

Abstract
Objective:
Cochlear implantation is the most important treatment currently available for profound sensorineural hearing loss. The aim of this study was to investigate the etiology of hearing loss in patients with cochlear implantation, and to compare outcomes.
Methods:
Japanese hearing loss patients who received cochlear implants (CIs) or electric acoustic stimulation (EAS) in Shinshu University hospital (n = 173, prelingual onset: 92, postlingual onset: 81) participated in this study. Invader assay followed by the targeted exon-sequencing of 63 deafness genes using Massively parallel DNA sequencing (MPS) was applied. For prelingual patients, additional imaging examination, cCMV screening, and pediatric examination were performed for precise diagnosis.
Results:
Genetic screening successfully identified the causative mutation in 60% of patients with prelingual onset hearing loss and in 36% of those with postlingual hearing loss. Differences in the kinds of genes identified were observed between the two groups. Although there were marked variations in the outcome of cochlear implantation, patients with specific deafness gene mutations showed relatively good results.
Conclusion:
The present study showed genetic etiology is a major cause of hearing loss in CI/EAS patients. Patients possessing mutations in a number of deafness genes known to be expressed within inner ear have achieved satisfactory auditory performance, suggesting that the identification of the genetic background facilitates the prediction of post-CI performance. MPS is a powerful tool for the identification of causative deafness genes in patients receiving cochlear implantation. Therefore, determination of the involved region inside/outside of the cochlea by identification of the responsible gene is essential.
Keywords: ACTG1, CDH23, COCH, Cochlear implantation, CRYM, DFNA5, DFNB31, Etiology, GJB2, LOXHD1, MYO7A, MYO6, MYO15A, Next-generation sequencing, OTOF, SLC26A4, TMPRSS3
Genetic factors, the most common etiology in severe-to-profound hearing loss, are one of the key determinants of cochlear implantation (CI) and electric acoustic stimulation (EAS) outcomes. If the genetic background involves an “intracochlear” etiology, there is potential for good outcomes (1). Therefore, it is important to identify the involved region inside/outside the cochlea by identifying the responsible gene. Our recent series of studies on satisfactory auditory performance after receiving CI/EAS in patients with specific deafness gene mutations indicates that genetic testing would be helpful in predicting CI/EAS outcomes, as well as in deciding treatment choices (2–8). However, because of the extreme genetic heterogeneity of deafness, the clinical application of genetic information continues to entail a number of difficulties. More than 80 genes have already been reported to be associated with nonsyndromic hereditary hearing loss. Recent advances in targeted exon-sequencing of selected genes using massively parallel DNA sequencing (MPS) technology have enabled the successful identification of the causative mutations in relatively rare genes (9). In the present study, to obtain a more complete picture, MPS was applied to a large cohort of CI/EAS patients to 1) clarify their genetic background and 2) compare their etiology and outcomes.
SUBJECTS AND METHODS
Subjects
A total of 173 consecutive patients who received CI or EAS at Shinshu University Hospital were enrolled in this study. Among them, hearing loss was prelingual onset in 92 and postlingual onset in 81 patients (male: 92, female: 81).
Of the 92 prelingual patients, hearing loss was congenital onset in 81 (62 were identified during newborn hearing screening), because of meningitis in 3, progressive because of a CDH23 mutation in 2, and caused by cCMV infection in 3 patients. Age at surgery ranged from 8 months to 58 years (mean = 63.3 mo, median = 31 mo).
For the 81 postlingual patients, the onset age ranged from 7 to 78, and age at surgery ranged from 25 to 89 years.
Written informed consent was obtained from the subjects (or from their next of kin, caretaker, or guardian in the case of minors/children) before enrollment.
This study was approved by the Shinshu University Ethical Committee.
Genetic Screening
Two-step screening (Invader assay followed by MPS analysis) was applied for all patients. In Japan, the cost (approximately $US320) of genetic testing for deafness using this two-step screening is currently fully covered by social health insurance.
Invader Assay
First, we screened for 46 known mutations in 13 known deafness genes using the Invader assay, which was followed by direct sequencing as necessary (10). At least one deafness gene mutation was found in 29.5% of the subjects (10). This method of simultaneous screening for multiple deafness mutations using the Invader assay, and then direct sequencing where necessary, enables us to detect deafness mutations in an efficient and practical manner. In Japan, genetic testing for deafness using the Invader assay has been covered by social health insurance since 2012.
MPS Analysis
The detailed methodological protocol was described elsewhere (9).
Amplicon Library Preparation
Amplicon libraries were prepared according to the manufacturer's instructions using an Ion AmpliSeq Custom Panel (Applied Biosystems, Life Technologies, Carlsbad, CA) for 63 genes that reportedly cause nonsyndromic hearing loss. The detailed protocol is described elsewhere (2). The amplicon libraries were diluted to 20 pM and equal amounts in six libraries for six patients were pooled for one sequence reaction.
Emulsion PCR and Sequencing
Emulsion PCR and sequencing were performed according to the manufacturer's instructions. The detailed protocol is described elsewhere (2). MPS was performed with an Ion Torrent Personal Genome Machine (PGM) using an Ion PGM 200 Sequencing Kit and an Ion 318 Chip (Life Technologies).
