

Abstract
This unit provides a protocol for indirect immunofluorescence, which is a method that provides information about the locations of specific molecules and the structure of the cell. Antibody molecules for a specific target molecule are exposed to the cell or tissue being investigated. The binding of these molecules is detected by incubating the sample with a secondary antibody specific for immunoglobulin molecules and conjugated to fluorophore. This provides both a visible signal and amplification of the signal and the results are observed with a fluorescence microscope.
This unit describes the widely used and powerful technique of localization of proteins in cells by immunofluorescence (see Basic Protocol). The location can be determined by double labeling with an antibody directed against a protein of known location. The technique can be used as a supplement to immunolocalization by electron microscopy and subcellular fractionation (Chapter 3). It allows not only identification of the antigen distribution in the cell, but also a survey of the dynamic aspects of protein movements in the cell—on and off membranes, into and out of the nucleus, and through membrane traffic pathways.
BASIC PROTOCOL
IMMUNOFLUORESCENCE LABELING OF CULTURED CELLS
The following is a basic “generic” method for localizing proteins and other antigens by indirect immunofluorescence. The method relies on proper fixation of cells to retain cellular distribution of antigen and to preserve cellular morphology. After fixation, the cells are exposed to the primary antibody directed against the protein of interest, in the presence of permeabilizing reagents to ensure antibody access to the epitope. Following incubation with the primary antibody, the unbound antibody is removed and the bound primary antibody is then labeled by incubation with a fluorescently tagged secondary antibody directed against the primary antibody host species. For example, incubation with a mouse IgG primary antibody might be followed by incubation with a RITC (rhodamine isothiocyanate)–labeled goat anti–mouse IgG secondary antibody. After removal of the secondary antibody, the specimen is ready for viewing on the fluorescence microscope. Once the conditions for observing specific immunolocalization have been identified for a given antibody and cell type, double labeling with two antibodies can be employed to compare localizations. To do this, primary antibody incubation can contain two antibodies generated in two species (e.g., mouse and rabbit), followed by incubation with two secondary antibodies coupled to different fluorophores. In addition, the availability of isotype-specific anti-mouse secondary antibodies allows simultaneous immunolocalization using two mouse monoclonal antibodies of different isotypes (e.g., IgG1 vs. IgG2a). This expands the number of proteins that can be immunolocalized. Care should be taken, however, that the two (or three) antibody combinations, especially the secondary antibodies, do not cross-react.
Materials
Cells of interest, growing in tissue culture
2% formaldehyde (see recipe)
Phosphate-buffered saline (PBS; see recipe), pH 7.4
PBS/FBS: PBS, pH 7.4, containing 10% fetal bovine serum (FBS)
0.1% (w/v) saponin in PBS/FBS: prepare fresh from 10% (w/v) saponin stock solution (APPENDIX 2A; store stock up to 2 months at 4°C or in aliquots up to 1 to 2 years at −20°C)
Primary antibody
Controls: preimmune serum (if using rabbit polyclonal antibody) or antigen added in excess to primary antibody
Secondary antibodies (against Ig of species from which primary antibody was obtained) coupled to fluorophore: e.g., RITC (rhodamine isothiocyanate), FITC (fluorescence isothiocyanate) or Alexa dyes (488, 594, 647, etc)
Mounting medium (see recipe)
10-cm diameter tissue culture dishes
12-mm no. 1 round glass coverslips, sterilized by autoclaving or soaking in 70% ethanol
12-well tissue culture plates
150-mm diameter petri dishes
Watchmaker’s forceps
Microscope slides
Nail polish
Fluorescence microscope with 63× oil-immersion lens
Additional reagents and equipment for trypsinization of cells (unit 1.1)
NOTE: All solutions and equipment coming into contact with live cells must be sterile, and aseptic technique should be used accordingly.
NOTE: All culture incubations should be performed in a humidified 37°C, 5% CO2 incubator unless otherwise specified. Some media (e.g., DMEM) may require altered levels of CO2 to maintain pH 7.4.
- For adherent cells, 1 to 2 days prior to experiment trypsinize cells and seed onto 10-cm culture dishes, each containing 15 to 20 sterilized coverslips, so that on day of experiment cells are 20% to 50% confluent.
