

Brentuximab vedotin is an antibody-drug conjugate composed of a CD30-directed monoclonal antibody covalently linked to the antimicrotubule agent monomethyl auristatin E (MMAE). The recommended dose is 1.8 mg/kg administered as an intravenous infusion over 30 minutes every 3 weeks. The scientific review of the application leading to approval in the European Union is summarized in the present report.
Keywords: Brentuximab vedotin, Adcetris, Hodgkin lymphoma, Systemic anaplastic large cell lymphoma, European Medicines Agency
Abstract
Background.
On October 25, 2012, a conditional marketing authorization valid throughout the European Union (EU) was issued for brentuximab vedotin for the treatment of adult patients with relapsed or refractory CD30+ Hodgkin lymphoma (HL) and for the treatment of adult patients with relapsed or refractory systemic anaplastic large cell lymphoma (sALCL). For HL, the indication is restricted to treatment after autologous stem cell transplantation (ASCT) or after at least two previous therapies when ASCT or multiagent chemotherapy is not a treatment option.
Materials and Methods.
Brentuximab vedotin is an antibody-drug conjugate (ADC) composed of a CD30-directed monoclonal antibody (recombinant chimeric IgG1) that is covalently linked to the antimicrotubule agent monomethyl auristatin E (MMAE). Binding of the ADC to CD30 on the cell surface initiates internalization of the MMAE-CD30 complex, followed by proteolytic cleavage that releases MMAE. The recommended dose is 1.8 mg/kg administered as an intravenous infusion over 30 minutes every 3 weeks.
Results.
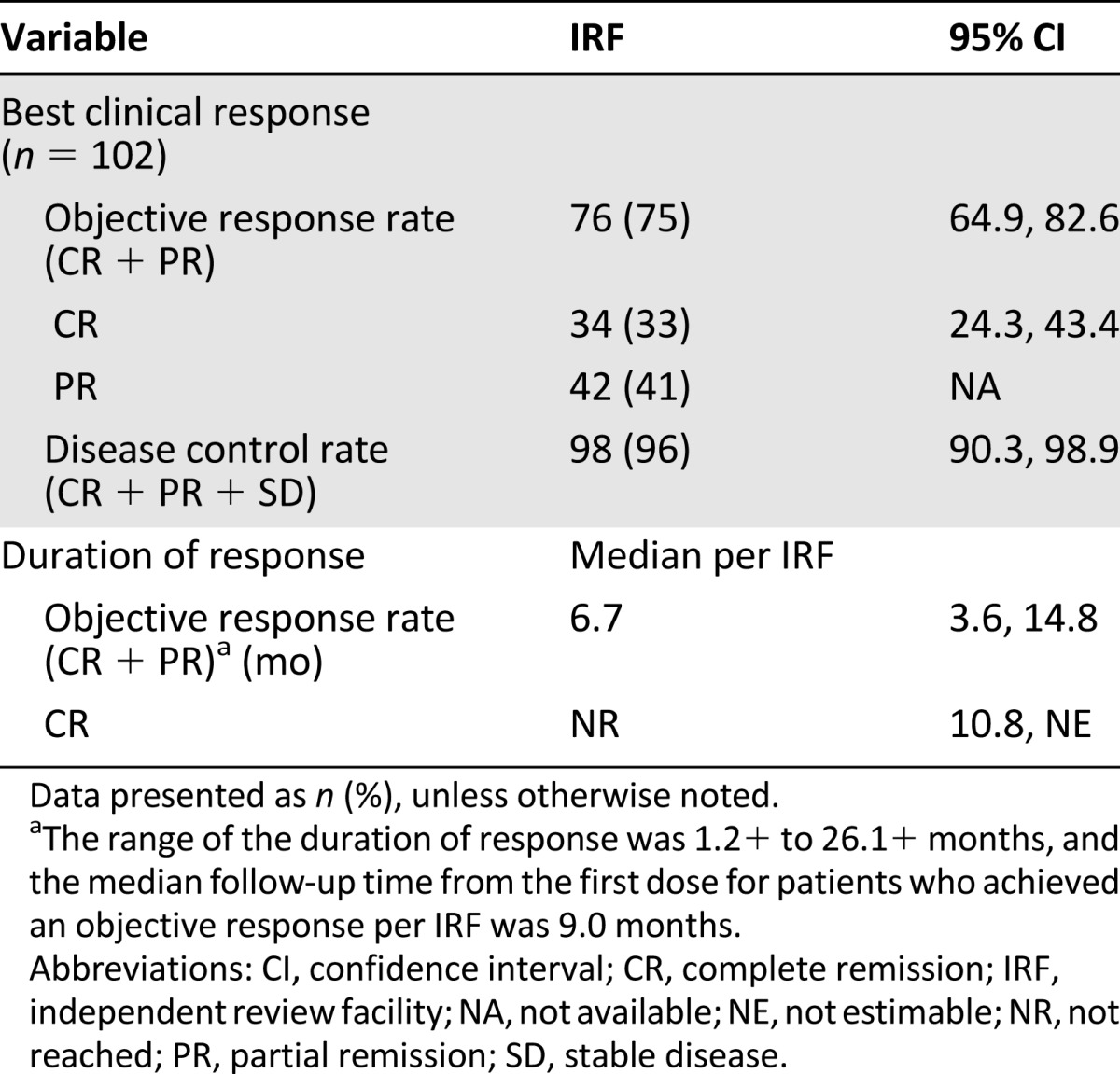
Brentuximab vedotin as a single agent was evaluated in two single-arm studies. Study SG035-003 included 102 patients with relapsed or refractory HL. An objective response was observed in 76 patients (75%), with complete remission in 34 (33%). Study SG035-004 included 58 patients with relapsed or refractory sALCL. An objective response was observed in 50 patients (86%), with complete remission in 34 (59%). The most frequently observed toxicities were peripheral sensory neuropathy, fatigue, nausea, diarrhea, neutropenia, vomiting, pyrexia, and upper respiratory tract infection.