Base call and Data Analysis
Sequence data were mapped against the human genome sequence (build GRCh37/hg19) with the Torrent Mapping Alignment Program. After sequence mapping, variant regions were piled up with Torrent Variant Caller plug-in software. After variant detection, effects were analyzed using ANNOVAR software (11,12). Missense, nonsense, insertion/deletion, and splicing variants were then selected from the identified variants. Variants were further selected if they were less than 1% of 1) the 1,000 Genome database (http://www.1000genomes.org/) (13), 2) the 6,500 exome variants in the Exome Variant Server (http://evs.gs.washington.edu/EVS/) (14), 3) the dataset of 1,208 Japanese exome variants in the Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/index.html) (15), and 4) 269 in-house Japanese normal hearing controls.
To predict the pathogenicity of missense variants, the following functional prediction software was used: PhyloP (16), Sorting Intolerant from Tolerant (SIFT) (17), Polymorphism Phenotyping (PolyPhen2) (18), LRT (19), MutationTaster (20), and GERP++ (21).
Candidate mutations were confirmed by Sanger sequencing and the responsible mutations were identified by segregation analysis using samples from the patients’ family members.
CT Imaging and Pediatric Consultation
All patients underwent examination by computed tomography at a slice thickness of 1 mm through the temporal bone to check for the presence of cochlear, vestibular, or inner ear canal malformation. Children with associated symptoms who were suspected of a syndromic disease underwent pediatric consultation and a diagnosis of the coexisting syndrome was made.
Diagnostic Testing for Congenital Cytomegalovirus Infection
For prelingual patients in whom no genetic mutation was detected, examination for congenital cytomegalovirus (cCMV) infection was performed using CMV-DNA quantitative PCR (qPCR) analysis. Before qPCR analysis, total DNA including genomic DNA and CMV DNA was extracted from preserved dried umbilical cords. As a positive control, we used preserved umbilical cords from patients with symptomatic congenital CMV infection. As a negative control, preserved umbilical cords from five healthy children without sensorineural hearing loss were used. Detailed methods are described elsewhere (22).
Outcome of CI
The implant was stimulated for the first time at least 3 to 4 weeks after the operation, and the evaluation of auditory and speech perception skills included the measurement of the aided free-field thresholds for adult patients.
In the prelingual patients, a LittlEARS auditory questionnaire (an assessment of early auditory development in young children) (23,24) was completed by the parents and audiologists. LittlEARS consists of 35 questions, each scored as 1 = yes, or 0 = no. For the postlingual patients, the Japanese monosyllable perception test (67-S test) and word perception test were applied. Assessment was performed at preoperation, and at 3, 6, and 12 months after implantation. We then compared the differences in outcomes for cochlear implantation between the various etiologies, and the distribution patterns of the LittlEARs auditory questionnaire scores were analyzed.
For postlingual patients, the distribution patterns of the Japanese monosyllable and word perception test results were examined and the statistical differences between the group with specific gene mutations and the other etiology group analyzed using Student's t test. We further divided all patients into two groups (the good outcome group and the moderate-poor outcome group) with the borderline set at 40% for the Japanese monosyllable perception test and at 60% for Japanese word perception test. Various factors, including age, sex, hearing loss threshold, and etiology, were compared.
RESULTS
Causes of Hearing Loss in Prelingual CI Patients
Of the 92 patients, causative mutations in deafness genes were identified for 55 patients in 49 families (59.8%). Five patients (5%) were diagnosed with cCMV infection on the basis of viral DNA diagnostic testing using dried umbilical cord samples. One patient was diagnosed with congenital rubella syndrome. CT imaging identified anomalies in five patients, including three patients with inner ear malformations (IP-2) and two patients with stenosis of the internal auditory canal. Nine patients (9.8%) were diagnosed with syndromic hearing loss through pediatric consultation, including three with Waardenburg syndrome, two with Usher syndrome (one with CDH23 biallelic mutations, and one with PCDH15 biallelic mutations), one with Down syndrome, one with Noonan syndrome, one with CHARGE syndrome, and one with Jervell and Lange-Nielsen syndrome (with KCNQ1 biallelic mutations). The etiologies of the prelingual CI patients in this study are summarized in Fig. 1A.
FIG. 1.
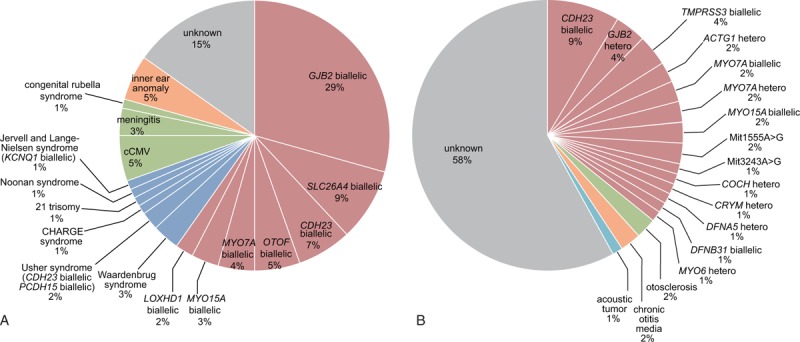
Etiology of CI/EAS patients. (A) Prelingual CI/EAS patients (n = 88). Blue indicates syndromic hearing loss; gray, unkown; green, infection-induced hearing loss; orange, inner ear anomaly; pink, nonsyndromic hearing loss associated with specific gene mutations. (B) Postlingual CI/EAS patients (n = 77). Blue indicates acoustic tumor; gray, unkown; green, otosclerosis; orange, otitis media; pink, nonsyndromic hearing loss associated with specific gene mutations.
