

Abstract
Genetically encoded fluorescence resonance energy transfer (FRET) reporters are powerful tools for analyzing cell signaling and function at single-cell resolution in standard 2D cell cultures, but these reporters rarely have been applied to 3D environments. FRET interactions between donor and acceptor molecules typically are determined by changes in relative fluorescence intensities, but wavelength-dependent differences in light absorption complicate this analysis method in 3D settings. Herein we report fluorescence lifetime imaging microscopy (FLIM) with phasor analysis, a method that displays fluorescence lifetimes on a pixel-wise basis in real time to quantify apoptosis in breast cancer cells stably expressing a genetically encoded FRET reporter. This microscopic imaging technology allowed us to identify treatment-induced apoptosis in single breast cancer cells in environments ranging from 2D cell culture, spheroids with cancer and bone marrow stromal cells, and living mice with orthotopic human breast cancer xenografts. Using this imaging strategy, we showed that combined metabolic therapy targeting glycolysis and glutamine pathways significantly reduced overall breast cancer metabolism and induced apoptosis. We also determined that distinct subpopulations of bone marrow stromal cells control the resistance of breast cancer cells to chemotherapy, suggesting heterogeneity of treatment responses of malignant cells in different bone marrow niches. Overall, this study establishes FLIM with phasor analysis as an imaging tool for apoptosis in cell-based assays and living mice, enabling real-time, cellular-level assessment of treatment efficacy and heterogeneity.
Keywords: breast cancer, intravital microscopy, fluorescence lifetime imaging, optical imaging
Introduction
Apoptosis, a common form of programmed cell death, is fundamental to cancer biology and therapy (1). Resistance to apoptosis defines a hallmark feature of cancer initiation and progression, allowing cells to overcome cell-intrinsic and tissue-level safeguards against malignant transformation (2). Apoptosis also defines a common mechanism of cell death caused by most cancer chemotherapeutic drugs. In response to inciting events such as drug-mediated DNA damage, the blockade of pathways necessary for cell survival, or immunotherapy, cancer cells begin a well-characterized cascade of molecular events that involve the activation of caspases, a family of proteases (3). The apoptotic cascade culminates with the activation of a common effector molecule, caspase-3, which cleaves numerous intracellular substrates to produce chromatin condensation and other phenotypic changes during cell death. Therefore, imaging caspase-3 activity provides a noninvasive, real-time method for quantifying apoptosis in response to environmental stresses and drugs in cell-based assays and living mice.
As a direct result of the importance of apoptosis in cancer and cancer therapy, investigators have developed several different approaches to image caspase-3 activity or other markers of apoptosis, such as changes in cell membrane composition. These imaging strategies encompass modalities, including positon emission tomography, bioluminescence, photoacoustics, or magnetic resonance imaging, using either genetically engineered reporters or exogenous probes (4–7). Although these approaches have successfully detected apoptosis in animal models and even initial patient studies, these imaging modalities define apoptosis at population-level scales of resolution rather than individual cells. Bulk measurements of tumor responses to therapy cannot capture heterogeneous responses among subpopulations of cancer cells, a key determinant of treatment success or failure (8).
Fluorescence imaging allows for the detection and quantification of apoptosis in single cells, complementing and expanding upon the capabilities of whole-organism imaging techniques. For example, apoptosis has been imaged with genetically encoded reporters in which fluorescence resonance energy transfer (FRET) occurs between 2 different fluorescent proteins linked by the specific amino acid motif (aspartate, glutamate, valine, and aspartate, designated by the single-letter amino acid code DEVD) for cleavage by caspase-3 (9). The intact reporter holds the fluorescent proteins in close proximity, permitting energy transfer from the donor to the acceptor fluorescent protein. Caspase-3 cleavage separates the fluorescent proteins and eliminates FRET, which can be detected by optical imaging. Studies of apoptosis with caspase-3-based FRET reporters typically have been performed in cultured cells, although a limited number of studies have used intravital microscopy to quantify changes in FRET in living organisms (10, 11). These prior in vivo imaging studies of apoptosis have quantified FRET by changes in ratios of intensities of donor and acceptor fluorescent proteins. Although ratiometric imaging works well in cultured cells, fluorescence intensities in tissues are affected by greater absorption of shorter versus longer wavelengths of visible light (12). Therefore, ratios of fluorescence intensities may show depth-dependent changes independent of FRET, complicating the analysis of imaging data for caspase activation.
In addition to changes in fluorescence intensities, FRET interactions shorten the fluorescence lifetime of a donor fluorescent protein. Fluorescence lifetime refers to the amount of time a fluorescent molecule remains in the excited state before releasing a photon. Because fluorescence lifetime is independent of intensity as long as there is a signal detectable above background, measurements of this parameter are not affected by depth within tissue. We capitalized on advantages of fluorescence lifetime imaging microscopy (FLIM) to analyze caspase-3 activity with a genetically encoded FRET reporter as a marker of apoptosis in breast cancer cells in vitro and in living mice (13). To further improve our ability to detect changes in FRET from our caspase-3 reporter, we used frequency-domain FLIM, which derives changes in fluorescence lifetime based on phase delay and the modulation ratio of light emitted from a fluorescent molecule relative to the excitation light (14). Frequency-domain FLIM allows for the rapid acquisition of fluorescence lifetime images, an advantage that permits nearly real-time imaging of dynamic events in living systems. Using FLIM imaging of a caspase-3 reporter, we detected apoptosis in cells in 2D and 3D cultures and orthotopic breast tumor xenografts after treatment with chemotherapeutic drugs or experimental targeting of metabolism. Overall, this study demonstrates that FLIM can measure apoptosis at single-cell resolution in preclinical model systems, facilitating analyses of intercellular heterogeneity in responses to therapy that may improve the design of future treatment protocols.
