and potassium absorption in the isolated perfused rat kidney. Kidney Int 58:995–1003. [DOI] [PubMed] [Google Scholar]
- 21.de Weerth A, Jonas L, Schade R, Schoneberg T, Wolf G, Pace A, et al. (1998) Gastrin/cholecystokinin type B receptors in the kidney: Molecular, pharmacological, functional characterization, and localization. Eur J Clin Invest 28:592–601. [DOI] [PubMed] [Google Scholar]
- 22.Melis M, Krenning EP, Bernard BF, de Visser M, Rolleman E, de Jong M. (2007) Renal uptake and retention of radiolabeled somatostatin, bombesin, neurotensin, minigastrin and cck analogues: Species and gender differences. Nucl Med Biol 34:633–641. [DOI] [PubMed] [Google Scholar]
- 23.Gildea JJ, Shah IT, Van Sciver RE, Israel JA, Enzensperger C, McGrath HE, et al. (2014) The cooperative roles of the dopamine receptors, D1R and D5R, on the regulation of renal sodium transport. Kidney Int 86:118–126. 10.1038/ki.2014.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sunahara RK, Guan HC, O'Dowd BF, Seeman P, Laurier LG, Ng G, et al. (1991) Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature 350:614–619. [DOI] [PubMed] [Google Scholar]
- 25.Tiberi M, Caron MG. (1994) High agonist-independent activity is a distinguishing feature of the dopamine D1B receptor subtype. J Biol Chem 269: 27925–27931. [PubMed] [Google Scholar]
- 26.Yang Z, Asico LD, Yu P, Wang Z, Jones JE, Bai RK, et al. (2005) D5 dopamine receptor regulation of phospholipase D. Am J Physiol Heart Circ Physiol 288:H55–H61. [DOI] [PubMed] [Google Scholar]
- 27.Yang Z, Asico LD, Yu P, Wang Z, Jones JE, Escano CS, et al. (2006) D5 dopamine receptor regulation of reactive oxygen species production, NADPH oxidase, and blood pressure. Am J Physiol Regul Integr Comp Physiol 290:R96–R104. [DOI] [PubMed] [Google Scholar]
- 28.Sanada H, Jose PA, Hazen-Martin D, Yu PY, Xu J, Bruns DE, et al. (1999) Dopamine-1 receptor coupling defect in renal proximal tubule cells in hypertension. Hypertension 33:1036–1042. [DOI] [PubMed] [Google Scholar]
- 29.Bavithra S, Selvakumar K, Pratheepa Kumari R, Krishnamoorthy G, Venkataraman P, Arunakaran J. (2012) Polychlorinated biphenyl (PCBs)-induced oxidative stress plays a critical role on cerebellar dopaminergic receptor expression: ameliorative role of quercetin. Neurotox Res 21:149–159. 10.1007/s12640-011-9253-z [DOI] [PubMed] [Google Scholar]
- 30.Ma J, Dankulich-Nagrudny L, Lowe G. (2013) Cholecystokinin: an excitatory modulator of mitral/tufted cells in the mouse olfactory bulb. PLoS One 8: e64170 10.1371/journal.pone.0064170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta c(T)) method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- 32.Goetze JP, Eiland S, Svendsen LB, Vainer B, Hannibal J, Rehfeld JF. (2013) Characterization of gastrins and their receptor in solid human gastric adenocarcinomas. Scand J Gastroenterol 48:688–695. 10.3109/00365521.2013.783101 [DOI] [PubMed] [Google Scholar]
- 33.Gildea JJ, Wang X, Jose PA, Felder RA. (2008) Differential D1 and D5 receptor regulation and degradation of the angiotensin type 1 receptor. Hypertension 51:360–366. 10.1161/HYPERTENSIONAHA.107.100099 [DOI] [PubMed] [Google Scholar]