With regard to the mode of inheritance, all 49 families were compatible with autosomal recessive inheritance. Segregation analysis as well as prediction software indicated that the identified mutations were compatible with being the responsible gene. The most frequent causative gene was GJB2, and c.235delC was found to be the most frequent mutation. The second most frequent genes were SLC26A4 and CDH23 (8% respectively). Genetic screening by using MPS identified causative mutations in many rare genes, such as OTOF, MYO15A, LOXHD1, and PCDH15.
As CDH23 heterozygous mutations were identified in two patients, these patients were categorized as unknown etiology.
Clinical Findings and Outcomes for Prelingual CI Patients
Of the 92 prelingual patients, 62 were detected by newborn hearing screening, so that the majority of the patients received CI before 2 years of age. Twenty-one patients, however, received CI passed school age (6 y.o.) because of progressive of hearing loss or a problem with timing, and therefore spent a considerable time with impaired hearing.
Eighty-two out of 92 patients had congenital profound hearing loss, and five patients showed late-onset progressive hearing loss at around the age of 2. Most of the patients with SLC26A4, LOXHD1, and MYO15A mutations showed progressive hearing loss. In contrast, the hearing loss in patients with GJB2 and OTOF mutations was not progressive in nature.
As for the outcomes of CI, in 23 of the 92 pre-lingual patients, early auditory development was assessed using the LittlEARS auditory questionnaire before the operation and at 3, 6, and 12 months after CI. Although scores varied among the patients, the majority of patients with non-syndromic hearing loss with specific deafness gene mutations showed good and rapid development of hearing behavior (Fig. 2). Some of the patients, who had already achieved good behavior using hearing aids, showed high performance preoperation (Fig. 2). In contrast, syndromic hearing loss patients as well as the patients with inner ear anomalies showed comparatively poorer and slower development (Fig. 2). The patients with unknown etiology but without any syndrome or malformation showed relatively good outcomes (Fig. 2). In addition, the cCMV patients, who had already good scores, maintained their high scores (Fig. 2). The distribution patterns of the LittlEARs auditory questionnaire scores are shown in Fig. 3. No statistically significant differences were observed between the group with specific gene mutations for nonsyndromic hearing loss and the other etiology group (Student's t test).
FIG. 2.
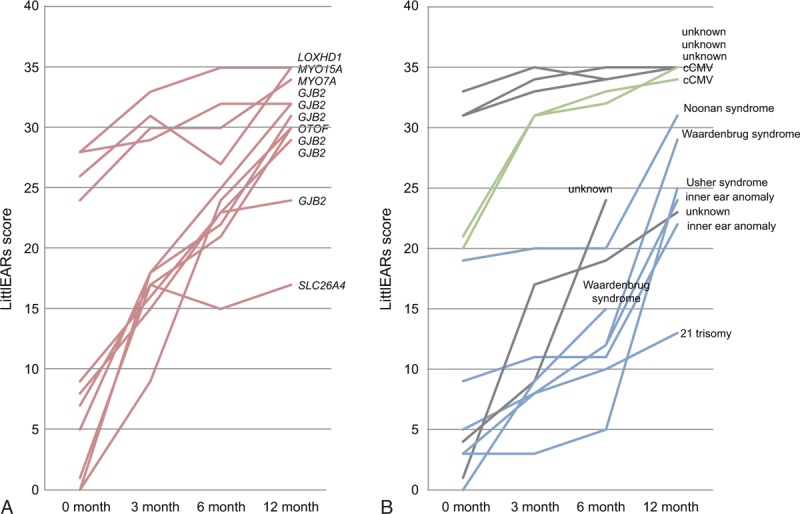
Early auditory development assessed using the LittlEARS auditory questionnaire. (A) Nonsyndromic hearing loss with specific gene mutations (n = 11). (B) Other etiology (n = 11). Blue indicates syndromic hearing loss; gray, unkown; green, infection-induced hearing loss; pink, nonsyndromic hearing loss associated with specific gene mutations.
FIG. 3.
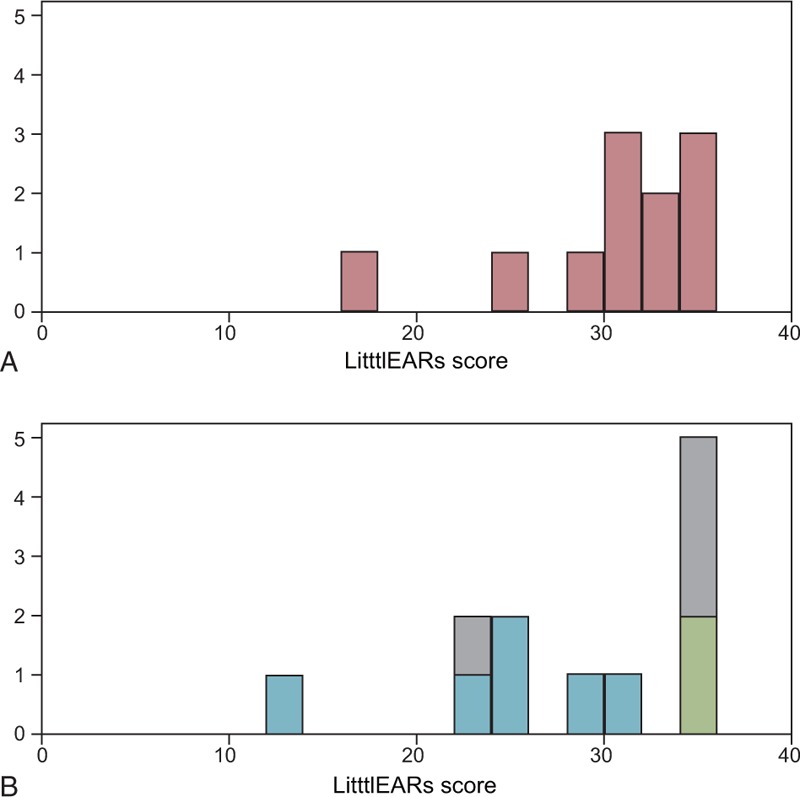
Distribution of auditory behavior assessment (LittlEARs) scores for prelingual CI patients. (A) Nonsyndromic hearing loss with specific gene mutations (n = 11). (B) Other etiology (n = 11). Blue indicates syndromic hearing loss; gray, unkown; green, infection-induced hearing loss; pink, nonsyndromic hearing loss associated with specific gene mutations.