Methods
Cells
We cultured HEK 293T cells (Thermo Fisher Scientific, Carlsbad, CA), MDA-MB-231 breast cancer cells (ATCC), and HS-5 and HS-27A bone marrow stromal cells (ATCC) in Dulbecco's modified eagle's medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) (HyClone; GE HealthCare, Logan, UT). For microscopy, we replaced the culture medium with phenol red-free DMEM (Thermo Fisher Scientific) supplemented with 1% FBS, 0.1 nM estrogen, 1% penicillin/streptomycin, 1% pyruvate, and 2.5% glutamine unless stated otherwise. Cells were cultured at 37°C in an incubator with 5% CO2.
Stable Cell Lines
To generate a lentiviral vector with a blasticidin selection marker, we amplified the blasticidin reading frame from pCDNA6 (Thermo Fisher Scientific) using the polymerase chain reaction (PCR) primers phosphorylated 5′-GTGGTTTTCCTTTGAAAAACACGATGATAATATGGCCAAGCCTTTGTCTC-3′ and 5′-CCAGACGCGTTCAATTAATTAGCCCTCCCACACATAACCAG-3′. We ligated the PCR product into the BmgB1 and MluI sites of lentiviral vector pLVX IRES puromycin (Clontech, Mountain View, CA), removing the puromycin cassette to generate pLVX IRES blasticidin. We amplified the fluorescent protein large Stokes shift (LSS)-mOrange (gift of V. Verkhusha, Albert Einstein College of Medicine) by PCR using the primers 5′-ATGCGCTAGCGCCACCATGGTGAGCAAGGGCGAGGAG-3′ and 5′-GCATGCGGCCGCTTACTTGTACAGCTCGTCCATGCCGC-3′. The PCR product was digested and ligated into corresponding NheI and NotI sites in pLVX IRES blasticidin. We produced lentiviruses by transiently transfecting HEK 293T cells (15). We selected MDA-MB-231 and HEK 293T cells stably expressing LSS-mOrange by selection with blasticidin. To generate stable cells expressing the caspase-3 reporter LSS-mOrange-DEVD-mKate2, we excised the FRET reporter cassette with NheI and NotI and ligated it into the corresponding sites in the PiggyBac transposon vector (Systems Bioscience, Mountain View, CA). We transfected HEK 293T or MDA-MB-231 cells with this vector and the transposase plasmid using calcium phosphate or FuGENE 6 (Promega, Madison, WI), respectively. We sorted for the top 30% of fluorescent cells by flow cytometry. This approach established a population of cells with relatively uniform levels of the apoptosis reporter while maintaining inherent heterogeneity known to exist within cancer cell lines.
2D Cell Culture Experiments
For selected experiments with HEK 293T cells, we transiently transfected cells with increasing concentrations of a plasmid encoding the proapoptotic protein Bcl-2-associated X (Bax) (gift of S. Galbán, University of Michigan) using calcium phosphate as described (15). We seeded cells into 6-well plates at 1.4 × 105 cells per well 1 day after transfection and imaged cells the following day. We seeded MDA-MB-231 cells at the same density. We treated cells with the following conditions 1 day before imaging studies: (1) 10 nM trametinib (Selleckchem, Houston, TX), 1 μM staurosporine (Cell Signaling Technology, Beverly, MA), or dimethyl sulfoxide vehicle control; (2) 40 mM dichloroacetate (DCA) (Sigma-Aldrich, St. Louis, MO); or (3) complete phenol red-free DMEM with 10% FBS, medium without glucose, medium without glutamine, or medium lacking both glucose and glutamine.
Metabolic Flux Studies
We seeded 10 × 103 MDA-MB-231 cells per well in 80 μL of complete medium in a Seahorse Bioscience (North Billerica, MA) XF96 cell culture 96-well plate. The next day we replaced the medium with 1 of 4 of the following different media and drug conditions:
Glutamine media: phenol red-free DMEM, 1% FBS, 0.1 nM estrogen, 1% penicillin/streptomycin, 1% pyruvate, and 2.5% glutamine
Glutamine media plus 40 mM DCA
No glutamine media: phenol red-free DMEM, 1% FBS, 0.1 nM estrogen, 1% penicillin/streptomycin, and 1% pyruvate
No glutamine media plus 40 mM DCA
The following day we performed a glycolysis stress test using the Seahorse Bioscience XF Analyzer. Before running the experiment we replaced the media with the Seahorse XF glycolysis stress test assay medium, excluding glutamine in conditions 3 and 4. After incubating in a CO2-free incubator for an hour, we added 10 mM glucose, 2 mM oligomycin, and 100 mM 2-deoxyglucose to the injection ports. After calibrating the analyzer, we ran the experiment for a total time of 80 minutes.
Spheroid Model
We seeded 3 × 103 total cells (20% MDA-MB-231 cells expressing LSS-mOrange or LSS-mOrange-DEVD-mKate2 mixed with 80% HS5 or HS27A cells) per well in complete phenol red-free medium in nonadhesive 384-well plates (Corning Inc., Corning, NY). The next day we replaced the medium with 20 μL of complete phenol red-free medium containing 100 nM trametinib or DMSO vehicle control and imaged cells 24 hours later.
Animal Studies
All animal experiments were approved by the University of Michigan Committee for the Care and Use of Animals. We implanted 5 × 105 cells (MDA-MB-231 cells expressing LSS-mOrange or LSS-mOrange-DVD-mKate2) in the mammary fat pads of ten 12- to 14-week-old female NSG (Jackson) mice as described (16). After implants formed 3- to 4-mm tumors by day 20, we treated the mice daily with 1 mg/kg trametinib or vehicle control in 50-μL doses by oral gavage (17). We imaged tumors by intravital microscopy after 8 and 14 consecutive days of treatment.
