

Abstract
The Wnt signaling pathway plays a key role in regulation of organ development and tissue homeostasis. Dysregulated Wnt activity is one of the major underlying mechanisms responsible for many diseases including cancer. We previously reported the FDA-approved anthelmintic drug Niclosamide inhibits Wnt/β-catenin signaling and suppresses colon cancer cell growth in vitro and in vivo. Niclosamide is a multi-functional drug that possesses important biological activity in addition to inhibition of Wnt/β-catenin signaling. Here, we studied the SAR of Wnt signaling inhibition in the anilide and salicylamide region of Niclosamide. We found that the 4′-nitro substituent can be effectively replaced by trifluoromethyl or chlorine and that the potency of inhibition was dependent on the substitution pattern in the anilide ring. Non-anilide, N-methyl amides and reverse amide derivatives lost significant potency, while acylated salicylamide derivatives inhibited signaling with potency similar to non-acyl derivatives. Niclosamide's low systemic exposure when dosed orally may hinder its use to treat systemic disease. To overcome this limitation we identified an acyl derivative of Niclosamide, DK-520 (compound 32), that significantly increased both the plasma concentration and the duration of exposure of Niclosamide when dosed orally. The studies herein provide a medicinal chemical foundation to improve the pharmacokinetic exposure of Niclosamide and Wnt-signaling inhibitors based on the Niclosamide chemotype. The identification of novel derivatives of Niclosamide that metabolize to Niclosamide and increase its drug exposure may provide important research tools for in vivo studies and provide drug candidates for treating cancers with dysregulated Wnt signaling including drug-resistant cancers. Moreover, since Niclosamide is a multifunctional drug, new research tools such as DK520 could directly result in novel treatments against bacterial and viral infection, lupus, and metabolic diseases such as type II diabetes, NASH and NAFLD.
Keywords: Niclosamide, Wnt signaling inhibitor, Small molecule, β-Catenin, Plasma concentration, Cancer, Disease treatment
1. Introduction
The Wnt signaling pathway plays a key role in tissue development and homeostasis and is dysregulated in many diseases.1–3 For example in colorectal cancer (CRC) more than 80% of all sporadic and hereditary cancers show hyperactivation of the pathway due to mutations in the adenomatous polyposis coli (APC) or the β-catenin gene.1,2 Given the importance of the Wnt signaling activity underlying tumor formation and metastasis, therapies against the Wnt signaling pathway are highly sought.1
A mechanistic understanding of the canonical Wnt/β-catenin signaling pathway has been studied over the past decades.4 Briefly, Wnt proteins are secreted glycoproteins that bind and activate the seven transmembrane receptor Frizzled and single trans-membrane receptors LRP5/6. Wnt binding to Frizzled and LRP5/6 results in activation of cytosolic proteins called Dishevelled (Dvl), leading to internalization of the Frizzled receptor.5 Downstream signaling events resulting from Wnt binding include the stabilization and translocation of cytosolic β-catenin proteins into the nucleus, activation of the transcription factor LEF/TCF and transcription of Wnt/β-catenin target genes. Overall, the Wnt signal transduction pathway consists of many protein–protein interactions that traditionally have been difficult to target with small drug-like molecules.1,6
Recent reports, including our own, have begun to provide insights into mechanisms and chemical starting points for inhibiting the Wnt pathway,7–13 though no drugs designed to target the pathway have been approved to date.13 Through a high-through-put drug screen we found Niclosamide (Fig. 1A), a drug approved by the FDA for human use as an anthelminthic therapy, promotes Frizzled internalization.7 Subsequent studies found Niclosamide downregulates Dishevelled and β-catenin and inhibits colon cancer cell growth in vitro and in vivo.7,9 Subsequently, other laboratories have confirmed that Niclosamide inhibits Wnt signaling.14–16 Our initial SAR studies indicated that inhibition of Wnt signaling by Niclosamide appeared unique among the structurally-related salicylanilide class of anthelmintic agents tested, and that the potency and Wnt/β-catenin functional response was dependent on small changes in the chemical structure of Niclosamide.8 In addition to our SAR studies, a limited SAR study of Niclosamide-mediated inhibition of S100A, a transcriptional target gene of β-catenin, was reported. Of the six derivatives of Niclosamide tested at 1 μM, none reduced expression of S100A.14
Figure 1.
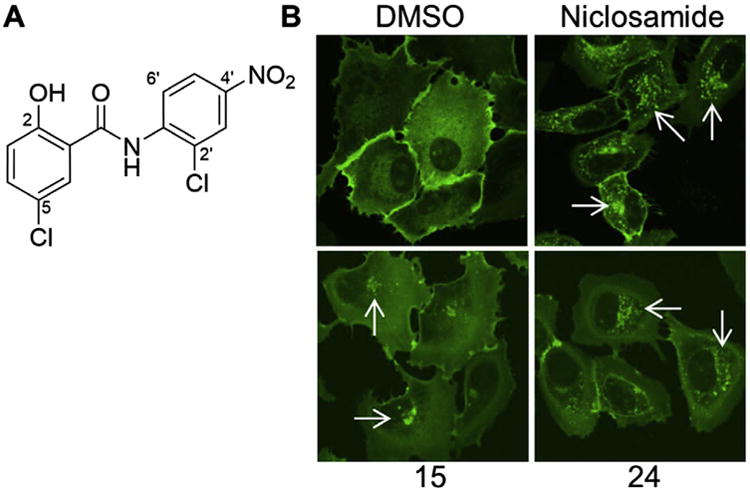
Frizzled1-GFP internalization by Niclosamide and related derivatives. (A) Structure of Niclosamide (B) confocal images of U2OS cells harboring Frizzled1-GFP treated with DMSO control, Niclosamide, compound 15, compound 24 for 6 h at 12.5 μM. Punctuate structures (white arrows) highlight internalized Frizzled1-GFP vesicles. Compounds 15 and 24 provided as representative examples of scoring. Scoring: DMSO control = 0; Niclosamide = 5; compound 15 = 1, and compound 24 = 3. See Table 1 for structures of 15 and 24.
Niclosamide is a multi-functional drug. It was used initially as an anthelmintic agent in livestock in the early 1960s before being approved by the FDA for use in humans in 1982 to treat tapeworm infections.17,18 Niclosamide is included in the World Health Organization's list of essential medicines19 and has been used to safely treat millions of patients. For such a widely used drug, its mechanism of action has not been well-delineated, although it has been reported to involve uncoupling of oxidative phosphorylation.20–22 In the years since its initial discovery, additional biological activities of Niclosamide have been identified that have served to generate considerable interest for its use to treat other human diseases. Among the additional biological activities discovered are antibacterial activity23 including antituberculous activity24 and drug resistant Staphylococcus aureus activity,25 antiviral activity,26,27 and anti-trypanosomal activity.28 Niclosamide has protonophore activity and has effects on metabolism, including activation of AMPK.26,29–31 In animal models of type 2 diabetes, Niclosamide improves glycemic control and delays disease progression reportedly through uncoupling of mitochondria oxidative phosphorylation.30,32 In recent years, Niclosamide has generated significant interest as a potential anticancer agent.9,14,16,33–39 Niclosamide inhibits the proliferation of tumor cell lines from multiple tumor types, for example, breast, colon, lung, prostate, ovary, blood and pancreas, over an IC50 range of 0.13–4 μM that overlaps with the IC50 of inhibition Wnt/β-catenin signaling,33 and has anti-cancer activity in drug resistant cancers.9,38,40 Of particular interest to cancer therapy, Niclosamide has been recently reported to inhibit key oncogenic signaling pathways33 in addition to Wnt,7,9,14,15 such as Notch,41 mTOR,29 NF-kB,34 and STAT-3.42
The increased interest in Niclosamide's inhibitory activity against key pathological signaling pathways has stimulated a search to improve its delivery and drug exposure to treat systemic disease. Whereas the pharmacokinetic properties of Niclosamide are appropriate for use in the gut as an anthelmintic agent, its low solubility, low bioavailability and poor pharmacokinetic profile results in low plasma exposure when dosed orally.9,18,27,43–45 Efforts to improve the solubility include the preparation of salt forms and derivatives containing water-solubilizing groups.18,46,47 In the search for STAT-3 inhibitors with improved solubility to treat cancer, amine water-solubilizing groups were added to the Niclosamide molecule.48 More recently, efforts to identify nanoparticle formulations for use in cancer have been reported.44,49 In one study in which the pharmacokinetic parameters of nanocrystals of Niclosamide were evaluated in vivo, the nanocrystal formulation did not significantly change the plasma concentration vs time profile when administered IV to rat, though increased tissue concentration at 2 h was noted.44 To our knowledge, no formulation of Niclosamide has been reported that increases its plasma concentration or the duration of exposure.
