

Abstract
Background
Dietary intake of specific non-digestible carbohydrates (including prebiotics) is increasingly seen as a highly effective approach for manipulating the composition and activities of the human gut microbiota to benefit health. Nevertheless, surprisingly little is known about the global response of the microbial community to particular carbohydrates. Recent in vivo dietary studies have demonstrated that the species composition of the human faecal microbiota is influenced by dietary intake. There is now potential to gain insights into the mechanisms involved by using in vitro systems that produce highly controlled conditions of pH and substrate supply.
Results
We supplied two alternative non-digestible polysaccharides as energy sources to three different human gut microbial communities in anaerobic, pH-controlled continuous-flow fermentors. Community analysis showed that supply of apple pectin or inulin resulted in the highly specific enrichment of particular bacterial operational taxonomic units (OTUs; based on 16S rRNA gene sequences). Of the eight most abundant Bacteroides OTUs detected, two were promoted specifically by inulin and six by pectin. Among the Firmicutes, Eubacterium eligens in particular was strongly promoted by pectin, while several species were stimulated by inulin. Responses were influenced by pH, which was stepped up, and down, between 5.5, 6.0, 6.4 and 6.9 in parallel vessels within each experiment. In particular, several experiments involving downshifts to pH 5.5 resulted in Faecalibacterium prausnitzii replacing Bacteroides spp. as the dominant sequences observed. Community diversity was greater in the pectin-fed than in the inulin-fed fermentors, presumably reflecting the differing complexity of the two substrates.
Conclusions
We have shown that particular non-digestible dietary carbohydrates have enormous potential for modifying the gut microbiota, but these modifications occur at the level of individual strains and species and are not easily predicted a priori. Furthermore, the gut environment, especially pH, plays a key role in determining the outcome of interspecies competition. This makes it crucial to put greater effort into identifying the range of bacteria that may be stimulated by a given prebiotic approach. Both for reasons of efficacy and of safety, the development of prebiotics intended to benefit human health has to take account of the highly individual species profiles that may result.
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-015-0224-3) contains supplementary material, which is available to authorized users.
Keywords: Bacteroidetes, Prebiotic, Colonic anaerobes, Faecalibacterium prausnitzii, Firmicutes, Inulin, Pectin, Propionate
Background
The human large intestine harbours up to 1014 bacteria, whose combined degradative and biochemical capabilities greatly exceed those of their host. The interplay between the host and its gut microbiota, via degradative activities that yield nutrients and metabolites and via interactions with the immune system, is highly complex, but the consequences for human health are increasingly recognised. Alterations in the gut environment and imbalances of intestinal homeostasis are associated with changes in microbiota composition during the development of gastrointestinal diseases [1], while microbiota composition is also thought to influence energy recovery from the diet, metabolic regulation, hormone signalling and systemic health [2, 3].
Although there is considerable variation in the gut microbiota even between healthy individuals, volunteer studies involving carefully controlled diets have now established that dietary intake exerts an important influence on the composition of the human gut microbiota [4–7]. These changes may reflect selective growth promotion by dietary components that provide the main energy sources for gut bacteria, as well as inhibitory effects resulting in particular from lipid intake and bile metabolism [5]. Human diets provide energy sources available to the large intestinal microbiota mainly in the form of non-digestible (ND) carbohydrates (plant fibre and resistant starch). Studies in which ND carbohydrate intake was varied in diets with matching macronutrient composition have established that the type of ND carbohydrate can alter microbiota composition, and that such changes occur within a few days [4]. Prebiotics based on ND carbohydrates, especially inulin and fructo-oligosaccharides, are already widely used with the aim of stimulating bacterial species and activities that are considered beneficial to health among the resident microbiota [8]. Most studies on prebiotics have focussed on selected target groups rather than the whole microbial community, but these also provide evidence for the selective stimulation of certain groups or genera within the human gut microbiota [9, 10–11].
It is not possible to establish from in vivo studies whether changes in microbiota composition resulting from dietary supplementation with a ND carbohydrate are due to direct stimulation of growth by the substrate, or alternatively to indirect effects such as the acidification of the colon that follows the ingestion of any fermentable fibre due to subsequent generation of short chain fatty acids (SCFAs) by the microbiota [11, 12]. This question can, however, be answered in vitro by using anaerobic pH-controlled continuous flow fermentors inoculated with human faecal microbiota. In previous work, we showed that, with ND polysaccharides (mainly starch) as a growth substrate, a one unit pH shift caused a major change in composition of the human colonic microbiota, with Gram-negative Bacteroides species predominating at pH 6.5, but Gram-positive Firmicutes increasing at pH 5.5 [11, 13]. We decided to use this approach to determine to what extent different ND carbohydrates affect the composition of the gut microbiota at a given controlled pH. Specifically, the experiments reported here compare the impact of inulin and pectin upon the microbiota. Inulin is a plant storage polysaccharide that consists of linear chains of fructose residues with a β-(2–1) linkage and is the basis for many existing prebiotics. Pectin comprises a highly complex set of plant cell wall polysaccharides that includes homogalacturonan and rhamnogalacturonan I and II, with side chains of arabinans, galactans, and arabinogalactans [14]. Our results show that inulin and pectin exert highly selective effects upon the gut microbiota not at the phylum level, but at the level of individual species, with very little overlap between the two substrates in the species promoted. Since pH is an important factor governing the competition between bacterial species [13] and luminal pH differs between the proximal and distal colon, it was important to obtain information across the physiologically relevant pH range. Responses are shown to be influenced quantitatively by the pH across the range 5.5–6.9. These findings have important consequences for our understanding of nutritional specialisation among human colonic bacteria and for predicting how diet composition, including the addition of prebiotics, can be used to manipulate microbiota composition.
Results
Influence of pH on competition for inulin and apple pectin in continuous culture
Continuous flow anaerobic fermentors were used to investigate the impact of single polysaccharide substrates upon the species composition of gut microbiota from healthy human volunteers. Two fermentors were run in parallel in each experiment; these received an identical faecal inoculum, but one was shifted in the sequence pH 5.5, 6.0, 6.4, 6.9 (upshift) and the other in the sequence pH 6.9, 6.4, 6.0, 5.5 (downshift), with pH shifts being applied at 3-day intervals (Fig. 1). Apple pectin or inulin was provided continuously as the sole added carbohydrate energy source through the supply of fresh medium at a rate of one turnover per day (see Methods). Separate experiments were conducted for each substrate with faecal inocula from three different healthy volunteer donors, with the same three donors being used for the two sets of experiments.
Fig. 1.
