

Abstract
Diabetes (DM) complicates 3%–10% of pregnancies, resulting in significant maternal and neonatal morbidity and mortality. DM pregnancies are also associated with vascular dysfunction, including blunted nitric oxide (NO) output, but it remains unclear why. Herein we examine changes in endothelial NO production and its relationship to Ca2+ signaling in endothelial cells of intact umbilical veins from control versus gestational diabetic (GDM) or preexisting diabetic subjects. We have previously reported that endothelial cells of intact vessels show sustained Ca2+ bursting in response to ATP, and these bursts drive prolonged NO production. Herein we show that in both GDM and DM pregnancies, the incidence of Ca2+ bursts remains similar, but there is a reduction in overall sustained phase Ca2+ mobilization and a reduction in NO output. Further studies show damage has occurred at the level of NOS3 protein itself. Since exposure to DM serum is known to impair normal human umbilical vein endothelial cell (HUVEC) function, we further studied the ability of HUVEC to signal through Ca2+ after they were isolated from DM and GDM subjects and maintained in culture for several days. These HUVEC showed differences in the rate of Ca2+ bursting, with DM > GDM = control HUVEC. Both GDM- and DM-derived HUVEC showed smaller Ca2+ bursts that were less capable of NOS3 activation compared to control HUVEC. We conclude that HUVEC from DM and GDM subjects are reprogrammed such that the Ca2+ bursting peak shape and duration are permanently impaired. This may explain why ROS therapy alone is not effective in DM and GDM subjects.
Keywords: calcium, diabetes, endothelium, nitric oxide, pregnancy
INTRODUCTION
In 2000 the prevalence of diabetes (DM) for all age-groups worldwide was estimated to be 2.8% [1]. DM complicates 3%–10% of pregnancies and results in significant maternal and neonatal morbidity and mortality. One of the many challenges with pregnancies complicated by DM is to maintain optimal maternal blood sugar homeostasis for both the mother and her fetus [2]. It is not surprising that there is a spectrum of clinical severity of DM in pregnancy that is correlated with the level of maternal hyperglycemia. DM in pregnancy can be further categorized as type 1, type 2, and gestational DM (GDM) [3]. A baby born to a mother with hyperglycemia due to poorly controlled blood sugar may be macrosomic (>90th% for gestational age) or growth restricted (<10% for gestational age), depending on the duration of maternal DM and underlying vascular disease [4]. As a result, even ‘normal'-sized babies born to women with DM complicated by vascular disease may in fact be pathologically growth restricted [5].
Normal pregnancy promotes changes in maternal nutrient metabolism to meet the increased energy needs of the mother and the growth of the fetus. This includes changes in glucose homeostasis that can also be described as an insulin-resistant state. In pregnancies complicated by GDM, there is a similar increase in basal endogenous insulin production as in normal glucose-tolerant pregnancies. While the data on changes in glucose metabolism in pregnant women with type 1 DM are sparse, Schmitz et al. [6] reported a 50% decrease in insulin sensitivity in late gestation. On a cellular level, endothelial dysfunction is also observed, most commonly as a reduced response to the vasodilators acetylcholine, bradykinin, histamine, and adenosine diphosphate, and this has been proposed to underlie the development of DM vascular disease [7].
In normal pregnancy, NO in particular serves as an important vasodilator necessary to maintain low vascular resistance in the face of otherwise increased cardiac output [8]. A failure to generally increase endothelial NO output has been implicated in a number of maternal hypertensive and vascular diseases of pregnancy, including DM [7]. It has been proposed that one particular issue with NO is its ability to react with reactive oxygen species (ROS) prevalent in the disease state, thereby resulting in the destruction of bioactive NO [7]. Clinical trials of antioxidants have attempted to overcome this problem, but such trials have not proven as effective as predicted [7], and the question therefore is why. One possibility is that we have been misled because NO itself has not been routinely measured in studies of DM subjects. A further possibility is that ROS scavenging of NO is not the only mechanism, and the Ca2+ activation signal to NOS3 is also being adversely affected, as we have recently shown occurs in pre-eclampsia (PE) [9]. Certainly ROS can affect several components of the Ca2+ signaling response, including transient receptor potential canonical (TRPC) channels operating in endothelial cells [10]. To that end we report our studies of Ca2+ signaling and NO output of intact umbilical vein endothelium in control versus GDM and DM subjects and also changes in Ca2+ signaling in the corresponding human umbilical vein endothelial cells (HUVEC) in primary culture in order to clarify these questions for the first time.
MATERIALS AND METHODS
This was a prospective cohort study approved by the Institutional Review Boards of the University of Wisconsin and Meriter Hospital. All study participants provided informed consent before participation. Study inclusion criteria were pregnant women with a singleton fetus with a diagnosis of DM (pregestational or gestational) with or without concomitant hypertension or vascular disease. Subjects with PE, however, were excluded to avoid superimposed confounding variables. Other study exclusion criteria were diagnoses of chorioamnionitis, presence of moderate/thick meconium, multiple gestation, and maternal age less than 18 yr. Subjects included a total n = 15: patients with diagnosis of pre-existing DM (n = 6, all type 1 DM), patients with diagnosis of GDM (n = 4) and control subjects (n = 5). Furthermore, cultured HUVEC were also prepared. All HUVEC were isolated from the umbilical cords per our previously published protocol [9], and cells were frozen for further analysis.
Characteristics of Study Subjects and Their Newborns
The clinical data for mothers enrolled in this study are shown in Table 1. Table 2 lists the parameters for the newborns of these subjects. All abbreviations for clinical parameters are also defined in Tables 1 and 2. Note that while birth-weight differences did not achieve significance between groups, body mass index (BMI) for GDM was significantly higher at P < 0.05 relative to DM subjects, as analyzed by one-way ANOVA (Student-Newman-Keuls test).
TABLE 1.
Maternal parameters.a

M, multiparous; P, primiparous; BMI, body mass index in kg/m2 determined at the time of delivery; Hgb A1C, glycosylated hemoglobin; DM med, diabetic medication (insulin infusion pump, subcutaneous insulin); GHTN, gestational hypertension; CHTN, chronic hypertension; BP, blood pressure; Incr LFTs, increased liver function tests; N/A, not available.
While birth weight did not achieve significance between groups, BMI for GDM was significant at P < 0.05 relative to DM subjects, one-way ANOVA (Student-Newman-Keuls test).
TABLE 2.
Delivery parameters for babies.a

CD, cesarean delivery; SVD, spontaneous vaginal delivery; h/o CD, history of cesarean delivery; IOL, induction of labor; NICU, neonatal intensive care unit; NRFWB, nonreassuring fetal well-being; FTP, failure to progress (FTP) in labor; N/A, not available.