As part of an effort to discover Wnt/β-catenin inhibitors based on the Niclosamide chemotype with improved potency, selectivity and pharmacokinetic properties for clinical evaluation, we continued our study of the structure–activity relationships of Niclosamide by focusing on the anilide and salicylamide regions of the molecule. Consistent with our earlier report, the SAR of inhibition of Wnt signaling was dependent on both the nature and the location of substitution.8 In particular we found that chloro or trifluoromethyl substituents can replace the nitro substituent, a major site of metabolism, and retain Wnt/β-catenin inhibitory activity, and found modifications that can significantly increase both the plasma concentration and the duration of exposure of Niclosamide in mice. Whereas dosing mice with Niclosamide results in low plasma concentrations and a short duration of exposure,9 DK-520 (compound 32), a derivative of Niclosamide had the unique property that it increased both the plasma concentration and the duration of exposure of Niclosamide in vivo with no observable adverse effects over three weeks of oral dosing. The ability of DK-520 to metabolize to Niclosamide in a manner that increases the exposure of Niclosamide when dosed orally may provide a valuable preclinical research tool to study Niclosamide in vivo in animal models of disease indicated by its known biological activity.
2. Material and methods
2.1. Compound synthesis
Anilide synthesis. General method. To a 100 mL flask equipped with a reflux condenser was added 5-chloro-2-hydroxybenzoic acid (1 equiv), the aniline derivative (1 equiv), and dry xylenes (stored over 3A molecular sieves, 40 mL per gram of 5-chloro-2-hydroxybenzoic acid) under an Argon atmosphere. The mixture was heated to reflux, and PCl3 (0.4 equiv) was added rapidly via syringe. The mixture was heated at reflux for 1 h and cooled to room temperature. Water (40 mL per gram of 5-chloro-2-hydroxybenzoic acid) was added and the resultant heterogeneous mixture stirred rapidly for 1 h. Saturated sodium bicarbonate was added to a final pH of 3–4, and the mixture stirred rapidly overnight. The solids were filtered and washed sequentially with water, toluene and hexane. Samples were analyzed by NMR, HPLC/mass spectrometry and TLC. Purification by crystallization or column chromatography on silica gel was performed when purity was less than 95% by LC. Additional experimental procedures and analytical data are provided in Supplemental data.
2.2. Frizzled internalization assay
The Fzd1-GFP assay was performed following a procedure similar to previously published work.7 Briefly, cells stably expressing Frizzled1-GFP plated in confocal dishes were treated, after 24 h, with 12.5 μM of test compound or DMSO for 6 h at 37°C followed by fixation with 4% paraformaldehyde. The cells were then examined by microscopy using a LSM 510-Meta confocal microscope (Carl Zeiss, Thornwood, NY, USA) equipped with 40× and 100× apo chromat objectives. YFP was excited using a 488-nm argon laser line. Images were processed using the LSM software Image Browser (CarlZeiss, Thornwood, NY, USA). Plates were read twice in blinded fashion using a 0–5 point scale. aPunctate similar to control = 0, trace amount of punctate greater than control = 1, moderate = 3, strong = 5.
2.3. TOPFlash reporter assay
Wnt-3A conditioned medium was prepared using L WNT-3A cells (ATCC® CRL-2647™) purchased from ATCC. Conditioned medium was obtained following published protocols (http://www.atcc.org/Products/All/CRL-2647.aspx#culturemethod).7 HEK293 cells were stably transfected with p8xTOPFlash, Renilla luciferase plasmid pRL-TK (Promega), and pLKO.1 as previously published. Briefly, stably transfected cells were seeded in 100 μl of cell growth medium/well in 96-well plates at 100% confluency. Fifty microliters of Wnt-3A conditioned medium containing the chemical compounds to be tested or DMSO was added to each well. After an 8 h treatment, the cells were washed once with PBS and lysed with 55 μl of Passive Lysis Buffer supplied in the Dual-Luciferase Reporter Assay kit (Promega, Madison, WI). Twenty-five microliters of cell lysate was used for measuring luciferase activity in a 96-well plate reader (FluoStar Optima, BMG Labtech, Chicago, IL).
2.4. Western blot
Western blots were performed following a procedure similar to previously published work.9 Briefly, HCT-116 cells were grown to about 80% confluency in poly-d-lysine coated six-well plates 48 h before treatment and followed by 2.0 μM compounds or DMSO incubation for 18 h in growth medium. After treatment, the cytosolic fraction was isolated as described.9 Immunoblots using antibodies to β-catenin (E-5, Santa Cruz Biotechnology catalog number sc-7963) was used to detect β-catenin protein levels in the cytosol, and immunoblots using antibodies to β-actin (C-4, Santa Cruz Biotechnology, catalog number SC-47778) was used for loading controls.
2.5. Cell proliferation assay
The colon cancer cell line HCT116 was used in the cell proliferation assay. The cells were plated in 100 μl of growing medium/well in 96-well plates at 5000 cells per well and treated with compounds from 0.04 to 10 μM for 72 h, after which point the cells were analyzed by colorimetric MTS assay (Promega, Madison, WI, USA).
2.6. Pharmacokinetic analysis
For Niclosamide dosed IV, Niclosamide was dissolved in a solvent of 67% polyethylene glycol 400 and 33% N,N-dimethylacetamide at a concentration of 3.27 mg/ml and tail vein injected at a dose of 2.6 mg/kg of body weight. Blood samples were obtained at predose and at 0.08, 0.17, 0.33, 0.67, 1.5, 4, 8, 12 h after drug administration. For compound 34 dosed orally, compound 34 was dissolved in a solvent of 90% polyethylene glycol 300 and 10% 1-methyl-2-pyrrolidinone at a concentration of 20 mg/ml and gavaged at a dose of 200 mg/kg of body weight. Blood samples were obtained at predose and at 0.25, 0.5, 1, 2, 4, 8 h after drug administration. For DK-520 (compound 32) dosed orally, DK-520 was dissolved in corn oil at a concentration of 20 mg/ml and dosed at 200 mg/kg of body weight. Blood samples were obtained at predose and at 0.25, 0.5, 0.75, 1, 1.5, 4, 8, 12, 24, 48, and 72 h after drug administration. All the solvent reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). CD1 mice were used in the pharmacokinetic studies. Quantification of Niclosamide in mouse plasma was done by LC/MS–MS using methods similar to those previously published.9
2.7. In vivo toxicity study
NOD/SCID mice received oral administration of vehicle (corn oil) or DK-520 (32) (200 mg/kg in corn oil) 6 days a week for 3 weeks. The animals were observed throughout the period for any side effect. The body weight was measured the same day of the first dosing (week 0) and the day after the last dosing (week 3).
3. Results
In our previous studies and the studies herein, we relied on the Fzd1-GFP confocal microscopy (Fig. 1) and the Wnt-stimulated TOPFlash assays to interrogate the SAR of Niclosamide and related derivatives.8 Our previous SAR studies indicted the 4′-position of the nitro anilide portion of Niclosamide had a significant impact on Wnt-mediated biological activity and that modification at this position could produce molecules that either increased or decreased Wnt3A-mediated signaling.8 Given that nitro substituents often impart poor solubility and poor pharmacokinetic properties, and that reduction of the nitro group is a major metabolite of Niclosamide18, we interrogated the SAR in the anilide to identify inhibitors of Wnt signaling with improved drug properties.
