

Abstract
Polycystic ovary syndrome (PCOS) pathophysiology is poorly understood, due partly to lack of PCOS animal models fully recapitulating this complex disorder. Recently, a PCOS rat model using letrozole (LET), a nonsteroidal aromatase inhibitor, mimicked multiple PCOS phenotypes, including metabolic features absent in other models. Given the advantages of using genetic and transgenic mouse models, we investigated whether LET produces a similar PCOS phenotype in mice. Pubertal female C57BL/6N mice were treated for 5 wk with LET, which resulted in increased serum testosterone and normal diestrus levels of estradiol, similar to the hyperandrogenemia and follicular phase estrogen levels of PCOS women. As in PCOS, ovaries from LET mice were larger, polycystic, and lacked corpora lutea versus controls. Most LET females were acyclic, and all were infertile. LET females displayed elevated serum LH levels and higher Lhb mRNA in the pituitary. In contrast, serum FSH and Fshb were significantly reduced in LET females, demonstrating differential effects on gonadotropins, as in PCOS. Within the ovary, LET females had higher Cyp17, Cyp19, and Fsh receptor mRNA expression. In the hypothalamus, LET females had higher kisspeptin receptor mRNA expression but lower progesterone receptor mRNA levels. LET females also gained more weight than controls, had increased abdominal adiposity and adipocyte size, elevated adipose inflammatory mRNA levels, and impaired glucose tolerance, mirroring the metabolic phenotype in PCOS women. This is the first report of a LET paradigm in mice that recapitulates both reproductive and metabolic PCOS phenotypes and will be useful to genetically probe the PCOS condition.
Keywords: androgen, aromatase, female, GnRH, infertility, metabolism, obesity, ovary, PCOS, pituitary, reproduction
INTRODUCTION
Polycystic ovary syndrome (PCOS) affects approximately 10% of reproductive-aged women [1–4]. PCOS is a complex disorder encompassing multiple phenotypic parameters, with clinical diagnosis typically requiring at least two of the following features: polycystic ovaries, androgen excess (hyperandrogenemia), and chronic anovulation, with the latter two often comprising the predominant features [5]. Neuroendocrine hallmarks of PCOS include increased gonadotropin-releasing hormone (GnRH) pulse frequency, increased luteinizing hormone (LH), reduced follicle-stimulating hormone (FSH), and insensitivity to progesterone (P4)-negative feedback [6–8]. Along with hyperandrogenemia, disruptions in the neuroendocrine reproductive axis contribute to dysfunctional ovarian maturation and diminished fertility [1, 3, 4, 6]. PCOS is also often associated with metabolic dysfunction, including obesity and increased abdominal adiposity, abnormal fat cell morphology and function, insulin resistance, glucose intolerance, metabolic syndrome, and heightened risk for diabetes [4, 6, 9, 10].
The development of animal models to study PCOS has been an important research focus [11–14]; however, there is no consensus on the best experimental animal model to study PCOS. Prenatally androgenized rhesus monkey and sheep display many of the reproductive and metabolic phenotypes associated with PCOS [15, 16], but ethical and economic considerations limit the utility of nonhuman primate and large domestic animal models as does the current inability to genetically and transgenically manipulate these larger species to probe underlying mechanisms. In rodents, various strategies have attempted to induce PCOS, including exposure to androgens, estrogens, antiprogestins, and genetic modifications [17–19]. Yet, elucidating the etiology and underlying mechanisms of PCOS has proven challenging, due largely to the heterogeneity of the disease. Most rodent models of PCOS have only partially recapitulated the array of PCOS phenotypes. For example, in female rats and mice, chronic postnatal administration of dihydrotestosterone (DHT), from before puberty to adult age, induces anovulation, increased body weight and adiposity, enlarged fat cells, and insulin resistance [20, 21]. However, unlike PCOS women, these chronically DHT-treated rodents have normal or reduced ovarian weight, lower LH, reduced plasma testosterone (T), and reduced estradiol (E2) levels compared with controls, indicating that this model does not faithfully recapitulate the reproductive endocrine phenotype of PCOS. Likewise, another mouse model utilizing acute prenatal DHT treatment (prenatal androgen: PNA) only reproduces a subset of the PCOS phenotype [14, 22–25] and lacks metabolic or ovarian effects (no polycystic ovaries or differences from controls in glucose tolerance test [GTT], insulin tolerance test, or obesity, even on high fat diet [26]). Thus, existing models recapitulate only some features of PCOS, and few have both the endocrine and metabolic phenotypes.
Recently, a rat model of PCOS using letrozole (LET), a nonsteroidal aromatase inhibitor [27], was shown to exhibit multiple facets of PCOS. This model is based on the finding of lower estrogen/androgen ratios in follicular fluid of PCOS women, suggesting low aromatase activity [28], and the fact that genetic variants of the aromatase gene (CYP19), which converts androgens to estrogen, are associated with the development of PCOS and hyperandrogenism in women [29–32]. In female rats, continuous LET treatment initiated before puberty (at doses that do not fully deplete E2 levels) disrupted estrous cyclicity, increased ovarian weight, and caused anovulation, ovarian cysts, and atretic follicles in adulthood [20, 27]. LET treatment in rats also increased LH (but not FSH) and T levels [20, 27], as occurs in PCOS women. Importantly, besides these reproductive symptoms of PCOS, LET-treated female rats also developed some metabolic features of PCOS, including increased body weight and insulin resistance [27]. Thus, unlike other rodent PCOS models, the LET model in rats recapitulates both the reproductive and metabolic phenotypes associated with PCOS, most of which are apparent within 5 wk after starting LET treatment [27]. Despite this promising outcome in rats, one recent study surprisingly reported no effect of LET on multiple PCOS phenotypes in mice [19]. Thus, there has yet to be a similar LET model developed in mice, which would be extremely advantageous due to the availability of murine transgenic lines that can be used in combination with LET to study the pathogenesis of PCOS. Here, we present, for the first time, a new mouse model utilizing LET that strongly recapitulates both the reproductive and metabolic hallmarks of PCOS. Moreover, we show for the first time that this novel LET paradigm significantly blocks fertility and alters reproductive gene expression at multiple levels of the hypothalamic-pituitary-gonadal axis, providing some mechanistic insight into how the reproductive axis is disrupted in a PCOS-like condition.
MATERIALS AND METHODS
Animals and LET Treatment
Female C57BL/6N (Harlan Laboratories) mice were housed on a 12L:12D cycle with food and water available ad libitum. Mice were housed two females/cage. All the experiments were approved by the University of California San Diego Institutional Animal Care and Use Committee.
At 4 wk of age, prior to pubertal completion, females were subcutaneously implanted with a LET (50 μg/day) or placebo control (CON) pellet (n = 10/group). LET was purchased from Fitzgerald, and custom 60-day continuous release pellets were made by Innovative Research of America. The 50 μg/day dose was selected based on previous studies in rats giving a LET dose of 200 or 400 μg/day [27] and scaled appropriately to mice.
Estrous Cycle Assessment
The stage of estrous cyclicity was determined by light microscopic analysis over 14 days of the predominant cell type in vaginal epithelia smears obtained 4–5 wk after LET or control pellet implantation. Proestrus was characterized by the presence of mostly nucleated and some cornified epithelial cells, estrus as mostly cornified cells, and diestrus/metestrus as some cornified epithelial cells and primarily leukocytes.
Tissue Collection and Histology
After 5 wk of LET exposure, mice were anesthetized with isoflurane, weighed, blood collected via retro-orbital bleeding, and then rapidly decapitated (between 1000 and 1200 h). Brains and pituitaries were collected, frozen on dry ice, and stored at −80°C. Additionally, dissected ovaries and fat pads were weighed. One ovary and one ovarian fat pad and a portion of periuterine (parametrial) fat pad from each mouse was fixed in 4% paraformaldehyde at 4°C overnight and stored in 70% ethanol before histologic processing. The ovarian fat pad connects to and surrounds the ovary; the parametrial fat pad is the main, large, bilateral adipose tissue in the lower abdominal cavity, around the uterus and bladder. The other ovary and fat pad were dissected straight into RNAlater (Life Technologies) and stored at −80°C until processing for mRNA expression levels using quantitative PCR.