Conclusion.
The present report summarizes the scientific review of the application leading to approval in the EU. The detailed scientific assessment report and product information, including the summary of the product characteristics, are available on the European Medicines Agency website (http://www.ema.europa.eu).
Implications for Practice:
Brentuximab vedotin was approved in the European Union for the treatment of adult patients with relapsed or refractory CD30+ Hodgkin lymphoma or systemic anaplastic large cell lymphoma. For Hodgkin lymphoma, brentuximab vedotin should only be used after autologous stem cell transplantation or following at least two prior therapies when transplantation or multiagent chemotherapy is not a treatment option. In two studies involving 160 patients, partial or complete responses were observed in the majority of patients. Although there was no information on the survival of patients treated in the studies at the time of approval, the responses were considered a clinically relevant benefit.
Introduction
The standard of care for relapsed or refractory Hodgkin lymphoma (HL) is second-line chemotherapy followed by autologous stem cell transplantation (ASCT), which can induce long-term remission in approximately 50% of patients. Consensus is lacking about the type of salvage chemotherapy used before the transplant, as well as about the treatment strategies for nontransplant-eligible patients [1–3]. Treatment of advanced-stage HL has typically been associated with success rates of 60%–80% with anthracycline-based polychemotherapy and radiotherapy [4]. The overall long-term prognosis for patients whose disease has relapsed after ASCT is poor, with a median survival in the range of 24 months [5, 6]. HL is divided into two major subtypes, according to the immunohistological features and microscopic appearance of the malignant cells. The nodular lymphocyte predominant subtype (NLPHL) constitutes 5% of all HL cases and has a generally more indolent course than classic HL (cHL). Most NLPHL cases are CD30 negative. In contrast, CD30 expression is a standard feature of Reed-Sternberg (RS) cells in cHL.
Anaplastic large cell lymphoma (ALCL) is an aggressive non-Hodgkin lymphoma of T-cell origin. It has two distinct forms, systemic ALCL (sALCL) and a primarily cutaneous form. ALCL accounts for 2%–8% of all T-cell lymphomas. CD30 is invariably expressed on the surface of ALCL cells. In approximately 50%–80% of sALCL cases, the t(2;5)(p23;q35) chromosome translocation, prompting the anaplastic lymphoma kinase (ALK) gene on chromosome 2 to fuse with the nucleophosmin (NPM) gene on chromosome 5 (ALK-positive), is detected. ALK-positive patients are younger and have a better prognosis. Although 75%–85% of patients achieve an objective response (either a complete or partial response) with frontline anthracycline-based therapy, the 5-year failure-free survival after treatment was 60% in ALK-positive compared with 36% in ALK-negative patients. The 5-year overall survival rate was 70% in those with ALK-positive and 49% in those with ALK-negative ALCL [7]. ABVD (Adriamycin, bleomycin, vinblastine, and dacarbazine) and MACOP-B (methotrexate, doxorubicin, cyclophosphamide, vincristine, prednisone, and bleomycin) show comparable efficacy, with approximately one half of patients developing a relapse within 2 years [8]. No consensus has been reached regarding the treatment of relapsed or refractory disease. A second complete remission with standard salvage chemotherapy can be achieved in 30%–40% of patients [9–11]. Some patients might benefit from high-dose therapy with ASCT. However, the clinical benefit might be limited to only those with chemotherapy-sensitive disease [12–14].
CD30 is a member of the tumor necrosis factor receptor superfamily [15, 16]. It was originally identified on RS cells of HL (CD30-positive Hodgkin or RS cells express 5,000–10,000 molecules of CD30). However, it is also expressed on subsets of non-Hodgkin lymphoma, including ALCL and cutaneous T-cell lymphoma, and on rare solid tumors such as testicular carcinomas of the nongerminal type (embryonal carcinomas). In nonmalignant cells, CD30 is expressed on activated, but not resting, lymphocytes (T, B, and natural killer cells) and weakly on activated monocytes. After binding to its ligand CD153, CD30 can activate nuclear factor-κB, Jun amino-terminal kinase, and p38. Furthermore, CD30 is expressed in decidual cells in the pregnant uterus [17].
