

Abstract
RNA interference (RNAi) is an RNA-dependent gene silencing approach controlled by RNA-induced silencing complex (RISC). Here we represent a synthetic RISC-mimic nanocomplex, which can actively cleave its target RNA in a sequence-specific manner. With high enzymatic stability and efficient self-delivery to target cells, the designed nanocomplex can selectively and potently induce gene silencing without cytokine activation. The nanocomplexes targeting to multidrug resistance are able to not only bypass P-glycoprotein (Pgp) transporter due to their nano-size effect, but also effectively suppress the Pgp expression, thus resulting in successful restoration of drug sensitivity of OVCAR8/ADR cells to Pgp-transportable cytotoxic agents. This nanocomplex approach has the potential for both functional genomics and cancer therapy.
Keywords: Nanocomplex, Biomimetic, Gene regulation, Multidrug resistance, Gold nanoparticle
RNA interference (RNAi) is a fundamental gene regulation pathway in eukaryotic cells.[1] In this pathway, sequence-specific siRNA is able to target and cleave complementary mRNA through the action of an endonuclease-containing protein complex known as RNA-induced silencing complex (RISC). Exogenous siRNA-based RNAi techniques have the potential to provide powerful therapeutic approaches for different diseases, and a number of siRNA-based therapies are currently being evaluated in clinical trials.[2] However, these siRNA drugs highly rely on cellular RNAi machinery to take into effects, they can perturb endogenous gene regulation pathways mediated by endogenous microRNAs that also rely on these cellular machinery, thus resulting in potential side effects.[2b,3] In addition, synthetic siRNA drugs require an additional delivery system to facilitate transfection because naked siRNA molecules cannot freely penetrate through the cell membranes and are prone to be degraded by endogenous nucleases.[3,4] In this study, we report an alternative approach to RNAi for gene regulation based on a synthetic RISC-mimic nanocomplex that consists of a nanoparticle, non-sequence specific endoribonuclease, single-strand DNA oligonucleotides and receptor-binding ligands (Scheme 1A).
Scheme 1.

Schematic representation describing the design and function of targeted nanocomplexes. a) Synthetic process of targeted nanocomplexes. b) Cellular functions of targeted nanocomplex including self-delivery, gene silencing, and reverse of drug resistance. c) Designed MDR1 DNA sequences.
To build the robust nanocomplex, we used a gold nanoparticle (AuNP), a promising intracellular delivery agent,[5] as the backbone of the nanocomplex because it not only provides a large surface area to hold endoribonucleases and DNA oligonucleotides at close proximity, but also is of low cytotoxicity and has unique surface chemical properties for alkanethiol functionalization.[5,6] RNase A was chosen as the catalytic component of the nanocomplex because it does not degrade the DNA-based recognition component of the nanocomplex, and is one of the most robust and active endoribonucleases for non-sequence specific degradation of single-stranded RNAs.[7] In addition, it is well-documented that RNase A can effectively bind onto the surface of AuNPs through noncovalent adsorption.[8] DNA oligonucleotides are responsible for recognizing target RNA via Watson-Crick base pairing[9] and guiding the neighbouring endoribonuclease to cleave target mRNAs that contain complementary sequences, as well as protecting endoribonuclease against proteinase attack due to the dense package of DNAs on the nanocomplex.[10] Cys-tag EGF (ctEGF) is an epidermal growth factor receptor (EGFR)-binding ligand[11] for efficient delivery of the nanocomplex to target cells. Due to their unusual cooperative effects, the nanocomplexes exhibit resistance to enzymatic degradation, transfection agent-free delivery to target cells, and more importantly, induction of sequence-specific gene silencing independent of cellular RNAi machinery (Scheme 1B).
To evaluate the function and efficacy of the EGFR-targeted nanocomplex for sequence-specific gene silencing, multidrug resistance (MDR) was chosen as a target model system. MDR is a major obstacle to the success of cancer chemotherapy.[12] One important mechanism of MDR is the active efflux of drugs by multidrug transporters. P-glycoprotein (Pgp), encoded by multidrug resistance protein 1 (MDR1) gene, is the most commonly encountered membrane transporter in the clinic;[13] and it is overexpressed in most cancer cells in response to a number of chemotherapeutic drugs.[12] Specific inhibition of Pgp expression could help restore the drug sensitivity of MDR cells.