FLIM and Data Analysis
We imaged orthotopic mammary tumors by 2-photon microscopy using an upright Olympus FV1000-MPE and a 25×, 1.05 numerical aperture objective as we reported previously (18). Using an 80-MHz pulsed scanning laser tuned to 820 nm (Mai Tai-DeepSee, Spectra-Physics, Santa Clara, CA) and 572/15-nm emission filter, we captured FLIM images using the FastFLIM frequency domain instrument (ISS, Champaign, IL) with a 256 × 256 pixel image size and 100 microsecond per pixel scan speed over 15-30 acquisition frames.
We processed FLIM data using VistaVision software (ISS). This software represents fluorescence lifetime on a phasor plot by transforming the fluorescence intensity decay of each image pixel to a point coordinate on the phasor plot (19). To eliminate noise, we set the minimum count threshold at 10 counts and used the smoothing operation. We used the “Zoom” operation to select regions of interest and the “Go to Peak” operation to objectively determine the average phasor values for individual cells.
Histology
After completing the intravital imaging studies, we fixed tumors in 10% formalin for histology with hematoxylin and eosin and immunohistochemistry for cleaved caspase-3 (Cell Signaling Technology) performed through the University of Michigan Unit for Laboratory Animal Medicine Core. A person unaware of treatment groups quantified numbers of cells positive for cleaved caspase-3 in 3 to 6 randomly selected fields per tumor.
Statistics
We compared differences between experimental groups with a t test that used GraphPad Prism 6 (San Diego, CA), with P < .05 defining statistically significant differences. We performed experiments a minimum of 2 times.
Results
Caspase Reporter Shows Changes in Fluorescence Lifetime after Incubation with Proapoptotic Stimuli
To develop a reporter for single-cell FLIM analysis of apoptosis in vitro and in vivo, we used a FRET reporter based on the fusion of fluorescent proteins LSS-mOrange and mKate2 with an intervening caspase-3 cleavage site (amino acids DEVD) (13). In cell-based assays, this reporter has previously been shown to indicate apoptotic activity via changes in FRET after cleaving caspase-3 (Figure 1A).
Figure 1.

Validation of caspase-3 reporter activity. (A) Schematic of FRET with the caspase-3 reporter. Intact LSS-mOrange-DEVD-mKate2 produces a shorter fluorescence lifetime of LSS-mOrange, whereas cleavage at the DEVD site in the reporter prolongs the fluorescence lifetime of LSS-mOrange. (B) Representative images of 293T cells transiently transfected with control (LSS-mOrange) or functional caspase-3 reporter plus increasing concentrations of a plasmid for Bax. Control cells were cotransfected with the highest amount of empty vector. Representative images are shown 24 hours after transfection with pixels pseudo-colored in red or yellow corresponding to shorter or longer fluorescence lifetimes, respectively. Pixels not highlighted in red or yellow correspond to autofluorescence. (C) Each image pixel is represented as an individual coordinate in the phasor plot, where coordinates nearer to the origin have a longer lifetime. The red region of interest represents the shorter intact reporter lifetime, and the yellow region of interest represents the longer cleaved reporter lifetime. (D) Graph shows the percentages of cleaved reporter (yellow in panel C) in each representative image. The bar for each condition defines aggregate pixels from multiple cells within a single image field, with lifetimes corresponding to fluorescence from the cleaved reporter, which accounts for absence of error bars.
To validate reporter function, we transiently transfected HEK-293T cells with the functional caspase-3 reporter or control LSS-mOrange fluorescent protein plus increasing concentrations of Bax, a proapoptotic molecule. We quantified reporter cleavage by FLIM after overnight incubation. To quantitatively assess differences in apoptotic activity, we used phasor analysis, which plots fluorescence lifetimes of individual image pixels as a graphical display (Figure 1C). The semicircle on the phasor plot denotes lifetimes with a single exponential decay, whereas pixels containing molecules with a multiexponential fluorescence lifetime are displayed within the semicircle. Fluorescence lifetimes with shorter or longer values appear on the right and left, respectively. We defined a region of interest (ROI) for the longer fluorescence lifetime of LSS-mOrange after caspase-3 cleavage of the reporter and loss of FRET with mKate 2 based on data from cells transfected with LSS-mOrange alone and without Bax. Cells transfected with the caspase-3 reporter without Bax defined the ROI for the shorter fluorescence lifetime of LSS-mOrange in the intact reporter. For each experimental condition, we then assigned lifetimes for each pixel to either cleaved (yellow circle) or intact (red circle) reporter ROIs. We then highlighted corresponding pixels in sample images with the same color scheme to show the localization of the cleaved and intact reporters. By our FLIM analysis, cells with greater apoptotic activity are expected to exhibit more extensive reporter cleavage and thus a greater percentage of yellow-highlighted image pixels.
Comparisons between cells transfected with control LSS-mOrange or with a functional reporter established a resolvable difference in fluorescence lifetimes of intact and cleaved reporters with the cleaved, or unfused, reporter exhibiting a longer fluorescence lifetime (Figure 1B). Compared with control cells transfected with a functional reporter, FLIM images of cells additionally transfected with increasing concentrations of Bax showed greater percentages of yellow-highlighted pixels (Figure 1D), indicating more reporter cleavage and greater apoptosis. As an auxiliary indicator of apoptosis, cells transfected with higher concentrations of Bax also showed more spherical morphology and reduced adherence to the plate. These results validate a functional reporter and image-analysis method for single-cell FLIM analysis of apoptosis.