The applicant company (Takeda Global Research and Development Centre Europe Ltd., London, U.K., http://www.takeda.com) submitted an application for marketing authorization to the European Medicines Agency (EMA) for brentuximab vedotin. Brentuximab vedotin (also known as SGN-35 [Fig. 1]), is a CD30-directed antibody-drug conjugate (ADC) composed of a chimeric monoclonal anti-CD30 antibody (cAC10) covalently linked, via an enzyme-cleavable linker, to the anti-mitotic small molecule monomethyl auristatin E (MMAE). After binding to surface CD30, the conjugate undergoes endocytosis. MMAE becomes active after the linker between MMAE and the antibody is cleaved by the lysosome. MMAE interferes with tubulin polymerization and consequently disrupts the formation of the mitotic spindle in dividing cells. The ensuing mitotic arrest eventually leads to cell death.
Figure 1.

Schematic diagram of SGN-35. The SGN-35 antibody-drug conjugate (ADC) is composed of three parts: (a) antibody: the cAC10 chimeric anti-human CD30 monoclonal antibody; (b) linker: a protease-cleavable linker composed of a maleimidocaproyl attachment group, a valine-citrulline dipeptide, and a PABC spacer; and (c) drug: MMAE (SGD-1010), a pentapeptide consisting of methyl valine, valine, dolaisoleuine, dolaproine, and norephedrine. In the resulting ADC, SGD-1010 is linked to the antibody via a thioether. Reprinted, with permission, from European Medicines Agency [26].
Abbreviations: MMAE, monomethyl auristatin E; PABC, p-aminobenzylcarbamate.
The EMA recommended the granting of a conditional marketing authorization for brentuximab vedotin for the treatment of adult patients with relapsed or refractory CD30+ HL after ASCT or after at least two previous therapies when ASCT or multiagent chemotherapy is not a treatment option and for the treatment of adult patients with relapsed or refractory sALCL. The recommended dose is 1.8 mg/kg administered as an intravenous infusion (IV) over 30 minutes every 3 weeks.
The present report summarizes the scientific review of the application leading to approval of brentuximab vedotin in the EU. The detailed scientific assessment report and product information (including the summary of the product characteristics) for this product is available on the EMA website (http://www.ema.europa.eu).
Nonclinical Aspects
On binding of the ADC to CD30 and subsequent internalization, brentuximab vedotin was shown to traffic to lysosomes within 4 hours of treatment by colocalization with the lysosomal-associated membrane protein 1 (LAMP-1) using immunofluorescence microscopy. By subcellular fractionation using brentuximab vedotin conjugated to 14C-labeled MMAE, it was shown that the drug is released intracellularly. This release occurred at 37°C and was dependent on lysosomal activity. By mass spectrometry, the released drug was identified as MMAE.
In vitro, after intracellular release, free MMAE appeared extracellularly over time. By this mechanism, free toxin might get into the circulation and cause toxicity in bystander cells [18].
Myelotoxicity was the toxicity associated with repeat-dose brentuximab vedotin administration to rats and monkeys. The bone marrow findings were generally reversible.
Brentuximab vedotin was genotoxic, consistent with the pharmacologic disruption of microtubules by MMAE. The linker portion of brentuximab vedotin, includes maleimide, which is mutagenic [19].
Repeat-dose toxicity studies in rats indicated the potential for brentuximab vedotin to impair male reproductive function and fertility. Testicular atrophy and degeneration were partially reversible after a 16-week treatment-free period. Brentuximab vedotin caused embryo-fetal lethality in pregnant female rats.
The precise mechanism of testicular toxicity in rats is not known, and uncertainty exists on the presence of CD30 in human spermatogonia/early spermatocytes. It remains, therefore, unclear whether the testicular toxicity would also occur in humans. This has not been studied. Even so, men treated with brentuximab vedotin should not father a child during treatment nor until 6 months after treatment.
Clinical Pharmacology
Pharmacokinetics
The pharmacokinetics (PK) of brentuximab vedotin was evaluated in phase I studies and in a population pharmacokinetic analysis of data from 314 patients. In all clinical trials, brentuximab vedotin was administered as an IV infusion.
A multiexponential decline in brentuximab vedotin serum concentrations was observed, with a terminal half-life of approximately 4–6 days. After multiple-dose administration of brentuximab vedotin, a steady state was achieved by 21 days, consistent with the terminal half-life estimate. The exposures were approximately dose proportional. Minimal to no accumulation of brentuximab vedotin was observed with multiple doses at every 3-week schedule.
The median maximum concentration, area under the curve, and time to maximum concentration of MMAE after a single infusion with 1.8 mg/kg of the ADC was approximately 4.97 ng/ml, 37.03 ng/ml per day, and 2.09 days, respectively. MMAE exposure decreased after multiple doses of brentuximab vedotin, with approximately 50%–80% of the exposure of the first dose observed at subsequent infusions. In the first cycle, higher MMAE exposure was associated with an absolute decrease in neutrophil count.