- Nonadherent cells can be coaxed into adhering to the coverslips by precoating the coverslip with poly-L-lysine (see recipe). Apply 10 to 20 μl of suspended cells to each coverslip, let sit 10 min, then proceed with fixation (step 3).
- Alternatively, cells can be attached to coverslips using a cytocentrifuge by following the manufacturer’s instructions.
On day of experiment, transfer each coverslip individually to a well of a 12-well tissue culture dish containing 1 ml culture medium. Subject cells to the desired experimental conditions (e.g., treat with various drugs, inhibitors, or temperatures prior to fixation and immunostaining).
Aspirate medium and add 1 ml of 2% formaldehyde to each well. Allow cells to fix at room temperature for 10 min.
- Aspirate the formaldehyde fixative and wash coverslips twice, each time by adding 1 ml PBS, pH 7.4, letting stand 5 min, then aspirating the PBS. Add 1 ml PBS/FBS to the fixed coverslips and let stand 10 to 20 min to block nonspecific sites of antibody adsorption.
- NOTE: Throughout the procedure, do not let cells dry out.
- In 1.5-ml microcentrifuge tubes dilute primary antibodies in 0.1% saponin/PBS/FBS.
- Typically affinity-purified antibodies are diluted in the range of 1 to 10 μg/ml, and rabbit antisera are diluted between 1:100 and 1:1000. If using a commercial antibody, follow suggested dilutions from manufacturer.
- During initial characterization, it is wise to try a range of dilutions of antibody.
- Prepare controls containing only 0.1% saponin/PBS/FBS or (if available) containing preimmune antiserum (if rabbit polyclonal antibody is being used) or specific (primary) antibody with the antigen added in excess.
- Controls are often the most important part of an immunofluorescence experiment.
- Microcentrifuge antibody dilutions and control solutions 5 min at maximum speed, room temperature, to bring down aggregates in pellet.
- Pipet the antibody solution from above the aggregate pellet.
Place a 10 × 10–cm piece of Parafilm in the bottom of a 150-mm petri dish. In a grid pattern that replicates the 12 wells used to incubate the coverslips, label the appropriate place on the Parafilm for each coverslip with a marker.
- Apply a 25-μl drop of appropriate primary antibody solution to each numbered section. Carefully remove each coverslip from the 12-well plate with watchmaker’s forceps, blot the excess fluid by touching the edge to a Kimwipe, then invert the coverslip over the appropriate 25-μl drop, making sure that the side with the cells is down. Place the top on the petri dish and incubate 1 hr at room temperature.
- Sometimes proper labeling will require a longer incubation time or the petri dish will be incubated >1 hr for convenience. For longer incubations, add some wetted Kimwipes to the dish to maintain a humid atmosphere. The incubation can be extended overnight at 4°C, if necessary.
- NOTE: It is important to always be aware of which side of the coverslip the cells are on. Cells should be facing up when in the 12-well plate, but facing down when placed on the antibody. Picking up coverslips with the forceps is awkward at first but becomes easier with practice.
Carefully pick up each inverted coverslip and flip it over so that it is cell-side-up, then place in a well of a 12-well plate. Wash each coverslip three times to remove unbound antibody, each time by adding 1 ml PBS/FBS, letting stand 5 min, then aspirating the solution.
- Dilute fluorophore-conjugated secondary antibodies in 0.1% saponin/PBS/FBS. Mix, then microcentrifuge as in step 7 to remove aggregates.
- Typically, commercial preparations are diluted between 1:100 and 1:500.
Prepare an incubation chamber as in step 8. Apply 25 μl of appropriate secondary antibody solution to each numbered section and invert coverslip over drop as in step 8. Cover petri dish and protect from light with aluminum foil or place chamber in drawer. Incubate 1 hr at room temperature.
Wash coverslips as in step 10. After removal of last PBS/FBS wash, add 1 ml PBS.
- Label slides and place 1 drop of mounting medium onto slide. Pick up coverslip from well, gently blot off excess PBS by touching the edge to a Kimwipe, then invert coverslip, cell-side-down, onto drop. Gently blot mounted coverslip with paper towel, then seal edge of coverslip onto slide by painting the edge with a rim of nail polish. Let dry.