- 34.Mizuta K, Zhang Y, Xu D, Mizuta F, D'Ovidio F, Masaki E, et al. (2013) The dopamine D1 receptor is expressed and facilitates relaxation in airway smooth muscle. Respir Res 14:89 10.1186/1465-9921-14-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Armando I, Yu P, Escano C, Mueller SC, Asico L, et al. (2008) Dopamine 5 receptor mediates Ang II type 1 receptor degradation via a ubiquitin-proteasome pathway in mice and human cells. J Clin Invest 118:2180–2189. 10.1172/JCI33637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takinami Y, Yuki H, Nishida A, Akuzawa S, Uchida A, Takemoto Y, et al. (1997) YF476 is a new potent and selective gastrin/cholecystokinin-B receptor antagonist in vitro and in vivo. Aliment Pharmacol Ther 11:113–120. [DOI] [PubMed] [Google Scholar]
- 37.Sordal O, Waldum H, Nordrum IS, Boyce M, Bergh K, Munkvold B, et al. (2013) The gastrin receptor antagonist netazepide (YF476) prevents oxyntic mucosal inflammation induced by Helicobacter pylori infection in Mongolian gerbils. Helicobacter 18:397–405. 10.1111/hel.12066 [DOI] [PubMed] [Google Scholar]
- 38.Beyer C. (1993) Creatine measurement in serum and urine with an automated enzymatic method. Clinical chemistry 39:1613–1619. [PubMed] [Google Scholar]
- 39.Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A. (2011) Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics 12:14 10.1186/1471-2164-12-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asico L, Zhang X, Jiang J, Cabrera D, Escano CS, Sibley DR, et al. (2011) Lack of renal dopamine D5 receptors promotes hypertension. J Am Soc Nephrol 22:82–89. 10.1681/ASN.2010050533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dahl LK, Heine M, Thompson K. (1972) Genetic influence of renal homografts on the blood pressure of rats from different strains. Proc Soc Exp Biol Med 140:852–856. [DOI] [PubMed] [Google Scholar]
- 42.Coffman TM. (2014) The inextricable role of the kidney in hypertension. J Clin Invest 124:2341–2347. 10.1172/JCI72274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hall JE, Granger JP, do Carmo JM, da Silva AA, Dubinion J, George E, et al. (2012) Hypertension: physiology and pathophysiology. Compr Physiol 2:2393–2442. 10.1002/cphy.c110058 [DOI] [PubMed] [Google Scholar]
- 44.Zhuo JL, Li XC. (2013) Proximal nephron. Compr Physiol 3:1079–1123. 10.1002/cphy.c110061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeng C, Yang Z, Wang Z, Jones J, Wang X, Altea J, et al. (2005) Interaction of angiotensin II type 1 and D5 dopamine receptors in renal proximal tubule cells. Hypertension 45:804–810. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Guo F, Guo H, Wang H, Zhang Z, Liu X, et al. (2010) The paradox of dopamine and angiotensin II-mediated Na(+), K(+)-ATPase regulation in renal proximal tubules. Clin Exp Hypertens 32:464–468. 10.3109/10641963.2010.496516 [DOI] [PubMed] [Google Scholar]
- 47.Gildea JJ, Wang X, Shah N, Tran H, Spinosa M, Van Sciver R, et al. (2012) Dopamine and angiotensin type 2 receptors cooperatively inhibit sodium transport in human renal proximal tubule cells. Hypertension 60:396–403. 10.1161/HYPERTENSIONAHA.112.194175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aperia A. (2012) 2011 Homer Smith Award: To serve and protect: classic and novel roles for Na+, K+ -adenosine triphosphatase. J Am Soc Nephrol 23:1283–1290. 10.1681/ASN.2012010102 [DOI] [PubMed] [Google Scholar]
- 49.Sharon P, Karmeli F, Rachmilewitz D. (1981) PGE2 mediates the effect of pentagastrin on intestinal adenylate cyclase and Na-K-ATPase activities. Prostaglandins 21 Suppl:81–87. [DOI] [PubMed] [Google Scholar]