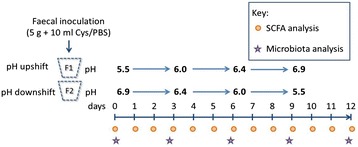
Diagram showing the design of the fermentor experiments used in this study. Single substrates (inulin or apple pectin) were supplied at a concentration of 0.5 %. In each experiment, two vessels were run in parallel (F1 and F2), with the same faecal inoculum and substrate. Fermentor 1 was shifted in the sequence pH 5.5, 6.0, 6.4, 6.9 (upshift) and fermentor 2 in the sequence pH 6.9, 6.4, 6.0, 5.5 (downshift). Samples were collected daily for short chain fatty acid analysis and DNA extraction was performed at days 0, 3, 6, 9, and 12 for qPCR and Illumina MiSeq-based sequencing of 16S rRNA gene amplicons. For each substrate, separate experiments were performed using faecal inocula from three different healthy volunteers (D1, D2 and D3)
Impact of pH changes
Changes in microbiota composition were assessed by analysis of bacterial 16S rRNA genes using qPCR and Illumina MiSeq sequencing of barcoded amplicons (see Methods). Data from pH upshift and pH downshift regimes for all donors are shown in Fig. 2 for broad bacterial groups. With inulin as a substrate there was a significant effect of pH upon the proportion of Bacteroidetes (from sequence data, ANOVA, P = 0.0025) and of Bacteroides plus Prevotella (as estimated by qPCR, ANOVA, P = 0.00037) (Fig. 2a,e). The percentage of Bacteroidetes sequences increased significantly between pH 5.5 and pH 6.9 both in F1 upshift (P = 0.019) and in F2 downshift (P = 0.031) fermentors (Fig. 2e). This is consistent with findings from previous in vitro studies in which starch was the major substrate supplied [11, 13]. pH had the opposite effect on the proportion of Actinobacteria sequences (ANOVA P = 0.0078), which decreased significantly at pH 6.9 compared with pH 5.5 both in F1 (P = 0.036) and F2 (P = 0.014) fermentors.
Fig. 2.

Effect of pH on microbial community composition. 16S rRNA gene-targeted qPCR data are shown for Bacteroides spp. (a) with inulin and (b) with apple pectin as energy sources and for Lachnospiraceae (c) with inulin and (d) with pectin. These refer to 12 fermentor runs (F1 (upshift), F2 (downshift) fermentors run in parallel for donors D1, D2 and D3 with pectin or inulin) *P <0.05, **P <0.001 (ANOVA, see Methods). Results from Illumina MiSeq sequencing of 16S rRNA gene amplicons are shown in (e) at the phylum level for F1 and F2 fermentors for each substrate; these revealed significant effects of pH when analyzed by ANOVA (see text). MiSeq data represent merged data from the same 12 fermentor runs, but also include four additional (repeat) runs for D2 inulin (F1, F2) and D3 pectin (F1, F2). Combined phylum and family level results from the sequence data are also shown for each substrate in Additional file 1: Figure S1. A list of operational taxonomic units obtained from analysis of 16S rRNA gene amplicon sequences for all samples is given in Additional file 2: Table S1
With pectin as a substrate, pH again had a significant effect on the percentage of Bacteroidetes sequences (ANOVA P = 0.0068); for this substrate, however, an increase in percentage Bacteroidetes sequences between pH 5.5 and pH 6.9 was seen only for the F2 downshift fermentors (P = 0.001) and not for the F1 upshift fermentors (Fig. 2e). Possible explanations for this intriguing effect of pH order are considered in the Discussion. Sequence analysis did not show a significant effect of pH upon percentage Firmicutes or Proteobacteria sequences for either substrate, although qPCR indicated a significant decrease in Lachnospiraceae between pH 5.5 and 6.9 with inulin as a substrate in F1 fermentors (Fig. 2c).
Responses at the operational taxonomic unit (OTU) level
When analyzed at the phylum or family level (Fig. 2e, Additional file 1: Figure S1), the Illumina sequence data do not suggest a major effect of the polysaccharide substrate supplied upon community composition. When viewed at the OTU level (at 97 % sequence similarity), however, a high degree of specificity is evident in the response to each substrate (Fig. 3). Careful manual curation of the OTUs allowed us to generate close approximations to species in many cases, although not all recognized species could be separated (e.g. B. vulgatus/B. dorei) while some less defined species (notably F. prausnitzii) were represented by several OTUs (Additional file 2: Table S1). Two OTUs derived from Bacteroides (related to B. uniformis and B. caccae, P = 0.001 and 0.001, respectively) became strongly enriched in the inulin fermentors (Fig. 4, Additional file 3: Table S2), whereas six different OTUs (B. vulgatus/dorei, B. stercoris, B. eggerthii, B. cellulosilyticus/intestinalis, B. ovatus, and B. thetaiotaomicron, P = 0.001, 0.001, 0.003, 0.001, 0.001, and 0.001, respectively) became enriched in the pectin fermentors. The impact of pH is clearly seen in the inulin fermentors for B. uniformis and B. caccae (Fig. 3a), for which the proportional representation increased with increasing pH. Within the Firmicutes, one OTU in particular, derived from Eubacterium eligens, and a less abundant, uncultured relative of Roseburia (OTU0016), were significantly enhanced by pectin relative to inulin (P = 0.001 for both OTUs). Firmicutes OTUs that benefited from the provision of inulin included Anaerostipes hadrus (OTU0018, P <0.002; Fig. 4, Additional file 3: Table S2, Additional file 4: Table S3). Based on the linear discriminant analysis effect size (LEfSe) method, several other Firmicutes, including one F. prausnitzii OTU (OTU0037, P <0.001), were also increased with inulin (Additional file 4: Table S3).
Fig. 3.