Causes of Hearing Loss in Postlingual CI/EAS Patients
The cause of hearing loss was identified in 34 out of 81 postlingual patients (42.0%) in this study (Fig. 1B). In 29 patients (35.8%), the hearing loss was caused by mutations in deafness genes, whereas two patients (5%) were diagnosed with otosclerosis, two patients developed hearing loss as a consequence of chronic otitis media, and one patient experienced acoustic neuroma. This patient had some residual hearing and CI was, therefore, indicated for this patient.
The mode of inheritance varied among the 81 patients (22 families were compatible with autosomal dominant inheritance, 7 families were compatible with autosomal recessive inheritance, and 27 patients were sporadic pattern). Mutations were identified on the basis of genetic screening, and segregation analysis and prediction software suggested them to be the responsible mutations. In total, 13 causative genes were identified in this group, although no major genes, such as GJB2 in the prelingual group, were identified. The most common causative gene was CDH23 (9%), followed by MYO7A (4%), TMPRSS3 (4%), MYO15A (2%), DFNB31 (1%), ACTG1 (2%), DFNA5 (1%), MYO6 (1%), and CRYM (1%). Mitochondrial 3243A >G (3%) and 1555A>G mutation (1%) were also involved in the postlingual CI/EAS patients.
Clinical Findings and Outcomes for Postlingual CI/EAS Patients
Outcomes of CI were shown to vary among the postlingual hearing loss patients (Fig. 4). With regard to the distribution patterns of the Japanese monosyllable and word perception test results, the CI outcomes showed a multipeaked distribution (Fig. 5). No statistically significant differences were observed between the group with specific gene mutations and the other etiology group. A comparison of the good outcome group and the moderate-poor outcome group (with the borderline set at 40% for the Japanese monosyllable perception test and at 60% for the Japanese word perception test) revealed that 1) the good outcome group was significantly younger (mean age = 52.79 for monosyllable test, 54.23 for word test) than the moderate-poor outcome group (mean age = 66.88 for monosyllable test, 64.85 for word test), and 2) there were significant differences in etiology; i.e., 40% for monosyllable test and 43% for word test of the good outcome patients were found to have specific gene mutations whereas only 27% for monosyllable test and 23 % for word test of the poorer outcome group had the same specific gene mutations (Table 1).
FIG. 4.

Japanese word perception test results for postlingual CI patients. (A) Nonsyndromic hearing loss with specific gene mutations (n = 22). (B) Unknown or other etiology (n = 50). Blue indicates acoustic tumor; gray, unkown; green, otosclerosis; orange, otitis media; pink, nonsyndromic hearing loss associated with specific gene mutations.
FIG. 5.

Distribution of Japanese perception test results for postlingual CI patients. (A) Monosyllable. (B) Word. Blue indicates acoustic tumor; gray, unkown; green, otosclerosis; orange, otitis media; pink, non-syndromic hearing loss associated with specific gene mutations.
TABLE 1.
Differences between patients with a good CI outcome and those with a moderate-poor CI outcome
| Monosyllable Test (12 mo) | n | Age at Surgery (mean) | Genetic Mutation |
| > = 40 | 48 | 52.79 | 40% |
| <40 | 26 | 66.88 | 27% |
| Word Test (12 mo) | n | Age at Surgery (mean) | Genetic Mutation |
| > = 60 | 47 | 54.23 | 43% |
| <60 | 37 | 64.85 | 23% |
DISCUSSION
Etiology
Because of the extreme genetic heterogeneity of deafness, beyond screening for common genes such as GJB2, it has been difficult to identify the responsible gene in individual CI/EAS patients, especially in a clinical setting. However, recent advances in NGS may afford a breakthrough as targeted exon-sequencing of selected deafness genes using MPS technology has enabled the successful identification of causative mutations in relatively rare genes. In fact, the current series using MPS successfully discovered rare causative genes among the enrolled CI/EAS patients. These genes have not usually been screened and, therefore, mutations in these genes have not been clinically diagnosed using a conventional approach. MPS, however, has the potential to identify such rare genes/mutations.