In this nanocomplex, an alkanethiol-terminated anti-MDR1 DNA sequence (Scheme 1C) was designed as the recognition component. This DNA sequence consists of a 15-bp-long adenine (A15) tether and a 21-bp-long fragment complementary to the region (nt 1001–1021) in the MDR1 mRNA. The A15 tether was used as a spacer to increase the efficiency of the hybridization between a nanocomplex and its complementary mRNA.[9a] Nanocomplex was synthesized using a two-step method (Scheme 1A). AuNPs (15 ± 0.7 nm in diameter) were first functionalized with RNase A in a carbonate buffered solution (pH 9.6) to obtain RNase-Au conjugates, followed by further modification with anti-MDR1 DNA oligonucleotides (or mutant sequence with seven mismatches). DNA-functionalized AuNPs without RNase A (denoted as DNA-Au NPs) were used as the control.
The resulting nanocomplexes are highly dispersible and stable in phosphate buffered saline (PBS) at pH 7.4. The UV-Vis spectra showed that the surface plasmon resonance (SPR) band of the nanocomplexes experienced a 7-nm red-shift as compared to that of AuNPs (Figure S1A), suggesting a change in the dielectric constant of the surrounding environment of the AuNPs due to the sequential loading of RNase A and DNA molecules. In contrast, DNA-Au NPs only had a 4-nm red shift of the SPR band. Furthermore, the width of the SPR band of the nanocomplexes remained nearly unchanged, indicating that the nanocomplexes were not aggregated, which was congruent with TEM analysis (Figure S1). Dynamic light scattering (DLS) measurements also showed that the nanocomplexes were discrete colloidal particles with a diameter of 49 ± 1.8 nm (Table S1). In addition, the nanocomplexes exhibited a weaker negative surface charge than DNA-Au NPs at pH 7.4 (−23 ± 1.6 mV vs. −30 ± 2.0 mV, Table S2), further indicating successful functionalization of the positively charged RNase A molecules on the surface of AuNPs. On average, the anti-MDR1 nanocomplexes were estimated to carry 13.9 ± 0.7 RNase A and 85 ± 4 DNA oligos (Figure S2 and Table S2).
We first assessed the target specificity of the anti-MDR1 nanocomplex using an in vitro RNase activity assay through electrophoresis analyses. MDR1 mRNA segment (nt 400–1369) acted as the target substrate, and the control substrate was a firefly luciferase (Fluc) mRNA segment (nt 1–1653) that does not contain complementary sequences to the anti-MDR1 DNA oligonucleotides. As shown in Figure 1, the anti-MDR1 nanocomplex did not show any measurable cleavage activity on the Fluc mRNA, but it did cleave the MDR1 mRNA target into two major fragments with a size of about 370 nt and 600 nt. As a result, the RNA cleavage site fully matched the predicted position where the MDR1 mRNA binds to the nanocomplexes via DNA/RNA hybridization (Scheme 1B). In addition, the sizes of these two RNA fragments almost perfectly matched the sizes of those corresponding MDR1 mRNA fragments cut by RNase H that specifically degrades the RNA of RNA/DNA hybrids (Figure 1A),[14] suggesting that the anti-MDR1 nanocomplex induced site-specific RNA cleavage (Scheme 1B). In contrast, the negative controls (DNA-Au, ctrl DNA-Au and ctrl nanocomplex) showed no cleavage activity against either the MDR1 or Fluc mRNAs, whereas unbound RNase A non-specifically degraded both mRNA substrates into short fragments, which appeared as broad smear bands. Together, these results demonstrate that the anti-MDR1 nanocomplexes exhibit remarkable target specificity and display a RISC-like function—cleaving the target RNAs in a sequence- and site-specific manner.
Figure 1.

RNase activity tests for assessing the target selectivity of nanocomplex and its ability to resist the degradation by proteinase. In these tests, the concentration of nanocomplexes (or a control: DNA-Au, control DNA-Au, control nanocomplexes and NC-Ls) was fixed at 0.02 nM and the concentrations of RNase A and proteinase K were 0.28 and 10 nM, respectively. The products of these tests were analyzed by using electrophoresis in a 2% formaldehyde agarose gel, and RNA bands were stained by using SYBR Green II. a) MDR1 mRNA segment (nt 400–1369) as the target substrate. b) Fluc mRNA segment (nt 1–1653) as the control substrate. Abbreviations: Ctrl, blank control; PK, proteinase K; and NC-L, nanocomplex with low DNA dense coverage.