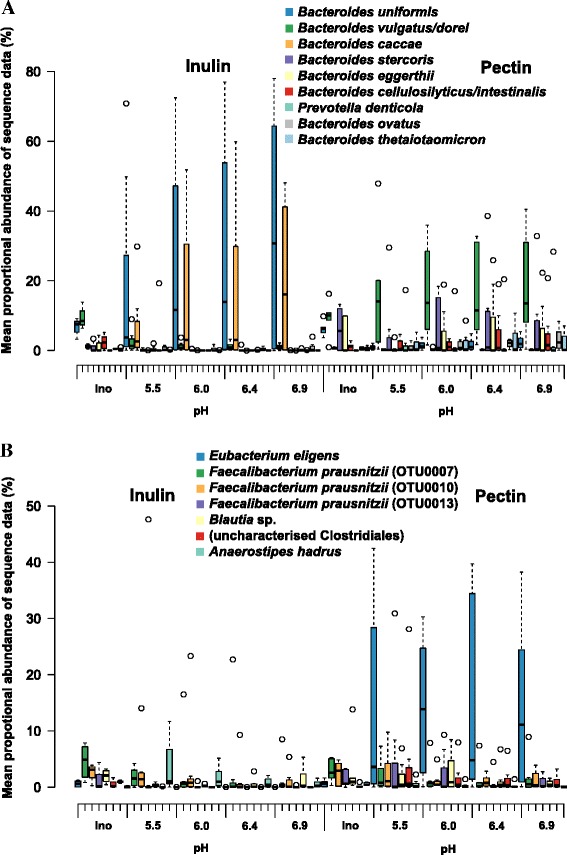
Bacteroidetes (a) and Firmicutes (b) changes across pH at the operational taxonomic unit (OTU) level. Merged data derived from Illumina MiSeq sequencing of 16S rRNA gene amplicons are shown from a total of 16 fermentor runs (F1 (upshift), F2 (downshift) fermentors run in parallel for donors D1, D2 and D3 with pectin or inulin as substrates, plus additional repeat runs for D2 inulin (F1, F2) and D2 pectin (F1, F2)). Ino = inoculum. Corresponding OTU numbers can be found in Additional file 3: Table S2 and a list of OTUs obtained from analysis of 16S rRNA gene amplicon sequences for all samples is given in Additional file 2: Table S1. Centre lines show the medians; box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, outliers are represented by dots
Fig. 4.

Mean proportional abundance of the top 20 most abundant operational taxonomic unit (OTUs) in inulin and pectin fermentor samples. Based on the 16 fermentor runs described in Fig. 3. Corresponding OTU numbers can be found in Additional file 3: Table S2 and the complete list of OTUs obtained from analysis of 16S rRNA gene amplicon sequences for all samples is given in Additional file 2: Table S1. Mean values and standard deviations are shown for the three donors. * Significant P value after Benjamini-Hochberg correction for false discovery rate (Metastats analysis)
Interspecies competition within individual microbiota
Changes within individual experiments are shown in Fig. 5 for selected OTUs. These illustrate the complexity of the competitive interactions that occur between bacterial strains within these microbial communities, and the influence of variation in composition between individual microbial communities. It can be seen (Fig. 5, Additional file 2: Table S1) that the two Bacteroides species found to be promoted by inulin did not co-exist in the same fermentor samples, with B. uniformis being dominant in the D1 and D3 experiments and B. caccae in the D2 experiments. Interestingly, both B. uniformis and B. caccae were detected in all three inocula. This suggests that differences at the strain level are also important, e.g. the D2, but not D1 or D3, inocula harboured a B. caccae strain that can outcompete the B. uniformis strains present during growth on inulin.
Fig. 5.

Microbial competition within individual fermentor runs. Changes in Bacteroides species and in the Firmicutes bacteria F. prausnitzii and Eubacterium eligens are shown for 12 fermentors (as described in Fig. 2a–d) that involved inocula from donors D1, D2 and D3. The sequence of imposed pH changes is shown for each run. These plots show the proportional abundance of OTUs determined from analysis of 16S rRNA gene amplicon sequences (Additional file 2: Table S1). Three F. prausnitzii OTUs and one E. eligens OTU were detected in inocula from all three donors, while a fourth F. prausnitzii OTU0013 was also detected in D1 and D3 inocula. Similarly, four Bacteroides OTUs (B. uniformis, B. vulgatus/dorei, B. caccae and B. ovatus) were present in inocula from all three donors. Of the remaining four Bacteroides OTUs shown, B. cellulosilyticus/intestinalis was not detected in D1 inocula, B. thetaiotaomicron and B. stercoris were not detected in D1 and D2 inocula, and B. eggerthii was not found in D3 inocula
The situation was more complex in the pectin-fed fermentors. B. vulgatus/dorei was apparently co-dominant with B. eggerthii in the D1 fermentors, with B. cellulosilyticus/intestinalis in the D2 fermentors and with B. stercoris in the D3 fermentors, with some indication of co-existence of co-dominant species at the same time-points. While B. vulgatus/dorei was detected in inocula from all three donors, B. stercoris and B. thetaiotaomicron were detected only in D3 and B. eggerthii and B. cellulosilyticus/intestinalis were not detected in D3 and D1 inocula, respectively. Thus, the emergence of different co-dominant OTUs appears to reflect the variable abundance of different Bacteroides species and strains in the inocula (Fig. 5, legend). Eubacterium eligens was able to compete with the Bacteroides species for pectin at certain pH values in all experiments, but was particularly successful in experiments with the D2 inoculum. In some of the downshift (F2) fermentors the dominance of the Bacteroides spp. was clearly curtailed or abolished at pH 5.5, and this was mirrored by a sharp increase in the abundance of F. prausnitzii OTUs at this pH. This phenomenon can be seen for D1_F2_Inulin, D2_F2_Inulin, and D1_F2_Pectin (Fig. 5d, e, j).
Targeted qPCR detection of selected groups (Fig. 2a–d) and species (Additional file 5: Figure S2, Additional file 6: Figure S3) further confirmed the findings obtained from amplicon sequencing. E. eligens was undetectable in inulin fermentors but was prominent in pectin fermentors, especially for D2, while the increase in F. prausnitzii, noted above, in certain fermentor communities at particular time points was again apparent (Additional file 5: Figure S2, Additional file 6: Figure S3).
Impacts on community diversity
The diversity of the fermentor community was reduced by comparison with the faecal inoculum in all experiments (P <0.001), but remained stable over time between days 3 and 12 (Shannon indices of between 2 and 3 compared with around 4 for the inoculum). Diversity was apparently unaffected by the imposed pH shifts, but Shannon diversity indices were significantly higher for pectin-fed than inulin-fed fermentors (P = 0.001; Fig. 6).
Fig. 6.