Two previous studies have described the genetic backgrounds of cochlear implant patients, although both studies used selected samples (25,26). A definitive genetic diagnosis was made in 20.6% (37/180) of CI children by the screening of four common deafness-associated genes, GJB2, SLC26A4, the mitochondrial 12S rRNA gene, and OTOF (Wu et al. (25)). More recently, using targeted resequencing of 204 candidate deafness genes and a phenotype-driven candidate gene approach, causative variants were found in 54.8% (51/93) of cochlear implantees (Park et al. (26)). Both studies suggested that genetic causes account for an important proportion of CI patients. The present study is the first to clarify the genetic epidemiology in a more comprehensive way using 1) a consecutive (nonbiased) large cohort of samples, 2) both pre- and postlingual patients, and 3) an updated genetic screening system (Invader assay followed by MPS-based screening).
In this study, two-step genetic screening (Invader assay followed by MPS-based screening) successfully identified causative mutations in 59.8% of congenital hearing loss patients, and 35.8% of postlingual hearing loss patients with cochlear implantation. For the prelingual CI/EAS patient group, in particular, genetic screening together with additional imaging examination, cCMV screening, and pediatric examination was able to detect successfully the etiology of deafness in 85% of the patients. The present high diagnostic rate is expected to have a great impact, with such epidemiological data being essential for decision making with regard to the decision to implement CI/EAS, the prediction of outcomes, and the provision of appropriate future intervention.
As shown in Fig. 1, the most common etiology was genetic (59.8%), followed by cCMV infection (5%), inner ear malformation (5%), meningitis (3%), and congenital rubella (1%). Among the genetic causes, the most frequent causative gene was GJB2 (29%), followed by SLC26A4 (9%), CDH23 (7%), MYO7A (4%), OTOF (5%), MYO15A (3%), LOXHD1 (2%). The present results indicated that these deafness genes are typical deafness genes indicative for CI in the prelingual group. A further 9% of the patients were diagnosed with syndromic deafness on the basis of associated symptoms.
All of the identified genes are known to be localized and function in the inner ear (27). GJB2, the most common cause of congenital deafness worldwide, encodes the gap junction protein connexin 26, which is essential for potassium recirculation and other metabolite transport. SLC26A4 is a common cause of deafness associated with an enlarged vestibular aqueduct. Pendrin protein, which is encoded by SLC26A4, acts as a transporter of chloride, bicarbonate, and iodide ions in the spiral prominence. Pendrin also contributes to pH homeostasis and the mineralization process in the organ of Corti and vestibule. Cadherin 23, which is encoded by CDH23, is a component of the tip link and transient lateral links of the stereocilia. MYO7A, which encodes unconventional myosin VIIA, acts as a component of the USH complex (including CDH23, SANS, USH1C, and MYO7A) in the tip link of the stereocilia. MYO15A directly binds to WHRN to form the MYO15A-WHRN-EPS8 complex in the stereocilia, which is essential for stereocilia elongation. LOXHD1 is also involved in the regulation of stereocilia elongation and mutations in LOXHD1 cause “fused stereocilia” and “membrane ruffling” at the apical surface of hair cells. CDH23, MYO7A, MYO15A, and LOXHD1 are all important for the development and maintenance of the stereocilia and have important roles in mechano-electro-transduction. OTOF is the most common cause of auditory neuropathy spectrum disorder and encodes the protein otoferin, which is involved in the late step of synaptic vesicle exocytosis as the major Ca2+ sensor for the ribbon synapse of inner hair cells.
Among the postlingual CI/EAS patients, the etiology was detectable in approximately 40% of patients. The most frequent etiology was genetic (35.8%), followed by otosclerosis (2%), otitis media (2%), and acoustic neuroma (1%). Interestingly, although genetic causes were the most common, a number of different kinds of causative genes, including various rare genes, were found to be involved in postlingual deafness. Only a small number of patients could be diagnosed by Invader assay, with the majority of the rare genes identified by MPS.
The most common causative gene was CDH23 (9%), followed by MYO7A (4%), TMPRSS3 (4%), MYO15A (2%), DFNB31 (1%), ACTG1 (2%), DFNA5 (1%), MYO6 (1%), and CRYM (1%). In the postlingual CI/EAS patients, mitochondrial 3243A>G (1%) and 1555A>G mutation (2%) were also found to be involved. Compared with the prelingual group, many dominant genes, such as MYO7A, ACTG1, DFNA5, MYO6, and CRYM, as well as mitochondrial genes reported to cause progressive hearing loss, were found to be involved.
These genes are also localized and play important roles in the inner ear (27). TMPRSS3 encodes transmembrane protease serine 3, which is involved in the maturation of the epithelial amiloride-sensitive sodium channel (ENaC) and K+ channel (KCNMA1). DFNB31 (WHRN) encodes the scaffolding protein whirlin, which directly binds to SANS, EPS8, and MYO15A, and is colocalized in the tip link of the stereocilia. ACTG1 encodes cytoskeletal nonmuscle actin protein gamma. This protein is localized in the F-actin gap region of the stereocilia. MYO6, which is expressed in the cuticular plate region of IHC and OHCs, is involved in stereocilia formation, and may have an important role as a stereocilia anchor. Mu-crystallin is encoded by CRYM and directly binds to thyroid hormone (T3) with high affinity in the presence of NADPH. CRYM in complex with NADPH transports T3 into the nucleus and activates T3-dependent transcription.
Interestingly, CDH23, MYO7A, and MYO15A were found in both the pre- and postlingual groups, indicating these genes may express variable phenotypes.