For histological analysis, fixed ovaries were serially sectioned at 12 μm and then stained with hematoxylin and eosin. Corpora lutea and cysts were counted from two sections randomly selected from the middle of each ovary. In each case, counts were made by an investigator blind to the treatment group. Fixed parametrial and ovarian fat pads were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. Slides were scanned on a Hamamatsu Nanozoomer scanner, and adipocyte number and area were quantified using ImageJ with the MRI Adipocyte Tools macros. Macrophage infiltration was assessed on additional adipose sections by immunohistochemistry using antibody F4/80 (1:100, clone CI:A3-1; Serotec) with 3,3′-diaminobenzidine detection.
Hormone Assays
All the hormone levels were measured by the University of Virginia Ligand Core Facility. Serum LH and FSH were measured by a mouse multiplex assay (reportable range 0.24–30.0 ng/ml and 2.4–300 ng/ml, respectively). Serum T was measured with radioimmunoassay (range 5.0–1075 ng/dl). Serum E2 was measured using a mouse enzyme-linked immunosorbent assay (range 3.0–300 pg/ml).
Quantitative Real-Time PCR of Ovary, Pituitary, and Adipose Genes
Total RNA from pituitary, ovary, and parametrial adipose tissue was isolated using an RNeasy Mini kit (Qiagen). Genomic DNA was eliminated from samples using a Turbo DNA-free kit (Ambion). Complementary DNA was made by reverse transcription of total RNA using an iScript cDNA synthesis kit (Bio-Rad Laboratories). Complementary DNA products were detected using iQ SYBR Green Supermix (Bio-Rad Laboratories) on a quantitative real-time-PCR iQ5 real-time detection system (Bio-Rad Laboratories). Data were analyzed by the 2−ΔΔCt method by normalizing the ovarian gene of interest to L19 and pituitary or adipose genes of interest to Gapdh. Data are represented as mean fold change compared with control ± SEM. Primer sequences used are listed in Supplemental Table S1 (available online at www.biolreprod.org).
Quantitative PCR Analysis of Reproductive Genes in the Brain
Frozen brains were dissected and micropunches taken from the medial basal hypothalamus (MBH) and the anterior hypothalamus/preoptic area (POA). Total RNA from POA and MBH samples was isolated using an RNeasy Mini kit (Qiagen), and cDNA was made by reverse transcription of total RNA using an iScript cDNA synthesis kit (Bio-Rad Laboratories). Complementary DNA products were analyzed in duplicate with real-time PCR using a CFX Connect system (Bio-Rad Laboratories) and Quantitect SYBR Green PCR kit (Qiagen). Standard curves were generated for each product using cloned cDNAs for Gnrh, Kiss1r, Pgr, and Gapdh to quantify the abundance of cDNA in each sample, in line with previously published methods [33]. For standard curves, a dilution series of cloned templates ranging from 10 to 108 copies were used. In order to ensure the presence of a single product, a dissociation curve was performed after each run. Data were collected from threshold values using the automatic function of the Bio-Rad MyIQ software and are reported as mRNA copy number.
Fertility Assessment
A separate cohort of female mice similarly implanted with LET or CON at 4-wk-old was used to assess fertility. At 8 wk of age, 4 wk after pellet implantation, LET and CON females (n = 5–6/group) were paired with adult C57BL/6N breeder males (∼3-mo-old). Breeder males were removed after 10 days, and females were assessed for presence of litters and time to first litter. All females were paired again with a new breeder male and similarly assessed for fertility at 20 wk of age, ∼10 wk after the LET pellet had fully exhausted (the LET pellets last for ∼7 wk).
GTT
Mice were fasted for 6 h with free access to water. Mice were tested for GTT independent of cycle stage. Basal blood glucose was then measured using a handheld glucometer (One Touch UltraMini; LifeScan, Inc.) just before (time 0) a single intraperitoneal injection of glucose (2g/kg body weight in sterile saline) and subsequently at 15, 30, 45, 60, 90, and 120 min postadministration (n = 10/group).
Statistical Analysis
All the data are expressed as the mean ± SEM for each group. Group differences for all data were analyzed by Student t-test except for differences in percent of animals in a group cycling or displaying fertility, which were analyzed using chi-squared test, and glucose tolerance, which was analyzed with repeated measures ANOVA followed by post hoc comparisons for individual time points. Statistical significance was set at P < 0.05.
RESULTS
Circulating Reproductive Hormones Are Altered in LET Mice
LET treatment differentially affected circulating levels of the two gonadotropins. LET female mice displayed significantly elevated serum LH levels (P < 0.05 vs. CON, Fig. 1A). In contrast, serum FSH was significantly reduced by 3-fold in LET females versus CON mice (P < 0.05, Fig. 1B). Coinciding with the elevated LH, serum T was robustly increased by 4-fold by LET (P < 0.05, Fig. 1C), mimicking a normal T range for adult male mice and mirroring the increased androgen levels in PCOS women. In contrast, circulating E2 levels were not significantly altered by LET and remained in the normal diestrous range (Fig. 1D).
FIG. 1.
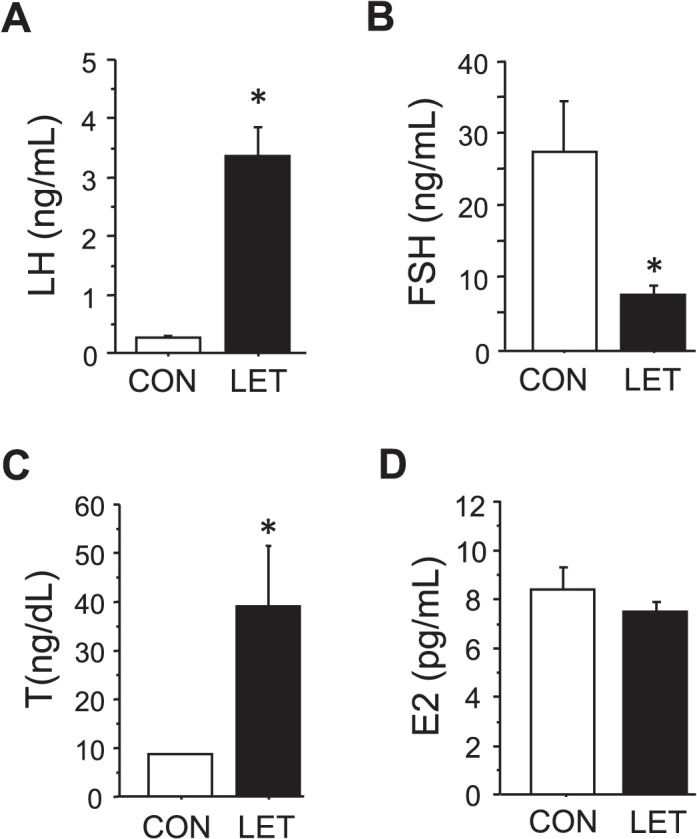
Altered endocrine profile in LET-treated female mice. Mean serum levels of LH (A), FSH (B), T (C), and E2 (D) in LET and CON females after 5 wk of treatment. Significantly different from CON (*P < 0.05); n = 10/group.
Reproductive mRNA Expression in the Pituitary Is Altered in LET Females
Along with elevated serum LH, LET females had significantly higher Lhb mRNA levels in the pituitary versus CON females (P < 0.05, Fig. 2A). Likewise, pituitary Fshb mRNA levels mirrored the decrease in circulating FSH, being significantly lower in LET females (P < 0.05, Fig. 2B). Pituitary Gnrhr (encoding GnRH receptor) mRNA was also higher in LET mice (P < 0.05, Fig. 2C). Conversely, pituitary Kiss1r and Pgr (encoding the kisspeptin receptor and progesterone receptor, respectively) levels were not different between LET and CON (Fig. 2).
FIG. 2.