This RISC-like function of the anti-MDR1 nanocomplex is attributed to the remarkable cooperative effect between the DNA and RNase A components of the nanocomplex. The densely packed DNA on the nanocomplex likely blocked the access of non-complementary RNAs to the nanocomplex-bearing RNase molecules through steric hindrance and repulsive coulomb interactions. Besides, these DNAs can also bind to target RNAs via base pairing and bring them to the RNase molecules on the nanocomplex, resulting in endonucleolytic cleavage of these RNAs into two fragments at positions close to the binding site (Scheme 1B). Therefore, the DNA surface density should be critical for the RNA target specificity of nanocomplexes. Indeed, the anti-MDR1 nanocomplexes with a low DNA surface coverage (i.e., NC-Ls, 14.1 ± 1.1 RNase A and 45 ± 6 DNA oligos) did not exhibit target specificity and cut both the MDR1 and Fluc mRNAs in a sequence non-specific manner (Figure 1).
Given that RNase A molecules can potentially be degraded by proteinases in the cells, we also examined the in vitro resistance of nanocomplexes against proteinase K compared with unbound RNase A and NC-Ls (Figure 1). The result showed that unbound RNase A and NC-Ls lost their activity almost completely after proteinase K treatment at 37 °C for 1 h. In contrast, nearly no measurable change was observed in the nanocomplex activity after an identical proteinase K treatment. We attributed the resistance to proteinase degradation of the nanocomplex to the fact that the RNase molecules on the nanocomplex were protected by the tightly packed oligonucleotides (Scheme 1A). It is expected that the ability to resist enzymatic degradation should enhance the stability of these nanocomplexes in the cells.
To investigate if the ctEGF-modified nanocomplexes preferentially bind to EGFR-overexpressing cells, we synthesized EGFR-targeted (6 ± 1 ctEGF per AuNP,) and non-targeted nanocomplexes through changing the initial molar ratio of DNA and ctEGF-modified DNA oligonucleotides to 12:1 and 12:0, respectively (see Experimental Section 2 and 3.3 in the Supporting Information for detailed synthesis). Three cell lines were chosen including EGFR-negative HCT116, EGFR-positive OVCAR8 and OVCAR8/ADR cells. EGFR expression levels in each type of cells were confirmed by western blot (Figure S3). As shown in Figure 2, the results from inductively coupled plasma mass spectrometry (ICP-MS) showed that the targeted nanocomplexes were effectively internalized by EGFR-positive cell lines (OVCAR8 and OVCAR8/ADR), ca. 6-fold higher than that of the non-targeted nanocomplexes, whereas HCT116 cells did not show a significant difference for targeted and non-targeted nanocomplex. In addition, all cells incubated with non-targeted nanocomplex only exhibited a background level of cell-associated particles. It is likely that nonspecific, scavenger-mediated endocytosis is the dominant internalization mechanism for non-targeted nanocomplexes.[15] However, the targeted nanocomplexes are prone to bind to cell surface EGF receptors and activate the energy-dependent endocytosis pathway, further significantly increasing the intracellular uptake of the nanocomplex. Furthermore, 50-nm hydrodynamic diameter of the nanocomplex might also be an optimal factor that maximizes the uptake rate of the targeted nanocomplex in specific cells.[16] Collectively, the data suggest that the nanocomplexes would preferably bind to the target cells with target moieties on the surface. The cytotoxicity of the nanocomplex was tested by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay in the OVCAR8/ADR cell as an example. The results indicated that the targeted nanocomplexes showed almost no detectable cytotoxicity or side effects to cells at the concentrations ranging from 0.01 to 0.5 nM even for 48 h incubation (Figure S5), and confirmed that the nanocomplex could be used for intracellular gene regulation.
Figure 2.
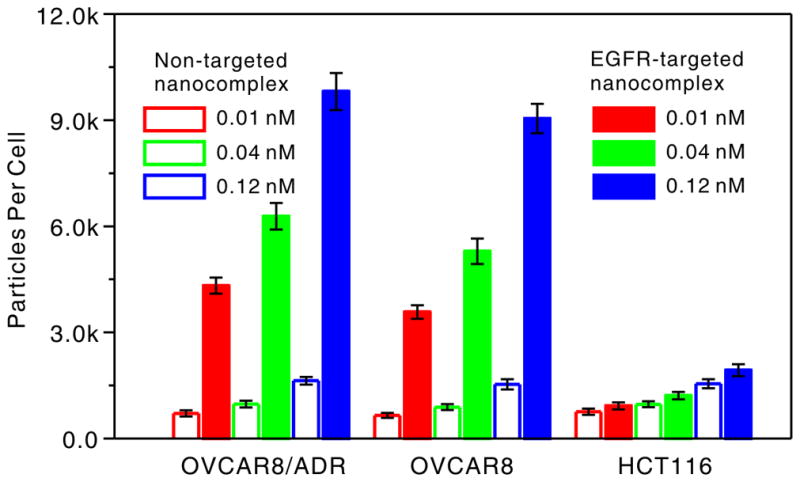
Cell type-dependent cellular uptake of EGFR-targeted and non-targeted nanocomplexes after 5 h incubation at 37 °C.