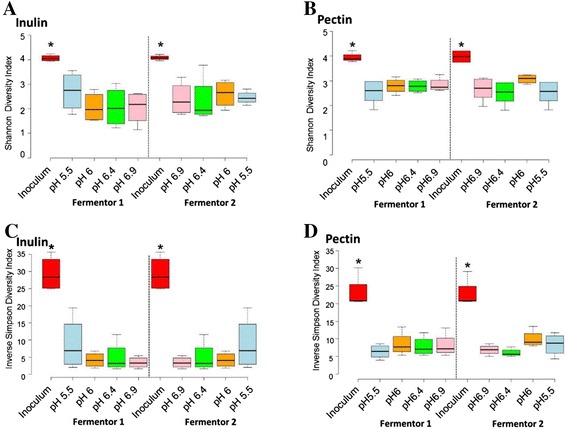
Bacterial diversity across pH range in inulin and pectin fermentors using both the Shannon index (a, b) and inverse Simpson index (c, d). Centre lines show the medians; box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, outliers are represented by dots. * Indicates significant difference between the faecal inocula and all fermentor pH conditions (ANOVA, P <0.001). Diversity estimates were derived from 16S rRNA gene OTU analysis of the 16 fermentor runs described in Fig. 3 and shown in Additional file 2: Table S1
Short chain fatty acids (SCFA)
SCFA concentrations were measured for all time points and the mean values across the whole pH range shown in Additional file 7: Table S4. This revealed that the proportion of Bacteroides spp. and Prevotella spp. present in the fermentor community (as determined by qPCR) was positively correlated with the concentration of propionate measured at the same time point (R2 = 0.558; Additional file 8: Figure S4A). The butyrate concentration was only weakly correlated to the combined abundance of two prominent groups of butyrate-producing bacteria, F. prausnitzii and Roseburia spp. (R2 = 0.278), which may reflect the known contribution of other Firmicutes to butyrate production [15] (Additional file 8: Figure S4B). SCFA production from pectin at low pH was apparently unaffected by the direction of the pH change, but altered SCFA proportions were observed with inulin as a substrate at pH 5.5 after 12 days in the downshift fermentors (with a significant decrease in the proportion of acetate, P = 0.02; Fig. 7). Family level changes derived from the MiSeq data were in line with the shifts in microbiota composition reported earlier in Fig. 2 (Fig. 7, legend).
Fig. 7.
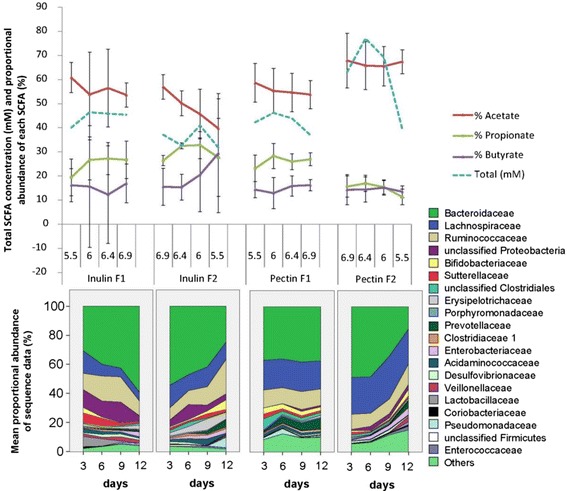
Short chain fatty acids (SCFA) in upshift and downshift fermentors. Mean SCFA values (means and standard deviations) and proportional abundance of bacterial families based on sequence analysis of 16S rRNA gene amplicons are shown for the 16 fermentor runs described in Fig. 3. Significant changes in % SCFA (from ANOVA) are discussed in the text. ANOVA revealed significant decreases in % Bacteroidaceae between pH 6.9 and pH 5.5 in inulin fermentors F1 (P = 0.015) and F2 (P = 0.012), but with pectin only for the F2 (downshift) fermentors (P = 0.0001). % Bifidobacteriaceae and % Lachnospiraceae increased significantly at pH 5.5 compared with pH 6.9 in F2 inulin (P = 0.007) and F1 inulin (P = 0.025) fermentors, respectively
Discussion
Several different approaches can be employed to understand and predict the selective influence of ND carbohydrates upon the gut microbiota. Analysis of carbohydrate active enzyme (CAZyme, Carbohydrate Active Enzymes database [16, 17], URL http://www.cazy.org/) complements from individual genomes [18] and growth tests on isolated species [19] may be indicative, but cannot predict how different organisms will compete and interact within the complex intestinal community. At the other extreme, in vivo feeding experiments cannot readily distinguish effects mediated via the gut environment, for example, changes in gut transit or pH, from the direct selective effects of the substrate. Furthermore, in vivo nutritional studies do not provide precise control over the substrates available to the microbiota, since endogenous substrates such as mucin and other food components will always be present. The approach that we have adopted here of using in vitro continuous flow fermentors to study the human intestinal microbiota as a microcosm has the considerable advantage of allowing precise control over both substrate supply and pH.
Detailed investigations into polysaccharide utilization have so far been mainly undertaken for Bacteroides spp. among predominant human gut anaerobes [20], although more recently studies have also been conducted in the Firmicutes representative Eubacterium rectale [21]. In Bacteroides spp., genes responsible for the degradation of particular polysaccharides are organized at polysaccharide utilization loci (PULs) that also encode transport functions and transcriptional regulators. Individual Bacteroides genomes typically possess large numbers of PULs (over 80) that are concerned with degradation of different host- and diet-derived polysaccharides [22, 23]. Variations in PUL distribution between species have been demonstrated to correlate with the ability to utilize particular polysaccharides in vitro and in gnotobiotic animal models [23–25]. Our results show that all of the eight most abundant Bacteroides OTUs present in the initial faecal microbiota gave a strong differential response in the mixed fermentor communities, being stimulated either by inulin (in two cases) or by pectin (in six cases). Similar selectivity was evident for many Firmicutes. Among the top 38 OTUs (each representing 0.5 % or more of the total sequences), five Firmicutes OTUs increased in abundance with inulin, while a further five Firmicutes OTUs increased with pectin (Additional files 3 and 4: Tables S2 and S3).
Previous studies on cultured isolates indicated that pectin utilization is widespread among Bacteroides spp., but relatively uncommon among human colonic Firmicutes [26, 27]. Nevertheless, the strong and highly specific enrichment of the pectin-utilizing Firmicutes species E. eligens [26, 27] could provide the basis for an effective prebiotic strategy while enrichment of one F. prausnitzii OTU by pectin was also observed here. Inulin stimulated the Firmicutes species F. prausnitzii and A. hadrus (strains related to the isolate SS2/1) as has also been reported in an in vivo human study [10, 15]. Both species are butyrate-producing bacteria that offer potential benefits to health, via anti-inflammatory action in the case of F. prausnitzii [28], and via conversion of D-lactate to butyrate in the case of A. hadrus [29, 30]. In addition to promoting particular Bacteroidetes and Firmicutes OTUs, inulin also promoted some Proteobacteria, including Sutterella wadsworthensis. This may warrant further investigation as S. wadsworthensis has been isolated not only from healthy individuals but also from gastrointestinal disease states [31].