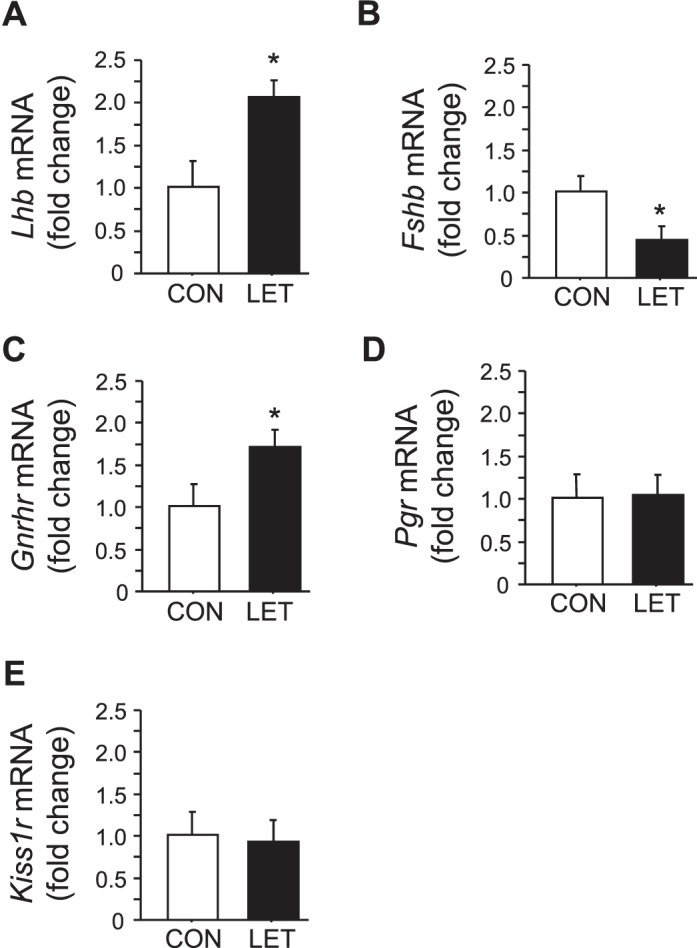
Altered pituitary gene profile in LET-treated female mice. Mean mRNA expression levels of Lhb (A), Fshb (B), Gnrhr (C), Pgr (D), and Kiss1r (E) in the pituitary of LET and CON females after 5 wk of treatment. Significantly different from CON (*P < 0.05); n = 7/group.
LET Impairs Ovarian Morphology and mRNA Expression Levels
Uterine weights were not significantly different between treatments at 5 wk of LET treatment (54.3 ± 10.7 mg vs. 67.0 ± 9.7 mg for LET and CON, respectively). However, the ovaries of LET mice were dramatically altered in multiple ways that mimicked PCOS. Ovary weights were significantly increased in LET versus CON females (P < 0.05, Fig. 3, A and B). The ovaries from LET, but not CON, female mice were also polycystic (Fig. 3, A and C). LET ovaries appeared to have more antral follicles than CON ovaries, consistent with PCOS ovaries [34]. Many of the LET ovarian follicles showed a disorganized granulosa cell compartment with irregular granulosa cell layer thickness, characteristic of atretic antral follicles (Fig. 3A). Furthermore, there were almost no large secondary follicles in LET ovaries, suggesting that their secondary follicles mature precociously to antral follicles. The hyperplastic and thickened theca cells, commonly seen in PCOS ovaries as compared with normal ovaries, were not distinctly observed in the ovaries of LET mice. Importantly, ovaries from LET mice lacked corpora lutea (CL), while CON female ovaries had multiple CL, indicative of ovulation (Fig. 3, A and D). Moreover, the vast majority of LET females were acyclic, being mostly arrested in diestrus, whereas only one CON female was acyclic (P < 0.05, Fig. 3E). Analysis of estrous cycle stages revealed that LET females spent significantly more time in diestrus and less time in proestrus and estrus than CON females (P < 0.05 for each stage, Fig. 3F).
FIG. 3.

Perturbed ovarian morphology and estrous status in LET-treated female mice. A) Representative images of ovaries from CON (top) and LET (bottom) female mice depicting presence or absence of cysts and corpus luteum (CL). Higher magnification images on the right depict the disorganized follicles of the LET mice. CF, cystic follicle; HC, hemorrhagic cyst. Mean ovarian weights (B), number of cystic follicles (C), and number of CL (D) were all significantly different between LET and CON females (n = 3). The percent of females that were acyclic (constant diestrus or constant estrous) (E) and relative amount of time spent in each cycle stage (F) were also significantly different between LET and CON females (n = 10/group). Significantly different from CON (*P < 0.05).
Within the ovary, LET mice had significantly higher expression of Cyp17, a key steroidogenic enzyme involved in androgen synthesis (P < 0.05, Fig. 4A). In addition, Fshr and Cyp19 (encoding FSH receptor and aromatase, respectively) mRNA levels were also significantly increased in LET female ovaries (P < 0.05, Fig. 4, B and C). In contrast, ovarian Lhr, Pgr, and Amh (LH receptor, progesterone receptor, and anti-Müllerian hormone, respectively) mRNA expression in LET females were identical to CON mice (Fig. 4, D–F).
FIG. 4.

Altered ovarian gene profile in LET-treated female mice. Mean mRNA expression levels of Cyp17 (A), Cyp19 (B), Fshr (C), Lhr (D), Pgr (E), and Amh (F) in the ovaries of LET and CON females after 5 wk of treatment. Significantly different from CON (*P < 0.05); n = 7/group.
Reproductive mRNA Expression in the Hypothalamus Is Altered in LET Mice
To begin to assess whether any of the alterations observed in circulating hormones or pituitary mRNA levels reflect upstream changes in reproductive brain circuits, we evaluated expression of several relevant genes in the hypothalamus. In the POA, Gnrh and Pgr mRNA expression levels were not different between LET and CON females (Fig. 5, A and C). However, in LET females, Kiss1r mRNA levels were elevated in the POA (where GnRH neurons reside), though this difference fell just short of reaching statistical significance (P < 0.059, Fig. 5B). LET females also had significantly lower Pgr mRNA expression in the MBH, a region known for mediating negative feedback effects of P4 on the reproductive axis (P < 0.05, Fig. 5D).
FIG. 5.
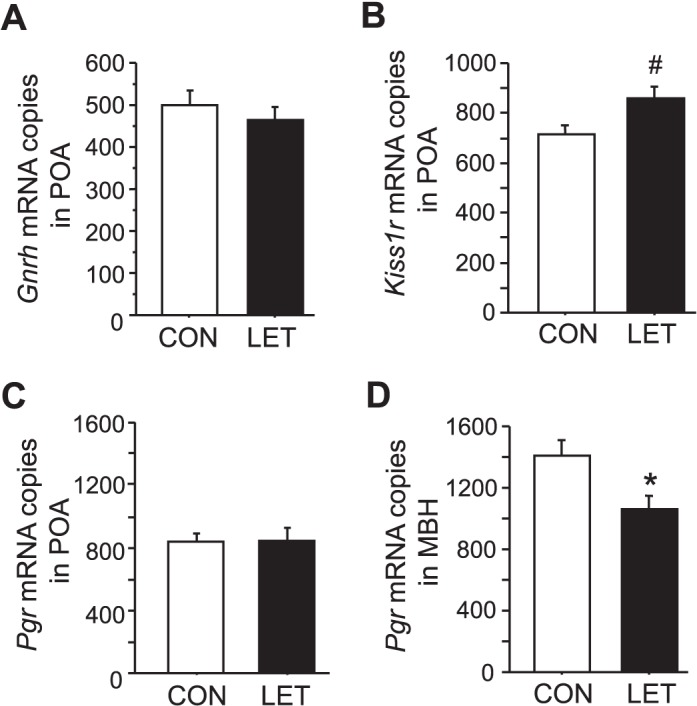
Brain profile of reproductive-related genes in LET-treated female mice. Mean mRNA expression levels of Gnrh in the anterior hypothalamus/preoptic area (POA) (A), Kiss1r in the POA (B), Pgr in the POA (C), and Pgr in the medial basal hypothalamus (MBH) (D) in brains of LET and CON females after 5 wk of treatment. Significantly different from CON (*P < 0.05); trend for group difference (#P = 0.059); n = 7/group.
LET Females Are Infertile but This Reproductive Deficit Is Reversible
To assess functional consequences of the LET paradigm on reproduction, we evaluated fertility in LET and CON females beginning 4 wk after initiation of LET treatment. All the CON females (5/5) successfully gave birth to a litter following pairing with a breeder male for 10 days. In contrast, none of the LET females (0/6) successfully gave birth (P < 0.05, Fig. 6A). To evaluate whether the LET effect is permanent or can be reversed, all females were paired again with another male 10 wk after the LET pellet expired (∼17 wk after LET implantation). In this case, 100% of both LET and CON females (6/6 and 5/5, respectively) gave birth to a litter, with no group differences in time to birth or litter size (Fig. 6, B and C). Similarly, circulating T levels were no longer significantly elevated in these females 10 wk after the LET pellet had exhausted (10.6 ± 1.4 ng/dl vs. 8.0 ± 1.4 ng/dl for LET and CON, respectively).