Birth weights for each group were not significantly different.
The age range of the control subjects was evenly distributed across all age-groups from 18 to 45 yr old; most were multiparous, and all were Caucasian. The average BMI in this group was 33.75 ± 3.77 kg/m2, which is considered class I obesity. The average gestational age (GA) at delivery was 38.8 ± 0.45 wk. None of the subjects had the following: gestational hypertension (GHTN), chronic hypertension (CHTN), severe range blood pressure (greater than 160/110 mm Hg), increased liver function tests (LFT), thrombocytopenia (platelets less than 100 000), retinopathy, or seizures. All control group babies were delivered via cesarean delivery (CD) for the history of prior CD. The average weight of the babies at birth was 3553.00 ± 342.08 g. The average Apgar score at 1 min was 9 ± 0, and the average Apgar score at 5 min was 9.33 ± 0.58. None of the babies in this group required admission to the neonatal intensive care unit (NICU).
The majority of the DM subjects were in the 38- to 45-yr-old age range; most were primiparous, and all were Caucasian. The average BMI in this group was 39.2 ± 6.61 kg/m2, which is considered class II obesity. The average Hgb A1C was 5.8 ± 0.42%. The average GA at delivery was 38.2 ±1.10 wk. The subjects used either SQ insulin injections or an insulin pump for glucose control, and the average blood sugar in the 24 h prior to delivery was 112.68 ± 13.74 mg/dl. All subjects had less than 300 mg of urinary protein excretion on their most recent 24-h urine protein collection analyses. None of the subjects had the following: GHTN, CHTN, severe range blood pressure (greater than 160/110 mm Hg), increased LFT, thrombocytopenia (platelets less than 100 000), retinopathy, or seizures. All DM babies were delivered via CD for a variety of indications: history of prior CD, nonreassuring fetal heart tracing, failure to progress (FTP) in labor and breech presentation. The average weight of the babies at birth was 3535.40 ± 662.84 g. The average Apgar score at 1 min was 7.6 ± 2.7, and the average Apgar score at 5 min was 8.4 ± 0.89. None of the babies in this group required admission to the NICU.
The age range of GDM subjects was evenly distributed across all age-groups from 18 to 45 yr old; most were primiparous, and most were Caucasian. The average BMI in this group was 45.0 ± 4.83 kg/m2, which is considered class III obesity. The average Hgb A1C was 5.77 ± 0.45%. The average GA at delivery was 37.5 ± 4.43 wk. The subjects used either SQ insulin injections or the American Diabetic Association diet for glucose control, and the average blood sugar in the 24 h prior to delivery was 108 ± 25.53 mg/dl. From the information available, one subject underwent a 24-h urine evaluation and had less than 300 mg of urinary protein excretion on the most recent 24-h urine protein collection analysis. None of the subjects had the following: GHTN, CHTN, severe range blood pressure (greater than 160/110 mm Hg), increased LFT, thrombocytopenia (platelets less than 100 000), retinopathy, or seizures. All GDM babies, except one, were delivered via CD for a variety of indications: history of prior CD, nonreassuring fetal well-being, and scheduled induction of labor. The average weight of the babies at birth was 3847.75 ± 481.83 g. The average Apgar score at 1 min was 9 ± 0, and the average Apgar score at 5 min was 9.25 ± 0.5. None of the babies in this group required admission to the NICU.
Materials
The endothelial cell agonist ATP (disodium salt) and all other general laboratory chemicals were purchased from Sigma-Aldrich Corp. (St. Louis, MO) to at least American Chemical Society standards unless stated otherwise. Minimum essential medium (MEM) and Medium 199 (M199), Fura-2-AM, DAF2-DA, Pluronic F-127 and all other general cell culture reagents were purchased from Life Technologies, Inc. (Grand Island, NY). Fetal bovine serum used in culture medium was from Life Technologies, and endothelial cell growth supplement (ECGS) was from Upstate (now EMD Millipore, Billerica, MA). Heparin was the sodium salt form, grade 1A, from porcine intestinal mucosa (Sigma; catalog no. H3149-1MU). Glass-bottom microwell dishes (35 mm) for Ca2+ imaging studies were from MatTek Corporation (Ashland, MA). All routine Western blot supplies were purchased from Bio-Rad (Hercules, CA) unless otherwise stated.
Physiologic Buffers
Umbilical cords were collected in PBS pH 7.4 for transport on ice to the laboratory. For nonsterile condition, acute experiments on vessels and cells, our standard buffer was Krebs buffer (125 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1 mM KH2PO4, 6 mM glucose, 2 mM CaCl2, 25 mM HEPES pH 7.4). For cell isolations, the standard wash buffer and centrifugation buffer was M199 with added 1% penicillin/streptomycin solution and 4 μg/ml gentamycin (M199/P/S/G). For cell digestion, this M199/P/S/G was used with added 0.5% BSA (fraction V; Sigma), and this in turn was sterilized at the time of preparation by 0.45-μm filtration. For HUVEC cell culture, standard growth medium was based on MEM with 20% serum and added penicillin, streptomycin, and gentamycin as above as used for UAEC preparation [11]. HUVEC, however, also requires for consistent growth the addition of ECGS (3.75 mg/100 ml) and heparin (0.1 mg/ml). This standard growth medium is referred to as HEH (human, ECGS, heparin) medium.
Simultaneous Monitoring of [Ca2+]i and NO in Intact UV Endothelium
Methods of dual imaging of [Ca2+]i response to vasodilation and simultaneous NO production in intact umbilical vein endothelium (UV endo) preparations were the same as used previously utilized to study the vasculature of normal and PE pregnancies by this group [9]. After fine dissection, vessels were loaded with both 10 μM DAF2-DA (to detect NO production) and 10 μM Fura-2-AM (a [Ca2+]i sensor) with 0.05% Pluronic F-127 in standard Krebs buffer (see above) for 90 min, after which the loading dye medium was replaced, and dye hydrolysis was allowed to proceed for 30 min. The vessel chamber was mounted on an inverted microscope (Diaphot 150; Nikon, Melville, NY) and imaged with a 20× phase/fluor objective focused on the endothelium. Individual endothelial cells were identified, and once their outline was defined, they were monitored using dual excitation (switching at 340 and 380 nm) for Fura-2 and 485-nm excitation for DAF2 (high-speed Lambda 10-2; Sutter Instruments, Novato, CA). Emission was measured at 535 nm. The fluorescence images were then recorded in real time by a PixelFly camera (Cooke, Romulus, MI). Imaging and analysis software (InCyt Im3; Intracellular Imaging, Cincinnati, OH) was used to acquire, digitize, and store all images and data for offline analysis. Intracellular NO was calculated as the relative measure of fluorescence (F/F0), and intracellular free [Ca2+]i was calculated in real time by comparison to a ratiometric standard curve established using buffers of known free [Ca2+].