In vivo data in animals and humans have suggested that only a small fraction of MMAE released from brentuximab vedotin is metabolized. The levels of MMAE metabolites have not been measured in human plasma. At least one metabolite of MMAE has been shown to be active in vitro. The role of the active metabolites is unclear. Given the importance of MMAE (and potentially some of its metabolites) for the toxicity of brentuximab vedotin, the applicant company will investigate MMAE metabolism further in a phase I study.
Brentuximab vedotin is eliminated by catabolism, with a typical estimated clearance of 1.457 L/day. The elimination of MMAE was limited by its rate of release from the ADC. The typical apparent clearance and half-life of MMAE was 19.99 L/day and 3–4 days, respectively. An excretion study was undertaken in patients who had received a dose of 1.8 mg/kg brentuximab vedotin. Approximately 24% of the total MMAE, as part of the amount of ADC administered during a brentuximab vedotin infusion, was recovered in both urine and feces over a 1-week period. Of the recovered MMAE, approximately 72% was recovered in the feces. A lesser amount of MMAE (28%) was excreted in the urine.
The liver is a major route of elimination of the unchanged active metabolite MMAE. Limited pharmacokinetic data are available from patients with hepatic impairment. The kidney is a route of excretion of the unchanged active metabolite MMAE. The population PK analysis indicated that MMAE clearance might be affected by moderate and severe renal impairment. MMAE clearance was reduced about twofold in patients with severe renal impairment (i.e., creatinine clearance <30 mL/min). The effect of hepatic and renal impairment on the elimination of MMAE is under investigation. In terms of pharmacodynamic drug interactions, when combined with bleomycin, brentuximab vedotin caused pulmonary toxicity, and the concurrent use of the two is contraindicated.
Clinical Efficacy and Safety
Clinical Efficacy
The pivotal efficacy study for the HL indication was study SG035-003 [20]. This was a single-arm, multicenter, clinical trial to evaluate the efficacy and safety of brentuximab vedotin as a single agent in patients with relapsed or refractory HL.
The eligibility criteria included relapsed or refractory HL, previous ASCT, and histologically documented CD30-positive disease by central review. Also, the patients could not have been treated previously with brentuximab vedotin or received an allogeneic transplantation. Patients with congestive heart failure or known cerebral/meningeal disease were also ineligible.
Brentuximab vedotin at 1.8 mg/kg was to be administered via outpatient IV infusion on day 1 of each 21-day cycle. This dosing regimen was based on the results from study SG035-0001, a dose-escalation study of SGN-35 in patients with relapsed/refractory CD30-positive hematologic malignancies; the maximum tolerated dose was identified as 1.8 mg/kg every 3 weeks [21]. Dose delays (up to 3 weeks each) and a one-level dose reduction to 1.2 mg/kg were allowed for toxicity. Patients could continue the study treatment until disease progression or unacceptable toxicity developed. Patients who achieved stable disease or better were to receive a minimum of 8, but no more than 16, cycles of study treatment. The primary efficacy endpoint was the overall objective response rate (ORR) according to the Revised Response Criteria for Malignant Lymphoma [22], as determined by an independent review facility.