For further analyses of caspase-3 activity in cell-based assays and living mice, we generated MDA-MB-231 breast cancer cells stably expressing the imaging reporter. We then tested these cells in standard 2D cultures for inducing apoptosis by treatment with trametinib, a clinically approved MEK inhibitor, or staurosporine, a nonspecific kinase inhibitor known to induce apoptosis (20). Overnight treatment with these compounds produced modest cleavage of the reporter compared with a very low level of caspase-3 activity in cells treated with vehicle control (Figure 2A, B). These data indicate that trametinib functions at least in part by activating caspase-3 and apoptosis in cancer cells.
Figure 2.

Trametinib activates caspase-3 in MDA-MB-231 cells. (A) Representative images of stable MDA-MB-231 reporter cells treated overnight with 10 nM trametinib, 1 μM staurosporine (positive control), or vehicle control. Fluorescence lifetimes in each pixel are pseudo-colored red or yellow corresponding to intact or cleaved reporters, respectively. (B) Graph shows the percentages of highlighted pixels in each representative image with fluorescence lifetimes of LSS-mOrange corresponding with caspase-3 cleavage of the reporter. The bar for each condition defines aggregate pixels from multiple cells within a single image field with lifetimes corresponding to fluorescence from the cleaved reporter, which accounts for absence of error bars.
Metabolic Stresses Initiate Apoptosis in Breast Cancer Cells
Metabolic reprogramming is a hallmark feature of cancer, providing motivation for identifying and targeting metabolic processes that are required for survival of cancer cells (2). We used stable MDA-MB-231 reporter cells to analyze caspase-3 activity in response to metabolic stress conditions. After overnight incubation in normal growth medium or medium deprived of glucose and/or glutamine, FLIM analysis showed minimal reporter cleavage in both full media and glutamine-deprived growth conditions. However, we observed significantly greater cleavage of the caspase-3 reporter in conditions without glucose, particularly when combined with the loss of glutamine (Figure 3A, B; P < .05 and P < .01 for glucose deprivation and glutamine/glucose deprivation conditions, respectively). These results show that glucose deprivation activates caspase-3 in MDA-MB-231 breast cancer cells, suggesting that blocking glycolysis may initiate apoptosis in these cells.
Figure 3.
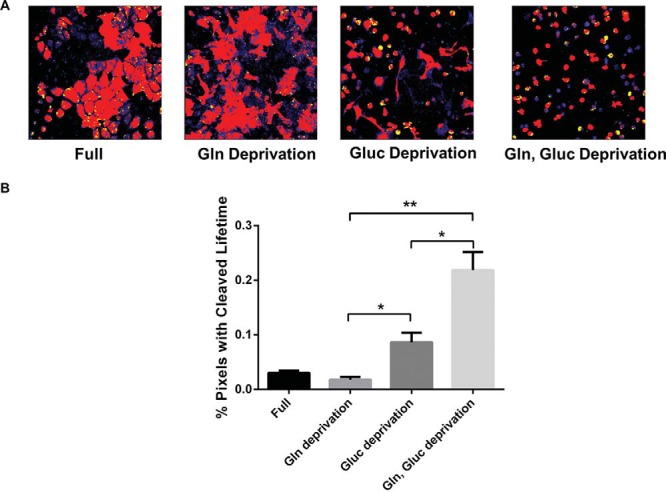
Glucose deprivation initiates apoptosis in MDA-MB-231 breast cancer cells. (A) Representative images of MDA-MB-231 reporter cells assayed in full media or media deprived of glucose (Gluc) and/or glutamine (Gln). (B) Graph shows mean values + SEMs (n = 4 per condition) of the percentages of pixels corresponding to a longer lifetime of LSS-mOrange after cleavage at the caspase-3 site (*P < .05; **P < .01).
Because metabolic reprogramming facilitates the survival of cancer cells, concurrent targeting of key metabolic processes may enhance the efficacy of cancer therapy. To query this possibility, we studied the effect of DCA on cancer cells under simultaneous metabolic stress. DCA is a chemical that inhibits pyruvate dehydrogenase kinase and funnels pyruvate toward mitochondrial metabolism to create excess reactive oxygen species (ROS), thus inducing apoptosis (21). Because glutamine serves as a precursor of α-ketoglutarate, a metabolite that eliminates ROS, glutamine-deprived media could enhance the apoptotic effect of DCA by insufficiently neutralizing DCA-driven ROS accumulation. FLIM analysis of stable MDA-MB-231 reporter cells treated with 40 mM DCA in full media or media deprived of glutamine showed significantly more yellow-highlighted pixels in the absence of glutamine (Figure 4A, B; P < .05), indicating greater reporter cleavage. These results show that simultaneous glutamine deprivation enhances the DCA effect on caspase-3 activity, supporting ongoing efforts to develop drugs that target steps in glycolysis and glutaminolysis in cancer cells.
Figure 4.
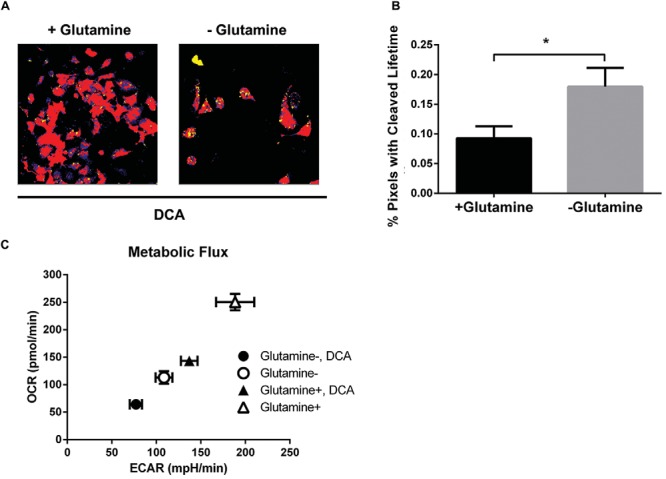
Deprivation of glutamine enhances the effects of DCA on caspase-3 activity and metabolic flux. (A) Representative images of transduced MDA-MB-231 cells treated with 40 mM DCA in full media or media deprived of glutamine. (B) Graph shows mean values + SEMs (n = 7-9 per condition) of the percentages of pixels with the fluorescence lifetime of unfused LSS-mOrange after caspase-3 cleavage (*P < .05). (C) Graph shows mean values + SEMs of ECARs and OCRs, where more metabolically active cells exhibit higher ECARs and OCRs (n = 20 per condition).