FIG. 6.
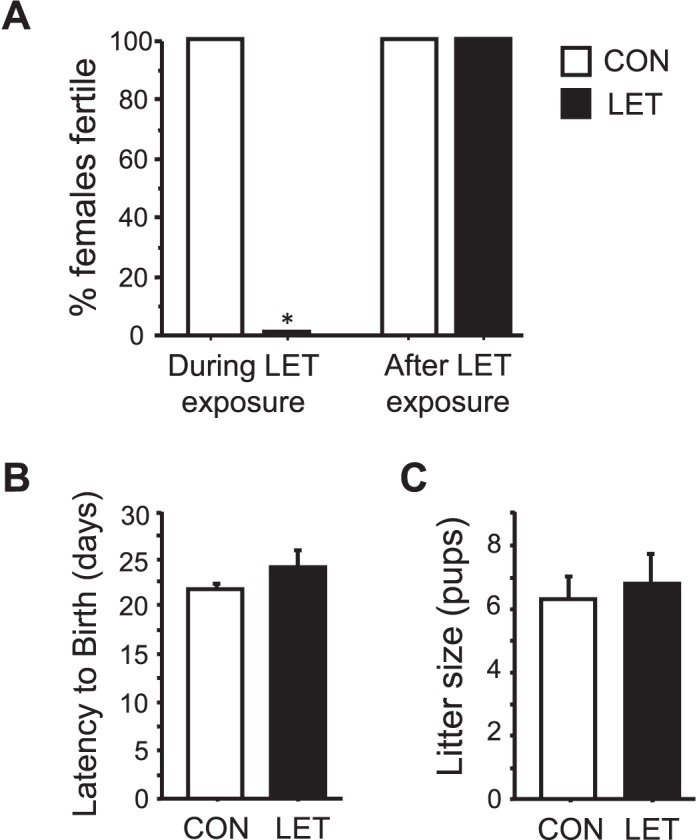
Fertility is completely blocked in LET females. A) Percent of females successfully giving birth after being paired with a male for 10 days. B) Mean time to birth after introduction of a male breeder partner during the second fertility pairing after LET pellet had expired. C) Mean number of pups per litter in LET and CON mice during the second fertility pairing after LET pellet had expired. Significantly different from CON (*P < 0.05); n = 5–6/group.
Body Weight and Adiposity Are Increased in LET Females
Because PCOS women often present with metabolic disorders, we investigated whether the LET-induced PCOS mouse model also exhibits a metabolic phenotype. LET mice gained more weight than CON females (by 1.5-fold) over the 5-wk treatment period (P < 0.05, Fig. 7, A and B). LET females also had markedly higher abdominal adipose weights (ovarian and parametrial fat depots) (P < 0.05, Fig. 7C) and dramatically increased adipocyte cell size in both fat pads (P < 0.05, Fig. 7, D and E).
FIG. 7.

Altered body weight and adiposity in LET female mice, reminiscent of obesity in many PCOS women. Mean body weights at end of study (A) and percent change in body weight change during the 5 wk treatment (B) are greater in LET than CON females (n = 10/group). Mean abdominal adipose weights (parametrial fat depot) (C) are also elevated in LET females (n = 10/group). Mean adipocyte cell size is also markedly increased in both the ovarian (D) and parametrial (E) fat pads (n = 4/group). Significantly different from CON (*P < 0.05). Original magnification ×200.
Inflammatory Markers and Glucose Tolerance Are Altered in LET Females
Given that inflammation is a recognized contributor to obesity and impaired glucose control, we examined the parametrial adipose tissue for the presence of inflammatory markers and macrophages. Messenger RNA expression levels of several inflammatory factors and cytokines, including IL-1β, IL-6, Mcp-1, and Tnfα, were significantly elevated in adipose tissue of LET females (P < 0.05, Fig. 8A). Staining for macrophage infiltration in ovarian fat tissue using the F4/80 antibody (which binds a glycoprotein expressed by murine macrophages) also revealed the notable presence of macrophages in adipose tissue of LET mice but not in CON females (Fig. 8B).
FIG. 8.

Increased markers of inflammation in adipose tissue of LET-treated female mice, reminiscent of similar increases in inflammation in PCOS women. A) Messenger RNA expression of inflammatory factors and cytokines is significantly elevated in parametrial adipose tissue of LET versus CON females (n = 6/group). LET female adipose tissue also contains noticeable macrophage infiltration (brown stain for macrophage-specific protein denoted by arrows) not observed in adipose tissue of CON females (B). Significantly different from CON (*P < 0.05). Original magnification ×200.
Glucose homeostasis is typically impaired in obesity and metabolic syndrome, including in PCOS women. We found that basal glucose levels were elevated in LET females (P < 0.05, Fig. 9A). Moreover, LET females displayed glucose intolerance, with significant delays in their ability to clear glucose after intraperitoneal injection (P < 0.05, Fig. 9B).
FIG. 9.

Glucose tolerance is impaired in LET-treated female mice. Mean basal glucose levels are elevated in LET females (A). LET females also display glucose intolerance in a GTT, with significant delays in the clearance of glucose after intraperitoneal injection (B) and overall greater area under the curve (AUC) during this test (C). Significantly different from CON (*P < 0.05); n = 10/group.
DISCUSSION
PCOS is a complex disorder encompassing multiple phenotypic parameters, including neuroendocrine and ovarian impairments, and a high prevalence of metabolic perturbation, such as obesity and glucose intolerance [8–10]. The development of rodent models to study mechanisms of PCOS has proven challenging due to the heterogeneity of the disease, with most models only partially recapitulating the many phenotypes of PCOS [14, 20, 22–24, 26], and few having both the reproductive and metabolic phenotypes. Recently, a rat model of PCOS using LET recapitulated multiple features of PCOS, but similar models in mice, which can be manipulated with genetic and transgenic technologies, are currently lacking. Here, we report, for the first time, a new LET mouse model that impressively recapitulates many of the reproductive and metabolic components of the human PCOS phenotype. In addition, we demonstrate for the first time several notable changes in reproductive mRNA expression in the hypothalamus and pituitary and inflammatory genes in adipose tissue not formerly studied in previous LET studies, which may relate to mechanisms enhancing GnRH/LH secretion and obesity, as occurs in PCOS women.
Neuroendocrine Phenotype
We found that LET female mice have significantly elevated circulating androgens, a hallmark of women with PCOS [6, 35]. Our observed increase in androgen levels in LET mice were of similar range and magnitude as has been reported for increased androgen levels in PCOS women [36, 37]. In PCOS, hyperandrogenemia is driven by upstream increases in LH secretion, which we also observed in LET mice, both at the level of pituitary Lhb mRNA expression and circulating LH levels. Additionally, at the ovarian level, alterations in steroidogenic enzymes, mainly increased activity of Cyp17 (the rate-limiting enzyme in androgen biosynthesis), synergize with the elevated LH signaling to promote high production of ovarian androgens in PCOS women and was also evident in our LET mice (discussed further below). Highlighting the fundamental role of androgen signaling in PCOS, clinical studies determined that treatment with the AR antagonist, flutamide, can reverse many symptoms of PCOS [38, 39]. Furthermore, as occurs in rodents on high-fat diet [40], obesity in women promotes increased androgen secretion [41], which exacerbates the PCOS pathology in cases in which it is accompanied by metabolic dysfunction.
In addition to hyperandrogenemia, persistently rapid GnRH pulsatility is a neuroendocrine hallmark of PCOS [42], and this favors pituitary synthesis and secretion of LH over FSH and higher LH-to-FSH ratios. Inadequate FSH levels in PCOS women contribute to impaired follicular development, while elevated LH levels augment ovarian androgen production [6]. Although GnRH expression is unchanged in LET mice, the LET mouse model faithfully recapitulates both of the gonadotropin phenotypes, with higher pituitary Lhb mRNA and circulating LH and lower Fshb mRNA and circulating FSH. The circulating gonadotropin abnormalities likely reflect changes in GnRH secretion patterns caused by upstream alterations in sex steroid feedback onto the GnRH pulse generator. This altered steroid feedback in women with PCOS may be influenced by both abnormal steroidogenesis and dysfunctional sex steroid-signaling mechanisms. For example, E2 administration decreases FSH secretion in normal women but fails to lower FSH in PCOS women [43, 44]. The LET mouse paradigm may prove useful to study the mechanisms of altered sex steroid feedback in a PCOS-like scenario.