We then studied the intracellular activity of the targeted anti-MDR1 nanocomplexes in OVCAR8/ADR cells, which overexpress the MDR1 gene (Figure S6). The cells were treated once with the targeted nanocomplex (or a control) at varying concentrations for 5 h at 37 °C, followed by incubation with fresh growth medium for another 48 h, and then harvested and processed for an mRNA and protein assay through quantitative real-time polymerase chain reaction (qRT-PCR) and western blot. As shown in Figure 3A, no measurable reduction in the MDR1 mRNA level was observed in the treatments using the targeted DNA-Au NPs at concentrations of 0.01, 0.04 and 0.12 nM. The inability of these conjugates to cause significant antisense effects on MDR1 replication is likely due to their low concentrations in our experiments.[9a,17] In contrast, the targeted anti-MDR1 nanocomplex with the identical dosages induced dramatic decrease of MDR1 mRNA level in a dose-dependent manner. As additional negative controls, neither ctrl DNA-Au NPs nor ctrl nanocomplex led to measurable anti-MDR1 effects (Figure 3A), which further proved that the anti-MDR1 nanocomplexes exhibited excellent intracellular target specificity. In addition, no changes were observed in the mRNA level of TNF-α and IFN-β genes in the interferon signaling pathway in OVCAR8/ADR cells treated with the targeted anti-MDR1 nanocomplexes, indicating that the nanocomplex-induced anti-MDR1 effect observed herein was not caused by cytokine activation (see Figure S7).
Figure 3.

Anti-MDR1 effects of the targeted anti-MDR1 nanocomplex in OVCAR8/ADR cells. a) qRT-PCR analyses of MDR1 mRNA expression in the OVCAR8/ADR cells treated with targeted nanocomplex or controls at varying doses: 0.01 nM (red), 0.04 nM (green) and 0.12 nM (blue), or non-targeted nanocomplex with 0.12 nM. Each bar presents the mean and standard deviation derived from three independent experiments; Student’s t test, ns = non-significance: P > 0.13, *: P< 0.006, **: P = 0.058. b) Western blot analyses of Pgp expression in the OVCAR8/ADR cells after treatment with nanoparticles at a concentration of 0.12 nM except for the targeted anti-MDR1 nanocomplexes at different concentrations of 0.01, 0.04 and 0.12 nM, which was probed with anti-Pgp antibody and anti-β-actin antibody, respectively.
Western blot showed consistent results with RT-PCR at the protein level. No detectable effect was observed in the cells treated with negative controls, whereas the cells displayed a significant decrease in Pgp protein level upon the targeted nanocomplex treatment (Figure 3B). Conversely, the cells treated with the non-target nanocomplexes of 0.12 nM showed no significant change, which is consistent with the RT-PCR result. Altogether, these results clearly demonstrate that the targeted anti-MDR1 nanocomplexes can more efficiently induce an intracellular MDR1 gene knockdown, and consequently decreased Pgp protein level in cultured cells than the native nanocomplexes with the same components.
To assess whether the nanocomplex-induced suppression of Pgp could sensitize OVCAR8/ADR cells to Pgp-transportable cytotoxic agents, such as doxorubicin (DOX), we compared the drug sensitivity of the nanocomplex-treated cells to that of the untreated cells using the MTT assay. As shown in Figure 4 and Table S3, the IC50 value decreased significantly as a function of the targeted nanocomplex concentration, indicating that the targeted nanocomplex treatment indeed led to the enhanced drug sensitivity of OVCAR8/ADR cells. For example, the IC50 value of cells after 0.12 nM of the nanocomplex treatment is 23 times lower than that of cells without the nanocomplex treatment. In contrast, the non-targeted nanocomplexes and the targeted ctrl nanocomplexes had no significant effect on the drug sensitivity of OVCAR8/ADR cells. It is the reason that the efficient suppression of the Pgp transporter by the targeted anti-MDR1 nanocomplex result in DOX accumulation in cells, thus facilitating the killing of cancer cells. In addition, the targeted anti-MDR1 nanocomplex reversed resistance to Pgp-transportable drugs, but did not affect the sensitivity to hydroxyurea, a non-Pgp substrate (Table S3).[18] These results further suggest that the suppression of Pgp expression mediated by the anti-MDR1 nanocomplexes is specific.