The SAR of the anilide region could be readily interrogated by the synthesis of derivatives prepared via the coupling of substituted anilines with 5-chloro-2-hydroxybenzoic acid (Scheme 1).
Scheme 1.

Synthesis of anilide derivatives.
Preparation of Niclosamide derivatives in which the nitro group was replaced with bioisosteric groups such as acetoyl and benzoyl (compound 4, 7), methyl sulphone (compound 6) or amide (compounds 5, 8) resulted in a significant loss of activity in both the Fzd1-GFP internalization and Wnt-3A-stimulated TOPFlash assay (Table 1, Fig. S1, Supplemental information). We then explored substituents that mimicked the electron-withdrawing characteristics of the nitro group. Substitution of the 4′-NO2 group with a 4′-CF3 group (compounds 9 and 10) produced compounds that internalized Fzd1-GFP and inhibited Wnt3A-stimulated signaling in the TOPFlash assay with potency similar to Niclosamide and had better solubility in organic solvents. Moving the CF3 substituent to the 3′-position (compound 11) led to a loss of activity, similar to previous findings with 3′-NO2 substitution.8 In contrast to compounds with 4′-NO2 substitution (Niclosamide 1 and compound 3), compounds with 4′-CF3 substitution (compounds 9 and 10) containing a 2′-Cl substituent, vs a 2′-H substituent had comparable potency. Replacement of 2′-Cl with 2′-F in the 4′-CF3 series (compound 12) resulted in similar activity to compounds with 2′-Cl, and 2′-H substitution (compounds 9, 10). Substitution of the 4′-NO2 group in Niclosamide with a 4′-Cl group (compound 16) resulted in a compound that internalized Fzd-1-GFP and inhibited Wnt3A-stimulated signaling with potency comparable to Niclosamide. The potency of compounds with two chlorine substituents was influenced by the substitution pattern. Compound 16 in which the anilide was substituted with chlorine at the 2′- and 4′-postion had potency similar to Niclosamide. Compounds with 2′,5′-dichloro substitution (compound 17), 3′,5′-dichloro substitution (compound 18) and 3′,4′-dichloro substitution (compound 19) were generally less potent, while 2′,6′ -dichloro substitution (compound 20) produced a compound that was inactive in both assays up 12 μM, the highest concentration tested. Dichloro derivatives tended to be more potent than mono-chloro derivatives (compounds 13, 14, 15). Compounds in which a chlorine atom was replaced with a fluorine atom produced compounds that were less potent than the similarly substituted chloro derivative (compounds 21, 22, 24, 25). Here also, the 2′,6′-difluoro substituted anilide derivative (compound 23) was not active up to 12 μM, supporting the notion that the substitution pattern of halogen in the anilide ring is important for potency. Whereas these studies did not identify molecules more potent than Niclosamide, they did identify compounds with similar potency and better overall handling and solubility properties than Niclosamide in solvents used in synthesis and purification. In particular, compounds in which the nitro substituent was replaced with a trifluoromethyl group (compounds 9, 10) or a chlorine group (compound 16) were similar in potency to Niclosamide in both the FZD1-GFP internalization and the Wnt3A-stimulated β-catenin TOPFlash transcription assays (Table 1 and Fig. 2A). The IC50 of inhibition in the TOPFlash assays for Niclosamide and compounds 9, 10 and 16 are 0.34 ± 0.08, 0.56 ± 0.21, 0.29 ± 0.06, and 0.42 ± 0.1 μM, respectively.
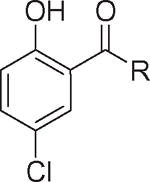
Table 1. Structure–activity relationships in the anilide ring.
| |||||
|---|---|---|---|---|---|
|
| |||||
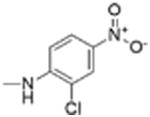
| Compound number | Structure R | Fzd1-GFP Internalization @12.5 μMa | Inhibition Wnt/β-catenin transcription TopFlash IC50 (μM) ± SE | ClogP | pKa NHb |
| 1 |
|
5 | 0.34 ± 0.08 | 3.1 | 11.2 |
| 2 |
|
0 | 11.81 | 3.3 | 13.2 |
| 3 |
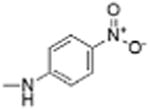
|
5 | 0.99 ± 0.32 | 2.7 | 12.1 |
| 4 |
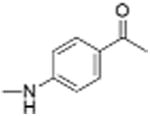
|
0 | 6.42 ± 0.51 | 3.0 | 12.5 |
| 5 |
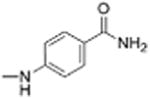
|
0 | >12 | 1.9 | 12.7 |
| 6 |
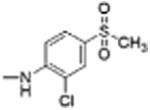
|
0 | 7.66 | 2.6 | 11.4 |
| 7 |
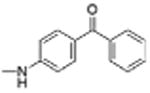
|
0 | 15.04 | 4.2 | 12.5 |
| 8 |
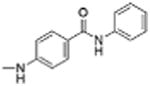
|
0 | 32.89 | 4.1 | 12.6 |
| 9 |
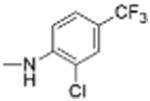
|
5 | 0.56 ± 0.21 | 4.8 | 11.9 |
| 10 |
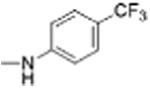
|
5 | 0.29 ± 0.06 | 4.5 | 12.4 |
| 11 |
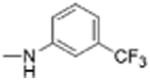
|
5 | 0.89 ± 0.32 | 4.7 | 12.5 |
| 12 |
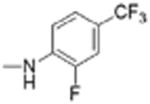
|
5 | 0.55 ± 0.11 | 4.5 | 11.8 |
| 13 |
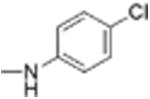
|
3 | 1.30 ± 0.18 | 4.0 | 12.8 |
| 14 |
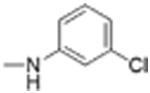
|
5 | 1.65 ±0.12 | 4.0 | 12.6 |
| 15 |
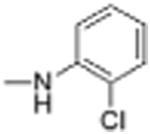
|
1 | 3.05 ± 0.87 | 3.9 | 12.3 |
| 16 |
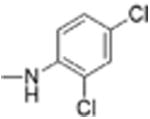
|
5 | 0.42 ± 0.10 | 4.5 | 11.9 |
| 17 |
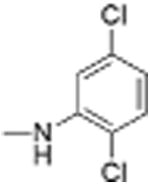
|
5 | 0.75 ± 0.13 | 4.4 | 11.7 |
| 18 |
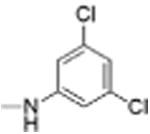
|
5 | 0.70 ± 0.13 | 4.5 | 12.0 |
| 19 |
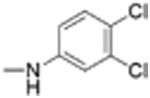
|
5 | 0.73 ± 0.13 | 4.5 | 12.2 |
| 20 |
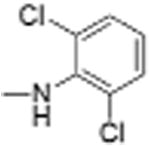
|
0 | >12 | 4.3 | 11.4 |
| 21 |
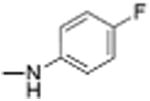
|
0 | 3.84 ± 0.29 | 3.9 | 13.1 |
| 22 |
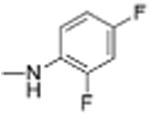
|
2 | 4.02 ± 0.79 | 4.0 | 12.6 |
| 23 |
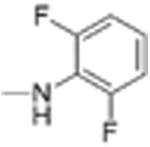
|
ND | >12 | 3.9 | 12.1 |
| 24 |
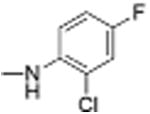
|
3 | 1.41 ± 0.21 | 4.4 | 12.2 |
| 25 |
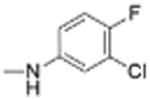
|
5 | 1.21 ± 0.17 | 4.4 | 12.5 |
Internalization of Frizzled1-GFP stably expressed in U2OS cells was determined by confocal microscopy. Cells were treated with compounds for 6 h, fixed, and scored visually by the amount of punctate observed versus DMSO control. Inhibition of Wnt3A-stimulated Wnt/β-catenin transcription was determined by TOPFlash normalized to renilla control. ClogP and pKa values were calculated in Maestro 9.3 (Schrodinger, Inc.) using Qikprops and Epik.