Many studies have shown that inulin has a bifidogenic effect in vivo [9, 32–34] and there is also a report suggesting that pectin can be bifidogenic [35]. Interestingly, we found no evidence in these experiments for an overall stimulation of bifidobacteria by inulin or pectin either from sequence analysis, even though B. pseudocatenulatum was detected as one of the 10 most abundant OTUs, or from qPCR monitoring of the Bifidobacterium genus. Since the representation of bifidobacteria was greatest at pH 5.5 in the inulin fermentors (Fig. 7), it therefore seems likely that the promotion of bifidobacteria by inulin that is observed in vivo depends critically on acidification in the colonic lumen that results from SCFA production [12]. At pH values closer to neutrality our evidence suggests that other bacteria will tend to out-compete bifidobacteria for inulin. It is also known that the chain length of inulin is critical in determining its utilization by isolated gut anaerobes [19], which can complicate the comparison of different studies. Many isolated bifidobacteria cannot utilize long chain inulin, although they are able to benefit from fructo-oligosaccharide breakdown products via cross-feeding in co-cultures [36]. In general, metabolic cross-feeding involving partial degradation products, fermentation products and growth factors, but also inhibitory interactions, can be expected to have played a role in the community changes occurring in these studies [37, 38].
The overall decrease in microbiota diversity in the fermentor community relative to the faecal inoculum (seen from Fig. 6) is assumed to result mainly from the supply of a single substrate as energy source. By contrast, typical human diets supply a very wide variety of types of fibre and a wide array of host-derived glycans and proteins (especially mucins). Another important factor is likely to be the relative stability of conditions within the fermentor compared with the fluctuations that occur in vivo as a result of periodic meals.
Our experiments also show that pH exerts a strong influence on competition between bacteria from different phyla or families that share the ability to utilize the same polysaccharide. This effect was particularly clear for the inulin fermentors, where the proportion of Bacteroidetes 16S rRNA gene copies fell from around 60 % at pH 6.9 to around 30 % at pH 5.5. This suppression of Bacteroides spp. at slightly acidic pH appears to reflect growth inhibition by SCFAs at pH values below 6 [11, 13]. Interestingly, this trend was not evident for the pectin upshift fermentors where E. eligens was apparently able to compete with pectin-utilizing Bacteroides spp. across the pH range. The correlation between Bacteroides representation in the community and the proportion of propionate that was seen here has also been noted for faecal samples from an in vivo study [6]. This is assumed to reflect the dominant role of the succinate pathway, which is found mainly in the Bacteroidetes among human colonic bacteria, in the formation of propionate from carbohydrates [39]. Thus, there was considerable functional redundancy with respect to propionate formation, as substrate-driven changes in the dominant species of Bacteroidetes appeared to have little influence. On the other hand, the species level changes seen here could potentially have many other effects on the host, including those via immune signalling and metabolite transformation and which will warrant investigation in future studies.
We observed some asymmetry in the response of the microbial community to low pH in parallel downshift and upshift fermentors that received the same inoculum. A possible explanation for this lies in the initial reduction in community diversity in the fermentors, discussed above. Theoretical modelling suggests that diversity will also have decreased at the strain level as strains that have optimal characteristics for pH tolerance and substrate utilization are selected [40]. Thus, the community at the start of the pH 5.5 phase in the downshift fermentors was considerably less diverse than the inoculum that initiated the pH 5.5 phase (P <0.005) in the upshift fermentors. This lower diversity may have led to the absence of Bacteroides strains tolerant of pH 5.5 at the end of the downshift runs, thus allowing the growth of more low-pH tolerant competitors such as F. prausnitzii [27, 40] and perhaps explaining the observed ‘blooms’ in F. prausnitzii that were seen in several fermentor runs (Fig. 5). Interestingly, sudden shifts in the ratio of Bacteroides to F. prausnitzii have also been reported in vivo in human subjects [41].
In summary, we have shown that two ND carbohydrates, inulin and pectin, promote very different community profiles when supplied as sole energy sources to human colonic microbial communities under conditions of controlled pH and turnover. Notably, these differences were found to lie at the species level rather than at the phylum or family level. We found very little overlap between the species stimulated by the two substrates, implying that evolution in the highly competitive environment of the large intestinal microbiota has favoured a high degree of nutritional niche specialization at the species level. At the same time, it is evident that a number of phylogenetically distant organisms belonging to different phyla have evolved convergently to gain the ability to utilize the same substrates. With a simple homopolymer (inulin) as a substrate, a single species was found to dominate the community at a given pH, but several species were apparently able to co-exist on pectin at the same pH. It is likely that the extreme chemical complexity of pectin [42] creates multiple nutritional niches which can explain the greater microbiota diversity seen in the pectin-fed relative to the inulin-fed communities. Our experiments also show that the species that become dominant with a given substrate can vary for individual microbial communities, and are likely to depend on the precise mix of competing strains within each microbiota.
Conclusions
In view of these and other recent findings, dietary manipulation through supplementation with ND carbohydrates has considerable potential for modifying the composition of the human colonic microbiota, with the aim of benefiting health. However, our findings show that it is crucial to define the profile of bacterial species promoted by an intended prebiotic, as even closely related species within the human colonic microbiota have evolved distinct substrate preferences. Bacterial populations that become promoted are likely to include species from outside the group originally targeted, and may potentially include harmful as well as beneficial organisms and also species whose effects on the host are unknown. Our work also provides a compelling rationale for pursuing ‘synbiotic’ approaches, whereby prebiotic substrates are specifically matched with beneficial microbes that are capable of utilizing them in order to enhance their colonization and activities in vivo. This could involve specific combinations of prebiotics and probiotics, or target populations of resident bacteria. First, however, it is necessary to identify which species within the complex gut microbiota are likely to respond to a given dietary manipulation. The approaches described here can thus help us to focus the necessary research into the health consequences of dietary manipulation to the most relevant bacterial species.
Methods
Simulated human colonic fermentor studies
Two single-stage fermentor systems, with a working volume of 250 mL, were set-up as described previously using a medium based on that of Macfarlane et al. [43], containing 0.6 % (w/v) peptide [13]. In the present experiments, however, the only added carbohydrate energy source was either apple pectin (Sigma) or inulin (Oliggo-Fiber DS2, avDP <10, Cargill Inc.) at 0.5 % (w/v). The fermentor culture vessels were maintained under a stream of CO2 at a constant temperature of 37 °C using a thermal jacket. Both the medium reservoir and fermentor culture vessel were mixed by internal stirrer bars powered by external stirring units. The volume of the culture was kept constant at 250 mL with a constant flow of fresh medium at a turnover of 250 mL/day. A SCFA mix was added initially in the apple pectin fermentors, containing 33 mM acetate, 9 mM propionate and 1 mM each of iso-butyrate, iso-valerate and valerate. The pH of the fermentor vessels were monitored and controlled using a pH controller that delivers either 0.1 M HCl or 0.1 M NaOH solutions to maintain the pH at 5.5 ± 0.2, 6.0 ± 0.2, 6.4 ± 0.2 and 6.9 ± 0.2 for a period of 3 days at each pH. One fermentor vessel (fermentor 1) started from the low pH (5.5) at day 0 with pH increasing over time (upshift) (Fig. 1). The other (fermentor 2) started from the highest pH (6.9) at day 0 with pH decreasing over time (downshift). Inocula were from fresh faecal samples and were prepared by mixing 5 g of faeces in 10 mL of 50 mM phosphate buffer (pH 6.5) under O2 free CO2 containing 0.05 % cysteine, using gentle MACS™ M Tubes (MACS Miltenyl Biotec). Faecal samples were donated by three healthy adult volunteers who had no history of colonic disease and had consumed no drugs known to influence the microbiota for at last 3 months prior to the sampling date. All volunteers were omnivores, one male (64 years old) and two female (53 and 42 years old).