Figure 4.
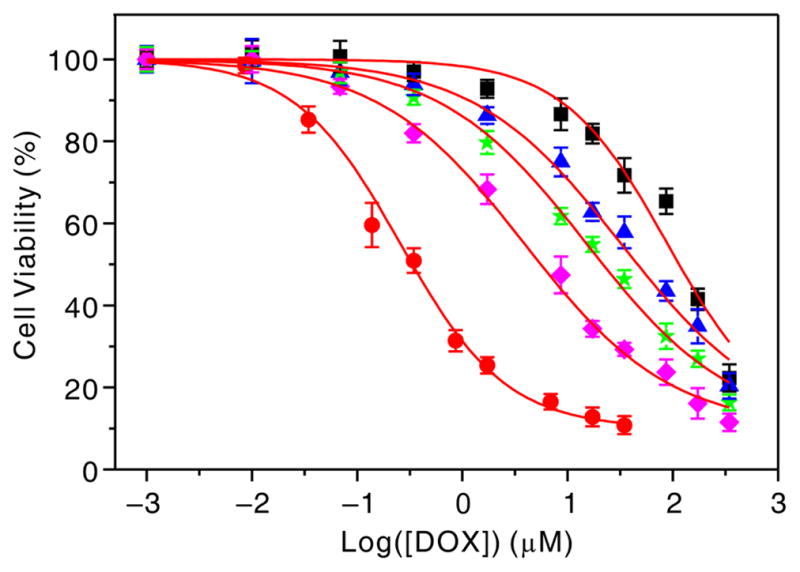
Effect of the targeted anti-MDR1 nanocomplexes on sensitivity of OVCAR8/ADR cells to doxorubicin: OVCAR8 cells (red), OVCAR8/ADR cells without (black) and with targeted nanocomplex treatment of 0.01 nM (blue), 0.04 (green) and 0.12 nM (pink). Each point represents the average value from three independent experiments.
In summary, the results presented herein provide unambiguous evidence that the synthetic nanocomplexes not only show resistance to enzymatic degradation, excellent target selectivity, specific self-delivery, and non-cytotoxicity, but also exhibit unique cooperative RISC-like gene silencing function without interferon response. Since the established function of the nanocomplex is independent of cellular RNAi machinery, this work paves a way to construct a novel class of nanoparticle-based intracellular machinery. Furthermore, the conceptual power of this nanocomplex follows from its programmability: by changing the sequence of the DNA oligos, it is possible to change the identity of the gene that is targeted for knockdown. Therefore, this nanocomplex approach has the potential to become an alternative tool for functional genomics or as a therapeutic agent for cancers and other diseases associated with abnormal protein expression.
Supplementary Material
Footnotes
This work was supported by National Natural Science Foundation (81171399, 81371596 and 81101077), CSC Foundation (2011832173), National Significant New Drugs Creation Program (2012ZX09505-001-001), National Basic Research Program of China (973 program, 2013CB733802 and 2014CB744503), Jiangsu Province Foundation (BK2011166, BE2012622, BL2012031, and BM2012066), Outstanding Professional Fund of Health Ministry in Jiangsu Province (RC2011095 and H201028) and the Intramural Research Program (IRP) of NIBIB /NIH.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx
Contributor Information
Zhongliang Wang, Key Laboratory of Nuclear Medicine, Ministry of Health, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine, Wuxi 214063, China. Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bio-engineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland 20892, United States. Center for Molecular Imaging and Translational Medicine, School of Public Health, Xiamen University, Xiamen, 361005, China.
Zhe Wang, Key Laboratory of Nuclear Medicine, Ministry of Health, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine, Wuxi 214063, China. Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bio-engineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland 20892, United States. Center for Molecular Imaging and Translational Medicine, School of Public Health, Xiamen University, Xiamen, 361005, China.
Dingbin Liu, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bio-engineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland 20892, United States.
Xuefeng Yan, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bio-engineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland 20892, United States.