- 50.Kopin AS, Lee YM, McBride EW, Miller LJ, Lu M, Lin HY, et al. (1992) Expression cloning and characterization of the canine parietal cell gastrin receptor. Proc Natl Acad Sci USA 89:3605–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wank SA, Harkins R, Jensen RT, Shapira H, de Weerth A, Slattery T. (1992) Purification, molecular cloning, and functional expression of the cholecystokinin receptor from rat pancreas. Proc Natl Acad Sci USA 89:3125–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wank SA, Pisegna JR, de Weerth A. (1992) Brain and gastrointestinal cholecystokinin receptor family: Structure and functional expression. Proc Natl Acad Sci USA 89:8691–8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ewart HS, Klip A. (1995) Hormonal regulation of the Na(+)-K(+)-atpase: Mechanisms underlying rapid and sustained changes in pump activity. Am J Physiol 269:C295–C311. [DOI] [PubMed] [Google Scholar]
- 54.Bastie MJ, Delvaux M, Dufresne M, Saunier-Blache JS, Vaysse N, Ribet A. (1988) Distinct activation of Na+-H+ exchange by gastrin and CCK peptide in acini from guinea pig. Am J Physiol 254:G25–G32. [DOI] [PubMed] [Google Scholar]
- 55.Delvaux M, Bastie MJ, Chentoufi J, Cragoe EJ Jr, Vaysse N, Ribet A. (1990) Amiloride and analogues inhibit Na(+)-H+ exchange and cell proliferation in AR42J pancreatic cell line. Am J Physiol 259:G842–G849. [DOI] [PubMed] [Google Scholar]
- 56.Brown DA, Melvin JE, Yule DI. (2003) Critical role for NHE1 in intracellular pH regulation in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol 285:G804–G812. [DOI] [PubMed] [Google Scholar]
- 57.Ahn W, Kim KH, Lee JA, Kim JY, Choi JY, Moe OW, et al. (2001) Regulatory interaction between the cystic fibrosis transmembrane conductance regulator and HCO3- salvage mechanisms in model systems and the mouse pancreatic duct. J Biol Chem 276:17236–17243. [DOI] [PubMed] [Google Scholar]
- 58.Zeng C, Armando I, Luo Y, Eisner GM, Felder RA, Jose PA. (2008) Dysregulation of dopamine-dependent mechanisms as a determinant of hypertension: Studies in dopamine receptor knockout mice. Am J Physiol Heart Circ Physiol 294:H551–H569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang MZ, Yao B, Wang S, Fan X, Wu G, Yang H, et al. (2011) Intrarenal dopamine deficiency leads to hypertension and decreased longevity in mice. J Clin Invest 121:2845–2854. 10.1172/JCI57324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang YR, Yuan ZY. (2010) Dopamine-mediated inhibition of renal Na+/K+-atpase in HK-2 cells is reduced by ouabain. Clin Exp Pharmacol Physiol 37:613–618. 10.1111/j.1440-1681.2010.05364.x [DOI] [PubMed] [Google Scholar]
- 61.Gildea JJ, Shah I, Weiss R, Casscells ND, McGrath HE, Zhang J, et al. (2010) HK-2 human renal proximal tubule cells as a model for G protein-coupled receptor kinase type 4-mediated dopamine 1 receptor uncoupling. Hypertension 56:505–511. 10.1161/HYPERTENSIONAHA.110.152256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Yuan Z, Ge H, Ren Y. (2010) Effects of long-term ouabain treatment on blood pressure, sodium excretion, and renal dopamine D(1) receptor levels in rats. J Comp Physiol B 180:117–124. 10.1007/s00360-009-0391-z [DOI] [PubMed] [Google Scholar]
- 63.Periyasamy SM, Liu J, Tanta F, Kabak B, Wakefield B, Malhotra D, et al. (2005) Salt loading induces redistribution of the plasmalemmal Na/K-ATPase in proximal tubule cells. Kidney Int 67:1868–1877. [DOI] [PubMed] [Google Scholar]