Punctate similar to control = 0, trace amount of punctate greater than control = 1, moderate = 3, strong = 5.
Calculated pKa values in water are reported. The calculated value of the pKa of the –OH group is 6.8 in each example listed. The calculated pKa of the NH is the salicylamide NH without ionization of the –OH group. The estimated error of the pKa calculation for the OH group is ±1 log unit, and ±2 for the NH group. ND: not determined.
Figure 2.
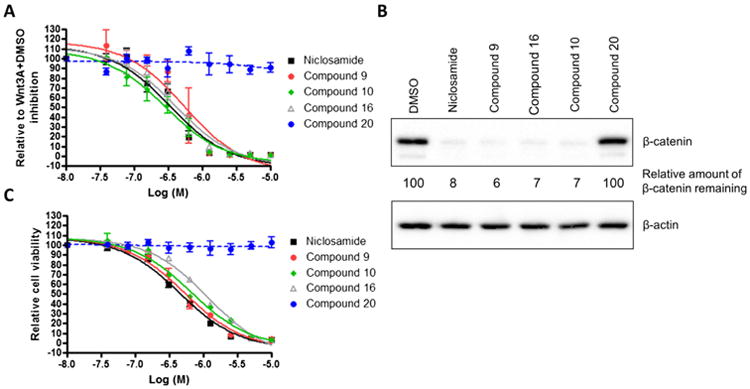
Niclosamide and its derivatives inhibit Wnt3A/β-catenin signaling and proliferation of HCT-116 cells. (A) Inhibition of Wnt signaling in TOPFlash assay. HEK293 cells stably expressing TOPFlash luciferase reporter and Renilla luciferase were treated with Wnt3A-conditioned medium in the presence of DMSO or compounds from 0.04 to 10 μM. The TOPFlash reporter activity of Wnt3A with DMSO treatment was set as 100%. Data were fit using GraphPad Prism (mean ± SEM, n ≥2). (B) Reduction of cytosolic β-catenin levels. HCT-116 cells were treated with 2.0 μM compounds for 18 h, lysed in hypotonic buffer. Lysates were eluted by SDS electrophoresis on acrylamide, transferred, and probed with antibodies to β-catenin as described in Section 2. β-Actin was used as a loading control. (C) Inhibition of proliferation of colon cancer cell HCT116. Cells were treated with DMSO or compounds from 0.04 to 10 μM for 72 h, and cell viability was measured using the MTS assay. The cell viability with DMSO treatment was set at 100%. Data were fit using GraphPad Prism (mean ± SEM, n = 3).
To further characterize the Wnt inhibitory and the cancer cell anti-proliferation activity of these compounds, we evaluated these compounds along with an inactive derivative (compound 20, Table 1) in HCT-116 colorectal cancer cell culture. Inhibition of Wnt signaling was determined by analyzing the reduction of cytosolic β-catenin by Western blot (Fig. 2B). Upon treating cells with 2.0 μM of compound for 18 h in culture, β-catenin levels were significantly decreased only by the compounds that showed activity in the FZD1-GFP and Wnt-3A stimulated TOPFlash assay. The amount of cytosolic β-catenin remaining relative to DMSO control without compound was: Niclosamide: 8%; compound 9: 6%; compound 10: 7%; and compound 16: 7%. The amount of cytosolic β-catenin remaining for the inactive derivative, compound 20, equaled 100% of the amount of the DMSO control. Anti-proliferation activity was also measured by MTS assay (Fig. 2C). In this assay, the inactive derivative, compound 20, had no effect up to 10 μM. In contrast, the compounds that were active in the FZD1-GFP internalization assay, the Wnt3A-stimulated TOPFlash assay and the β-catenin Western blot assay were also active in the MTS assay. The IC50 of inhibition of the active compounds, Niclosamide, compounds 9, 10 and 16 are 0.45 ± 0.05, 0.54 ± 0.08, 0.67 ± 0.09 and 1.18 ± 0.14 μM, respectively.
Given the site of the major metabolite of Niclosamide had been removed in compound 9 and 16, and the fact that Niclosamide was efficacious in xenograft models despite exposure well below the IC50 of inhibition of Wnt signaling in cell culture during the majority of the dosing period,9 we next asked whether these compounds would have better exposure that could improve the anti-tumor responses in vivo. Upon searching the literature we found that the PK properties for both compounds in rat had been reported.27 In these studies, the authors report that the bioavailability of compound 9 and 16 was similar to Niclosamide (%F = 15, 12 and 10, respectively). In each molecule, compound 9, 16, and Niclosamide, the clearance was moderate (39, 27, and 20 mL/Kg/min, respectively), the Volumes of Distribution were low (Vss = 1.1, 0.3, 0.9 L/kg, respectively), and the t1/2 values were relatively short (3.7, 2.6, 6.7 h, respectively). Based on this data, we reasoned that factors other than simply metabolism of the nitro group may need to be addressed to improve the PK properties of compounds in the Niclosamide chemotype.
It is well known that pharmacokinetic performance, including oral bioavailability, is influenced by multiple parameters such as solubility, permeability, metabolic stability, pKa, logP, hydrogen bond donors, among others.50,51 In light of the published pharmacokinetic data we reasoned that the salicylamide group might also play an important role in limiting the exposure of the inhibitors. The phenolic–OH group is a potential site of glucuronidation and clearance. Moreover, the calculated pKa of the –OH group (pKa = 6.8)52 indicates that the molecule would be substantially ionized at the pH of intestinal fluid (pH = 4–8), and the pH of blood (pH = 7.4). Ionization of the –OH group, would be expected to limit exposure by reducing the permeability of the molecule and limit the volume of distribution, both of which could be expected to reduce exposure. In addition, the salicylamide moiety possesses two hydrogen bond donors, albeit with the potential for intramolecular hydrogen bonding, that could be expected to reduce permeability.51 Based on this reasoning, we next turned out attention to the SAR of the amide linker and the phenol that make up the salicylamide functionality.
Molecules to interrogate the SAR of the salicylamide were prepared as outlined in Scheme 2 or purchased from chemical vendors. We found that reversing the orientation of the amide resulted in the complete loss of activity (compound 26) (Table 2, Fig. S1, Supplemental information). Insertion of a carbon atom between the nitrogen atom of the amide and the aromatic ring also reduced the potency of inhibition (Compounds 28, 29), as did replacement of the amide NH substituent with an N-Me sub-stituent (compound 27). Replacing the amide with a sulphonamide substituent produced a compound (30) that inhibited Wnt signaling in the TOPFlash assay in the low micromolar range and induced Fzd1-GFP internalization very weakly. Our previous work indicated that 2-methoxy substituted derivatives were not active inhibitors of Wnt signaling.8 Thus, in an effort to reduce phase II metabolism and ionization of the –OH group and reduce the number of hydrogen bond donors, we explored 4′-CF3 and 4′-NO2 anilide derivatives in which the phenol was converted to ester (compound 31, 32) and carbonate (compound 33). In addition, we explored the activity of compounds in which both –OH and –NH groups were modified by tying the phenol and the amide together in a 6-membered ring oxazine-dione motif (compounds 34, 35, and 36), a modification that removes two hydrogen bond donors. Upon testing these compounds, we were delighted to find that both the 4′-CF3 and the 4′-NO2 anilide derivatives produced a robust punctate pattern in the Fzd1-GFP internalization assay and inhibited Wnt-signaling in the TOPFlash assay (Table 2, Fig 3A). The IC50 of inhibition in the TOPFlash assay for Niclosamide and compounds 32, 33 and 34 are: 0.34 ± 0.08, 0.23 ± 0.06, 0.34 ± 0.12 and 0.33 ± 0.1 μM, respectively.
Scheme 2.