Our data highlight several possibilities for contributing factors to the elevated LH secretion in LET mice. First, pituitary Gnrhr mRNA is significantly increased in LET females, potentially providing a pituitary-based mechanism for enhanced GnRH signaling in this condition. Second, LET mice have elevated Kiss1r levels in the anterior hypothalamus and POA region, where GnRH neurons reside. This raises the possibility that enhanced kisspeptin signaling to GnRH neurons, related to elevated kisspeptin receptor mRNA levels, may help drive increased GnRH secretion, a key trait in PCOS women. Future studies will determine to what extent kisspeptin signaling is altered and involved in the LET PCOS-like condition. Regardless, enhanced kisspeptin activation of GnRH secretion may act in concert with the observed elevations in GnRH receptor mRNA levels in the pituitary to achieve exaggerated LH synthesis and secretion in the PCOS condition. Lastly, P4-mediated negative feedback on GnRH circuits may be reduced in LET mice, allowing for greater GnRH/LH secretion. PCOS women have decreased GnRH pulse generator sensitivity to P4-mediated negative feedback [45]. P4 similarly exerts inhibitory feedback effects on gonadotropin secretion in animal models, presumably by acting on GnRH circuits [46–52]. However, the brain location(s) where P4 acts to repress GnRH and LH secretion (negative feedback) are not fully known but may involve neurons in the arcuate nucleus/MBH region, which express progesterone receptors and are implicated in negative feedback [53]. Interestingly, we found reduced Pgr mRNA levels in the MBH of LET females, but normal Pgr mRNA levels in more anterior brain areas. This MBH-specific decrease in Pgr mRNA levels may cause reduced progesterone receptor protein levels and could be related to diminished capability to respond to P4-negative feedback signals in the PCOS condition, thereby promoting enhanced GnRH secretion. Because mRNA levels do not always represent protein levels [54, 55], future studies using fixed tissues will need to determine if the alterations in mRNA expression observed in our study reflect similar alterations in protein levels.
Ovarian Phenotype and Fertility
In addition to changes in circulating gonadotropins and GnRH secretion, the ovaries of PCOS women are typically larger, polycystic, anovulatory, and have changes in multiple steroidogenic enzymes and hormone receptors. The ovaries of the LET mouse model faithfully recapitulate these PCOS phenotypes. LET ovaries are enlarged and contain multiple cysts but lack corpora lutea, signifying absence of ovulatory activity. In addition, mRNA levels of Fshr, Cyp17, and Cyp19 were also significantly increased in ovaries of LET females, mirroring similar changes in PCOS ovaries [56]. Consistent with these findings, androgen administration to monkeys increases FSH receptor mRNA [57], and FSH binding to granulosa cells from ovaries of PCOS women is increased compared with size-matched normal follicles [58]. We speculate that the higher Cyp19 (aromatase) mRNA levels in LET females are due to both elevated FSH receptor and elevated T, the latter of which enhances FSH receptor action on Cyp19 expression and itself stimulates Cyp19 [57, 59]. Whether levels of Cyp19 in ovaries from PCOS women are different is currently unclear and requires more data for definitive conclusions [60]. However, genetic variants of the CYP19 gene have been associated with the development of PCOS and altered androgenic profile in women [29, 31]. We also found that ovarian Amh mRNA levels were unchanged in the LET mice, though we did not assess protein levels. Circulating AMH protein levels have been reported to be higher in some but not all PCOS women [61], leaving this issue of the AMH phenotype in PCOS unclear at present.
Along with the above alterations to ovarian morphology and gene expression, we determined that LET females have dysfunctional estrous cyclicity, being mostly arrested in diestrus and rarely exhibiting proestrus. To assess functional consequences of such impairments, we evaluated fertility in females beginning 4 wk after initiation of LET treatment. In contrast to CON females that all gave birth after pairing with a male, none of the LET females gave birth, demonstrating infertility in this PCOS model. However, the effects of LET on fertility were reversible because all LET females were able to give birth to normal-sized litters once the LET pellet was exhausted (tested 10 wk later). This reversibility of the LET model will allow for comprehensive analyses in future studies before and after LET treatment.
Interestingly, circulating E2 was not dramatically lower in LET females at the time of killing, being in the normal diestrus range at 5 wk of treatment. This was confirmed by multiple E2-sensitive anatomical and molecular measures indicating that E2 was not significantly lowered: 1) both uterine weights and Pgr mRNA levels in several tissues were unchanged (both are reduced by lower E2 levels), and 2) FSH levels and Fsh mRNA expression were reduced rather than elevated (FSH is normally increased with lower E2 levels [62–64]). It is possible that E2 levels are initially decreased by LET but eventually return to normal levels as a long-term consequence of the dramatic increases observed in Cyp19 gene expression (aromatase) and T synthesis (the primary substrate for E2). Importantly, the normal levels of E2 along with high T observed in our LET mice mirror these hormone levels in women with PCOS, in which T is elevated and E2 is at normal follicular phase levels. These findings suggest that many reproductive and metabolic phenotypes observed in the LET mice (and perhaps PCOS) do not reflect lower circulating E2 and may instead be attributable to elevated androgens.
Metabolic Phenotype
Along with the reproductive phenotype, many women with PCOS have metabolic abnormalities, including peripheral insulin resistance, hyperinsulinemia, and abdominal adiposity that increase their risk of developing type 2 diabetes and cardiovascular disease [9, 65, 66]. Metabolic dysfunction has been shown to occur predominantly in PCOS women with hyperandrogenism and ovulatory dysfunction, regardless of their body mass index [67, 68]. This association is weaker for women with hyperandrogenism and polycystic ovaries, while women with ovulatory dysfunction and polycystic ovaries, but no evidence of androgen excess, typically lack a metabolic phenotype. These studies highlight the potential role of hyperandrogenemia in the development of the PCOS metabolic syndrome. Additionally, hyperinsulinemia in women with PCOS has been proposed to contribute to the metabolic phenotype by increasing ovarian androgen production and increasing androgen bioactivity through decreased sex hormone-binding globulin production [69, 70]. Obesity also exacerbates insulin resistance and, thus, hyperinsulinemia and androgen production in women with PCOS [71].
Here, we show that the LET mouse model recapitulates several metabolic phenotypes observed in PCOS women. LET females weigh more, have increased adipose tissue weight, larger adipocytes, elevated inflammatory gene mRNA expression in adipose tissue, and increased macrophage infiltration of adipose tissue. The LET mice also have elevated fasting glucose and were moderately glucose intolerant. Such metabolic impairments were notable after just 5 wk of LET treatment and mirror the reported phenotype of female rats treated with LET for a longer period of 10 wk. Unlike the LET model, other paradigms previously used to induce PCOS in mice have typically reported a lack of consistent metabolic phenotype, suggesting that the LET paradigm may be useful for studying PCOS in a context of metabolic dysfunction, as occurs in many women with the disorder.
Comparison with Other Mouse PCOS Models
Several other models have been used to mimic PCOS in rodents, the most prevalent being the PNA model whereby females are exposed to elevated DHT levels late in gestation. While the PNA model nicely resembles several notable features of PCOS, there are several aspects that are not recapitulated and that differ from LET mice. Ovaries of PNA mice have a reduced number of, but not absent, CL, indicating some ovulation [19, 24]. Polycystic ovaries are not present in PNA mice, and their ovaries are smaller or normal sized [19, 24, 25], opposite to the larger ovaries present in PCOS women and LET mice. Serum LH in PNA mice is elevated in some reports but not others [19, 25, 72], and FSH levels, which are lower in both PCOS women and LET mice, are normal in PNA mice [19, 25]. Circulating T in PNA female mice is either normal (i.e., low) [19, 23, 25] or moderately elevated [24, 26, 72], differing from the larger degree of hyperandrogenemia present in both PCOS women and LET mice. PNA mice also do not exhibit the same marked metabolic defects observed in LET mice or LET rats. Although PNA mice have elevated fasting glucose, slightly impaired glucose tolerance, and a minor increase in adipocyte size [23, 25], there is no significant alteration in body weight, adiposity, or insulin resistance [19, 23–26]. Moreover, another study reported that glucose tolerance was normal in PNA mice [26]. It has been suggested that the absence of significant metabolic dysfunction or obesity in PNA mice signifies it as a lean PCOS model [73] and could be useful for representing the condition in some PCOS women who lack metabolic impairments [74]. Alternatively, the PNA model may represent a milder form of PCOS across the PCOS spectrum. Table 1 summarizes the key phenotypes of the LET mouse model compared with PCOS women, the PNA mouse model, and the postnatal DHT rodent model [19–21], the latter of which lacks multiple aspects of the reproductive phenotype while presenting some of the metabolic phenotype.