HUVEC Cell Preparation
The umbilical cord segments (approximately 4 cm from an unclamped area of the cord) were prepared, and a blunt-end needle was placed into the vein lumen. After thoroughly rinsing the vessel free of blood using ∼10 ml M199/P/S/G, the needle in the vessel was then tied off; the vessel was flushed one more time and incubated for 15 min at 37°C. The vessel was then removed from the incubator and inflated with M199/P/S/G/0.5% BSA containing 2 mg/ml collagenase B (Roche Molecular Biochemicals, Indianapolis, IN) through a Luer-Lock three-way tap before clamping off for digestion. Digestion was allowed to proceed for 25 min at 37°C before flushing the collagenase solution and endothelial cell sheets from the inner surface of the vessels using 10 ml M199/P/S/G/0.5% BSA medium. Recovered cells were pelleted by centrifugation (2 min, 300 × g) and resuspended in HEH media before plating to three wells of a six-well plate (Falcon Primaria, Fisher Scientific, Chicago, IL). Medium was changed after 4 h to remove unattached cells (including the few vascular smooth muscle cells present). Cells were grown to ∼70% confluence, when they were selectively trypsinized from the dish and replated to 3 × 60-mm dishes. Cells were grown to 80%–90% confluence before recovery and freezing (cells from two dishes in medium plus 10% dimethyl sulfoxide) or one dish passaged to a single T75 flask. Cells were grown to 95% confluence and trypsinized for freezing in 35 cryovials at passage 3. Each vial was then used for plating to four to six glass-bottom dishes for fluorescent imaging.
In the development of this method for the preparation of early passage HUVEC, purity was extensively evaluated. All cells showed uniform uptake of Alexa 484 Low Density Lipoprotein and morphology was consistent in monolayers, all consistent with the absence of contaminating vascular smooth muscle or fibroblast cells. Attempts to purify cells further using fluorescence-activated cell sorting did not alter or improve cell purity. All cell preparations uniformly responded to endothelial agonists ATP and Bradykinin with a rapid elevation in [Ca2+]i. Cells also responded to vascular endothelial growth factor (J. Krupp and D. Boeldt, unpublished data) in much the same way as ovine uterine artery endothelial cell preparations investigated using identical methodology [12].
Western Blot
At the time of vessel imaging, parallel sections were also digested with collagenase and flushed with M199/P/S/G without BSA. Cells isolated using same method above were then pelleted by centrifugation and immediately solubilized in 50 μl protein lysis buffer (50 mM HEPES pH 7.5, 4 mM Na4P2O7-10 H2O, 100 mM NaCl, 10 mM EDTA, 10 mM NaF, 2 mM Na3[VO4]2, 1% Triton X-100, 5 μg/ml leupeptin, 5 μg/ml aprotinin, and 1.0 mM phenylmethylsulfonylfluoride) and stored at −70°C. Samples were later sonicated, and protein concentration was determined using the bicinchoninic acid assay (Sigma) run against BSA protein standards. Western analyses, as described by Sullivan et al. [11] and Yi et al. [13], were then performed using 10 μg cell protein per lane on 7.5% polyacrylamide gels followed by transfer to Immobilon polyvinylidene fluoride membrane (100 V, 2 h). Total NOS3 was detected using anti-eNOS antibody (1:1000; #610297; BD BioSciences, San Jose, CA) and rabbit anti-mouse IgG horseradish peroxidase (HRP)-conjugated F(ab′)2 secondary antibody (1:2000; #AQ160P; EMD Millipore). Total GJA1 was detected using anti-Cx43 antibody (1:5000; #C6219; Sigma) and goat anti-rabbit HRP-conjugated secondary antibody (1:3000; #7074; Cell Signaling Technology, Danvers, MA).
[Ca2+]i Imaging in HUVEC
Cultured HUVEC stored frozen at passage 3 were plated to one well of a six-well dish in HEH media on Day 1 and trypsinized at 95% confluence as described above (Day 3) to 6 × 35-mm glass-bottom dishes in HEH media. Cells were imaged at 95% confluence (Day 7). On the day of imaging, HUVEC were incubated in 10 μM of membrane-permeable Fura-2 AM/0.05% Pluronic acid F127 dissolved in 1 ml HEH media for 1 h at 37°C. The cells were washed with Krebs buffer, and 1.8 ml Krebs buffer were added to each dish for an additional 30 min at room temperature to allow complete ester hydrolysis. All of the subsequent steps were also performed at room temperature. The dish was placed in the field of view, and Fura-2 loading was verified by viewing at 380-nm UV excitation on a Nikon inverted microscope as used for UV endo above. The data for approximately 80 noncontiguous cells were then recorded simultaneously for an initial 5-min period to obtain baseline [Ca2+]i prior to the addition of 100 μM ATP. The cells were incubated with agonist for a further 30 min. From the ratio of emission at 340- and 380-nm excitation wavelengths, the intracellular [Ca2+]i was calculated by comparison to a ratiometric [Ca2+]i standard curve using InCyt Im2 software.
Statistical Analyses
Results were reported as the mean ± SEM. For all studies by Western blot analysis, mean data from at least n = 5 subjects per group were analyzed by Student t-test and ANOVA; results were considered significant at P < 0.05. For imaging experiments on freshly isolated tissue, vessels from subjects in each group were imaged as required. Recordings from at least 20 individual cells per vessel segment were made for each treatment and each subject. Each treatment in [Ca2+]i imaging of Fura-2-loaded HUVEC in primary culture was replicated on multiple dishes as indicated with multiple individual cells monitored simultaneously on each dish (typically <80). Student t-test or one-way ANOVA were used to analyze the data as appropriate, and P < 0.05 was considered significant. For measures of a response at any given time, tracings were aligned for this purpose with the time of agonist addition being zero.
RESULTS
Ex Vivo Studies of Altered Function of UV Endo at the Level of Ca2+ Signaling and NO
Pregnancy has long been known to promote greater vasodilatory function in the uterine arteries in particular but also the systemic vasculature in general. Studies in endothelial cells from both maternal arterial and venous beds have shown that more sustained Ca2+ signaling is observed in response to a variety of agonists in pregnancy, and such sustained bursting is mirrored in the endothelium of the fetal vasculature [14, 15]. Representative recordings from individual cells of the intact UV endo from normal pregnancy are shown in Figure 1. Typically, individual cells of the UV endo from control subjects showed from two to four [Ca2+]i bursts, and sustained NO output continued as long as these bursts were seen (Fig. 1A). As a control, cells were also stimulated with ionomycin to raise [Ca2+]i independent of receptor and fully activate the available NOS3 pool. Ionomycin treatment promoted a sustained elevation of [Ca2+]i as well as a more robust and sustained increase in NO, as expected (Fig. 1D).