DNA extraction
Each fermentor sample collected was immediately processed using the FastDNA Spin kit (MP Biomedicals). The samples (460 μL) were placed in lysing matrix E tubes and 978 μL of sodium phosphate buffer and 122 μL MT buffer were added to each tube, which was processed following the manufacturer’s instructions. The DNA was eluted in 50 μL FastPrep elution buffer. Faecal DNA was quantified by Nanodrop mass spectrophotometry.
PCR amplification and Illumina MiSeq sequencing
The extracted DNA was used as a template for PCR amplification of the V1-V2 region of bacterial 16S rRNA genes using the barcoded fusion primers MiSeq-27F (5′-AATGATACGGCGACCACCGAGATCTACACTATGGTAATTCCAGMGTTYGATYMTGGCTCAG-3′) and MiSeq-338R (5′-CAAGCAGAAGACGGCATACGAGAT-barcode-AGTCAGTCAGAAGCTGCCTCCCGTAGGAGT-3′), which also contain adaptors for downstream Illumina MiSeq sequencing. Each of the samples was amplified with a unique (12 base) barcoded reverse primer.
Initial PCR amplification was undertaken with New England BioLabs Q5 High-fidelity DNA Polymerase, utilizing a per-reaction mix of DNA template (1 μL), 5X Q5 Buffer (5 μL), 10 mM dNTPs (0.5 μL), 10 μM F Primer (1.25 μL), 10 μM R Primer (1.25 μL), Q5 Taq (0.25 μL) and sterile, deionised water (15.75 μL) to a final volume of 20 μL. PCR cycling conditions were as follows: 2 minutes at 98 °C; 20 cycles of 30 s at 98 °C, 30 s at 50 °C, 120 s at 72 °C; end with 5 mins at 72 °C then holding temperature at 10 °C. Quadruplicate PCR reactions per DNA sample were set up. Following confirmation of adequate and appropriately sized products, the quadruplicate reactions were pooled and ethanol precipitated. The pooled amplicons were then quantified using a Qubit 2.0 Fluorometer (Life Technologies Ltd) and a sequencing mastermix was created using equimolar concentrations of DNA from each sample. Sequencing was carried out on an Illumina MiSeq machine, using 2 × 250 bp read length, at the Wellcome Trust Sanger Institute (Cambridgeshire, UK). Sequence data has been deposited in the European Nucleotide Archive and is available under study accession number ERP010892, and sample accession numbers ERS782738 to ERS782828 (Additional file 2: Table S1).
The data obtained from Illumina MiSeq sequencing was analyzed using the mothur software package [44] and their MiSeq SOP [45]. Forward and reverse reads generated by sequencing were assembled into paired read contigs. Resulting contigs that were shorter than 270 bp, longer than 480 bp, contained ambiguous bases, or contained homopolymeric stretches of longer than 7 bases, were all removed. Unique sequences were then grouped together and aligned against the SILVA reference database. Pre-clustering (diffs = 3) was carried out to reduce the impact of sequencing errors and the OTUs were generated at a 97 % similarity cut-off level. Taxonomic classifications for each OTU from phylum to genus level were assigned using the RDP reference database (release 10) [46]. To assign species-level classifications, representative sequences for each OTU were generated using the “get.oturep” command in mothur and then searched against the NCBI Nucleotide database using MegaBLAST. Species-level classifications were assigned to OTUs where there was a greater than 99 % similarity to a reference sequence derived from a cultured isolate, and no similarity at greater than 97 % to any other cultured species in order to allow for the 3 % variation within each OTU. For OTUs where there were more than one species within a 3 % similarity margin “spp.” or multiple species names were used to indicate that we could be capturing more than one characterized species within a given OTU. Chimeric molecules created during PCR amplification as well as reads from chloroplast, mitochondria, archaea, eukaryote and unknown sequences were removed from the dataset [47]. The resulting dataset had a total of 1,721,766 sequences with a range of 8767–27679 sequences per sample. All samples were rarefied to 8767 to ensure equal sequencing depth for all comparisons. The final OTU-level results are shown in Additional file 2: Table S1. Metastats analysis [48] was used to determine any OTUs that were significantly different between two sample cohorts, and P values were corrected with the Benjamini-Hochberg method [49] to allow for the false discovery rate over multiple comparisons. In addition, significant differences across all cohorts were identified using LEfSe analysis [50]. The Shannon diversity index and inverse Simpson diversity index were used to calculate bacterial diversity per sample.
Quantitative real-time PCR (qPCR)
qPCR was performed in duplicate with iTaqTM Universal SYBR® Green Supermix (Bio-Rad) in a total volume of 10 μL with primers at 500 nM and 5 ng of DNA in optical-grade 384-well plates sealed with optical sealing tape. Amplification was performed with a CFX384TM Real-time System (Bio-Rad) with the following protocol: one cycle of 95 °C for 3 min, 40 cycles of 95 °C for 5 s and annealing temperature as per Additional file 9: Table S5 for 30 s, 1 cycle of 95 °C for 10 s, and a stepwise increase of the temperature from 65 °C to 95 °C (at 5 s per 0.5 °C) to obtain melt curve data. As described previously [10], standard curves consisted of 10-fold dilution series of amplified bacterial 16S rRNA genes from reference strains. Samples were amplified with universal primers against total bacteria and specific primers against Bifidobacterium spp., Bacteroides spp., Prevotella spp., Clostridial cluster XIVa spp. (Lachnospiraceae), F. prausnitzii, A. hadrus, E. eligens and E. rectale/Roseburia spp. (Additional file 9: Table S5). The abundance of 16S rRNA genes was determined from standard curves and specific bacterial groups were expressed as a percentage of total bacteria as determined by the universal primers. The detection limit was determined with negative controls containing only herring sperm DNA. qPCR were performed in duplicates and the data were analysed using BioRad CFX manager software.