Synthesis of salicylamide derivatives. Reagents and conditions: (i) Route A: PCl3, ArNHMe, Δ, xylene; route B: (1) (COCl)2, ArCH2NH2, pyr, 0–5 °C; (ii) ArCOCl, pyr, 0–5 °C; (iii) (a) ClSO2H, (b) ArNH2, pyr, 0–5 °C; (c) PhSNa, DMF, Δ. (iv) R3COCl, pyr, THF; (v) triphosgene, NEt3, THF.
Table 2. Structure–activity relationships in the salicylamide motif.
| Compound number | Structure | Fzd1-GFP internalization @12.5 μMa | Inhibition Wnt/β-catenin transcription TopFlash IC50 (μM) ± SE | Clog P | pKa OH | pKa NHb |
|---|---|---|---|---|---|---|
| 26 |
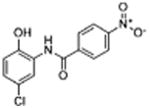
|
ND | >12 | 2.2 | 8.3 | 12.1 |
| 27 |
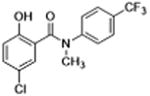
|
0 | >12 | 4.5 | 8.4 | – |
| 28 |
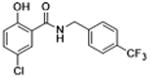
|
0 | 15.10 ± 1.40 | 4.6 | 8.4 | 12.0 |
| 29 |
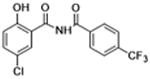
|
0 | >12 | 4.2 | 7.4 | 11.9 |
| 30 |
|
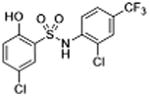
1 | 2.57 ± 0.27 | 3.8 | 6.6 | 6.2 |
| 31 |
|
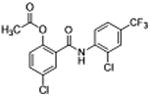
5 | 0.32 ± 0.03 | 4.2 | – | 9.6 |
| 32 (DK-520) |
|
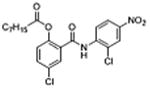
5 | 0.23 ± 0.06 | 4.3 | – | 9.4 |
| 33 |
|
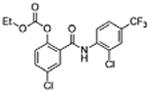
5 | 0.34 ±0.12 | 4.8 | – | 10.5 |
| 34 |
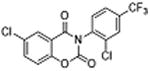
|
5 | 0.33 ±0.10 | 4.3 | – | – |
| 35 |
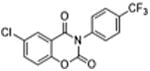
|
5 | 0.32 ± 0.03 | 3.9 | – | – |
| 36 |
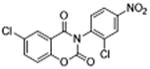
|
ND | 0.37 ±0.13 | 2.6 | – | – |
Internalization of Frizzled1-GFP stably expressed in U2OS cells was determined by confocal microscopy. Cells were treated with compounds for 6 h, fixed, and scored visually by the amount of punctate observed versus DMSO control. Inhibition of Wnt3A-stimulated Wnt/β-catenin transcription was determined by TOPFlash normalized to renilla control. ClogP and pKa values were calculated in Maestro 9.3 (Schrodinger, Inc.) using Qikprops and Epik.
Punctate similar to control = 0, trace amount of punctate greater than control = 1, moderate = 3, strong = 5.
Calculated pKa values in water are reported. The calculated pKa of the NH is the salicylamide NH without ionization of the –OH group. The estimated error of the pKa calculation for the OH group is ±1 log unit, and ±2 for the NH group. ND: not determined.
Figure 3.
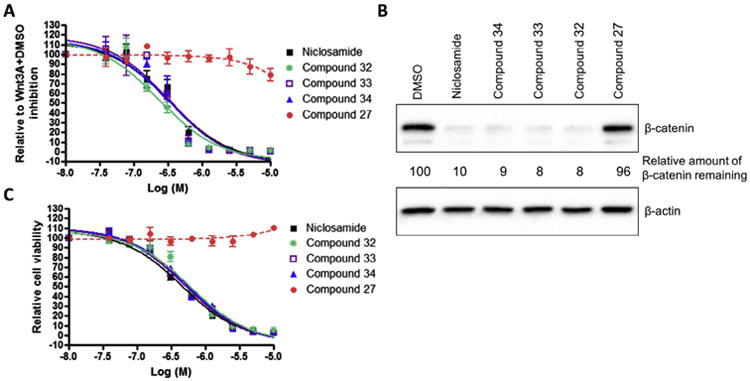
Salicylamide derivatives inhibit Wnt3A-stimulated β-catenin signaling and proliferation of HCT-116 cells. (A) Inhibition of Wnt signaling in TOPFlash assay. HEK293 cells stably expressing TOPFlash luciferase reporter and Renilla luciferase were treated with Wnt3A-conditioned medium in the presence of DMSO or compounds from 0.04 to 10 μM. The TOPFlash reporter activity of Wnt3A with DMSO treatment was set as 100%. Data were fit using GraphPad Prism (mean ± SEM, n ≥ 2). (B) Reduction of cytosolic β-catenin levels. HCT-116 cells were treated with 2.0 μM compounds for 18 h, lysed in hypotonic buffer. Lysates were eluted by SDS electrophoresis on acrylamide, transferred, and probed with antibodies to β-catenin as described in Section 2. β-Actin was used as a loading control. (C) Inhibition of proliferation of colon cancer cell HCT116. Cells were treated with DMSO or compounds from 0.04 to 10 μM for 72 h, and cell viability was measured using the MTS assay. The cell viability with DMSO treatment was set at 100%. Data were fit using GraphPad Prism (mean ± SEM, n = 3).
Similar to earlier studies, we selected the active derivatives, compound 32, 33, and 34 and a less active derivative, compound 27 (Table 2), for further evaluation in HCT-116 cells (Fig. 3). As anticipated by the FZD1-GFP and TOPFlash data, the active compounds inhibited Wnt/β-catenin signaling as measured by the reduction of cytosolic β-catenin (Fig. 3B). Upon treating cells with 2.0 μM compound for 18 h in culture, β-catenin levels were significantly decreased by the active compounds. The amount of cytosolic β-catenin remaining relative to DMSO control without compound were: Niclosamide: 10%; compound 32: 8%; compound 33: 8%; and compound 34: 9%. The amount of cytosolic β-catenin remaining for the less active derivative, compound 27, was 96%, nearly equal to the amount remaining in the DMSO control. Likewise, only the active compounds inhibited proliferation of HCT-116 cells in culture by MTS assay, with potency comparable to Niclosamide (Fig. 3C). The IC50 of inhibition in the MTS assay for Niclosamide and compounds 32, 33 and 34 are: 0.45 ± 0.05, 0.61 ± 0.11, 0.51 ± 0.06 and 0.55 ± 0.07 μM, respectively.
Based on the data in the Wnt/β-catenin signaling assays, the proliferation assay, and the structural modifications that eliminate the 4′nitro group, ionization of the –OH group and the removal of two hydrogen bond donors, we next asked whether compound 34 improved exposure in mice compared to Niclosamide at 200 mg/kg, the dose used in previous tumor xenograft studies.9 Upon dosing a solution of compound 34 orally at 200 mg/kg we were disappointed to find the plasma exposure of the compound was still comparable to Niclosamide,9 and that exposure was not significantly improved. Based on the hypothesis compound 34 could be metabolized to an active metabolite (compound 9) that would reduce plasma levels of compound 34, we also analyzed plasma for this potential metabolite. Indeed, we could detect compound 9 in the plasma samples from mice dosed with compound 34, but unfortunately, the levels of this active metabolite was still low and only comparable to the levels obtained by dosing Niclosamide to mice at 200 mg/kg9 (Fig. S2, Supplemental information).