Outcomes
In the prelingual group, the majority of patients with nonsyndromic hearing loss and with specific deafness gene mutations showed good and rapid development of auditory behavior (Fig. 2A). This is in line with the general hypothesis that good outcomes can be expected if the etiology is located within the cochlea (Fig. 2A). According to a previous study, the children with mutations had better auditory nerve responses (CAP scores) than did the children without mutations (25).
In contrast, a number of children in the other etiology group showed moderate-poor CI outcomes. The etiology in these poorer outcome patients involved inner ear/cochlear nerve malformation or syndromic hearing loss (Down syndrome, Noonan syndrome, or Waardenburg syndrome) (Fig. 2B). We compared the distribution patterns of LittlEARs auditory questionnaire scores (Fig. 3), but no statistically significant differences were observed between the two groups, probably because the other etiology group also contained patients showing good outcomes. One example is the patient with unknown etiology but without any malformation or syndrome (Fig. 2B), and another is a cCMV patient who had already recorded good scores before receiving CI because of progressive hearing loss (Fig. 2B).
In the postlingual group, performance after CI varied due to a number of factors; however, the majority of patients showed a good overall outcome after implantation (Fig. 4, A and B).
To identify differences in outcome between patients with specific nonsyndromic deafness gene mutations and those with hearing loss of other etiology, including many unknown patients, we compared the distribution patterns of the Japanese monosyllable perception test results and Japanese word perception test results (Fig. 5). As a result, CI patients with specific nonsyndromic deafness gene mutations tended to show better outcomes than did patients with other etiologies, although this difference was not statistically significant.
Interestingly, with regard to the distribution patterns of the Japanese monosyllable perception test results and Japanese word perception test results (Fig. 5), the CI outcomes showed a multipeaked distribution. These results suggested that patients do not form a homogeneous group. This distribution pattern is commonly observed in both monosyllable and word tests. We divided all patients into two groups (the good outcome group and moderate-poor outcome group) with the borderline set at 40% for the Japanese monosyllable perception test and at 60% for the Japanese word perception test. A comparison of outcomes and various factors, including age, sex, hearing loss threshold, and etiology, revealed that the good outcome group was significantly younger than the moderate-poor outcome group. These results indicated that younger patients can expect a better CI outcome than older patients. In addition to age, significant differences were also observed for etiology; i.e., 40 to 43% of the good outcome patients were found to have specific gene mutations, whereas only 23 to 27% of the poorer outcome group had the same specific gene mutations (Table 1).
With regard to the relationship between etiology and outcome in CI/EAS patients, a large number of articles have focused on the outcomes in patients with GJB2(28–39), but not many articles have described outcomes in CI/EAS patients with associated uncommon gene mutations. A series of studies have demonstrated that CI has brought about tremendous improvements in auditory skills as well as in speech production development in patients with profound hearing loss resulting from GJB2 mutations (28–39). Further, although some literature described comparable results, no articles have reported poorer outcomes for patients with GJB2 mutations.
There have been fewer reports on the outcomes for CI for other genes, although some reports have described good performances in CI/EAS patients with associated SLC26A4(25,39), OTOF(25,40,41), MYO6(8,42), MYO15A(3,4), TECTA(3), CDH23(2), COCH(5,43), MYH9(44,45), and TMPRSS3 mutations (1,3,6,25,46,47). There have also been some reports describing poorer outcomes in patients with POU3F4 mutations, which are known to cause inner ear anomalies (48–51).
Further, the outcomes of CI for patients with TMPRSS3 mutations seem controversial. A majority of patients with TMPRSS3-associated hearing loss (13 out of 15 based on a literature review) were reported to show good outcomes for CI, whereas two patients reported by Eppsteiner et al. showed a poorer performance (1). We evaluated the improvement in speech discrimination and perception scores (using the 67S Japanese monosyllable test) preoperatively and at 12 months after initial EAS stimulation in three patients with TMPRSS3 mutations who underwent EAS and 27 other patients, and confirmed that they showed relatively good outcomes and were good candidates for CI/EAS (6). Our recent gene expression study, in which the Tmprss3 gene was found to be predominantly expressed within the cochlea (Nishio et al, submitted), supports our clinical data.
CONCLUSION
The present results suggest that a variety of genes may be involved in hearing loss in CI/EAS patients. In the present study, patients with these mutations showed relatively good auditory performance after receiving CI/EAS. Therefore, although many factors may influence outcomes, genetic background can be included as useful in predicting performance after implantation.
Acknowledgments
This study was grant aided by a Health and Labour Sciences Research Grant for Research on rare and intractable diseases and Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labour and Welfare of Japan (S.U.), grant aided by Practical Research Project for Rare / Intractable Disease from Japan Agency for Medical Research and development (AMED), and by a Grant-in-Aid for Scientific Research (A) from the Ministry of Education, Science and Culture of Japan (S.U.).
Footnotes
The authors disclose no conflicts of interest.
This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-No Derivatives License 4.0 (CCBY-NC-ND), where it is permissible to download and share the work provided it is properly cited. The work cannot be changed in any way or used commercially.