FIG. 1.

Representative single cell tracings from UV endo. Representative tracings of DAF-2 and Fura-2 output recorded from cells located in the intact endothelium of umbilical vein segments (UV endo) are shown. A representative recording from control subject is shown in A. Note that UV endo was treated with the physiologic agonist ATP (100 μM) at the time shown by the arrow (time 0). The comparable data are also shown for a vessel segment from a GDM subject (B) or a DM subject (C). As a control to bypass the receptor signaling apparatus and indicate total functional NOS3 in each cell, Ca2+ ionophore (ionomycin 5 μM) was also added as indicated at time 0. D) The ionomycin response of a vessel segment from control subjects. Recording in all cases was continued for 30 min.
Previously, we have shown in PE subjects that the repeated Ca2+ burst pattern is blunted in duration, and it is this failure of Ca2+ bursting that contributes greatly to impaired NO output. In contrast, individual cells of UV endo from DM and GDM pregnancies treated with ATP continued to show bursting for durations similar to that of UV endo from normal pregnancy (Fig. 1, A–C). While representative traces give some illustration of the data, they are not a summary of the whole vessel response. The overall combined data was summarized further in two ways to allow comparisons with statistical analyses. The percentage of cells that showed each successive Ca2+ burst is shown in Figure 2. Clearly, there was no change from control in the percentage of cells showing detectable bursts in DM or GDM pregnancies, but this does not take into account the size of the individual bursts, only their rate. As an alternative summary, the overall average of all cell tracings for [Ca2+]i and NO in response to ATP or ionomycin was calculated for each treatment group, as shown in Figure 3. While this no longer shows individual bursts, the resulting average takes into account both the rate and the size of individual bursts. This form of average tracing analysis shows the overall mean [Ca2+]i above basal control and thus indicates if the respective patient groups can maintain elevated [Ca2+]i for any length of time following the initial [Ca2+]i peak of release from the endoplasmic reticulum. While the initial Ca2+ peaks were of similar height, the average sustained phase [Ca2+]i levels were clearly different, with UV endo from control patients maintaining elevated [Ca2+]i in response to ATP above basal for longer than was observed in UV endo from GDM or DM patient populations. Likewise, the overall sustained NO output seen in control UV endo appeared greater than that in UV endo from GDM and DM subjects. Quantification of these apparent differences were then performed to establish significance in each case. Figure 4, A and B, summarizes the combined average levels of peak [Ca2+]i above basal at 600 sec (where the maximum differences are noted) and at 900 sec (where there has previously been such measures in uterine artery) [13] and subsequent maximal NO output at 1200–1500 sec in each case for ATP stimulation. Such analysis showed that the reduced average Ca2+ per cell at 600 sec in GDM- and DM-derived UV endo was significantly less than in control and paralleled closely the reduced overall NO output in each case. The reduction in both [Ca2+]i at 600 sec and NO at 1200–1500 sec in GDM subjects was not different from that in DM subjects.
FIG. 2.
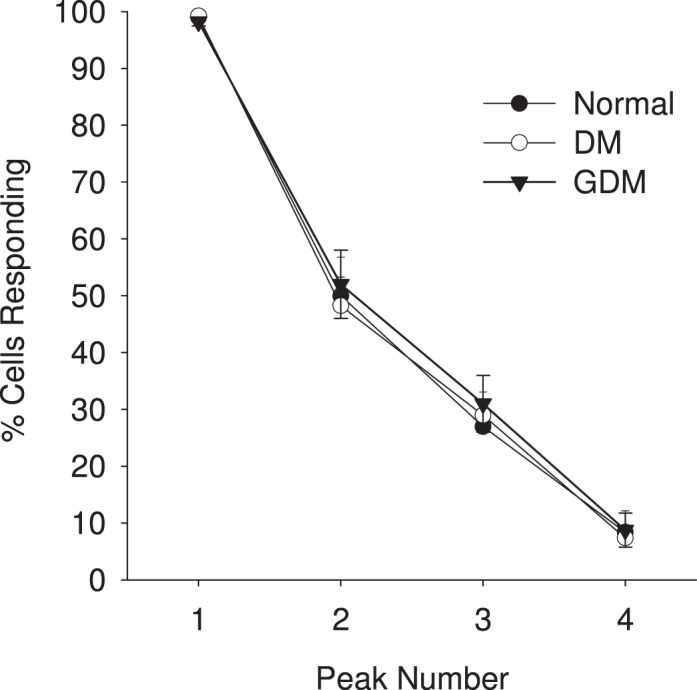
Percent (%) of UV endo cells showing each successive burst after stimulation with 100 μM ATP. Vessels from each treatment group as indicated in Figure 1 were assessed for [Ca2+]i bursts and the data plotted as shown for each group. Incidence of [Ca2+]i bursts was determined for 40 or more individual cells per image field. Data are means ± SEM of such analysis of six to nine vein segments per treatment group. Percentage of cells responding in the vessel segment with each successive Ca2+ burst peak are shown. One-way ANOVA was used to analyze the data. No significant differences (P < 0.05) in occurrence of bursts were observed in UV endo from control versus GDM versus DM UV endo, but this does not take into account burst size; see average tracings in Figure 3.
FIG. 3.

Average Ca2+ and NO tracings from UV endo cells. Overall combined average tracings from UV endo loaded with DAF-2 DA and Fura-2 AM dissected from the cords from DM, GDM and control subjects are shown. Vessel segments were treated with the physiologic agonist ATP (100 μM; column A) or a receptor-independent Ca2+ ionophore (ionomycin 5 μM; column B) that were added as indicated on the X axis as time 0. Data were first calculated as an average trace of all responding cells for each vessel segment, and then combined mean ± SEM was calculated from six to nine UV segments per group. UV endo from control patients maintain an elevated average [Ca2+]i level in response to ATP longer than is observed in GDM or DM patient populations, and a sustained NO output is also seen to a greater level in control UV endo than in those from GDM and DM subjects (for quantification and statistical analysis, see Fig. 4).
FIG. 4.