Having replaced the nitro group, and blocked the site of ionization and glucuronidation, we reasoned that the parameter now limiting oral exposure was likely to be poor solubility in aqueous media. Given these inhibitors are somewhat hydrophobic with clogPs of 4–5, we questioned whether formulation in oil-based vehicles, such as corn oil, might be better vehicles for PK studies.53,54 To test these concepts, we decided to study DK-520 (compound 32), a derivative of Niclosamide because this compound was freely soluble in corn oil, and because it could metabolize to Niclosamide, a compound for which we had established and well-developed methods to accurately measure its plasma concentration. Upon dosing a solution of DK-520 to mice at 200 mg/kg in corn oil, we were delighted to find DK-520 provided significantly increased plasma exposure of Niclosamide (Fig. 4) when compared to published studies of 200 mg/kg Niclosamide dosed orally.9 The Cmax, the AUC and the duration of exposure of Niclosamide obtained by dosing DK-520 were all increased (Fig. S3, Supplemental information), even though on a molar basis, the amount of DK-520 dosed was approximately 1/3 less than the amount of Niclosamide dosed. In fact, the plasma levels of Niclosamide obtained by dosing DK-520 at 200 mg/kg were above the IC50 of inhibition of Wnt signaling in the TOPFlash assay for nearly 24 h, whereas the reported plasma levels of Niclosamide dosed as a solution at 200 mg/kg were only above the IC50 of Wnt inhibition in the TOPFlash assay for less than 1 h.9 Moreover, DK-520 dosed orally at 200 mg/kg was well tolerated when administered daily for three weeks, as judged by body weight and observing the behavior of the mice (Fig. S4, Supplemental information).
Figure 4.
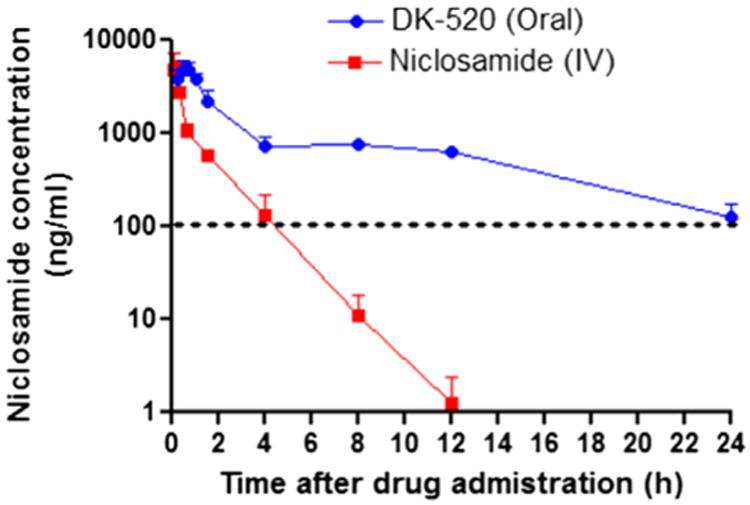
Plasma concentration of Niclosamide. CD1 mice were dosed with DK-520 (32) (p.o., 200 mg/kg of body weight) or Niclosamide (IV, 2.6 mg/Kg). For oral dosing of DK-520, blood samples were drawn serially at predose and at 0.25, 0.5, 0.75, 1, 1.5, 4, 8, 12, and 24 h after drug administration, n = 4 per time point. For IV dosing of Niclosamide, blood samples were obtained at predose and at 0.08, 0.17, 0.33, 0.67, 1.5, 4, 8, 12 h after drug administration, n = 2 per time point. Quantification of Niclosamide in mouse plasma was done by LC/MS–MS9 and reported as ng/mL. The dotted line indicates the IC50 of Niclosamide inhibition of Wnt/β-catenin signaling in the TOPFlash assay. The data were presented as mean ± SEM.
4. Discussion
In our earlier studies, we demonstrated that Niclosamide inhibits Wnt/β-catenin signaling in vitro and in vivo in models of CRC.7,9 In our in vivo tumor models, oral administration of Niclosamide inhibited tumor growth and decreased levels of Dvl and β-catenin in tumors implanted subcutaneously despite poor exposure to Niclosamide during the dosing period. In humans, low systemic exposure of Niclosamide given orally could mitigate its utility to treat disseminated metastatic disease.
To aid the translation of our initial findings toward a clinical application, we conducted additional SAR studies seeking to identify agents with increased potency, selectivity, and pharmacokinetic exposure. Since bio-reduction of the nitro group is a major metabolite of Niclosamide, and that nitro groups can impart poor solubility and reduce oral exposure, we focused initially on the SAR of the anilide ring. Consistent with our previous report, activity of these derivatives in the FZD1-GFP internalization assay and Wnt3A-stimulated β-catenin TOPFlash gene transcription were in general agreement. Molecules that produced more potent inhibition in the Wnt3A-stimulated β-catenin TOPFlash gene transcription assay also produced stronger responses in the FZD1-GFP internalization assay. In the anilide ring, we found that the potency of inhibition was dependent on both the nature and the location of substitution. Replacement of the electron-withdrawing 4′-nitro group in Niclosamide or in compound 3 with electron-withdrawing substituents such as acetoyl, benzoyl, amide, and sulphonyl groups were, in general, all inactive in the FZD1-GFP internalization assay and were weak inhibitors in the Wnt3A-stimulated TOPFlash assay. Given reports that Niclosamide uncouples oxidative phosphorylation, perhaps by acting as a protonophore, we calculated the clogP and the pKa of the OH and NH groups, SAR parameters associated protonophore activity.31 Here, active and inactive molecules were in a similar clogP range, had identical OH pKa values, and variable NH pKa values. In contrast, to the above derivatives, replacement of the 4′-nitro group with electron-withdrawing 4′-trifluoromethyl or 4′-chloro groups yielded molecules of similar potency to Niclosamide. Moving the 4′-trifluoromethyl or -chloro substituent to other positions in the ring generally yielded compounds of less potency. The clogP values of the trifluoromethyl or chloro substituted compounds were values higher than Niclosamide, the calculated pKa of the OH groups were identical, and the pKa of the NH protons varied. The impact of the structural changes and differences in calculated properties on protonophore activity and uncoupling of oxidative phosphorylation activity requires further investigation. With respect to inhibition of Wnt/β-catenin signaling, as anticipated by the FZD1-GFP internalization and TOPFlash assay data, compounds with similar activity to Niclosamide provided similar reduction in cytosolic β-catenin by Western blot and similar reduction in cell proliferation by MTS assay in HCT-116 cells. In the salicylamide region, reversing the orientation of the amide, inserting an atom between the amide nitrogen and the aryl ring, methylation of the amide, and replacing the amide with a sulphonamide group all reduced activity. Acylation of the phenolic OH group or tying it back to the amide via a six-membered oxazine-dione ring motif produced compounds of similar potency to Niclosamide in the FZD1-GFP internalization and TOPFlash assays and again yielded a similar reduction in cytosolic β-catenin by Western blot and similar reduction in cell proliferation by MTS assay in HCT-116 cells. The cell-based format of these assays could result in the metabolism of the compounds in the assay to yield compounds with a phenolic OH group. Additional studies are needed to better understand the SAR of these derivatives.
Given the need to improve the systemic exposure of Niclosamide given orally, we also interrogated structural features in the anilide and salicylamide for their impact on plasma exposure. Using medicinal chemical principles known to influence absorption and pharmacokinetic properties, we evaluated specific changes to the chemical structure. From these studies we discovered DK-520 a derivative of Niclosamide, can be metabolized in vivo and dramatically increase both the plasma concentration of Niclosamide and its duration of exposure. Functionally, DK-520 and Niclosamide had similar Wnt/β-catenin inhibitory activity and effects on cell proliferation in our assays. When DK-520 was dosed orally to mice, Niclosamide was detected in plasma in concentrations that exceeded the IC50 of inhibition of Wnt/β-catenin signaling in the TOPFlash assay for as long as 24 h after a 200 mg/kg dose. Importantly, this high level of exposure to Niclosamide did not result in toxicity as judged by changes in body weight or by observing the mice behavior when dosed at 200 mg/kg over three weeks. Previously we and others9,18,27,30 reported that Niclosamide is cleared rapidly when dosed by the oral, intraperitoneal (IP), or intravenous (IV) route. To our knowledge, this is the first report to describe the ability to increase the systemic drug exposure of Niclosamide in plasma and extend its duration of exposure. Further studies are needed to define the plasma, tissue and tumor levels of the parent compound DK-520, tissue and tumor levels of the metabolite Niclosamide, and the mechanism responsible for its increased absorption and conversion to Niclosamide. Regardless of the mechanism, DK-520 provides a significant level and duration of plasma exposure to the drug Niclosamide to enable in vivo assessment of Niclosamide in animal models of disease. Given the extent of systemic exposure, DK-520 and related derivatives may provide exposure suitable to evaluate inhibition of Wnt/β-catenin signaling by Niclosamide in the clinic.