Short chain fatty acid (SCFA) analysis
SCFA formation was assessed in fermentor samples by gas chromatography as described previously [51]. Briefly, following derivatisation of the samples using N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide, the samples were analysed using a Hewlett Packard gas chromatograph fitted with a fused silica capillary column with helium as the carrier gas.
Statistical analyses
SCFA, MiSeq and qPCR data from these experiments were analysed by ANOVA with donor, fermentor, and donor and time within fermentor as random effects, and with pH, run (upshift or downshift), and their interaction as fixed effects. When an effect was significant (P <0.05) mean values were then compared by post-hoc t-test based on the output from the ANOVA analysis.
Acknowledgements
We would like to thank Donna Henderson for carrying out GC analysis, and Paul Scott, Iraad Bronner and the Wellcome Trust Sanger Institute’s core sequencing team for carrying out the Illumina MiSeq sequencing of 16S rRNA genes. We are grateful to Grietje Holtrop (BioSS) for advice on statistical analysis. Freda Farquharson kindly contributed her expertise with training on qPCR. The Rowett Institute of Nutrition and Health receives financial support from the Scottish Government Rural and Environmental Sciences and Analytical Services (RESAS) and this work was partly funded by the Biotechnology and Biological Sciences Research Council (BBSRC-CASE) and Cargill, Inc. AWW, JP and 16S rRNA gene sequencing costs were funded by the Wellcome Trust (grant number 098051).
Additional files
Effect of pH on microbial community composition at the (A) phylum and (B) family level. (DOCX 240 kb)
Proportional abundance of each operational taxonomic unit (OTU; in %) per sample (97 % OTU cut-off). (XLSX 1538 kb)
Metastats analysis of the top 38 most abundant operational taxonomic unit (OTUs; those accounting for more than 0.5 % of total proportional abundance) between pectin and inulin fermentors. (DOCX 41 kb)
Linear discriminant analysis effect size of different sample cohorts – faecal inoculum, pectin and inulin. (DOCX 40 kb)
Changes in the human faecal microbial community composition with apple pectin as monitored by qPCR. (DOCX 938 kb)
Changes in the human faecal microbial community with inulin monitored by qPCR. (DOCX 932 kb)
Summary table of mean short chain fatty acid (SCFA) concentrations (mM) in pectin and inulin fermentors across all pH values. (DOCX 38 kb)
Correlation of metabolite concentrations with key propionate and butyrate producers as determined by qPCR in both apple pectin and inulin fermentors. (DOCX 43 kb)
List of qPCR primers used in this study including information on primer target, sequence, annealing temperature and references. (DOCX 39 kb)
Footnotes
Competing interests
JM and DB are employed by Cargill R&D Europe, who provided partial support for this work via a BBSRC Case PhD studentship to WSFC. WSFC, AWW, PL, JP, SHD and HJF have no conflict of interest to declare. Cargill, Inc. is a producer of food ingredients. This does not alter our adherence to BMC Biology’s policies on sharing data and materials.
Authors’ contributions
HJF, SHD and AWW devised the study. WSFC and SHD performed the experiments. WSFC, AWW, SHD and HJF analyzed data and prepared Figures and Tables. JP, DB, JV and PL provided critical resources and support. HJF, WSFC, SHD and AWW wrote the paper. All authors read and approved the final manuscript.
References
- 1.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474(7351):298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 2.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541–6. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 3.Flint HJ, Scott KP, Louis P, Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9(10):577–89. doi: 10.1038/nrgastro.2012.156. [DOI] [PubMed] [Google Scholar]
- 4.Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5(2):220–30. doi: 10.1038/ismej.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–63. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salonen A, Lahti L, Salojärvi J, Holtrop G, Korpela K, Duncan SH, et al. Impact of diet and individual variation on intestinal microbiota composition and fermentation products in obese men. ISME J. 2014;8(11):2218–30. doi: 10.1038/ismej.2014.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martínez I, Lattimer JM, Hubach KL, Case JA, Yang J, Weber CG, et al. Gut microbiome composition is linked to whole grain-induced immunological improvements. ISME J. 2013;7(2):269–80. doi: 10.1038/ismej.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macfarlane GT, Macfarlane S. Fermentation in the human large intestine its physiologic consequences and the potential contribution of prebiotics. J Clin Gastroenterol. 2011;45:S120–7. doi: 10.1097/MCG.0b013e31822fecfe. [DOI] [PubMed] [Google Scholar]
- 9.Bouhnik Y, Raskine L, Simoneau G, Vicaut E, Neut C, Flourie B, et al. The capacity of nondigestible carbohydrates to stimulate fecal bifidobacteria in healthy humans: a double-blind, randomized, placebo-controlled, parallel-group, dose–response relation study. Am J Clin Nutr. 2004;80(6):1658–64. doi: 10.1093/ajcn/80.6.1658. [DOI] [PubMed] [Google Scholar]
- 10.Ramirez Farias C, Slezak K, Fuller Z, Duncan A, Holtrop G, Louis P. Effect of inulin on the human gut microbiota: stimulation of Bifidobacterium adolescentis and Faecalibacterium prausnitzii. Br J Nutr. 2009;101(4):541–50. doi: 10.1017/S0007114508019880. [DOI] [PubMed] [Google Scholar]
- 11.Duncan SH, Louis P, Thomson JM, Flint HJ. The role of pH in determining the species composition of the human colonic microbiota. Environ Microbiol. 2009;11(8):2112–22. doi: 10.1111/j.1462-2920.2009.01931.x. [DOI] [PubMed] [Google Scholar]
- 12.Bown RL, Gibson JA, Sladen GE, Hicks B, Dawson AM. Effects of lactulose and other laxatives on ileal and colonic pH as measured by a radiotelemetry device. Gut. 1974;15(12):999–1004. doi: 10.1136/gut.15.12.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker AW, Duncan SH, Leitch E, Child MW, Flint HJ. pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl Environ Microbiol. 2005;71(7):3692–700. doi: 10.1128/AEM.71.7.3692-3700.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schols HA, Voragen AGJ. Complex pectins: structure elucidation using enzymes. Prog Biotechnol. 1996;14:3–19. doi: 10.1016/S0921-0423(96)80242-5. [DOI] [Google Scholar]
- 15.Louis P, Young P, Holtrop G, Flint HJ. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ Microbiol. 2010;12(2):304–14. doi: 10.1111/j.1462-2920.2009.02066.x. [DOI] [PubMed] [Google Scholar]
- 16.Cantarel BI, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37(Suppl 1):D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(D1):D490–5. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaoutari AE, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol. 2013;11(7):497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- 19.Scott KP, Martin JC, Duncan SH, Flint HJ. Prebiotic stimulation of human colonic butyrate-producing bacteria and bifidobacteria in vitro. FEMS Microbiol Ecol. 2014;87(1):30–40. doi: 10.1111/1574-6941.12186. [DOI] [PubMed] [Google Scholar]