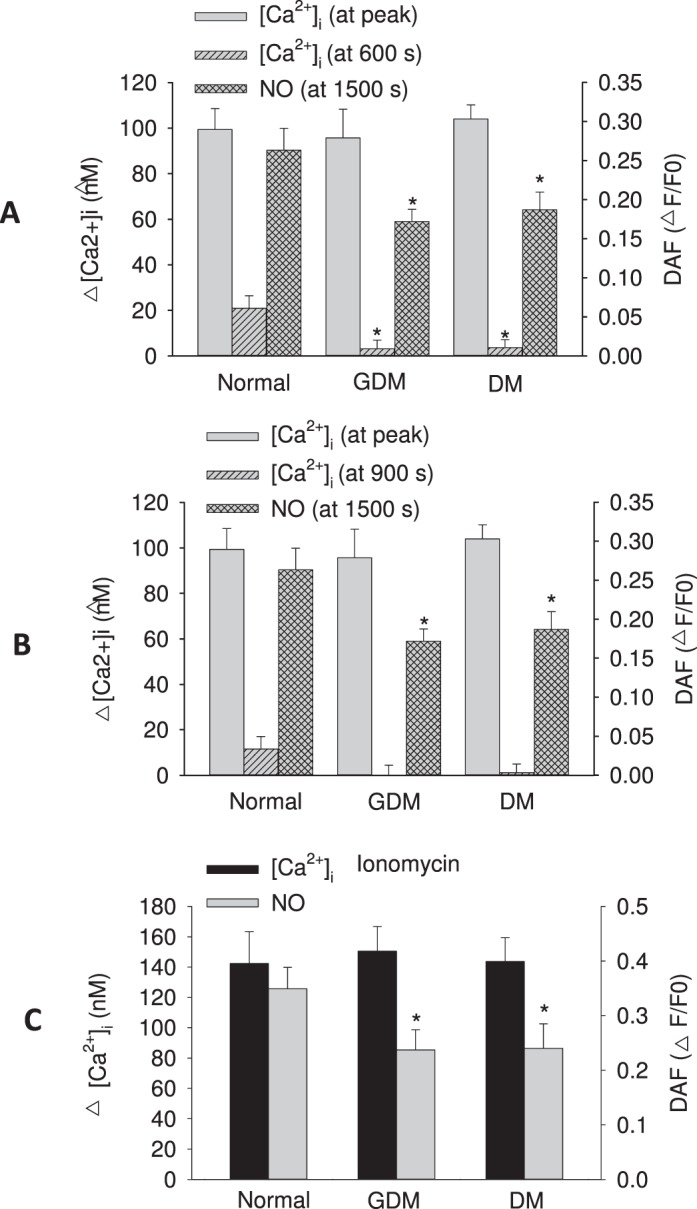
Quantification of overall average Ca2+ and NO responses from UV endo (from Fig. 3). Quantification of average ATP (100 μM) stimulated change in [Ca2+]i above basal (basal was typically in the range ∼80–100 nM) measured as the initial peak maximum and the sustained phase [Ca2+]i at 600 sec (A) or 900 sec (B) for all UV endo (control, DM and GDM). Data are shown alongside measures of total NO produced (measured at 1200–1500 sec). For comparison, the response to receptor-independent Ca2+ ionophore (ionomycin 5 μM) stimulation is also shown in C. Data are calculated from the same UV endo data shown in Figure 3 and represent combined means ± SEM of data from six to nine vein segments per group. Student t-test or one-way ANOVA were used to analyze the data as appropriate. In all cases, significant differences from control are indicated (*P < 0.05).
We also monitored the size of the activatable pool of NOS3 per cell by averaging NO output per cell in response to ionomycin. Figure 4C shows that a reduction of NO output in response to ionomycin was observed in cords from GDM and DM subjects relative to controls, and this was similar to the reduction in response to ATP. Such a finding infers that a reduced size of activatable NOS3 pool itself is indeed a contributing problem, perhaps as a consequence of direct damage to NOS3 by toxic ROS known to be perpetuated by the hyperglycemic environment of DM. In view of this finding, additional Western blot analyses were undertaken for NOS3 and for GJA1 (connexin 43) gap junction protein. Western blot data for GDM and DM subjects demonstrate a similar level of NOS3 expression (normalized to HSP90AA1) when compared to controls (Fig. 5). Although for GJA1 it appeared that the level in DM subjects may be lower, this was not found to be significant compared to control. These results suggest that it was not a deficiency of protein expression that caused the reduction of NOS3 activity in response to ATP or indeed ionomycin challenge in intact vessels, and such a finding is entirely consistent with prior ROS damage to the cytosolic NOS3 protein pool in vivo, thereby reducing the activatable NOS3 pool.
FIG. 5.
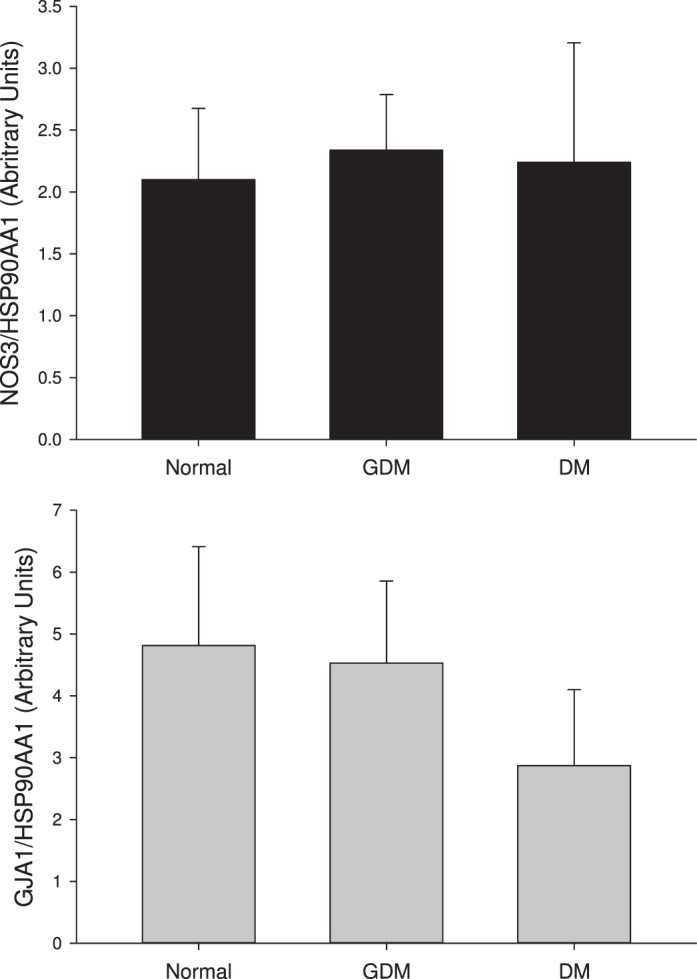
NOS3, GJA1 and HSP 90 expression levels in UV endo cells. Western blot analyses for NOS3 as well as for GJA1 gap junction protein (Cx43) and a control marker heat shock protein 90 (HSP90AA1) for n = 3–6 subjects per group. Western blot data for control, DM and GDM subjects demonstrate a normal range of expression, depicted by the distribution of the control data. While scatter of the data is large, and thus statistical analysis of this limited data set has little value, it is clear that immunodetectable NOS3 levels for DM and GDM subjects were not substantially reduced (differences between groups not significant).