Fu Wang, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bio-engineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland 20892, United States.
Gang Niu, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bio-engineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland 20892, United States.
Min Yang, Email: yangmin@jsinm.org, Key Laboratory of Nuclear Medicine, Ministry of Health, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine, Wuxi 214063, China.
Xiaoyuan Chen, Email: shawn.chen@nih.gov, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bio-engineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland 20892, United States.
References
- 1.a) Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]; b) Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 2.a) Castanotto D, Rossi JJ. Nature. 2009;457:426–433. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Moazed D. Nature. 2009;457:413–420. doi: 10.1038/nature07756. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu G, Swierczewska M, Lee S, Chen X. Nano Today. 2010;5:524–539. doi: 10.1016/j.nantod.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Nat Rev Drug Discov. 2011;10:521–535. doi: 10.1038/nrd3499. [DOI] [PubMed] [Google Scholar]; e) Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]; b) Kurreck J. Angew Chem. 2009;121:1404–1426. [Google Scholar]; Angew Chem Int Ed. 2009;48:1378–1398. doi: 10.1002/anie.200802092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Whitehead KA, Langer R, Anderson DG. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tseng YC, Mozumdar S, Huang L. Adv Drug Delivery Rev. 2009;61:721–731. doi: 10.1016/j.addr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Rana S, Bajaj A, Mout R, Rotello VM. Adv Drug Delivery Rev. 2012;64:200–216. doi: 10.1016/j.addr.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA. Angew Chem. 2010;122:3352–3366. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xia Y, Li W, Cobley CM, Chen J, Xia X, Zhang Q, Yang M, Cho EC, Brown PK. Acc Chem Res. 2011;44:914–924. doi: 10.1021/ar200061q. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lee N, Choi SH, Hyeon T. Adv Mater. 2013;25:2641–2660. doi: 10.1002/adma.201300081. [DOI] [PubMed] [Google Scholar]
- 6.Pan Y, Neuss S, Leifert A, Fischler M, Wen F, Simon U, Schmid G, Brandau W, Jahnen-Dechent W. Small. 2007;3:1941–1949. doi: 10.1002/smll.200700378. [DOI] [PubMed] [Google Scholar]
- 7.Alberts B. Molecular biology of the cell. 5. Garland Science; 2008. [Google Scholar]
- 8.Hayat MA. Colloid Gold: Principles, Methods, and Applications. Academic Press; San Diego: 1989. [Google Scholar]
- 9.a) Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS, Mirkin CA. Science. 2006;312:1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]; b) Tan LH, Xing H, Chen H, Lu Y. J Am Chem Soc. 2013 doi: 10.1021/ja408033e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Seferos DS, Prigodich AE, Giljohann DA, Patel PC, Mirkin CA. Nano Lett. 2009;9:308–311. doi: 10.1021/nl802958f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen T, Wu CS, Jimenez E, Zhu Z, Dajac JG, You M, Han D, Zhang X, Tan W. Angew Chem. 2013;125:2066–2070. doi: 10.1002/anie.201209440. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2013;52:2012–2016. [Google Scholar]
- 11.Li W, Niu G, Lang L, Guo N, Ma Y, Kiesewetter DO, Backer JM, Shen B, Chen XY. Eur J Nucl Med Mol Imaging. 2012;39:300–308. doi: 10.1007/s00259-011-1969-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 13.Duhem C, Ries F, Dicato M. Oncologist. 1996;1:151–158. [PubMed] [Google Scholar]
- 14.Cerritelli SM, Crouch RJ. FEBS J. 2009;276:1494–1505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giljohann DA, Seferos DS, Patel PC, Millstone JE, Rosi NL, Mirkin CA. Nano Lett. 2007;7:3818–3821. doi: 10.1021/nl072471q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Albanese A, Tang PS, Chan WC. Annu Rev Biomed Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]; b) Chen J, Corbin IR, Li H, Cao W, Glickson JD, Zheng G. J Am Chem Soc. 2007;129:5798–5799. doi: 10.1021/ja069336k. [DOI] [PubMed] [Google Scholar]; c) Wang Z, Chui WK, Ho PC. Pharm Res. 2009;26:1162–1171. doi: 10.1007/s11095-009-9837-y. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Liu H, Yang SH, Wang T, Liu C, Cao YC. Proc Natl Acad Sci USA. 2012;109:12387–12392. doi: 10.1073/pnas.1207766109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu H, Hait WN, Yang JM. Cancer Res. 2003;63:1515–1519. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.