The baseline characteristics and efficacy results from relapsed or refractory HL patients are summarized in Tables 1 and 2. The applicant company also provided an intraindividual comparison of investigator-based progression-free survival (PFS) after brentuximab vedotin with the investigator-based PFS after the most recent previous therapy. An equal to longer PFS for brentuximab vedotin compared with that after the previous chemotherapy regimen was observed in 60 of 102 HL patients (59%).
Table 1.
Summary of baseline patient and disease characteristics in the phase II study of relapsed or refractory Hodgkin lymphoma patients (study SG035-003) (n = 102)
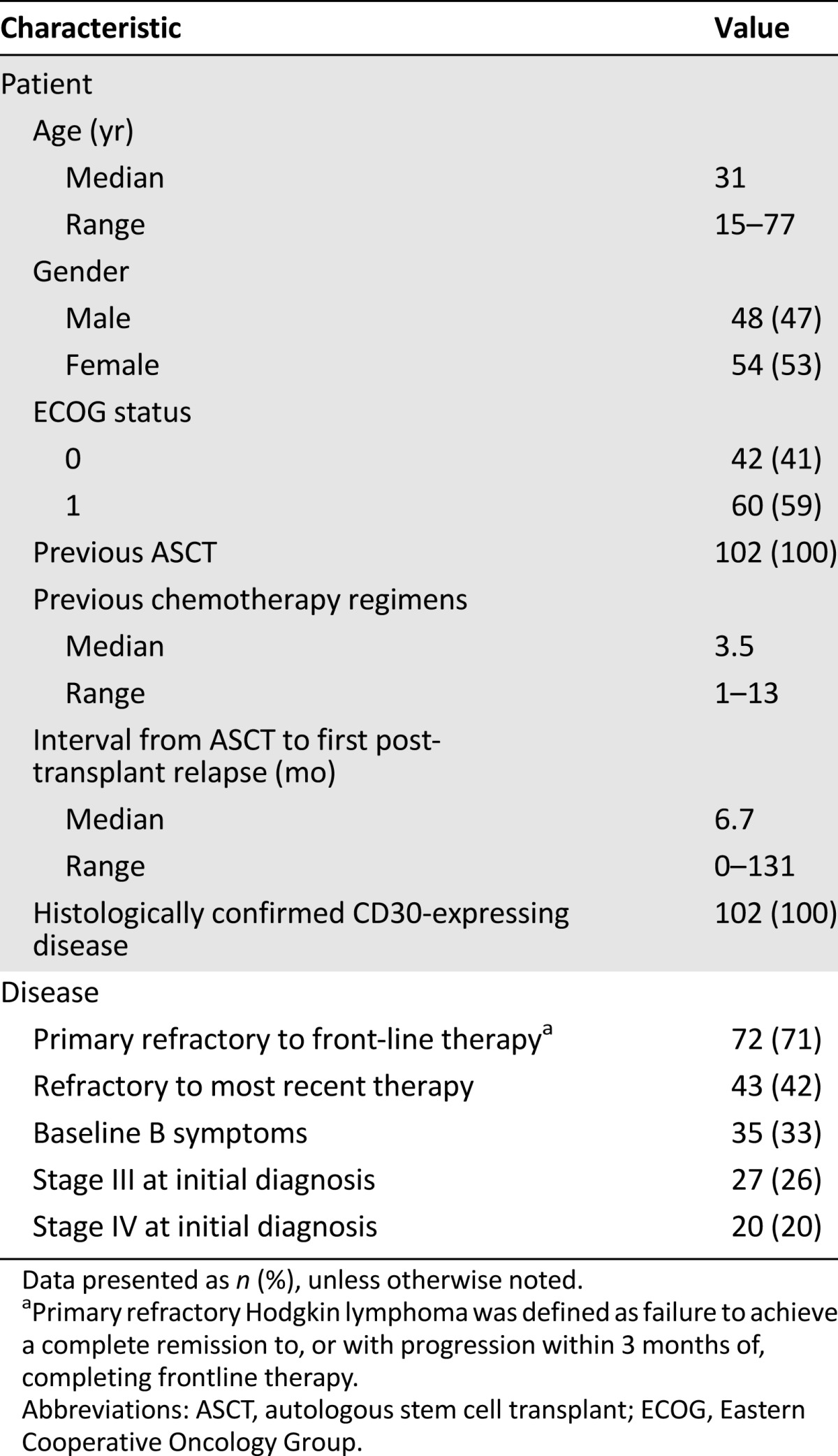
Table 2.
Efficacy results in relapsed or refractory Hodgkin lymphoma (study SG035-003)
Supportive efficacy data in the ASCT-naïve HL indication were derived from 59 patients from the phase I studies SG035-001 and SG035-002 [23], a Japanese study (TB-BC010088), and Named Patient Programs. The most common first-line previous therapies were ABVD (66%) and BEACOPP (bleomycin, etoposide, Adriamycin, cyclophosphamide, Oncovin, procarbazine, prednisone)-based regimens (10%). The most common second-line therapies were ESHAP (etoposide, methylprednisolone, high-dose cytarabine, and cisplatinum; 19%); radiotherapy (17%); and ICE (ifosfamide, carboplatin, and etoposide; 13.5%). The baseline characteristics and efficacy results from ASCT-naïve HL patients are summarized in Tables 3 and 4.
Table 3.
Basic demographic characteristics for the 59-patient population with relapsed or refractory HL without previous ASCT (phase I studies SG035-001 and SG035-002, a Japanese study [TB-BC010088] and Named Patient Programs)

Table 4.
Response data for Hodgkin lymphoma (HL) patients with relapsed or refractory HL without previous autologous stem cell transplantation (phase I studies SG035-001 and SG035-002, Japanese study TB-BC010088, and Named Patient Programs)

The pivotal efficacy study for the sALCL indication was study SG035-004. It was a single-arm, multicenter clinical trial to evaluate the efficacy and safety of brentuximab vedotin as a single agent in patients with relapsed or refractory sALCL [24]. The eligibility criteria included relapsed or refractory systemic ALCL, previous front-line chemotherapy with curative intent and histologically documented CD30-positive disease.
Documented ALK status was required. Patients could not have previously received an allogeneic stem cell transplantation. Patients with a current diagnosis of primary cutaneous ALCL were not eligible. However, patients whose disease had transformed to systemic ALCL were eligible. The baseline characteristics and efficacy results from the relapsed or refractory systemic ALCL patients are summarized in Tables 5 and 6.
Table 5.
Summary of baseline patient and disease characteristics in the phase II relapsed or refractory sALCL study (study SG035-004) (n = 58)

Table 6.
Efficacy results in relapsed or refractory sALCL patients treated with 1.8 mg/kg of brentuximab vedotin every 3 weeks (study SG035-004)

Clinical Safety
The applicant submitted safety data from 6 studies with 357 patients with CD30+ hematologic malignancies who had received at least 1 dose of brentuximab vedotin. The median duration of treatment in study SG035-003 was 27 weeks (range, 3–56), and the median number of cycles administered per patient was 9 (range, 1–16). The median treatment duration with brentuximab vedotin in study SG035-004 was 20 weeks (range, 3–51). A median of 6 cycles (range, 1–16) was administered per patient, and slightly fewer than 50% of patients received 7 or more cycles.