In Vitro Studies of Altered Programming in Primary Cultures of HUVEC
There are no studies to date that address the function and dysfunction of HUVEC obtained from DM and/or GDM patients. Herein, we show representative tracings for individual HUVEC prepared in primary culture from control, DM and GDM subjects. Similar to control HUVEC, Ca2+ signaling in the HUVEC of DM and GDM showed an initial [Ca2+]i peak followed by repeated bursts (Fig. 6) that were synchronized between cells (not shown). However, there was some suggestion that the repeated bursts were smaller and more rapid in cells from DM and GDM subjects. For this reason, the same analysis of the percentage of cells showing successive bursts and the overall average responses were calculated as before and are shown in Figure 7A. Of particular note, analysis of percentage of cells showing each burst confirmed that the number of cells showing sustained bursts was significantly higher in cells derived from DM subjects than in cells from control or GDM subjects, thus revealing the first clear difference between the effects of DM and GDM pregnancy. In spite of the more rapid bursts, the average tracing for all cells combined was still lower for HUVEC from DM and GDM pregnancy relative to controls (Fig. 7B). The reason is that although the rate of Ca2+ bursting was accelerated in HUVEC from DM > GDM cords, the actual burst shape is also subject to change. The observations of Tran et al. [16] tell us that NOS3 activation is initiated through the range 100–300 nM. The shape of the third burst corresponding to that activation phase (after correction for basal Ca2+ levels) is shown for each subject group in Figure 7C. Of particular note, the time spent in this activation range was far less for bursts in HUVEC from DM and GDM pregnancies than in HUVEC from control pregnancies, so less effective NO production would result, even in the face of more rapid bursts.
FIG. 6.
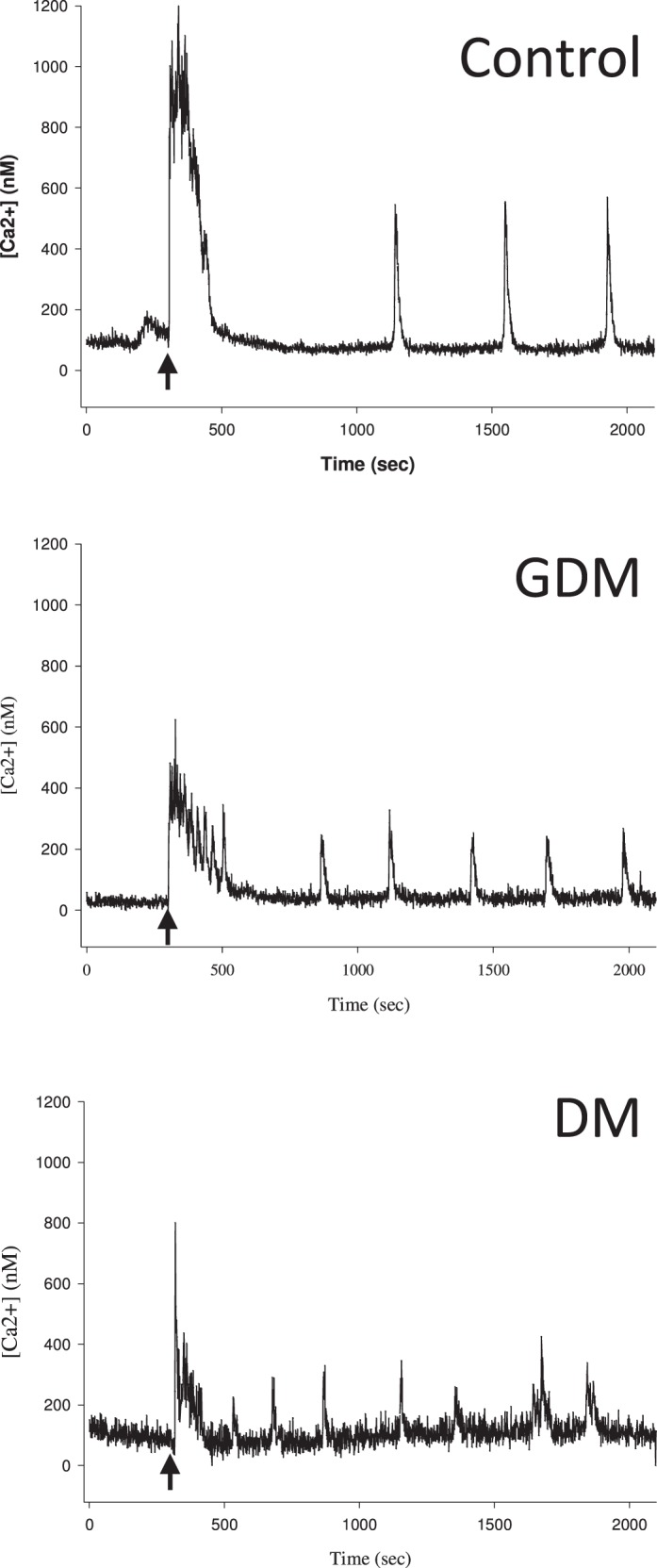
Representative Ca2+ tracings from HUVEC cells. Representative tracings from Fura-2-loaded primary HUVEC from each treatment group (control, DM and GDM subjects) are shown. Each representative tracing is from one cell from one patient in each group (control, DM and GDM). After recording basal [Ca2+]i levels to establish baseline, ATP (100 μM) was added at 300 sec (as indicated by arrow). Recording was then continued for 30 min. Of note, the DM and GDM tracing consistently showed more bursts but of smaller magnitude.
FIG. 7.

Average tracings and Ca2+ burst events from HUVEC cells. For each panel, data were first the average from ∼80 cells per dish on six combined dishes per patient, then these values were combined to give the overall combined mean and SEM for control: n = 5 patients; GDM: n = 4 patients; DM: n = 6 patients. Student t-test or one-way ANOVA were used to analyze the data as appropriate. Significance is as indicated (*P < 0.05). A) The percent of HUVEC showing each successive burst after stimulation with 100 μM ATP is shown for each subject group. B) The overall average tracing of primary HUVEC responses to ATP (100 μM) stimulation are shown, corrected for basal, which was typically in the range 30–65 nM prior to ATP addition. ATP (100 μM) was added at time 0. These results suggest that in spite of increased burst rate, both the initial peak height and the sustained phase average [Ca2+]i are still reduced in GDM and DM subjects. C) The average third burst elevation of Ca2+ above basal (set at 0) for combined cells from each patient donor was calculated over a period of ±30 sec from maximum, and then data from each patient were combined for each treatment group to give mean and SEM as shown. For symbols, key see A. The dashed lines represent the predicted activation range for NOS3 relative to this basal-corrected scale, as predicted by the data of Tran et al. [16].