REFERENCES
- 1.Eppsteiner RW, Shearer AE, Hildebrand MS, et al. Prediction of cochlear implant performance by genetic mutation: The spiral ganglion hypothesis. Hear Res 2012; 292:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Usami S, Miyagawa M, Nishio SY, et al. Patients with CDH23 mutations and the 1555A>G mitochondrial mutation are good candidates for electric acoustic stimulation (EAS). Acta Otolaryngol 2012; 132:377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miyagawa M, Nishio SY, Ikeda T, et al. Massively parallel DNA sequencing successfully identifies new causative mutations in deafness genes in patients with cochlear implantation and EAS. PLoS One 2013; 8:e75793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyagawa M, Nishio SY, Hattori M, et al. Mutations in the MYO15A gene are a significant cause of nonsyndromic hearing loss: Massively parallel DNA sequencing-based analysis. Ann Otol Rhinol Laryngol 2015; 124 Suppl 1:158S–168S. [DOI] [PubMed] [Google Scholar]
- 5.Tsukada K, Ichinose A, Miyagawa M, et al. Detailed hearing and vestibular profiles in the patients with COCH mutations. Ann Otol Rhinol Laryngol 2015; 124 Suppl 1:100S–110S. [DOI] [PubMed] [Google Scholar]
- 6.Miyagawa M, Nishio SY, Sakurai Y, et al. The patients associated with TMPRSS3 mutations are good candidates for electric acoustic stimulation. Ann Otol Rhinol Laryngol 2015; 124 Suppl 1:193S–204S. [DOI] [PubMed] [Google Scholar]
- 7.Miyagawa M, Nishio SY, Ichinose A, et al. Mutational spectrum and clinical features of patients with ACTG1 mutations identified by massively parallel DNA sequencing. Ann Otol Rhinol Laryngol 2015; 124 Suppl 1:84S–93S. [DOI] [PubMed] [Google Scholar]
- 8.Miyagawa M, Nishio SY, Kumakawa K, et al. Massively parallel DNA sequencing successfully identified seven families with deafness-associated MYO6 mutations: The mutational spectrum and clinical characteristics. Ann Otol Rhinol Laryngol 2015; 124 Suppl 1:148S–157S. [DOI] [PubMed] [Google Scholar]
- 9.Nishio SY, Usami S. Deafness gene variations in a 1120 nonsyndromic hearing loss cohort: Molecular epidemiology and deafness mutation spectrum of patients in Japan. Ann Otol Rhinol Laryngol 2015; 124 Suppl 1:49S–60S. [DOI] [PubMed] [Google Scholar]
- 10.Usami S, Nishio SY, Nagano M, et al. Deafness Gene Study Consortium. Simultaneous screening of multiple mutations by invader assay improves molecular diagnosis of hereditary hearing loss: A multicenter study. PLoS One 2012; 7:e31276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang X, Wang K. wANNOVAR: Annotating genetic variants for personal genomes via the web. J Med Genet 2012; 49:433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.NHLBI Exome Sequencing Project (ESP) Exome Variant Server. http://evs.gs.washington.edu/EVS/ Accessed February 10, 2015. [Google Scholar]
- 15.Narahara M, Higasa K, Nakamura S, et al. Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS One 2014; 9:e100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pollard KS, Hubisz MJ, Rosenbloom KR, et al. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010; 20:110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 18.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res 2009; 19:1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwarz JM, Rodelsperger C, Schuelke M, et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7:575–576. [DOI] [PubMed] [Google Scholar]
- 21.Cooper GM, Stone EA, Asimenos G, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005; 15:901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furutate S, Iwasaki S, Nishio SY, et al. Clinical profile of hearing loss in children with congenital cytomegalovirus (CMV) infection: CMV DNA diagnosis using preserved umbilical cord. Acta Otolaryngol 2011; 131:976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coninx F, Weichbold V, Tsiakpini L, et al. Validation of the LittlEARS((R)) Auditory Questionnaire in children with normal hearing. Int J Pediatr Otorhinolaryngol 2009; 73:1761–1768. [DOI] [PubMed] [Google Scholar]
- 24.May-Mederake B, Kuehn H, Vogel A, et al. Evaluation of auditory development in infants and toddlers who received cochlear implants under the age of 24 months with the LittlEARS) Auditory Questionnaire. Int J Pediatr Otorhinolaryngol 2010; 74:1149–1155. [DOI] [PubMed] [Google Scholar]
- 25.Wu CC, Liu TC, Wang SH, et al. Genetic characteristics in children with cochlear implants and the corresponding auditory performance. Laryngoscope 2011; 121:1287–1293. [DOI] [PubMed] [Google Scholar]
- 26.Park JH, Kim NK, Kim AR, et al. Exploration of molecular genetic etiology for Korean cochlear implantees with severe to profound hearing loss and its implication. Orphanet J Rare Dis 2014; 9:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishio SY, Hattori M, Moteki H, et al. Gene expression profiles of the cochlea and vestibular endorgans: Localization and function of genes causing deafness. Ann Otol Rhinol Laryngol 2015; 124 Suppl 1:6S–48S. [DOI] [PubMed] [Google Scholar]
- 28.Fukushima K, Sugata K, Kasai N, et al. Better speech performance in cochlear implant patients with GJB2-related deafness. Int J Pediatr Otorhinolaryngol 2002; 62:151–157. [DOI] [PubMed] [Google Scholar]