The most frequently observed adverse reactions in patients receiving this treatment were peripheral sensory neuropathy, fatigue, nausea, diarrhea, neutropenia, vomiting, pyrexia, and upper respiratory tract infection (Table 7). In the phase II studies, 15% of patients experienced a serious adverse drug reaction, including neutropenia, thrombocytopenia, constipation, diarrhea, vomiting, pyrexia, peripheral motor neuropathy and peripheral sensory neuropathy, hyperglycemia, demyelinating polyneuropathy, tumor lysis syndrome, and Stevens-Johnson syndrome. The serious adverse reactions that led to treatment discontinuation in 2 or more HL or sALCL patients were peripheral sensory neuropathy (6%) and peripheral motor neuropathy (2%). Six deaths occurred in the phase II studies within 30 days of the last dose of brentuximab vedotin. These deaths all occurred in patients with systemic ALCL and were considered unrelated to treatment.
Table 7.
Treatment-emergent adverse events occurring in ≥10% of patients in phase II studies

The median duration of grade 3 or 4 neutropenia was limited (1 week); 2% of patients had grade 4 neutropenia that lasted ≥7 days. Fewer than one half of the patients in the phase II population with grade 3 or 4 neutropenia had temporally associated infections, and most temporally associated infections were grade 1 or 2.
The percentage of patients with treatment-emergent upper respiratory tract infection was higher in HL patients (37%) relative to sALCL patients (12%). It is conceivable that this difference was due to impairment in cell-mediated immunity in the patients with classic HL [25].
Peripheral neuropathy was the most common adverse event, occurring in 55% of patients (39% sensory only, 3% motor only, and 12% with both), and led to treatment discontinuation in 12% and dose reductions in 10%. The peripheral neuropathy was typically an effect of cumulative exposure and was reversible in most cases. The incidence and severity of neuropathy and the nature of the events (i.e., sensory events were more common than motor events, and a distal pattern of development was more common than a proximal pattern of development) was similar to that observed with other microtubule inhibitor-based chemotherapy agents (including vinca alkaloids, taxanes, and epothilones). At the time of the initial approval, 2 confirmed cases and 1 suspect case of progressive multifocal leukoencephalopathy had been reported in the 2,000 patients treated worldwide.
The missing information included safety in pediatric populations, safety in the elderly, safety in patients with renal, hepatic, or cardiac impairment, and long-term safety. Pharmacovigilance activities, in addition to the use of routine pharmacovigilance, included monitoring the pharmacokinetic data in renal- and hepatic-impaired patients (study SGN-35-008b); a randomized, double-blind, placebo-controlled AETHERA study of single-agent brentuximab vedotin in patients at high risk of residual HL after transplantation; and a randomized, controlled trial to examine patients with newly diagnosed mature T-cell lymphoma, including 75% ± 5% of patients with sALCL (study SGN35-014). Important safety concerns identified since the initial approval include pancreatitis, pulmonary toxicity, toxic epidermal necrolysis, liver function test abnormalities, and its use for patients with renal and hepatic impairment, because such impairment could impede the clearance of the MMAE component of brentuximab vedotin.
Important safety concerns identified since the initial approval include pancreatitis, pulmonary toxicity, toxic epidermal necrolysis, liver function test abnormalities, and its use in patients with renal and hepatic impairment, because such impairment could impede the clearance of the MMAE component of brentuximab vedotin.
Benefit-Risk Assessment
The Committee for Medicinal Products for Human Use (CHMP) initially considered that the absence of a controlled trial was a major deficiency for a convincing demonstration of efficacy. A Scientific Advisory Group (SAG) for oncology was convened to provide recommendations regarding the interpretation of the efficacy results observed in the single-arm studies presented. The SAG advised that, despite the limited data, the observed antitumor activity of brentuximab vedotin was considered clinically relevant, because the high response rate and duration of response were associated with a reduction of B symptoms (i.e., fever, night sweats, and weight loss). Moreover, the high complete response rate and duration of response in HL might constitute a clinical benefit in allowing a relevant percentage of patients to undergo a subsequent ASCT; however, definitive data are lacking. The SAG also recommended that a further single-arm study of the response rate, duration of response, rate of second ASCT, and data from subpopulations might provide confirmation in terms of efficacy and safety in those with relapsed/refractory sALCL.
The SAG also recommended that a further single-arm study of the response rate, duration of response, rate of second ASCT, and data from subpopulations might provide confirmation in terms of efficacy and safety in those with relapsed/refractory sALCL.