TABLE 1.
Summary and comparison of letrozole (LET)-induced polycystic ovary syndrome (PCOS) mouse model with PCOS women and other mouse models.a

✓, yes; X, no; *, conflicting reports; ?, unknown, not yet reported.
Data from LET mice are from the present study.
Data from prenatal androgen mice (PNA, given dihydrotestosterone [DHT] prenatally) are from [19, 23–26, 72], and data from postnatal DHT are from [19–21]. CL, corpus luteum, T, testosterone, LH, luteinizing hormone, FSH, follicle-stimulating hormone, Cyp19, aromatase gene, Fshr, FSH receptor gene, P4, progesterone, Pgr, progesterone receptor gene, Lhb, luteinizing hormone beta polypeptide gene, Fshb, Follicle-stimulating beta polypeptide gene.
One previous study recently tested the ability of LET to induce PCOS in mice. That study similarly reported polycystic ovaries and irregular estrous cycles as well as elevated T [19]. However, unlike our present findings, the previous study reported normal serum LH and FSH levels in LET-treated mice along with normal ovarian weights, body weights, adipocyte size, and no metabolic dysfunction [19]. The reason for the absent hormonal and metabolic phenotype in the prior study is not immediately clear, especially given the strong phenotypes elicited in the present study. The Caldwell et al. study [19] used two LET doses (44 and 88 μg/day) similar to our dose of 50 μg/day, so the differences in outcomes between our study and theirs are unlikely to be due to dosing. There were other minor technical differences between the two studies, including different C57 mouse substrains (C57BL/6J vs. C57BL/6N), different ages and durations of LET exposure (90 days initiated at Postnatal Day 21 vs. 35 days initiated at Postnatal Day 28), and different sources of the LET compound (Fitzgerald versus Novartis Pharma), but it is unknown if any of these minor differences underlie the lack of a strong PCOS phenotype in the previous study. We note that our phenotype in mice nicely mirrors the phenotype previously obtained in rats as seen by Maliqueo et al. [27]. Moreover, we have now tested our LET treatment three different times in independent cohorts of mice, and we consistently observe both the reproductive and metabolic PCOS phenotypes. Thus, this present LET paradigm in mice may be a useful tool in future studies to genetically probe the mechanisms of the PCOS phenotype.
ACKNOWLEDGMENT
We thank Dana Skarra, Jason Meadows, Emily Rickert, Navdeep Chahal, Martina Belli, and Alissa Rivera for technical support and helpful comments on the manuscript.
Footnotes
Supported by NIH through a cooperative agreement as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research U54 HD012303 (U.C. San Diego) and U54 HD028934 (University of Virginia Ligand Assay and Analysis Core). It was also supported by NIH R01 HD065856 (A.S.K.), R01 HD072754 and R01 HD082567 (P.L.M.), and R01 HD067448 (V.G.T.). P.L.M. was also partially supported by P30 DK063491, P30 CA023100, K12 HD001259, and P42 ES101337. G.E.R. was partially supported by P42 ES101337 and T32 GM008666. T32 HD007203 partially supported K.P.T., S.J.S., and C.A.G.-K. K.P.T. was partially supported by F32 HD076606. S.J.S. was partially supported by F32 HD066849. C.A.G.-K. was partially supported by the Hartwell Foundation Fellowship. N.J.W. was partially supported by P30 DK063491, P30 CA023100, and U54 CA155435.
REFERENCES
- Zadwadski J, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: Toward a rational approach. In: Dunaif A, Givens JR, Haseltine FP, Merriam GR, editors. Polycystic Ovary Syndrome. Boston: Blackwell Scientific; 1992. pp. 377–384. In. (eds) [Google Scholar]
- Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81:19–25. doi: 10.1016/j.fertnstert.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, Janssen OE, Legro RS, Norman RJ, Taylor AE, Witchel SF. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report. Fertil Steril. 2009;91:456–488. doi: 10.1016/j.fertnstert.2008.06.035. [DOI] [PubMed] [Google Scholar]
- Fauser BC, Tarlatzis BC, Rebar RW, Legro RS, Balen AH, Lobo R, Carmina E, Chang J, Yildiz BO, Laven JS, Boivin J, Petraglia F, Wijeyeratne CN, Norman RJ, Dunaif A, Franks S, Wild RA, Dumesic D, Barnhart K. Consensus on women's health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil Steril. 2012;97:28–38. doi: 10.1016/j.fertnstert.2011.09.024. [DOI] [PubMed] [Google Scholar]
- Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS) Hum Reprod. 2004;19:41–47. doi: 10.1093/humrep/deh098. [DOI] [PubMed] [Google Scholar]
- Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol. 2011;7:219–231. doi: 10.1038/nrendo.2010.217. [DOI] [PubMed] [Google Scholar]
- Blank SK, McCartney CR, Helm KD, Marshall JC. Neuroendocrine effects of androgens in adult polycystic ovary syndrome and female puberty. Semin Reprod Med. 2007;25:352–359. doi: 10.1055/s-2007-984741. [DOI] [PubMed] [Google Scholar]
- Blank SK, McCartney CR, Marshall JC. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Hum Reprod Update. 2006;12:351–361. doi: 10.1093/humupd/dml017. [DOI] [PubMed] [Google Scholar]
- Moran LJ, Misso ML, Wild RA, Norman RJ. Impaired glucose tolerance, type 2 diabetes and metabolic syndrome in polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod Update. 2010;16:347–363. doi: 10.1093/humupd/dmq001. [DOI] [PubMed] [Google Scholar]
- Moran LJ, Lombard CB, Lim S, Noakes M, Teede HJ. Polycystic ovary syndrome and weight management. Women's Health (Lond Engl) 2010;6:271–283. doi: 10.2217/whe.09.89. [DOI] [PubMed] [Google Scholar]
- Foecking EM, Szabo M, Schwartz NB, Levine JE. Neuroendocrine consequences of prenatal androgen exposure in the female rat: absence of luteinizing hormone surges, suppression of progesterone receptor gene expression, and acceleration of the gonadotropin-releasing hormone pulse generator. Biol Reprod. 2005;72:1475–1483. doi: 10.1095/biolreprod.105.039800. [DOI] [PubMed] [Google Scholar]
- Abbott DH, Barnett DK, Levine JE, Padmanabhan V, Dumesic DA, Jacoris S, Tarantal AF. Endocrine antecedents of polycystic ovary syndrome in fetal and infant prenatally androgenized female rhesus monkeys. Biol Reprod. 2008;79:154–163. doi: 10.1095/biolreprod.108.067702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foecking EM, McDevitt MA, Acosta-Martinez M, Horton TH, Levine JE. Neuroendocrine consequences of androgen excess in female rodents. Horm Behav. 2008;53:673–692. doi: 10.1016/j.yhbeh.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SD, Moenter SM. Prenatal androgens alter GABAergic drive to gonadotropin-releasing hormone neurons: implications for a common fertility disorder. Proc Natl Acad Sci U S A. 2004;101:7129–7134. doi: 10.1073/pnas.0308058101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott DH, Tarantal AF, Dumesic DA. Fetal, infant, adolescent and adult phenotypes of polycystic ovary syndrome in prenatally androgenized female rhesus monkeys. Am J Primatol. 2009;71:776–784. doi: 10.1002/ajp.20679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan V, Veiga-Lopez A. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol. 2013;373:8–20. doi: 10.1016/j.mce.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters KA, Allan CM, Handelsman DJ. Rodent models for human polycystic ovary syndrome Biol Reprod 2012. 86 149. [DOI] [PubMed] [Google Scholar]