To further investigate the effects of metabolic stress on MDA-MB-231 cells, we performed metabolic flux studies to compare metabolic activity of cancer cells under each condition. Measurements of the extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) reported glycolysis and oxidative phosphorylation, respectively. Control cells grown in full medium were most metabolically active for both ECAR and OCR (Figure 4C). Although cells either treated with DCA or deprived of glutamine showed reduced metabolic flux, simultaneously treating both conditions dramatically reduced cellular metabolism as assessed by ECAR and OCR. These data further illustrate that glutamine deprivation enhances the effect of DCA in curbing cancer cell metabolic activity and promoting apoptosis in MDA-MB-231 breast cancer cells.
Fluorescence Lifetime Imaging Identifies Cell Death in 3D Spheroid Environments
Breast cancer commonly metastasizes to bone and bone marrow, causing considerable morbidity (22). Previous studies have reported that bone marrow stromal cells exhibit heterogeneous phenotypes (23–25). We used immortalized human bone marrow stromal cells HS5 and HS27A as representative examples of phenotypically distinct bone marrow stroma. These cell lines exhibit distinct profiles of cytokine expression, including chemokine (C-X-C) motif ligand 12, interleukin-6, and other molecules known to regulate the proliferation of hematopoietic stem and progenitor cells (26, 27).
To determine the effects of different bone marrow stromal environments on resistance to trametinib, we formed 3D spheroids composed of 10% stable MDA-MB-231 reporter cells with either HS5 or HS27A bone marrow-derived stromal cells. After cells formed compact spheroids, we added either trametinib or vehicle control for 24 hours before FLIM analysis. Control spheroids with either HS5 or HS27A cells showed minimal cleavage of the caspase-3 reporter in MDA-MB-231 cells, suggesting that breast cancer cells in a bone marrow stromal cell environment undergo limited spontaneous apoptosis. However, compared with untreated spheroids, FLIM analysis of both HS5 and HS27A spheroids treated with trametinib showed significantly greater percentages of apoptotic (yellow-highlighted) pixels (Figure 5A, B; P < .005 and P < .01, respectively), corresponding to more caspase-3 activation in MDA-MB-231 breast cancer cells. More notably, breast cancer cells showed significantly greater caspase-3 activation in HS27A spheroids compared with HS5 spheroids (P < .01), suggesting a possible role of bone marrow stromal cells in differentially regulating the effect of trametinib treatment.
Figure 5.

Bone marrow stromal cells in 3D culture differentially regulate the activation of caspase-3 in MDA-MB-231 cells. (A) Representative images of spheroids composed of MDA-MB-231 reporter cells with HS5 or HS27A bone marrow stromal cells treated overnight with 1 μM trametinib or vehicle control. Shorter and longer fluorescence lifetimes are depicted as red and yellow pixels, respectively. Pixels not highlighted correspond to autofluorescence and are pseudo-colored on a scale by counts. The table shows pixels corresponding to intact and cleaved caspase-3 reporter highlighted as red and yellow, respectively. τφ and τm define phase and modulation lifetimes from phasor analyses of FLIM data. (B) Graph shows mean values + SEMs (n = 4-5 per condition) of the percentages of pixels with the fluorescence lifetime of unfused LSS-mOrange after caspase-3 cleavage (**P < .01, ***P < .001).
Intravital Microscopy Identifies Treatment-Induced Cell Death In Vivo
To investigate FLIM for detecting apoptosis in vivo, we established orthotopic tumor xenografts of 231-LSS-Orange or 231-caspase-3 reporter cells, respectively. When tumors reached approximately 3 to 4 mm in diameter, we randomly assigned mice to treatment with the MEK inhibitor trametinib or vehicle control delivered daily. We imaged mice by intravital 2-photon microscopy, using FLIM to identify apoptotic cells based on changes in the lifetime of LSS-Orange in the caspase-3 reporter construct.
Mice with control 231-LSS-Orange tumors showed cancer cells with the expected fluorescence lifetime of unfused LSS-Orange, and treatment with trametinib did not affect the lifetime of this protein (data not shown). In animals with 231-caspase-3 reporter tumors, studies in mice treated with vehicle control predominantly showed pixels with the shorter fluorescent lifetime of LSS-Orange fused to mKate2, although cells did show low levels of cleaved caspase-3 by intravital microscopy (Figure 6A, B). We validated the presence of active caspase-3 in control mice by immunohistochemistry (Figure 6C). These results are consistent with prior studies that showed apoptotic cells even in growing tumors (28). By comparison, treatment with trametinib significantly increased the number of apoptotic 231-caspase-3 cells, as evidenced by a higher percentage of yellow-highlighted pixels corresponding to the cleaved reporter (Figure 6D). Immunohistochemistry for cleaved caspase-3, the active form of this enzyme, corroborated that treatment with trametinib increased the number of apoptotic cells in 231-caspase-3 tumors. Collectively, these data establish the ability to image caspase-3 activity at single-cell resolution by intravital 2-photon microscopy and FLIM.
Figure 6.