The CHMP concluded that in patients with relapsed or refractory CD30+ HL after ASCT or in patients with relapsed or refractory systemic ALCL, brentuximab vedotin showed efficacy in terms of a significant increase in ORR and PFS, despite the absence of confirmatory controlled data. The intraindividual comparisons of investigator-based PFS after brentuximab vedotin with investigator-based PFS after the most recent previous therapy revealed a relevant treatment effect for most post-ASCT HL and sALCL patient populations studied. Regarding the relapsed/refractory HL patients for whom ASCT or multidrug chemotherapy is not an option, antitumor activity was considered to be established by the response rates. Importantly, for all these indications, the chance to obtain a complete response and the option for a potentially curative stem cell transplantation is of major importance. Because brentuximab vedotin was proposed, in particular, as a late-line treatment, the suggested effects were considered of clinical relevance. Together with the acceptable safety profile and considering the overall grave prognosis of the patients involved, the CHMP judged that the benefit-risk balance of Adcetris was positive in the treatment of adult patients with relapsed or refractory CD30+ HL after ASCT or after at least 2 previous therapies when ASCT or multiagent chemotherapy was not a treatment option and in the treatment of adult patients with relapsed or refractory sALCL.
However, the CHMP considered a need existed to further confirm the positive benefit-risk in such populations. Therefore, a conditional marketing authorization was recommended to gather the missing information. Thus, further evidence on this medicinal product is awaited. The applicant company has committed to provide comprehensive clinical data from single-arm studies investigating the efficacy in HL patients with relapsed/refractory disease ineligible for ASCT and in the relapsed/refractory sALCL population. In addition, the CHMP considered that it is important to further confirm the safety in both the HL and the sALCL populations. As a specific obligation, the applicant company will conduct a postauthorization safety study to provide comprehensive safety data. Finally, updated overall survival data from the pivotal studies SG035-003, SG035-004 will be provided, with the aim of satisfying the conditions of the conditional marketing authorization. A similar approach was taken by the U.S. Food and Drug Administration, with marketing approval of Adcetris granted under accelerated approval.
The EMA will review new information on a yearly basis until all specific obligations have been fulfilled. Detailed information on this medicinal product is available on the website of the EMA (http://www.ema.europa.eu/).
Acknowledgments
The scientific assessment as summarized in this report is based on the marketing authorization application submitted by the applicant company and on important contributions from, among others, the rapporteur and co-rapporteur assessment teams, Committee for Medicinal Products for Human Use members, and additional experts. This publication is a summary of the European Public Assessment Report, available in the public domain, together with the summary of product characteristics, and other product information on the EMA website (http://www.ema.europa.eu). The authors remain solely responsible for the opinions expressed therein.
Author Contributions
Data analysis and interpretation: Iordanis Gravanis, Kyriaki Tzogani, Paula van Hennik, Pieter de Graeff, Petra Schmitt, Jan Mueller-Berghaus, Tomas Salmonson, Christian Gisselbrecht, Edward Laane, Lothar Bergmann, Francesco Pignatti
Manuscript writing: Iordanis Gravanis, Kyriaki Tzogani, Francesco Pignatti
Final approval of manuscript: Iordanis Gravanis, Kyriaki Tzogani, Paula van Hennik, Pieter de Graeff, Petra Schmitt, Jan Mueller-Berghaus, Tomas Salmonson, Christian Gisselbrecht, Edward Laane, Lothar Bergmann, Francesco Pignatti
Disclosures
The authors indicated no financial relationships.
References
- 1.Sureda A, Constans M, Iriondo A, et al. Prognostic factors affecting long-term outcome after stem cell transplantation in Hodgkin’s lymphoma autografted after a first relapse. Ann Oncol. 2005;16:625–633. doi: 10.1093/annonc/mdi119. [DOI] [PubMed] [Google Scholar]
- 2.Majhail NS, Weisdorf DJ, Defor TE, et al. Long-term results of autologous stem cell transplantation for primary refractory or relapsed Hodgkin’s lymphoma. Biol Blood Marrow Transplant. 2006;12:1065–1072. doi: 10.1016/j.bbmt.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Kuruvilla J. Standard therapy of advanced Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program. 2009:497–506. doi: 10.1182/asheducation-2009.1.497. [DOI] [PubMed] [Google Scholar]
- 4.Borchmann P, Haverkamp H, Diehl V, et al. Eight cycles of escalated-dose BEACOPP compared with four cycles of escalated-dose BEACOPP followed by four cycles of baseline-dose BEACOPP with or without radiotherapy in patients with advanced-stage Hodgkin’s lymphoma: Final analysis of the HD12 trial of the German Hodgkin Study Group. J Clin Oncol. 2011;29:4234–4242. doi: 10.1200/JCO.2010.33.9549. [DOI] [PubMed] [Google Scholar]