DISCUSSION
There are few studies published of HUVEC function in DM or indeed GDM subjects. A novel study by Salvolini et al. [17] sought to evaluate the effect of plasma from insulin-dependent DM pregnant patients on NOS3 activity, intracellular [Ca2+]i and Na+/K+-adenosine triphosphatase activity in cultured HUVEC from normal subjects [17]. Nonetheless, this investigative approach assumes that all dysfunction in diabetes is induced by circulating factors (including ROS), so simple correction of these factors or even their removal would automatically restore normal function. Such an assumption, however, may not be valid. We have shown that endothelial cells can become programmed according to the physiologic condition from which they were derived and continue to display altered Ca2+ signaling function even when maintained in vitro in normal culture medium (reviewed in Boeldt et al. [15]). Herein, we address for the first time in freshly isolated vessels whether the endothelial dysfunction in DM and indeed GDM subjects at the level of NO output are mediated by changes in Ca2+ signaling or alterations in NOS3 activity itself or both. We also examine if such alterations in function are retained in vitro as abnormal cell programming and if such alterations in programming are the same for cells from DM versus GDM subjects.
From our initial study of responses in endothelial cells of intact vessels, it can be concluded that in DM and GDM pregnancies, endothelial Ca2+ bursting is still observed, but overall Ca2+ mobilized is significantly less and corresponds with reduced NO output for each subject group. Even though Western analysis suggest that NOS3 protein expression is largely maintained, our studies of the NO response to ionomycin stimulation further reveal that functional damage has occurred to NOS3 itself. Western blotting certainly detects the presence of an amino acid sequence, but it does not indicate that the protein is capable of activation. Treatment of intact cells with ionomycin, however, elevates Ca2+ to levels beyond the maximum needed for full NOS3 activation regardless of phosphorylation state, thus indicating the size of the activatable pool. The finding that there were no observable differences in immunodetectable NOS3 but that DM- and GDM-derived vessels consistently showed reduced NO output relative to control is confirmation that DM and GDM results in reduction of the size of the activatable NOS3 pool, perhaps through direct damage to NOS3 by ROS, as is already known to be perpetuated by the hyperglycemic environment of DM [7].
Having established the finding that DM and GDM impairment of vessel function occurs at the level of Ca2+ signaling and at the level of NOS3 itself in UV endo, the question now arises if the cells were reprogrammed; that is, when removed from the in vivo “toxic” environment and thus having reduced ROS exposure, would they show normal Ca2+ signaling function, or would subtle changes in Ca2+ responses be observed that are suggestive of permanently altered functional programming? Given the recent publication by Tran et al. [16] that shows while NOS3 activity, even in its optimally phosphorylated state, is not significantly activated without elevation of [Ca2+]i in the range 100–300 nM inside the cell, permanent alteration of [Ca2+]i signaling in HUVEC could contribute to hypertension long term. This point is critical because such a finding would call into question whether DM therapy to combat ROS damage of NOS3 protein alone could ever recover full function. Certainly, clinical trials of antioxidants have yielded disappointing results [18–20]. Our previous studies with PE subjects have already shown that cells maintained in primary culture show the ability to display Ca2+ bursting patterns, but these patterns are altered compared to controls in a manner consistent with PE subjects being prone to hypertension later in life [9]. In this study, we also observe that the cells of DM but not GDM subjects show a similar acceleration of Ca2+ bursting previously observed in cells from PE subjects, and in both DM- and GDM-derived cells, the average Ca2+ burst response is reduced in size and width. As a result, the bursting would be less effective at fully activating NOS3 to a magnitude or for a duration comparable to that observed in control HUVEC.
While we have so far identified similar dysfunction in cells from GDM versus DM pregnancies, there are also some notable differences. Of particular note, while the differences in the overall mean [Ca2+]i response after ATP (100 μM) stimulation of HUVEC from both DM and GDM subjects were reduced relative to control, the dramatic increase in Ca2+ burst number per cell in DM but not GDM subjects suggests differences between these two conditions. This discovery in particular underlines the fact that although DM and GDM are two diseases that appear similar in terms of hyperglycemia as the common pathway, there are still programming differences at the level of Ca2+ signaling that may relate to the duration of the condition and likely impact on the magnitude of vasodilatory dysfunction long term, even when the glycemic condition is well controlled. Even if ROS-mediated damage to NOS3 is prevented using antioxidants, there may still be incomplete recovery of full functionality of the cells without addressing programing changes in Ca2+ signaling.
In conclusion, we have demonstrated in GDM and DM pregnancies that while Ca2+ bursts are still observed in endothelium of intact vessels compared to control, there is a reduction in overall Ca2+ mobilization and a significantly reduced pool of bioactive NOS3 in vessels from GDM and DM subjects. Acutely, the smaller activatable pool of NOS3 is a major contributor to the loss of NO output in vessels from both GDM and DM pregnancies. Nonetheless, there is evidence in intact vessels showing some degree of additional Ca2+ signaling dysfunction, and our further studies in HUVEC isolated from both GDM and DM cords and maintained in culture also show that such defects in Ca2+ burst signaling may have become programmed and thus could contribute to reduced NOS3 activation longer term. Certainly, there are also differences in Ca2+ signaling dysfunction in GDM versus DM subjects, with bursting rate becoming increased in HUVEC from DM but not GDM subjects. Nonetheless, regardless of the rate of bursting, if the reprograming of Ca2+ burst shape common to HUVEC from GDM and DM subjects is also occurring in the maternal vasculature, it would explain the greater ongoing hypertension observed in GDM or DM subjects after pregnancy. This seems likely given former studies by ourselves and others that suggest that endothelial cell Ca2+ signaling function depends on TRPC3 and likely TRPC6, which are common to both maternal uterine artery and umbilical vein endothelial cells [21, 22]. ROS is known to alter the function of endothelial cell TRPC proteins [10], and TRPC failure has been proposed to be a causative factor in the development of hypertension in a number of cardiovascular diseases [10]. Should such a link be established, the direct or even indirect targeting of TRPC for therapy may be a viable means to restore normal endothelial function. Given the subtle differences seen here in bursting patterns of HUVEC from GDM versus DM origin, it also remains to be seen if such therapy will be effective only in GDM subjects or equally effective in GDM and DM subjects long term.
Footnotes
This study was supported by an R&D Grant by the Department of Obstetrics and Gynecology, University of Wisconsin, Madison, SMPH. This work was also supported in part by the National Institutes of Health award HD069181. D.S.B. was supported by NIH T32 predoctoral training award (T32HD41921) and the University of Wisconsin School of Medicine and Public Health Shapiro Award.