Intravital microscopy reveals therapy-induced apoptosis in breast cancer cells. (A) Representative images of transduced MDA-MB-231 breast cancer cells in orthotopic tumors in mice treated with trametinib or vehicle control; pixels are pseudo-colored by fluorescence lifetime, with red and yellow defining the shortest and longest values, respectively. Pixels not highlighted correspond to autofluorescence and are pseudo-colored on a scale by counts. (B) Graph shows mean values + SEMs (n = 4-5 imaging fields per condition) of the percentages of pixels with the fluorescence lifetime of unfused LSS-mOrange after caspase-3 cleavage (*P < .05). (C) Representative images of tumor histology sections stained for cleaved caspase-3. (D) Graph shows mean values + SEMs of the cleaved caspase-3 cell count per field of view (**P < .01).
Discussion
Investigators have generated several FRET-based reporters for fluorescence microscopy of processes such as cell signaling molecules, kinases, and proteases (29). The vast majority of studies with FRET reporters, particularly genetically encoded reporters with fluorescent proteins, have been performed in 2D cell cultures. In this experimental context, FRET reporters have proven to be powerful tools for quantifying signaling pathways and biologic events such as apoptosis at the single-cell level. However, only a small number of studies have utilized this fluorescence technique for microscopy studies in 3D environments, including tissue-engineered constructs and living animals (30–32). Methods that facilitate FRET microscopy in 3D environments offer the potential to greatly enhance understanding of normal physiology, disease, and therapy because regulation, dynamics, and functional outputs of many cell-signaling pathways differ markedly between 2D and 3D settings (33–35).
As we have demonstrated, FLIM provides a facile approach for detecting FRET signals in 3D environments by eliminating quantification based on fluorescence intensities from donor and acceptor molecules. Particularly for fluorescent proteins emitting visible light, preferential absorption of shorter wavelength light in tissues can potentially produce depth-dependent shifts in detected ratios of donor and acceptor light independent of actual changes in FRET. Measuring changes in the fluorescence lifetime of a donor fluorophore in the presence or absence of a FRET interaction overcomes 1 key challenge to the standard use of FRET microscopy beyond 2D cultures. Unlike a time-domain analysis of fluorescence lifetimes, which requires exponential fitting of fluorescence decay, the phasor approach graphically displays fluorescence lifetimes on a pixel-by-pixel basis in real time (19). The phasor display format allows for the analysis of data based on a clustering of pixels, facilitating the identification of heterogeneous responses of FRET reporters on a cellular or even subcellular basis.
The ability to identify responses of subpopulations of cells within a tumor is especially advantageous for studies of tumor heterogeneity, which increasingly is recognized as a central challenge in cancer biology and therapy (36). Understanding the mechanisms of tumor heterogeneity and developing new approaches to overcome it require therapeutic approaches for analyzing tumors at cellular rather than whole-tumor resolution. The FLIM technique and reporter construct described in this study meet the need for cellular-level analysis of apoptosis in cell culture systems and in living mice. Using phasor analysis of FRET interactions, we identified subsets of cells undergoing apoptosis in complex multicellular, 3D environments. In preclinical models, FLIM imaging of the FRET reporter can be used to test the effects of existing drugs to induce apoptotic cell death in cancer cells, optimize the efficacy of dosing protocols and treatment combinations, and assess the effects of novel therapies targeting processes such as cancer metabolism. More generally, FLIM provides a sensitive, rapid method for quantifying multiple biologic processes and diseases other than apoptosis in cancer using endogenous fluorophores or FRET from different reporter constructs or imaging probes. Although genetically encoded reporters are restricted to preclinical studies, fluorescence imaging methods are advancing to clinical medicine for applications that include image-guided surgery and endoscopy, and initial studies show that FLIM is feasible in patients (37). Therefore, FLIM-based analysis techniques can potentially advance understanding of cancer, normal physiology, and other diseases in preclinical model systems and may be translated to focused clinical applications in image-guided detection and therapy.
As 1 application of the apoptosis reporter and FLIM technology, we investigated induction of apoptosis in response to metabolic stresses. Malignant cells reprogram metabolic pathways to survive and proliferate in different tumor environments, creating potential vulnerabilities for targeted therapy (38). Most types of cancer cells uptake extracellular glucose to a greater extent than normal tissues and preferentially utilize glycolysis even under aerobic conditions, a phenomenon known as the Warburg effect (39, 40). Consistent with past observations about the dependence of cancer cells on glycolysis, we demonstrated that culturing breast cancer cells without glucose significantly increased apoptosis. Although glutamine is also a major metabolic building block for cancer cells (41), cells grown without this amino acid showed only basal levels of apoptosis. However, depriving both glucose and glutamine synergistically increased apoptosis in breast cancer cells, suggesting that targeting multiple pathways is necessary to overcome metabolic plasticity of malignant cells. We also showed that treatment with the glycolysis inhibitor DCA plus glutamine deprivation induced apoptosis and decreased both glycolytic and oxidative metabolism. Because glutamine serves as a precursor of α-ketoglutarate, a metabolite that eliminates ROS, deprivation of glutamine could enhance the apoptotic effect of DCA-driven ROS (42). These data further support ongoing efforts to block both glycolysis and glutaminolysis for cancer therapy and highlight uses of imaging for identifying effective treatments targeting cancer cell metabolism (43).
In addition to metabolic stress, we used this imaging method to analyze the effects of bone marrow stromal cells on the resistance of cancer cells to chemotherapy, a critical determinant of treatment failure in breast cancer and many other malignancies (44). HS5 and HS27A bone marrow stromal cells, originally isolated and immortalized from the same normal volunteer, represent functionally distinct subpopulations of mesenchymal stromal cells regulating stem cell niches (45). These cell lines show distinct patterns of cytokine secretion with HS5 cells producing molecules that promote differentiation of hematopoietic stem and progenitor cells, whereas HS27A cells secrete cytokines typically associated with maintaining undifferentiated stem cells (27, 46). Because metastatic cancer cells may displace normal hematopoietic stem and progenitor cells from niche environments, HS5 and HS27A cells model different potential sites for cancer cells in bone marrow (47, 48). Notably, we discovered that trametinib, a clinically approved MEK inhibitor, produced significantly greater apoptosis of MDA-MB-231 cells in spheroids with HS27A relative to HS5 cells. We are currently investigating mechanisms underlying the differences in drug resistance between these 2 bone marrow stromal cell environments. These data emphasize the effects of different bone marrow stromal cell environments on treatment efficacy and the utility of analyzing these effects with single-cell FLIM in 3D environments.