- Franks S. Animal models and the developmental origins of polycystic ovary syndrome: increasing evidence for the role of androgens in programming reproductive and metabolic dysfunction. Endocrinology. 2012;153:2536–2538. doi: 10.1210/en.2012-1366. [DOI] [PubMed] [Google Scholar]
- Caldwell AS, Middleton LJ, Jimenez M, Desai R, McMahon AC, Allan CM, Handelsman DJ, Walters KA. Characterization of reproductive, metabolic, and endocrine features of polycystic ovary syndrome in female hyperandrogenic mouse models. Endocrinology. 2014;155:3146–3159. doi: 10.1210/en.2014-1196. [DOI] [PubMed] [Google Scholar]
- Manneras L, Cajander S, Holmang A, Seleskovic Z, Lystig T, Lonn M, Stener-Victorin E. A new rat model exhibiting both ovarian and metabolic characteristics of polycystic ovary syndrome. Endocrinology. 2007;148:3781–3791. doi: 10.1210/en.2007-0168. [DOI] [PubMed] [Google Scholar]
- van Houten EL, Kramer P, McLuskey A, Karels B, Themmen AP, Visser JA. Reproductive and metabolic phenotype of a mouse model of PCOS. Endocrinology. 2012;153:2861–2869. doi: 10.1210/en.2011-1754. [DOI] [PubMed] [Google Scholar]
- Roland AV, Moenter SM. Prenatal androgenization of female mice programs an increase in firing activity of gonadotropin-releasing hormone (GnRH) neurons that is reversed by metformin treatment in adulthood. Endocrinology. 2011;152:618–628. doi: 10.1210/en.2010-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland AV, Nunemaker CS, Keller SR, Moenter SM. Prenatal androgen exposure programs metabolic dysfunction in female mice. J Endocrinol. 2010;207:213–223. doi: 10.1677/JOE-10-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AM, Prescott M, Campbell RE. Estradiol negative and positive feedback in a prenatal androgen-induced mouse model of polycystic ovarian syndrome. Endocrinology. 2013;154:796–806. doi: 10.1210/en.2012-1954. [DOI] [PubMed] [Google Scholar]
- Caldwell AS, Eid S, Kay CR, Jimenez M, McMahon AC, Desai R, Allan CM, Smith JT, Handelsman DJ, Walters KA. Haplosufficient genomic androgen receptor signaling is adequate to protect female mice from induction of polycystic ovary syndrome features by prenatal hyperandrogenization. Endocrinology. 2015;156:1441–1452. doi: 10.1210/en.2014-1887. [DOI] [PubMed] [Google Scholar]
- Witham EA, Meadows JD, Shojaei S, Kauffman AS, Mellon PL. Prenatal exposure to low levels of androgen accelerates female puberty onset and reproductive senescence in mice. Endocrinology. 2012;153:4522–4532. doi: 10.1210/en.2012-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maliqueo M, Sun M, Johansson J, Benrick A, Labrie F, Svensson H, Lonn M, Duleba AJ, Stener-Victorin E. Continuous administration of a P450 aromatase inhibitor induces polycystic ovary syndrome with a metabolic and endocrine phenotype in female rats at adult age. Endocrinology. 2013;154:434–445. doi: 10.1210/en.2012-1693. [DOI] [PubMed] [Google Scholar]
- Naessen T, Kushnir MM, Chaika A, Nosenko J, Mogilevkina I, Rockwood AL, Carlstrom K, Bergquist J, Kirilovas D. Steroid profiles in ovarian follicular fluid in women with and without polycystic ovary syndrome, analyzed by liquid chromatography-tandem mass spectrometry. Fertil Steril. 2010;94:2228–2233. doi: 10.1016/j.fertnstert.2009.12.081. [DOI] [PubMed] [Google Scholar]
- Xita N, Lazaros L, Georgiou I, Tsatsoulis A. CYP19 gene: a genetic modifier of polycystic ovary syndrome phenotype. Fertil Steril. 2010;94:250–254. doi: 10.1016/j.fertnstert.2009.01.147. [DOI] [PubMed] [Google Scholar]
- Xita N, Chatzikyriakidou A, Stavrou I, Zois C, Georgiou I, Tsatsoulis A. The (TTTA)n polymorphism of aromatase (CYP19) gene is associated with age at menarche. Hum Reprod. 2010;25:3129–3133. doi: 10.1093/humrep/deq276. [DOI] [PubMed] [Google Scholar]
- Wang H, Li Q, Wang T, Yang G, Wang Y, Zhang X, Sang Q, Wang H, Zhao X, Xing Q, Shi J, He L, et al. A common polymorphism in the human aromatase gene alters the risk for polycystic ovary syndrome and modifies aromatase activity in vitro. Mol Hum Reprod. 2011;17:386–391. doi: 10.1093/molehr/gar007. [DOI] [PubMed] [Google Scholar]
- Ito Y, Fisher CR, Conte FA, Grumbach MM, Simpson ER. Molecular basis of aromatase deficiency in an adult female with sexual infantilism and polycystic ovaries. Proc Natl Acad Sci U S A. 1993;90:11673–11677. doi: 10.1073/pnas.90.24.11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Semaan SJ, Clifton DK, Steiner RA, Dhamija S, Kauffman AS. Regulation of Kiss1 expression by sex steroids in the amygdala of the rat and mouse. Endocrinology. 2011;152:2020–2030. doi: 10.1210/en.2010-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughesdon PE. Morphology and morphogenesis of the Stein-Leventhal ovary and of so-called “hyperthecosis”. Obstet Gynecol Surv. 1982;37:59–77. doi: 10.1097/00006254-198202000-00001. [DOI] [PubMed] [Google Scholar]
- Escobar ME, Ropelato MG, Ballerini MG, Gryngarten MG, Rudaz MC, Veldhuis JD, Barontini M. Acceleration of luteinizing hormone pulse frequency in adolescent girls with a history of central precocious puberty with versus without hyperandrogenism. Horm Res. 2007;68:278–285. doi: 10.1159/000104177. [DOI] [PubMed] [Google Scholar]
- Legro RS, Driscoll D, Strauss JF, III, , Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci U S A. 1998;95:14956–14960. doi: 10.1073/pnas.95.25.14956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmina E, Rosato F, Janni A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. 2006;91:2–6. doi: 10.1210/jc.2005-1457. [DOI] [PubMed] [Google Scholar]
- De Leo V, Lanzetta D, D'Antona D, la Marca A, Morgante G. Hormonal effects of flutamide in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1998;83:99–102. doi: 10.1210/jcem.83.1.4500. [DOI] [PubMed] [Google Scholar]
- Eagleson CA, Gingrich MB, Pastor CL, Arora TK, Burt CM, Evans WS, Marshall JC. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 2000;85:4047–4052. doi: 10.1210/jcem.85.11.6992. [DOI] [PubMed] [Google Scholar]
- Delemarre-van de Waal HA, van Coeverden SC, Engelbregt MT. Factors affecting onset of puberty. Horm Res. 2002;57(Suppl 2):15–18. doi: 10.1159/000058095. [DOI] [PubMed] [Google Scholar]
- Pasquali R. Obesity and androgens: facts and perspectives. Fertil Steril. 2006;85:1319–1340. doi: 10.1016/j.fertnstert.2005.10.054. [DOI] [PubMed] [Google Scholar]
- Burt Solorzano CM, Beller JP, Abshire MY, Collins JS, McCartney CR, Marshall JC. Neuroendocrine dysfunction in polycystic ovary syndrome. Steroids. 2012;77:332–337. doi: 10.1016/j.steroids.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebar R, Judd HL, Yen SS, Rakoff J, Vandenberg G, Naftolin F. Characterization of the inappropriate gonadotropin secretion in polycystic ovary syndrome. J Clin Invest. 1976;57:1320–1329. doi: 10.1172/JCI108400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird DT, Corker CS, Davidson DW, Hunter WM, Michie EA, Van Look PF. Pituitary-ovarian relationships in polycystic ovary syndrome. J Clin Endocrinol Metab. 1977;45:798–801. doi: 10.1210/jcem-45-4-798. [DOI] [PubMed] [Google Scholar]
- Pastor CL, Griffin-Korf ML, Aloi JA, Evans WS, Marshall JC. Polycystic ovary syndrome: evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 1998;83:582–590. doi: 10.1210/jcem.83.2.4604. [DOI] [PubMed] [Google Scholar]