- 20.Martens EC, Koropatkin NM, Smith TJ, Gordon JI. Complex glycan catabolism by the human gut microbiota: the bacteroidetes sus-like paradigm. J Biol Chem. 2009;284(37):24673–7. doi: 10.1074/jbc.R109.022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cockburn DW, Orlovsky NI, Foley MH, Kwiatkowski KJ, Bahr CM, Maynard M, et al. Molecular details of a starch utilization pathway in the human gut symbiont Eubacterium rectale. Mol Microbiol. 2015;95(2):209–30. doi: 10.1111/mmi.12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravcheev DA, Godzik A, Osterman AL, Rodionov DA. Polysaccharides utilization in human gut bacterium Bacteroides thetaiotaomicron: comparative genomics reconstruction of metabolic and regulatory networks. BMC Genomics. 2013;14:1. doi: 10.1186/1471-2164-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141(7):1241–52. doi: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonnenburg JL, Xu J, Leip DD, Chen C, Westover BP, Weatherford J, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307(5717):1955–9. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 25.Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci U S A. 2009;106(14):5859–64. doi: 10.1073/pnas.0901529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salyers AA, West SEH, Vercellotti JR, Wilkins TD. Fermentation of mucins and plant polysaccharides by anaerobic bacteria from human colon. Appl Environ Microbiol. 1977;34(5):529–33. doi: 10.1128/aem.34.5.529-533.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez-Siles M, Khan TM, Duncan SH, Harmsen HJM, Garcia-Gil LJ, Flint HJ. Cultured representatives of two major phylogroups of human colonic Faecalibacterium prausnitzii can utilize pectin, uronic acids, and host-derived substrates for growth. Appl Environ Microbiol. 2012;78(2):420–8. doi: 10.1128/AEM.06858-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux J, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105(43):16731–6. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen-Vercoe E, Daigneault M, White A, Panaccione R, Duncan SH, Flint HJ, et al. Anaerostipes hadrus comb. nov., a dominant species within the human colonic microbiota; reclassification of Eubacterium hadrum Moore et al. 1976. Anaerobe. 2012;18(5):523–9. doi: 10.1016/j.anaerobe.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Duncan SH, Louis P, Flint HJ. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl Environ Microbiol. 2004;70(10):5810–7. doi: 10.1128/AEM.70.10.5810-5817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukhopadhya I, Hansen R, Nicholl CE, Alhaidan YA, Thomson JM, Berry SH, et al. A comprehensive evaluation of colonic mucosal isolates of Sutterella wadsworthensis from inflammatory bowel disease. PLoS ONE. 2011;6(10):e27076. doi: 10.1371/journal.pone.0027076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouhnik Y, Raskine L, Champion K, Andrieux C, Penven S, Jacobs H, et al. Prolonged administration of low-dose inulin stimulates the growth of bifidobacteria in humans. Nutr Res. 2007;27(4):187–93. doi: 10.1016/j.nutres.2007.01.013. [DOI] [Google Scholar]
- 33.Meyer D, Stasse-Wolthuis M. The bifidogenic effect of inulin and oligofructose and its consequences for gut health. Eur J Clin Nutr. 2009;63(11):1277–89. doi: 10.1038/ejcn.2009.64. [DOI] [PubMed] [Google Scholar]
- 34.Costabile A, Kolida S, Klinder A, Gietl E, Buerlein M, Frohberg C, et al. A double-blind, placebo-controlled, cross-over study to establish the bifidogenic effect of a very-long-chain inulin extracted from globe artichoke (Cynara scolymus) in healthy human subjects. Br J Nutr. 2010;104(7):1007–17. doi: 10.1017/S0007114510001571. [DOI] [PubMed] [Google Scholar]
- 35.Olano-Martin E, Gibson GR, Rastall RA. Comparison of the in vitro bifidogenic properties of pectins and pectic-oligosaccharides. J Appl Microbiol. 2002;93(3):505–11. doi: 10.1046/j.1365-2672.2002.01719.x. [DOI] [PubMed] [Google Scholar]
- 36.Belenguer A, Duncan SH, Calder AG, Holtrop G, Louis P, Lobley GE, et al. Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl Environ Microbiol. 2006;72(5):3593–9. doi: 10.1128/AEM.72.5.3593-3599.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duncan SH, Scott KP, Ramsay AG, Harmsen HJM, Welling GW, Stewart CS, et al. Effects of alternative dietary substrates on competition between human colonic bacteria in an anaerobic fermentor system. Appl Environ Microbiol. 2003;69(2):1136–42. doi: 10.1128/AEM.69.2.1136-1142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flint HJ, Duncan SH, Scott KP, Louis P. Interactions and competition within the microbial community of the human colon: links between diet and health. Environ Microbiol. 2007;9(5):1101–11. doi: 10.1111/j.1462-2920.2007.01281.x. [DOI] [PubMed] [Google Scholar]
- 39.Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP, et al. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 2014;8(6):1323–35. doi: 10.1038/ismej.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kettle H, Donnelly R, Flint HJ, Marion G. pH feedback and phenotypic diversity within bacterial functional groups of the human gut. J Theor Biol. 2014;342:62–9. doi: 10.1016/j.jtbi.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 41.Lahti L, Salojärvi J, Salonen A, Scheffer M, De Vos WM. Tipping elements in the human intestinal ecosystem. Nat Commun. 2014;5:4344. doi: 10.1038/ncomms5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voragen AGJ, Coenen G, Verhoef RP, Schols HA. Pectin, a versatile polysaccharide present in plant cell walls. Struct Chem. 2009;20(2):263–75. doi: 10.1007/s11224-009-9442-z. [DOI] [Google Scholar]
- 43.MacFarlane GT, Hay S, Gibson GR. Influence of mucin on glycosidase, protease and arylamidase activities of human gut bacteria grown in a 3-stage continuous culture system. J Appl Bacteriol. 1989;66(5):407–17. doi: 10.1111/j.1365-2672.1989.tb05110.x. [DOI] [PubMed] [Google Scholar]
- 44.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kozich J, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl Environ Microbiol. 2013;79(17):5112–20. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–67. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinform. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5(4):e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57(1):289–300. [Google Scholar]
- 50.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:6. doi: 10.1186/gb-2011-12-S1-P6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richardson AJ, Calder AG, Stewart CS, Smith A. Simultaneous determination of volatile and non-volatile acidic fermentation products of anaerobes by capillary gas chromatography. Lett Appl Microbiol. 1989;9(1):5–8. doi: 10.1111/j.1472-765X.1989.tb00278.x. [DOI] [Google Scholar]