Overall, this research demonstrates the capabilities of FLIM in analyzing apoptosis at single-cell resolution using the same FRET reporter across standard 2D cultures, engineered 3D coculture environments, and living animal models. We demonstrated applications of this imaging approach to establish apoptosis under conditions of metabolic stress and stromal-mediated drug resistance in breast cancer, but the technology could be applied readily to quantify apoptosis in other cancers or disease processes. More generally, we expect FLIM with phasor analysis will enable investigators to transition single-cell FRET microscopy from 2D cultures to more complex environments, advancing understanding of disease processes and therapy in vivo.
Acknowledgments
This research was supported by National Institutes of Health grants R01CA170198 and 1S10RR028819. We thank Vladislav Verkhusha for the LSS-mOrange-mKate2 caspase-3 reporter plasmid.
Footnotes
- Bax
- Bcl-2-associated X
- DCA
- dichloroacetate
- DMEM
- Dulbecco's modified Eagle's medium
- ECAR
- extracellular acidification rate
- FBS
- fetal bovine serum
- FLIM
- fluorescence lifetime imaging microscopy
- FRET
- fluorescence resonance energy transfer
- LSS
- large Stokes shift
- MEK
- mouse embryonic kidney
- OCR
- oxygen consumption rate
- PCR
- polymerase chain reaction
- ROS
- reactive oxygen species
- ROI
- region of interest
References
- 1. Ouyang L, Shi Z, Zhao S, Wang F, Zhou T, Liu B, et al. : Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45(6):487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg R. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. [DOI] [PubMed] [Google Scholar]
- 3. Wong R. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coppola J, Ross B, Rehemtulla A. Noninvasive imaging of apoptosis and its application in cancer therapeutics. Clin Cancer Res. 2008;14(8):2492–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ye D, Shuhendler A, Pandit P, Brewer K, Tee S, Cui L, Tikhomirov G, Rutt B, Rao J. Caspase-responsive smart gadolinium-based contrast agent for magnetic resonance imaging of drug-induced apoptosis. Chem Sci. 2014;4(10):3845–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang Q, Cui H, Cai S, Yang X, Forrest M. In vivo photoacoustic imaging of chemotherapy-induced apoptosis in squamous cell carcinoma using a near-infrared caspase-9 probe. J Biomed Opt. 2011;16(11):116026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Witney T, Hoehne A, Reeves R, Ilovich O, Namavari M, Shen B, Chin FT, Rao J, Gambhir SS. A systematic comparison of 18F-C-SNAT to established radiotracer imaging agents for the detection of tumor response to treatment. Clin Cancer Res. 2015;21(17):3896–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dexter D, Leith J. Tumor heterogeneity and drug resistance. J Clin Oncol. 1986;4(2):244–257. [DOI] [PubMed] [Google Scholar]
- 9. Tyas L, Brophy V, Pope A, Rivett A, Tavare J. Rapid caspase-3 activation during apoptosis revealed using fluorescence-resonance energy transfer. EMBO Rep. 2000;1(3):266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamaguchi Y, Shinotsuka N, Nonomura K, Takemoto K, Kuida K, Yoshida H, Miura M. Live imaging of apoptosis in a novel transgenic mouse highlights its role in neural tube closure. J Cell Biol. 2011;195(6):1047–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garrod K, Moreau H, Garcia Z, Lemaitre F, Bouveri I, Albert ML, Bousso P. Dissecting T cell contraction in vivo using a genetically encoded reporter of apoptosis. Cell Rep. 2012;2(5):1438–1447. [DOI] [PubMed] [Google Scholar]
- 12. Jacques S. Optical properties of biological tissues: a review. Phys Med Biol. 2013;58(11):R37–R61. [DOI] [PubMed] [Google Scholar]
- 13. Shcherbakova D, Hink M, Joosen L, Gadella T, Verkhusha V. An orange fluorescent protein with a large Stokes shift for single-excitation multicolor FCCS and FRET imaging. J Am Chem Soc. 2012;134(18):7913–7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun Y, Day R, Periasamy A. Investigating protein-protein interactions in living cells using fluorescence lifetime imaging microscopy. Nat Protoc. 2011;6(9):1324–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith MC, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, Luker GD. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64(23):8604–8612. [DOI] [PubMed] [Google Scholar]
- 16. Luker KE, Mihalko LA, Schmidt BT, Lewin SA, Ray P, Shcherbo D, Chudakov DM, Luker GD. In vivo imaging of ligand receptor binding with Gaussia luciferase complementation. Nat Med. 2012;18(1):172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, Jakubosky D, Genovese G, Muller FL, Jeong JH, Bender RP, Chu GC, Flaherty KT, Wargo JA, Collins JJ, Chin L. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18(10):1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salomonnson E, Mihalko L, Verkhusha V, Luker K, Luker G. Cell-based and in vivo spectral analysis of fluorescent proteins for multiphoton microscopy. J Biomed Opt. 2012;17(9):96001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Digman M, Caiolfa V, Zamai M, Gratton E. The phasor approach to fluorescence lifetime imaging analysis. Biophys J. 2008;94(2):L14–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feng G, Kaplowitz N. Mechanism of staurosporine-induced apoptosis in murine hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2002;282(5):G825–34. [DOI] [PubMed] [Google Scholar]
- 21. Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2(31):31ra24. [DOI] [PubMed] [Google Scholar]