- 5.Crump M. Management of Hodgkin lymphoma in relapse after autologous stem cell transplant. Hematology Am Soc Hematol Educ Program. 2008:326–333. doi: 10.1182/asheducation-2008.1.326. [DOI] [PubMed] [Google Scholar]
- 6.Kewalramani T, Nimer SD, Zelenetz AD, et al. Progressive disease following autologous transplantation in patients with chemosensitive relapsed or primary refractory Hodgkin’s disease or aggressive non-Hodgkin’s lymphoma. Bone Marrow Transplant. 2003;32:673–679. doi: 10.1038/sj.bmt.1704214. [DOI] [PubMed] [Google Scholar]
- 7.Savage KJ, Harris NL, Vose JM, et al. ALK− anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: Report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008;111:5496–5504. doi: 10.1182/blood-2008-01-134270. [DOI] [PubMed] [Google Scholar]
- 8.Sibon D, Fournier M, Briere J, et al. Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte trials. J Clin Oncol. 2012;30:3939–3946. doi: 10.1200/JCO.2012.42.2345. [DOI] [PubMed] [Google Scholar]
- 9.Kewalramani T, Zelenetz AD, Teruya-Feldstein J, et al. Autologous transplantation for relapsed or primary refractory peripheral T-cell lymphoma. Br J Haematol. 2006;134:202–207. doi: 10.1111/j.1365-2141.2006.06164.x. [DOI] [PubMed] [Google Scholar]
- 10.Vose JM, Peterson C, Bierman PJ, et al. Comparison of high-dose therapy and autologous bone marrow transplantation for T-cell and B-cell non-Hodgkin’s lymphomas. Blood. 1990;76:424–431. [PubMed] [Google Scholar]
- 11.Rodriguez J, Munsell M, Yazji S, et al. Impact of high-dose chemotherapy on peripheral T-cell lymphomas. J Clin Oncol. 2001;19:3766–3770. doi: 10.1200/JCO.2001.19.17.3766. [DOI] [PubMed] [Google Scholar]
- 12.Gisselbrecht C, Bosly A, Lepage E, et al. Autologous hematopoietic stem cell transplantation in intermediate and high grade non-Hodgkin’s lymphoma: A review. Ann Oncol. 1993;4(suppl 1):7–13. doi: 10.1093/annonc/4.suppl_1.s7. [DOI] [PubMed] [Google Scholar]
- 13.Moskowitz CH, Bertino JR, Glassman JR, et al. Ifosfamide, carboplatin, and etoposide: A highly effective cytoreduction and peripheral-blood progenitor-cell mobilization regimen for transplant-eligible patients with non-Hodgkin’s lymphoma. J Clin Oncol. 1999;17:3776–3785. doi: 10.1200/JCO.1999.17.12.3776. [DOI] [PubMed] [Google Scholar]
- 14.Shipp MA, Abeloff MD, Antman KH, et al. International consensus conference on high-dose therapy with hematopoietic stem-cell transplantation in aggressive non-Hodgkin's lymphomas: Report of the jury. Ann Oncol. 1999;10:13–19. doi: 10.1023/a:1008397220178. [DOI] [PubMed] [Google Scholar]
- 15.Smith CA, Gruss HJ, Davis T, et al. CD30 antigen, a marker for Hodgkin’s lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell. 1993;73:1349–1360. doi: 10.1016/0092-8674(93)90361-s. [DOI] [PubMed] [Google Scholar]
- 16.Gruss HJ, Dower SK. Tumor necrosis factor ligand superfamily: Involvement in the pathology of malignant lymphomas. Blood. 1995;85:3378–3404. [PubMed] [Google Scholar]
- 17.Chiarle R, Podda A, Prolla G, et al. CD30 in normal and neoplastic cells. Clin Immunol. 1999;90:157–164. doi: 10.1006/clim.1998.4636. [DOI] [PubMed] [Google Scholar]
- 18.Okeley NM, Miyamoto JB, Zhang X, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16:888–897. doi: 10.1158/1078-0432.CCR-09-2069. [DOI] [PubMed] [Google Scholar]
- 19.Chemical Carcinogenesis Research Information System (CCRIS) [Internet]. CAS Registry No. 541-59-3, Maleimide [cited 2011 March 21]. U.S. National Library of Medicine (NLM); 1993. Available at http://toxnet.nlm.nih.gov/cgi-bin/sis/search2/f?./temp/~TPkH87:4. Accessed March 21, 2011.
- 20.Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol. 2012;30:2183–2189. doi: 10.1200/JCO.2011.38.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Younes A, Forero-Torres A, Bartlett NL, et al. Robust antitumor activity of the antibody-drug conjugate SGN-35 when administered every 3 weeks to patients with relapsed or refractory CD30-positive hematologic malignancies in a phase 1 study. Haematologica. 2009;94(suppl 2):0503a. [Google Scholar]
- 22.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 23.Forero-Torres A, Fanale M, Advani R, et al. Brentuximab vedotin in transplant-naive patients with relapsed or refractory Hodgkin lymphoma: Analysis of two phase I studies. The Oncologist. 2012;17:1073–1080. doi: 10.1634/theoncologist.2012-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J Clin Oncol. 2012;30:2190–2196. doi: 10.1200/JCO.2011.38.0402. [DOI] [PubMed] [Google Scholar]
- 25.Franzke A, Geffers R, Hunger JK, et al. Identification of novel regulators in T-cell differentiation of aplastic anemia patients. BMC Genomics. 2006;7:263. doi: 10.1186/1471-2164-7-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.European Medicines Agency. Assessment report for ADCETRIS 2012. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002455/WC500135054.pdf. Accessed at June 30, 2015.