- 29.Green GE, Scott DA, McDonald JM, et al. Performance of cochlear implant recipients with GJB2-related deafness. Am J Med Genet 2002; 109:167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsushiro N, Doi K, Fuse Y, et al. Successful cochlear implantation in prelingual profound deafness resulting from the common 233delC mutation of the GJB2 gene in the Japanese. Laryngoscope 2002; 112:255–261. [DOI] [PubMed] [Google Scholar]
- 31.Bauer PW, Geers AE, Brenner C, et al. The effect of GJB2 allele variants on performance after cochlear implantation. Laryngoscope 2003; 113:2135–2140. [DOI] [PubMed] [Google Scholar]
- 32.Sinnathuray AR, Toner JG, Geddis A, et al. Auditory perception and speech discrimination after cochlear implantation in patients with connexin 26 (GJB2) gene-related deafness. Otol Neurotol 2004; 25:930–934. [DOI] [PubMed] [Google Scholar]
- 33.Sinnathuray AR, Toner JG, Clarke-Lyttle J, et al. Connexin 26 (GJB2) gene-related deafness and speech intelligibility after cochlear implantation. Otol Neurotol 2004; 25:935–942. [DOI] [PubMed] [Google Scholar]
- 34.Cullen RD, Buchman CA, Brown CJ, et al. Cochlear implantation for children with GJB2-related deafness. Laryngoscope 2004; 114:1415–1419. [DOI] [PubMed] [Google Scholar]
- 35.Lustig LR, Lin D, Venick H, et al. GJB2 gene mutations in cochlear implant recipients: Prevalence and impact on outcome. Arch Otolaryngol Head Neck Surg 1004; 130:541–546. [DOI] [PubMed] [Google Scholar]
- 36.Taitelbaum-Swead R, Brownstein Z, Muchnik C, et al. Connexin-associated deafness and speech perception outcome of cochlear implantation. Arch Otolaryngol Head Neck Surg 2006; 132:495–500. [DOI] [PubMed] [Google Scholar]
- 37.Kawasaki A, Fukushima K, Kataoka Y, et al. Using assessment of higher brain functions of children with GJB2-associated deafness and cochlear implants as a procedure to evaluate language development. Int J Pediatr Otorhinolaryngol 2006; 70:1343–1349. [DOI] [PubMed] [Google Scholar]
- 38.Yoshida H, Takahashi H, Kanda Y, et al. Long term speech perception after cochlear implant in pediatric patients with GJB2 mutations. Auris Nasus Larynx 2013; 40:435–439. [DOI] [PubMed] [Google Scholar]
- 39.Yan YJ, Li Y, Yang T, et al. The effect of GJB2 and SLC26A4 gene mutations on rehabilitative outcomes in pediatric cochlear implant patients. Eur Arch Otorhinolaryngol: 2013; 270:2865–2870. [DOI] [PubMed] [Google Scholar]
- 40.Rouillon I, Marcolla A, Roux I, et al. Results of cochlear implantation in two children with mutations in the OTOF gene. Int J Pediatr Otorhinolaryngol 2006; 70:689–696. [DOI] [PubMed] [Google Scholar]
- 41.Zhang LP, Chai YC, Yang T, et al. Identification of novel OTOF compound heterozygous mutations by targeted next-generation sequencing in a Chinese patient with auditory neuropathy spectrum disorder. Int J Pediatr Otorhinolaryngol 2013; 77:1749–1752. [DOI] [PubMed] [Google Scholar]
- 42.Volk AE, Lang-Roth R, Yigit G, et al. A novel MYO6 splice site mutation causes autosomal dominant sensorineural hearing loss type DFNA22 with a favourable outcome after cochlear implantation. Audiol Neurootol 2013; 18:192–199. [DOI] [PubMed] [Google Scholar]
- 43.Vermeire K, Brokx JP, Wuyts FL, et al. Good speech recognition and quality-of-life scores after cochlear implantation in patients with DFNA9. Otol Neurotol 2006; 27:44–49. [DOI] [PubMed] [Google Scholar]
- 44.Nishiyama N, Kawano A, Kawaguchi S, et al. Cochlear implantation in a patient with Epstein syndrome. Auris Nasus Larynx 2013; 40:409–412. [DOI] [PubMed] [Google Scholar]
- 45.Pecci A, Verver EJ, Schlegel N, et al. Cochlear implantation is safe and effective in patients with MYH9-related disease. Orphanet J Rare Dis 2014; 9:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elbracht M, Senderek J, Eggermann T, et al. Autosomal recessive postlingual hearing loss (DFNB8): Compound heterozygosity for two novel TMPRSS3 mutations in German siblings. J Med Genet 2007; 44:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weegerink NJ, Schraders M, Oostrik J, et al. Genotype-phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J Assoc Res Otolaryngol 2011; 12:753–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee HK, Lee SH, Lee KY, et al. Novel POU3F4 mutations and clinical features of DFN3 patients with cochlear implants. Clin Genet 2009; 75:572–575. [DOI] [PubMed] [Google Scholar]
- 49.Stankovic KM, Hennessey AM, Herrmann B, et al. Cochlear implantation in children with congenital X-linked deafness due to novel mutations in POU3F4 gene. Ann Otol Rhinol Laryngol 2010; 119:815–822. [DOI] [PubMed] [Google Scholar]
- 50.Gong WX, Gong RZ, Zhao B. HRCT and MRI findings in X-linked non-syndromic deafness patients with a POU3F4 mutation. Int J Pediatr Otorhinolaryngol 2014; 78:1756–1762. [DOI] [PubMed] [Google Scholar]
- 51.Moteki H, Shearer AE, Izumi S, et al. De novo mutation in X-linked hearing loss-associated POU3F4 in a sporadic case of congenital hearing loss. Ann Otol Rhinol Laryngol 2015; 124 Suppl 1:169S–176S. [DOI] [PMC free article] [PubMed] [Google Scholar]