- 22. Suva L, Griffin R, Makhoul I. Mechanisms of bone metastases of breast cancer. Endocr Relat Cancer. 2009;16(3):703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheung W-C, Van Ness B. The bone marrow stromal microenvironment influences myeloma therapeutic response in vitro. Leukemia. 2001;15(2):264–271. [DOI] [PubMed] [Google Scholar]
- 24. Quail D, Joyce J. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. James S, Fox J, Afsari F, Lee J, Clough S, Knight C, Ashmore J, Ashton P, Preham O, Hoogduijn M, Ponzoni Rde A, Hancock Y, Coles M, Genever P. Multiparameter analysis of human bone marrow stromal cells identifies distinct immunomodulatory and differentiation-competent subtypes. Stem Cell Reports. 2015;4(6):1004–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bernad A, Kopf M, Kulbacki R, Welch N, Koehler G, Gutierrez-Ramos J. Interleukin-6 is required in vivo for the regulation of stem cells and committed progenitors of the hematopoietic system. Immunity. 1994;1(9):725–731. [DOI] [PubMed] [Google Scholar]
- 27. Graf L, Iwata M, Torok-Storb B. Gene expression profiling of the functionally distinct human bone marrow stromal cell lines HS-5 and HS-27a. Blood. 2002;100(4):1509–1511. [DOI] [PubMed] [Google Scholar]
- 28. Milross C, Mason K, Hunter N, Chung W, Peters L, Milas L. Relationship of mitotic arrest and apoptosis to antitumor effect of paclitaxel. J Natl Cancer Inst. 1996;88(18):1308–1314. [DOI] [PubMed] [Google Scholar]
- 29. Aoki K, Kamioka Y, Matsuda M. Fluorescence resonance energy transfer imaging of cell signaling from in vitro to in vivo: basis of biosensor construction, live imaging, and image processing. Develop Growth Differ. 2013;55:515–522. [DOI] [PubMed] [Google Scholar]
- 30. Atkin S, Patel S, Kocharyan A, Holtzclaw L, Weerth S, Schram V, Pickel J, Russell JT. Transgenic mice expressing a cameleon fluorescent Ca2+ indicator in astrocytes and Schwann cells allow study of glial cell Ca2+ signals in situ and in vivo. J Neurosci Methods. 2009;181:212–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsujino N, Yamanaka A, Ichiki K, Muraki Y, Kilduff T, Yagami K, Takahashi S, Goto K, Sakurai T. Cholecystokinin activates orexin/hypocretin neurons through the cholecystokinin A receptor. J Neurosci. 2005;25:7459–7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kamioka Y, Sumiyama K, Mizuno R, Sakai Y, Hirata E, Kiyokawa E, Matsuda M. Live imaging of protein kinase activities in transgenic mice expressing FRET biosensors. Cell Struct Funct. 2012;37(1):65–73. [DOI] [PubMed] [Google Scholar]
- 33. Rhee S, Jiang H, Ho C-H, Grinnell F. Microtubule function in fibroblast spreading is modulated according to the tension state of cell–matrix interactions. Proc Natl Acad Sci U S A. 2007;104(13):5425–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pick M, Ries C. Comparison of 3D and 2D tumor models reveals enhanced HER2 activation in 3D associated with an increased response to trastuzumab. Oncogene. 2009;28:461–468. [DOI] [PubMed] [Google Scholar]
- 35. Fallica B, Maffei J, Villa S, Makin G, Zaman M. Alteration of cellular behavior and response to PI3K pathway inhibition by culture in 3D collagen gels. PloS One. 2012;7(10):e48024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805(1):105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sun Y, Hatami N, Yee M, Phipps J, Elson D, Gorin F, Schrot RJ, Marcu L. Fluorescence lifetime imaging microscopy for brain tumor image-guided surgery. J Biomed Opt. 2010;15(5):0566022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Keijer J, van Dartel D. Reprogrammed metabolism of cancer cells as a potential therapeutic target. Curr Pharm Des. 2014;20(15):2580–2594. [DOI] [PubMed] [Google Scholar]
- 39. Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. [DOI] [PubMed] [Google Scholar]
- 40. Vander Heiden M, Cantley L, Thompson C. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Nat Rev Drug Discov. 2009;10:671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DeBerardinis R, Cheng T. Q's next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dai Y, Xiong X, Huang G, Liu J, Sheng S, Wang H, Qin W. Dichloroacetate enhances adriamycin-induced hepatoma cell toxicity in vitro and in vivo by increasing reactive oxygen species levels. PLoS One. 2014;9(4):e92962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ramsay E, Hogg P, Dikla P. Mitochondrial metabolism inhibitors for cancer therapy. Pharm Res. 2011;28(11):2731–2744. [DOI] [PubMed] [Google Scholar]
- 44. Meads M, Hazlehurst L, Dalton W. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519–2526. [DOI] [PubMed] [Google Scholar]
- 45. Roecklein B, Torok-Storb B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood. 1995;85:997–1005. [PubMed] [Google Scholar]
- 46. Iwata M, Sandstrom R, Delrow J, Stamatoyannopoulos J, Torok-Storb B. Functionally and phenotypically distinct subpopulations of marrow stromal cells are fibroblast in origin and induce different fates in peripheral blood monocytes. Stem Cells Dev. 2014;23(7):729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, Means TK, Luster AD, Scadden DT, Lin CP. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435(7044):969–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shiozawa Y, Pedersen E, Havens A, Jung Y, Mishra A, Joseph J, Kim JK, Patel LR, Ying C, Ziegler AM, Pienta MJ, Song J, Wang J, Loberg RD, Krebsbach PH, Pienta KJ, Taichman RS. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121(4):1298–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]