- The fixed, mounted, and nail polish–sealed coverslips can be stored in the dark for 6 months to 1 year at 4°C.
View specimen on fluorescence microscope using an 63× oil immersion objective.
REAGENTS AND SOLUTIONS
Use deionized or distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX.
Formaldehyde, 2%
In a chemical fume hood, dilute 2 ml 37% reagent-grade formaldehyde into 35 ml PBS (see recipe), pH 7.4.
As reagent-grade formaldehyde contains 11% methanol, as an alternative (or if necessary) make up formaldehyde from paraformaldehyde by dissolving 0.4 g paraformaldehyde powder in 10 ml H2O that has been heated to 60°C, then diluting 1:1 with 2× PBS, pH 7.4. It might be desirable to try both procedures for preparing the formaldehyde solution and see which gives better results.
Mounting medium
Use Fluormount G (Southern Biotechnology) or prepare 50% (w/v) glycerol and 0.1% (w/v) p-phenylenediamine in PBS (see recipe), pH 8.0.
Phosphate-buffered saline (PBS)
0.144 g KH2PO4
9.0 g NaCl
0.795 g Na2HPO4·7H2O
H2O to 1 liter
Adjust to desired pH with 1 M NaOH or 1 M HCl
Store indefinitely at room temperature
Poly-L-lysine-coated coverslips
Apply 25 μl of 1 mg/ml poly-L-lysine to each sterilized no. 1 coverslip in a hood and allow to stand ~10 min. Carefully rinse coverslips three times with water, then allow to air dry.
COMMENTARY
Background Information
The first use of fluorescently labeled antibodies to localize a protein in cells occurred over 50 years ago (see Coons, 1961, for a reminiscence). Since that time, the wide availability of numerous antibodies and improvements in indirect labeling methods and fluorophores has made immunofluorescent localization of proteins in cells both a routine and a vital component in any study. Immunofluorescence labeling is quite effective when combined with biochemical or ultrastructural studies because the technique is rapid and so many parameters can be assessed. Furthermore, in contrast to biochemical studies, which assume uniformity of the sample, immunofluorescence technique allows analysis of individual cell differences.
Immunofluorescence labeling has been employed in a variety of cell-biological studies, including the first description of peptide targeting sequences specifying retention of endoplasmic reticulum (ER) lumenal proteins (Munro and Pelham, 1987) and initial studies describing the dynamic membrane trafficking between the ER and Golgi complex (Lippincott-Schwartz et al., 1990). The first study describing the family of Rab GTPases used immunofluorescence to demonstrate distinct localization of the different Rabs to different organelles in the cell (Chavrier et al., 1990). Immunofluorescent localization of proteins encoded by novel genes, including those associated with human diseases and cancer (Nathke et al., 1996), provides critical information for determining the cellular function of these proteins. More recently, the immunolocalization of endogenous proteins serves as an important control when fluorescent chimeric proteins are being expressed to study the dynamic behavior of the protein of interest in living cells. Similar localization of the endogenous and over-expressed protein indicates the approach is valid.
Critical Parameters
To ensure success with immunofluorescence, three parameters are critical—fixation, permeabilization, and determination of the specificity of labeling. The fixation and permeabilization conditions must be assessed individually for each antibody and each cell type investigated. Refer to Griffiths (1993) for an excellent discussion on fixation and issues related to specificity of antibody labeling.
Different fixatives might be investigated to optimize preservation of the antigen, its distribution, and the morphology of other cellular constituents. For example, some epitopes are lost upon aldehyde fixation but preserved with alcohol fixation, and vice versa. Alternative fixatives to try include methanol at −20°C (5 min exposure) or formaldehyde fixation followed by a brief (1-min) exposure to methanol at 0°C. Methanol fixation is often quite effective for localizing cytoskeletal elements. Alcohols work by extracting lipids and precipitating remaining proteins, whereas aldehydes are cross-linking reagents that generally preserve membranous structures better (McCaffery and Farquhar, 1995).