Efforts are underway to identify the target responsible for inhibition of Wnt signaling and to understand the SAR that impacts the selectivity of Niclosamide. However, given Niclosamide is a multifunctional drug that (1) inhibits other important oncogenic signaling pathways such as mTOR,29 Notch,41 NF-kB34 and STAT-342 in addition to Wnt/β-catenin signaling, (2) is active in drug-resistant cancers,9,38,40 and (3) appears well-tolerated in vivo, greater selectivity may not give better anti-tumor effects. In other words, the current selectivity profile of Niclosamide may be clinically valuable in its own right such that the ability of Niclosamide to inhibit important anticancer targets such as mTOR, Notch, NF-kB and STAT-3 in addition to Wnt, may provide important therapeutic benefits versus more selective agents. Thus, a benefit of derivatives of Niclosamide such as DK-520 that increase the exposure of Niclosamide in vivo, is that they may behave in part, like pro-drugs that metabolize to produce the multi-functional drug Niclosamide. As prodrugs of Niclosamide they could be valuable agents to study clinically to treat a broad range of diseases in which Niclosamide has demonstrated biological activity in preclinical models.
5. Conclusion
SAR studies in the Niclosamide chemotype have identified structural features that impact inhibition of Wnt/β-catenin signaling and plasma exposure when dosed orally. Through these studies, we identified multiple active derivatives of Niclosamide including DK-520 (compound 32) that when dosed orally, can be metabolized to Niclosamide in vivo and produce high concentrations of Niclosamide in plasma and extend the duration of exposure to the drug Niclosamide. To our knowledge, this is the first report to identify derivatives of Niclosamide that increase plasma exposure to Niclosamide and extend its duration of exposure. The discovery of Niclosamide derivatives that improve systemic Niclosamide drug exposure overcomes a significant barrier to the clinical translation of Niclosamide to treat cancer. Moreover, it also allows the study of Niclosamide in vivo in other diseases for which Niclosamide has demonstrated biological activity. Thus, the findings described here may provide a breakthrough to a new class of Niclosamide-based drug candidates to treat disease associated with its multi-function activity ranging from cancer,33 particularly cancers with dysregulated Wnt signaling,1,9 drug-resistant cancers9,38,40 including resistant cancers due to up-regulation of genes involved in oxidative phosphorylation,55–57 to bacterial and viral disease,23–27 and lupus58 to metabolic diseases such as type II diabetes,30,32 non-alcoholic fatty liver disease (NAFLD), and non-alcoholic steatohepatitis (NASH).59,60
Supplementary Material
Acknowledgments
This work was funded in part by 5 R01 CA172570 (W.C.), BC123280 (W.C.), and Clinical Oncology Research Center Development Grant 5K12-CA100639-08 (R.A.M., M.C.). Wei Chen is a V foundation Scholar and an American Cancer Society Research Scholar. NIH 5P30-CA-014236-36 (I.S.). NMR instrumentation in the Duke NMR Spectroscopy Center was funded by the NIH, NSF, NC Biotechnology Center and Duke University. The authors gratefully acknowledge this support and the support of Anthony Ribeiro in the Duke NMR Center and Professor Eric Toone and the Duke Small Molecule Synthesis Facility.
Abbreviations
- APC
adenomatous polyposis coli
- CRC
colorectal cancer
- Dvl
Dishevelled
- LEF/TCF
lymphoid enhancer factor/T cell factor
- Fzd1
Frizzled1
- GFP
green fluorescent protein
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- SAR
structure–activity relationships
- Vss
volume of distribution steady-state
Footnotes
Supplementary data: Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2015.07.001.
References and notes
- 1.Barker N, Clevers H. Nat Rev Drug Discovery. 2006;5:997. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 2.Coombs GS, Covey TM, Virshup DM. Curr Drug Targets. 2008;9:513. doi: 10.2174/138945008784911796. [DOI] [PubMed] [Google Scholar]
- 3.Besancon R, Valsesia-Wittmann S, Puisieux A, de Fromentel CC, Maguer-Satta V. Curr Med Chem. 2009;16:394. doi: 10.2174/092986709787315531. [DOI] [PubMed] [Google Scholar]
- 4.MacDonald BT, Tamai K, He X. Dev Cell. 2009;17:9. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen W, ten Berge D, Brown J, Ahn S, Hu LA, Miller WE, Caron MG, Barak LS, Nusse R, Lefkowitz RJ. Science. 2003;301:1391. doi: 10.1126/science.1082808. [DOI] [PubMed] [Google Scholar]
- 6.Meireles LMC, Mustata G. Curr Top Med Chem. 2011;11:248. doi: 10.2174/156802611794072632. [DOI] [PubMed] [Google Scholar]
- 7.Chen MY, Wang JB, Lu JY, Bond MC, Ren XR, Lyerly HK, Barak LS, Chen W. Biochemistry. 2009;48:10267. doi: 10.1021/bi9009677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mook RA, Jr, Chen M, Lu J, Barak LS, Lyerly HK, Chen W. Bioorg Med Chem Lett. 2013;23:2187. doi: 10.1016/j.bmcl.2013.01.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osada T, Chen MY, Yang XY, Spasojevic I, Vandeusen JB, Hsu D, Clary BM, Clay TM, Chen W, Morse MA, Lyerly HK. Cancer Res. 2011;71:4172. doi: 10.1158/0008-5472.CAN-10-3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang SMA, Mishina YM, Liu SM, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi XY, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao WL, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F. Nature. 2009;461:614. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 11.Chen BZ, Dodge ME, Tang W, Lu JM, Ma ZQ, Fan CW, Wei SG, Hao WN, Kilgore J, Williams NS, Roth MG, Amatruda JF, Chen C, Lum L. Nat Chem Biol. 2009;5:100. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mallinger A, Crumpler S, Pichowicz M, Waalboer D, Stubbs M, Adeniji-Popoola O, Wood B, Smith E, Thai C, Henley AT, Georgi K, Court W, Hobbs S, Box G, Ortiz-Ruiz MJ, Valenti M, De Haven Brandon A, Te Poele R, Leuthner B, Workman P, Aherne W, Poeschke O, Dale T, Wienke D, Esdar C, Rohdich F, Raynaud F, Clarke PA, Eccles SA, Stieber F, Schiemann K, Blagg J. J Med Chem. 2015;58:1717. doi: 10.1021/jm501436m. and references within. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sebio A, Kahn M, Lenz H. Exp Opin Ther Targets. 2014;18:611. doi: 10.1517/14728222.2014.906580. [DOI] [PubMed] [Google Scholar]
- 14.Sack U, Walther W, Scudiero D, Selby M, Kobelt D, Lemm M, Fichtner I, Schlag Peter M, Shoemaker Robert H, Stein UJ. Natl Cancer Inst. 2011;103:1018. doi: 10.1093/jnci/djr190. [DOI] [PubMed] [Google Scholar]
- 15.Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y. PLoS ONE. 2011;6:e29290. doi: 10.1371/journal.pone.0029290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arend RC, Londono-Joshi AI, Samant RS, Li Y, Conner M, Hidalgo B, Alvarez RD, Landen CN, Straughn JM, Buchsbaum DJ. Gynecol Oncol. 2014;134:112. doi: 10.1016/j.ygyno.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Pearson RD, Hewlett EL. Ann Intern Med. 1985;102:550. doi: 10.7326/0003-4819-102-4-550. [DOI] [PubMed] [Google Scholar]