- 64.Hollon TR, Bek MJ, Lachowicz JE, Ariano MA, Mezey E, Ramachandran R, et al. (2002) Mice lacking D5 dopamine receptors have increased sympathetic tone and are hypertensive. J Neurosci 22:10801–10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ericsson P, Hakanson R, Rehfeld JF, Norlen P. (2010) Gastrin release: Antrum microdialysis reveals a complex neural control. Regul Pept 161:22–32. 10.1016/j.regpep.2010.01.004 [DOI] [PubMed] [Google Scholar]
- 66.McQuarrie EP, Traynor JP, Taylor AH, Freel EM, Fox JG, Jardine AG, et al. (2014) Association between urinary sodium, creatinine, albumin, and long-term survival in chronic kidney disease. Hypertension 64:111–117. 10.1161/HYPERTENSIONAHA.113.03093 [DOI] [PubMed] [Google Scholar]
- 67.Grimes CA, Riddell LJ, Campbell KJ, Nowson CA. (2013) Dietary salt intake, sugar-sweetened beverage consumption, and obesity risk. Pediatrics 131:14–21. 10.1542/peds.2012-1628 [DOI] [PubMed] [Google Scholar]
- 68.Larsen SC, Ängquist L, Sørensen TI, Heitmann BL. (2013) 24h urinary sodium excretion and subsequent change in weight, waist circumference and body composition. PLoS One 8:e69689 10.1371/journal.pone.0069689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang X, Luo Y, Escano CS, Yang Z, Asico L, Li H, et al. (2010) Upregulation of renal sodium transporters in d5 dopamine receptor-deficient mice. Hypertension 55:1431–1437. 10.1161/HYPERTENSIONAHA.109.148643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rusk IN, Cooper SJ. (1989) The selective dopamine D1 receptor agonist SK&F 38393: its effects on palatability- and deprivation-induced feeding, and operant responding for food. Pharmacol Biochem Behav 34:17–22. [DOI] [PubMed] [Google Scholar]
- 71.Garlicki J, Konturek PK, Majka J, Kwiecien N, Konturek SJ. (1990) Cholecystokinin receptors and vagal nerves in control of food intake in rats. Am J Physiol 258: E40–45. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
D5R and CCKBR mRNA (A and B) and protein (C) expressions in co-transfected HEK293-D5R-CCKBR cells. HDC: HEK293-D5R-CCKBR cell total mRNA; NC: negative control, HEK293 cell total mRNA; Blank: H2O; PC: positive control, plasmid including the human D5R or CCKBR gene. GAPDH (36kDa) is used for the correction of protein loading.
(TIF)
(A) Effects of fenoldopam (10−6 mol/L, 24 hours) and D1R/D5R antagonist Sch23390 (10-6mol/L, 24 hours) on CCKBR protein expression (n = 6, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). (B) Effects of gastrin (10-8mol/L, 24 hours) and CCKBR antagonist YF476 (10-8mol/L, 24 hours) on D5R protein expression (n = 6, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). All immunoblotting results are expressed as relative density units (DU) and normalized by GAPDH expression. Immunoblots of D5R, CCKBR, and GAPDH are shown in the inset.
(TIF)
White bar represents HK-2 cells; black bar represents NT cells. cAMP production is expressed as nanogram (ng) per liter of solution (n = 6, *P<0.05 vs others, one-way factorial ANOVA, Duncan’s test).
(TIF)
(A) HK-2 cells, (B) NT cells. Effects of fenoldopam (10−6 mol/L, 24 hours) and D5R antagonist LE-PM436 (10-6mol/L, 24 hours) on CCKBR protein expression (n = 5, *P<0.05 vs control, one-way factorial ANOVA, Duncan’s test). All immunoblotting results are expressed as relative density units (DU) and normalized by GAPDH expression. Immunoblots of CCKBR and GAPDH are shown in the inset.
(TIF)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.