Sometimes, even when material is appropriately fixed, the epitope is obscured and not accessible to antibody binding. Permeabilizing reagents are typically detergents that partially denature fixed proteins, exposing the epitope. In addition to saponin, which the authors include throughout the staining procedure, treatments after fixation with 0.2% to 0.5% Triton X-100 or SDS can also be tried. Often a short treatment with these reagents prior to antibody incubations is sufficient to expose the epitope. Special attention should be directed toward ensuring that the access to the epitope is the same even if the protein has undergone a translocation; for example proteins that shuttle between the cytoplasm and nucleus are not always equally accessible to antibody labeling (Pines, 1997).
The primary antibody should be purified to the extent that it recognizes only the protein of interest on immunoblots. Although affinity purification often resolves this, be aware that “affinity-purified” antibodies do not always result in antibodies that recognize only a single protein. Especially in the case of anti-peptide antibodies, multiple bands are sometimes labeled even after affinity purification. By immunoblotting on a gel (UNIT 6.2), it is easy to determine on the basis of molecular weight whether it is the protein of interest that is recognized. However, not all antibodies that can be used to detect a specific protein by immunoblotting work for immunofluorescence detection. Likewise, there are antibodies that recognize the protein by immunofluorescence that do not recognize the separated, denatured proteins transferred to nitrocellulose.
Next, it is important to be able to determine what constitutes “specific labeling” with the antibody. To assess this, control-stained slides (incubated with secondary antibody only or with preimmune serum) should first be viewed to determine what constitutes “nonspecific staining.” Ideally this is negligible, and, by contrast, the staining pattern obtained with the specific antibody is much brighter and more distinct. When initially characterizing immunolocalization using a particular antibody, a range of dilutions of the primary and secondary antibodies should be examined to determine optimal concentrations that minimize nonspecific staining and maximize specific staining. Minimal background staining from secondary antibodies can often be achieved by dilution or trying different secondary antibodies made in different host species or obtained from different commercial suppliers. Then, a dilution of the primary antibody is selected to optimally label the specific protein. At this point, further controls should be analyzed to ensure that the staining observed is due to the presence of the protein of interest. If a rabbit antiserum is used, preimmune serum should yield a pattern of staining similar to that of the secondary antibody alone. If the immunizing antigen is available, it can be added to the antibody dilution to compete for the specific staining; and it should yield “background” levels of fluorescence. Lastly, support for specific staining can be confirmed by decreased labeling in a cell type that does not express the protein of interest or in a cell where the protein has been depleted by siRNA, and by increased labeling in cells overexpressing the protein by transient transfection. Finally, the use of different antibodies to the protein, if available, can independently confirm the localization pattern.
Troubleshooting
If background staining with secondary antibodies is too high, there are several possible remedies. The secondary antibodies may be diluted further, or different secondary antibodies, different hosts, or different fluorophores may be tried. Also, it is possible to try other “sorbing” reagents. PBS/FBS is generally a good sorbing reagent that effectively blocks out nonspecific sites, but other agents can be used—e.g., 1% (w/v) BSA (immunoglobulin-free) or gelatin.
If no specific staining is observed, it is possible to increase the concentration of or the time of exposure to the primary antibody. One can also try alternative fixation and permeabilization regimens to visualize specific staining.
If specific staining is observed, but it is very dim, it is possible to increase the concentration of the primary and/or secondary antibodies. As long as background staining is low, the signal can be enhanced by using a fluorescence double-sandwich technique. To do this, following incubation with primary antibody (e.g., a mouse monoclonal antibody) and washing, incubate first in FITC-conjugated goat anti–mouse IgG and then in FITC-conjugated donkey anti–goat IgG. Caution should be observed that secondary antibodies do not alter the pattern of single antibody labeling.
Anticipated Results
Once conditions for specific localization have been optimized, the distribution of the protein in a variety of cells under a variety of conditions can be observed (see Fig. 4.3.1). Double labeling of two antigens allows the distribution of one protein to be compared with that of another protein in the same sample using two secondary antibodies attached to fluorophores that can be monitored in two different channels by flow cytometry. Thus, the location of a known protein, say an antibody to a Golgi-resident protein, can be used as a marker for the Golgi complex to see whether the antigen under study colocalizes with it (Fig. 4.3.1, panels A and B).