- 18.Andrews P, Thyssen J, Lorke D. Pharmacol Ther. 1983;19:245. doi: 10.1016/0163-7258(82)90064-x. [DOI] [PubMed] [Google Scholar]
- 19.WHO. The Selection and Use of Essential Medicines. World Health Organization; Geneva: 2007. [Google Scholar]
- 20.Weinbach EC, Garbus J. Nature. 1969;221:1016. doi: 10.1038/2211016a0. [DOI] [PubMed] [Google Scholar]
- 21.Frayha GJ, Smyth JD, Gobert JG, Savel J. Gen Pharmacol. 1997;28:273. doi: 10.1016/s0306-3623(96)00149-8. [DOI] [PubMed] [Google Scholar]
- 22.Swan GE. J S Afr Vet Assoc. 1999;70:61. doi: 10.4102/jsava.v70i2.756. [DOI] [PubMed] [Google Scholar]
- 23.Imperi F, Massai F, Ramachandran Pillai C, Longo F, Zennaro E, Rampioni G, Visca P, Leoni L. Antimicrob Agents Chemother. 2013;57:996. doi: 10.1128/AAC.01952-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piccaro G, Giannoni F, Filippini P, Mustazzolu A, Fattorini L. Antimicrob Agents Chemother. 2013;57:1428. doi: 10.1128/AAC.02154-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajamuthiah R, Fuchs BB, Conery AL, Kim W, Jayamani E, Kwon B, Ausubel FM, Mylonakis E. PLoS ONE. 2015;10:e0124595. doi: 10.1371/journal.pone.0124595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jurgeit A, McDowell R, Moese S, Meldrum E, Schwendener R, Greber UF. PLoS Pathog. 2012;8:e1002976. doi: 10.1371/journal.ppat.1002976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang YW, Yeh TK, Lin KT, Chen WC, Yao HT, Lan SJ, Wu YS, Hsieh HP, Chen CM, Chen CT. Yaowu Shipin Fenxi. 2006;14:329. [Google Scholar]
- 28.Merschiohann K, Steverding D. Exp Parasitol. 2008;118:637. doi: 10.1016/j.exppara.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Fonseca BD, Diering GH, Bidinosti MA, Dalal K, Alain T, Balgi AD, Forestieri R, Nodwell M, Rajadurai CV, Gunaratnam C, Tee AR, Duong F, Andersen RJ, Orlowski J, Numata M, Sonenberg N, Roberge MJ. Biol Chem. 2012;287:17530. doi: 10.1074/jbc.M112.359638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tao H, Zhang Y, Zeng X, Shulman GI, Jin S. Nat Med. 2014;20:1263. doi: 10.1038/nm.3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terada H. Environ Health Perspect. 1990;87:213. doi: 10.1289/ehp.9087213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin T. Diabetologia. 2008;51:1771. doi: 10.1007/s00125-008-1084-y. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Li PK, Roberts MJ, Arend RC, Samant RS, Buchsbaum DJ. Cancer Lett. 2014;349:8. doi: 10.1016/j.canlet.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin Y, Lu Z, Ding K, Li J, Du X, Chen C, Sun X, Wu Y, Zhou J, Pan J. Cancer Res. 2010;70:2516. doi: 10.1158/0008-5472.CAN-09-3950. [DOI] [PubMed] [Google Scholar]
- 35.Khanim FL, Merrick BAME, Giles HV, Jankute M, Jackson JB, Giles LJ, Birtwistle J, Bunce CM, Drayson MT. Blood Cancer J. 2011;1:e39. doi: 10.1038/bcj.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yo YT, Lin YW, Wang YC, Balch C, Huang RL, Chan MWY, Sytwu HK, Chen CK, Chang CC, Nephew KP, Huang T, Yu MH, Lai HC. Mol Cancer Ther. 2012 doi: 10.1158/1535-7163.MCT-12-0002. [DOI] [PubMed] [Google Scholar]
- 37.Londono-Joshi AI, Arend RC, Aristizabal L, Lu W, Samant RS, Metge BJ, Hidalgo B, Grizzle WE, Conner M, Forero-Torres A, Lobuglio AF, Li Y, Buchsbaum DJ. Mol Cancer Ther. 2014;13:800. doi: 10.1158/1535-7163.MCT-13-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, Gao AC. Clin Cancer Res. 2014;20:3198. doi: 10.1158/1078-0432.CCR-13-3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wieland A, Trageser D, Gogolok S, Reinartz R, Hofer H, Keller M, Leinhaas A, Schelle R, Normann S, Klaas L, Waha A, Koch P, Fimmers R, Pietsch T, Yachnis AT, Pincus DW, Steindler DA, Brustle O, Simon M, Glas M, Scheffler B. Clin Cancer Res. 2013;19:4124. doi: 10.1158/1078-0432.CCR-12-2895. [DOI] [PubMed] [Google Scholar]
- 40.Li R, Hu Z, Sun SY, Chen ZG, Owonikoko TK, Sica GL, Ramalingam SS, Curran WJ, Khuri FR, Deng X. Mol Cancer Ther. 2013;12:2200. doi: 10.1158/1535-7163.MCT-13-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang AM, Ku HH, Liang YC, Chen YC, Hwu YM, Yeh TS. J Cell Biochem. 2009;106:682. doi: 10.1002/jcb.22065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ren X. ACS Med Chem Lett. 2010;1:434. doi: 10.1021/ml100146z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.World_Health_Organization. WHO; Geneva: 2002. WHO Specifications and Evaluations for Public Health Pesticides. http://www.who.int/whopes/quality/en/Niclosamide.pdf. [Google Scholar]
- 44.Ye Y, Zhang X, Zhang T, Wang H, Wu B. Drug Dev Ind Pharm. 2014:1. doi: 10.3109/03639045.2014.954585. [DOI] [PubMed] [Google Scholar]
- 45.Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennernäs H, Hussain AS, Junginger HE, Stavchansky SA, Midha KK, Shah VP, Amidon GL. Mol Pharm. 2004;1:85. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 46.Ding K, Pei D, Zhou J. Application: CN Patent 2009–10036483, 101775032. 2010:8.
- 47.Grifasi F, Chierotti MR, Gaglioti K, Gobetto R, Maini L, Braga D, Dichiarante E, Curzi M. Cryst Growth Des. 1939;2015:15. [Google Scholar]
- 48.Chen H, Yang Z, Ding C, Chu L, Zhang Y, Terry K, Liu H, Shen Q, Zhou J. ACS Med Chem Lett. 2013;4:180. doi: 10.1021/ml3003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhushan B, Dubey P, Kumar SU, Sachdev A, Matai I, Gopinath P. RSC Adv. 2015;5:12078. doi: 10.1016/j.cis.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 50.Kwon Y. Handbook of Essential Pharmacokinetics, Pharmacodynamics and Drug Metabolism for Industrial Scientists. Springer; 2001. [Google Scholar]
- 51.Kerns EH, Di L. Drug-like Properties: Concepts, Structure, Design and Methods— From ADME to Toxicity Optimization. Elsevier-Academic Press; Burlington: 2008. [Google Scholar]
- 52.Limited physiochemical data exist for Niclosamide in the literature. In WHO documents the pKa of Niclosamide is reported to be 5.6, the logP at pH ≤4 = 5.95, and the logD at pH = 7 as 4.48. Calculated pKa values are used throughout the manuscript to allow comparison between compounds.
- 53.Chen XQ, Gudmundsson OS, Hageman MJ. J Med Chem. 2012;55:7945. doi: 10.1021/jm3006433. [DOI] [PubMed] [Google Scholar]
- 54.Strickley RG. Pharm Res. 2004;21:201. doi: 10.1023/b:pham.0000016235.32639.23. [DOI] [PubMed] [Google Scholar]
- 55.Strohecker AM, White E. Cancer Discovery. 2014;4:766. doi: 10.1158/2159-8290.CD-14-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, Arany Z, Widlund HR. Cancer Cell. 2013;23:302. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Patzold S, Villanueva J, Krepler C, Fukunaga-Kalabis M, Hoth M, Bastian BC, Vogt T, Herlyn M. Cancer Cell. 2013;23:811. doi: 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, Morel L. Sci Transl Med. 2015;7:a18. doi: 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Michelotti GA, Machado MV, Diehl AM. Nat Rev Gastroenterol Hepatol. 2013;10:656. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 60.Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI. Science. 2015;347:1253. doi: 10.1126/science.aaa0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.