- Levine JE, Chappell PE, Schneider JS, Sleiter NC, Szabo M. Progesterone receptors as neuroendocrine integrators. Front Neuroendocrinol. 2001;22:69–106. doi: 10.1006/frne.2001.0210. [DOI] [PubMed] [Google Scholar]
- Goodman RL, Bittman EL, Foster DL, Karsch FJ. The endocrine basis of the synergistic suppression of luteinizing hormone by estradiol and progesterone. Endocrinology. 1981;109:1414–1417. doi: 10.1210/endo-109-5-1414. [DOI] [PubMed] [Google Scholar]
- McPherson JC, III, , Costoff A, Mahesh VB. Influence of estrogen-progesterone combinations on gonadotropin secretion in castrate female rats. Endocrinology. 1975;97:771–779. doi: 10.1210/endo-97-4-771. [DOI] [PubMed] [Google Scholar]
- Leipheimer RE, Bona-Gallo A, Gallo RV. The influence of progesterone and estradiol on the acute changes in pulsatile luteinizing hormone release induced by ovariectomy on diestrus day 1 in the rat. Endocrinology. 1984;114:1605–1612. doi: 10.1210/endo-114-5-1605. [DOI] [PubMed] [Google Scholar]
- Leipheimer RE, Bona-Gallo A, Gallo RV. Influence of estradiol and progesterone on pulsatile LH secretion in 8-day ovariectomized rats. Neuroendocrinology. 1986;43:300–307. doi: 10.1159/000124544. [DOI] [PubMed] [Google Scholar]
- Goodman RL, Daniel K. Modulation of pulsatile luteinizing hormone secretion by ovarian steroids in the rat. Biol Reprod. 1985;32:217–225. doi: 10.1095/biolreprod32.2.217. [DOI] [PubMed] [Google Scholar]
- Pielecka J, Quaynor SD, Moenter SM. Androgens increase gonadotropin-releasing hormone neuron firing activity in females and interfere with progesterone negative feedback. Endocrinology. 2006;147:1474–1479. doi: 10.1210/en.2005-1029. [DOI] [PubMed] [Google Scholar]
- Cheng G, Coolen LM, Padmanabhan V, Goodman RL, Lehman MN. The kisspeptin/neurokinin B/dynorphin (KNDy) cell population of the arcuate nucleus: sex differences and effects of prenatal testosterone in sheep. Endocrinology. 2010;151:301–311. doi: 10.1210/en.2009-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003;4:117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson VL, Legro RS, Strauss JF, III, , McAllister JM. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol. 1999;13:946–957. doi: 10.1210/mend.13.6.0311. [DOI] [PubMed] [Google Scholar]
- Weil S, Vendola K, Zhou J, Bondy CA. Androgen and follicle-stimulating hormone interactions in primate ovarian follicle development. J Clin Endocrinol Metab. 1999;84:2951–2956. doi: 10.1210/jcem.84.8.5929. [DOI] [PubMed] [Google Scholar]
- Almahbobi G, Anderiesz C, Hutchinson P, McFarlane JR, Wood C, Trounson AO. Functional integrity of granulosa cells from polycystic ovaries. Clin Endocrinol (Oxf) 1996;44:571–580. doi: 10.1046/j.1365-2265.1996.724545.x. [DOI] [PubMed] [Google Scholar]
- Wu YG, Bennett J, Talla D, Stocco C. Testosterone, not 5alpha-dihydrotestosterone, stimulates LRH-1 leading to FSH-independent expression of Cyp19 and P450scc in granulosa cells. Mol Endocrinol. 2011;25:656–668. doi: 10.1210/me.2010-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakimiuk AJ, Weitsman SR, Brzechffa PR, Magoffin DA. Aromatase mRNA expression in individual follicles from polycystic ovaries. Mol Hum Reprod. 1998;4:1–8. doi: 10.1093/molehr/4.1.1. [DOI] [PubMed] [Google Scholar]
- Maas KH, Chuan SS, Cook-Andersen H, Su HI, Duleba A, Chang RJ. Relationship between 17-hydroxyprogesterone responses to human chorionic gonadotropin and markers of ovarian follicle morphology in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2015;100:293–300. doi: 10.1210/jc.2014-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glidewell-Kenney C, Weiss J, Hurley LA, Levine JE, Jameson JL. Estrogen receptor alpha signaling pathways differentially regulate gonadotropin subunit gene expression and serum follicle-stimulating hormone in the female mouse. Endocrinology. 2008;149:4168–4176. doi: 10.1210/en.2007-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw ND, Histed SN, Srouji SS, Yang J, Lee H, Hall JE. Estrogen negative feedback on gonadotropin secretion: evidence for a direct pituitary effect in women. J Clin Endocrinol Metab. 2010;95:1955–1961. doi: 10.1210/jc.2009-2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe SA, Adams B, Adams TE. Effect of phytoestrogens on basal and GnRH-induced gonadotropin secretion. J Endocrinol. 2013;219:243–250. doi: 10.1530/JOE-13-0158. [DOI] [PubMed] [Google Scholar]
- Wild RA, Carmina E, Diamanti-Kandarakis E, Dokras A, Escobar-Morreale HF, Futterweit W, Lobo R, Norman RJ, Talbott E, Dumesic DA. Assessment of cardiovascular risk and prevention of cardiovascular disease in women with the polycystic ovary syndrome: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) Society. J Clin Endocrinol Metab. 2010;95:2038–2049. doi: 10.1210/jc.2009-2724. [DOI] [PubMed] [Google Scholar]
- Wild S, Pierpoint T, McKeigue P, Jacobs H. Cardiovascular disease in women with polycystic ovary syndrome at long-term follow-up: a retrospective cohort study. Clin Endocrinol (Oxf) 2000;52:595–600. doi: 10.1046/j.1365-2265.2000.01000.x. [DOI] [PubMed] [Google Scholar]
- Barber TM, Wass JA, McCarthy MI, Franks S. Metabolic characteristics of women with polycystic ovaries and oligo-amenorrhoea but normal androgen levels: implications for the management of polycystic ovary syndrome. Clin Endocrinol (Oxf) 2007;66:513–517. doi: 10.1111/j.1365-2265.2007.02764.x. [DOI] [PubMed] [Google Scholar]
- Moghetti P, Tosi F, Bonin C, Di Sarra D, Fiers T, Kaufman JM, Giagulli VA, Signori C, Zambotti F, Dall'Alda M, Spiazzi G, Zanolin ME, et al. Divergences in insulin resistance between the different phenotypes of the polycystic ovary syndrome. J Clin Endocrinol Metab. 2013;98:E628–E637. doi: 10.1210/jc.2012-3908. [DOI] [PubMed] [Google Scholar]
- Barbieri RL, Makris A, Randall RW, Daniels G, Kistner RW, Ryan KJ. Insulin stimulates androgen accumulation in incubations of ovarian stroma obtained from women with hyperandrogenism. J Clin Endocrinol Metab. 1986;62:904–910. doi: 10.1210/jcem-62-5-904. [DOI] [PubMed] [Google Scholar]
- Plymate SR, Matej LA, Jones RE, Friedl KE. Inhibition of sex hormone-binding globulin production in the human hepatoma (Hep G2) cell line by insulin and prolactin. J Clin Endocrinol Metab. 1988;67:460–464. doi: 10.1210/jcem-67-3-460. [DOI] [PubMed] [Google Scholar]
- Stepto NK, Cassar S, Joham AE, Hutchison SK, Harrison CL, Goldstein RF, Teede HJ. Women with polycystic ovary syndrome have intrinsic insulin resistance on euglycaemic-hyperinsulaemic clamp. Hum Reprod. 2013;28:777–784. doi: 10.1093/humrep/des463. [DOI] [PubMed] [Google Scholar]
- Sullivan SD, Moenter SM. Prenatal androgens alter GABAergic drive to gonadotropin-releasing hormone neurons: implications for a common fertility disorder. Proc Natl Acad Sci U S A. 2004;101:7129–7134. doi: 10.1073/pnas.0308058101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland AV, Moenter SM. Reproductive neuroendocrine dysfunction in polycystic ovary syndrome: insight from animal models. Front Neuroendocrinol. 2014;35:494–511. doi: 10.1016/j.yfrne.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev. 2012;33:981–1030. doi: 10.1210/er.2011-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]