Figure 4.3.1.
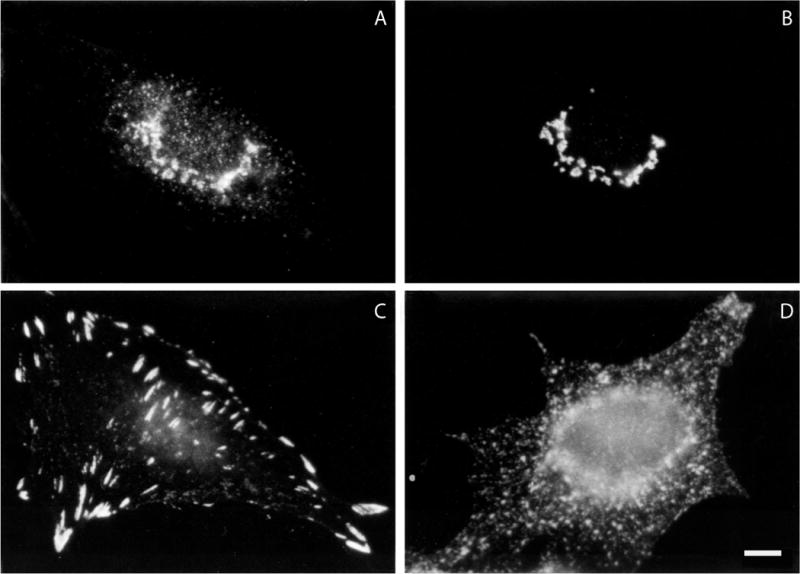
Examples of immunofluorescence labeling of formaldehyde-fixed cells. (A) and (B) Double labeling of a normal rat kidney cell with a mouse monoclonal antibody to (A) the β-COP component of coatomer and (B) a rabbit polyclonal antibody to mannosidase II. (C) Distribution of vinculin in a formaldehyde-fixed normal rat kidney cell using a mouse monoclonal antibody. (D) Distribution of transferrin receptor in formaldehyde-fixed HeLa cells using a mouse monoclonal antibody. Bar is equal to 10 μm.
Time Considerations
The entire procedure can be performed in ~3 hr. Fixed, washed coverslips can be stored at 4°C for several days prior to immunolabeling. Incubations with primary antibodies can be extended to overnight at 4°C if necessary (or if it is desirable to increase staining).
Footnotes
Disclaimer
This article was written by Julie Donaldson in her private capacity. No official support or endorsement by the NHLBI or NIH is intended and none should be inferred.
Literature Cited
- Chavrier P, Parton RG, Hauri HP, Simons K, Zerial M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62:317–329. doi: 10.1016/0092-8674(90)90369-p. [DOI] [PubMed] [Google Scholar]
- Coons AH. The beginnings of immunofluorescence. J Immunol. 1961;87:499–503. [PubMed] [Google Scholar]
- Griffiths G. Fine Structure Immunocytochemistry. Springer-Verlag; Heidelberg: 1993. [Google Scholar]
- Lippincott-Schwartz J, Donaldson JG, Schweizer A, Berger EG, Hauri HP, Yuan LC, Klausner RD. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell. 1990;60:821–836. doi: 10.1016/0092-8674(90)90096-w. [DOI] [PubMed] [Google Scholar]
- McCaffery JM, Farquhar MG. Localization of GTPases by indirect immunofluorescence and immunoelectron microscopy. Methods Enzymol. 1995;257:259–279. doi: 10.1016/s0076-6879(95)57031-4. [DOI] [PubMed] [Google Scholar]
- Munro S, Pelham HRB. A C-terminal signal prevents secretion of lumenal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- Nathke IS, Adams CL, Polakis P, Sellin JH, Nelson WJ. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J Cell Biol. 1996;134:165–179. doi: 10.1083/jcb.134.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J. Localization of cell cycle regulators by immunofluorescence. Methods Enzymol. 1997;283:99–113. doi: 10.1016/s0076-6879(97)83010-8. [DOI] [PubMed] [Google Scholar]