REFERENCES
- Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- Conway DL. Obstetric management in gestational diabetes. Diabetes Care. 2007;30(suppl 2):S175–S179. doi: 10.2337/dc07-s212. [DOI] [PubMed] [Google Scholar]
- Kuzuya T, Nakagawa S, Satoh J, Kanazawa Y, Iwamoto Y, Kobayashi M, Nanjo K, Sasaki A, Seino Y, Ito C, Shima K, Nonaka K, et al. Report of the Committee on the classification and diagnostic criteria of diabetes mellitus. Diabetes Res Clin Pract. 2002;55:65. doi: 10.1016/s0168-8227(01)00365-5. [DOI] [PubMed] [Google Scholar]
- Ehrlich SF, Crites YM, Hedderson MM, Darbinian JA, Ferrara A. The risk of large for gestational age across increasing categories of pregnancy glycemia Am J Obstet Gynecol 2011. 204 240 e241 e246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth C, Gazis A, James D. Associations of type 1 diabetes mellitus, maternal vascular disease and complications of pregnancy. Diabetic Med. 2007;24:1229. doi: 10.1111/j.1464-5491.2007.02254.x. [DOI] [PubMed] [Google Scholar]
- Schmitz O, Klebe J, Moller J, Arnfred J, Hermansen K, Orskov H, Beck-Nielsen H. In vivo insulin action in type 1 (insulin-dependent) diabetic pregnant women as assessed by the insulin clamp technique. J Clin Endocrinol Metab. 1985;61:877–881. doi: 10.1210/jcem-61-5-877. [DOI] [PubMed] [Google Scholar]
- De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH, Vanhoutte PM. Endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek SM, Magness RR, Conrad KP. Nitric oxide and pregnancy. Am J Physiol. 1997;272:R441. doi: 10.1152/ajpregu.1997.272.2.R441. [DOI] [PubMed] [Google Scholar]
- Krupp J, Boeldt DS, Yi FX, Grummer MA. Bankowski Anaya HA, Shah DM, Bird IM. The loss of sustained Ca(2+) signaling underlies suppressed endothelial nitric oxide production in preeclamptic pregnancies: implications for new therapy. Am J Physiol Heart Circ Physiol. 2013;305:H969–H979. doi: 10.1152/ajpheart.00250.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan HY, Huang Y, Yao X. TRP channels in endothelial function and dysfunction. Biochim Biophys Acta. 2007;1772:907–914. doi: 10.1016/j.bbadis.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Sullivan JA, Grummer MA, Yi FX, Bird IM. Pregnancy-enhanced endothelial nitric oxide synthase (eNOS) activation in uterine artery endothelial cells shows altered sensitivity to Ca2+, U0126, and wortmannin but not LY294002 – evidence that pregnancy adaptation of eNOS activation occurs at multiple levels of cell signaling. Endocrinology. 2006;147:2442. doi: 10.1210/en.2005-0399. [DOI] [PubMed] [Google Scholar]
- Boeldt DS, Grummer MA, Magness RR, Bird IM. Altered VEGF-stimulated Ca2+ signaling in part underlies pregnancy-adapted eNOS activity in UAEC. J Endocrinol. 2014;223:1–11. doi: 10.1530/JOE-14-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi FX, Boeldt DS, Gifford SM, Sullivan JA, Grummer MA, Magness RR, Bird IM. Pregnancy enhances sustained Ca2+ bursts and endothelial nitric oxide synthase activation in ovine uterine artery endothelial cells through increased connexin 43 function. Biol Reprod. 2010;82:66. doi: 10.1095/biolreprod.109.078253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird IM, Boeldt DS, Krupp J, Grummer MA, Yi FX, Magness RR. Pregnancy, programming and preeclampsia: gap junctions at the nexus of pregnancy-induced adaptation of endothelial function and endothelial adaptive failure in PE. Curr Vasc Pharmacol. 2013;11:712–729. doi: 10.2174/1570161111311050009. [DOI] [PubMed] [Google Scholar]
- Boeldt DS, Yi FX, Bird IM. eNOS activation and NO function: pregnancy adaptive programming of capacitative entry responses alters nitric oxide (NO) output in vascular endothelium – new insights into eNOS regulation through adaptive cell signaling. J Endocrinol. 2011;210:243. doi: 10.1530/JOE-11-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran QK, Leonard J, Black DJ, Nadeau OW, Boulatnikov IG, Persechini A. Effects of combined phosphorylation at Ser-617 and Ser-1179 in endothelial nitric-oxide synthase on EC50(Ca2+) values for calmodulin binding and enzyme activation. J Biol Chem. 2009;284:11892. doi: 10.1074/jbc.M806205200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvolini E, Rabini RA, Martarelli D, Moretti N, Cester N, Mazzanti L. A study on human umbilical cord endothelial cells: functional modifications induced by plasma from insulin-dependent diabetes mellitus patients. Metabolism. 1999;48:554. doi: 10.1016/s0026-0495(99)90049-5. [DOI] [PubMed] [Google Scholar]
- Farvid MS, Jalali M, Siassi F, Hosseini M. Comparison of the effects of vitamins and/or mineral supplementation on glomerular and tubular dysfunction in type 2 diabetes. Diabetes Care. 2005;28:2458–2464. doi: 10.2337/diacare.28.10.2458. [DOI] [PubMed] [Google Scholar]
- Koya D, Lee IK, Ishii H, Kanoh H, King GL. Prevention of glomerular dysfunction in diabetic rats by treatment with d-alpha-tocopherol. J Am Soc Nephrol. 1997;8:426–435. doi: 10.1681/ASN.V83426. [DOI] [PubMed] [Google Scholar]
- Winterbone MS, Sampson MJ, Saha S, Hughes JC, Hughes DA. Pro-oxidant effect of alpha-tocopherol in patients with type 2 diabetes after an oral glucose tolerance test – a randomised controlled trial. Cardiovasc Diabetol. 2007;6:8. doi: 10.1186/1475-2840-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford SM, Yi FX, Bird IM. Pregnancy-enhanced store-operated Ca2+ channel function in uterine artery endothelial cells is associated with enhanced agonist-specific transient receptor potential channel 3-inositol 1,4,5-trisphosphate receptor 2 interaction. J Endocrinol. 2006;190:385. doi: 10.1677/joe.1.06773. [DOI] [PubMed] [Google Scholar]
- Groschner K, Hingel S, Lintschinger B, Balzer M, Romanin C, Zhu X, Schreibmayer W. Trp proteins form store-operated cation channels in human vascular endothelial cells. FEBS Lett. 1998;437:101. doi: 10.1016/s0014-5793(98)01212-5. [DOI] [PubMed] [Google Scholar]