

The relative contributions of the molecular and physical characteristics of tissue microenvironments to cell responses to receptor tyrosine kinase inhibitors are not well understood. Polymer-based tissue culture substrata were used to isolate and study the contribution of matrix elastic modulus. YAP and TAZ, transcriptional coactivators and mechanotransducers of the Hippo pathway, are essential for mediating elastic modulus–dependent resistance to the HER2-targeted anticancer drug, lapatinib.
Abstract
Stiffness is a biophysical property of the extracellular matrix that modulates cellular functions, including proliferation, invasion, and differentiation, and it also may affect therapeutic responses. Therapeutic durability in cancer treatments remains a problem for both chemotherapies and pathway-targeted drugs, but the reasons for this are not well understood. Tumor progression is accompanied by changes in the biophysical properties of the tissue, and we asked whether matrix rigidity modulated the sensitive versus resistant states in HER2-amplified breast cancer cell responses to the HER2-targeted kinase inhibitor lapatinib. The antiproliferative effect of lapatinib was inversely proportional to the elastic modulus of the adhesive substrata. Down-regulation of the mechanosensitive transcription coactivators YAP and TAZ, either by siRNA or with the small-molecule YAP/TEAD inhibitor verteporfin, eliminated modulus-dependent lapatinib resistance. Reduction of YAP in vivo in mice also slowed the growth of implanted HER2-amplified tumors, showing a trend of increasing sensitivity to lapatinib as YAP decreased. Thus we address the role of stiffness in resistance to and efficacy of a HER2 pathway–targeted therapeutic via the mechanotransduction arm of the Hippo pathway.
INTRODUCTION
Human epidermal growth factor receptor 2 (HER2)–positive breast cancers account for ∼15–20% of breast cancers, have poor prognosis, and are less responsive to hormone treatment than HER2(–) breast cancers (Kun et al., 2003; Koboldt et al., 2012). Although HER2-targeted therapies are one of the success stories in breast cancer treatment (Kim et al., 2013), generating a durable drug response remains a challenge (Rexer and Arteaga, 2012). The tyrosine kinase inhibitor lapatinib is a potent inhibitor of catalytic activity of both epidermal growth factor receptor (EGFR) and HER2 (Medina and Goodin, 2008). Most tumor cells with elevated HER2 levels show high sensitivity to growth inhibition by lapatinib, but resistance often develops (Rusnak et al., 2007).
Several mechanisms of lapatinib resistance have been reported, including compensatory activation of parts of the HER network. Compensatory up-regulation of HER3 activation, driven by Akt activity, confers resistance to lapatinib in HER2-amplified breast cancer cell lines (Amin et al., 2010). Incomplete inhibition of EGFR results in heregulin-driven feedback, sustaining EGFR activation, which contributes to lapatinib resistance in HER2(+) breast cancers (Xia et al., 2013). Up-regulation of the membrane tyrosine kinase AXL sustains phosphoinositide 3-kinase (PI3K)/Akt signaling, conferring lapatinib resistance (Liu et al., 2009). Hepatocyte growth factor (HGF) activation of its cognate receptor MET also is associated with resistance to lapatinib in HER2(+) gastric cancers (Chen et al., 2012). Thus activation of redundant survival pathways can be induced, either intrinsically or extrinsically, by microenvironmental factors, such as growth factors.
The biophysical properties of tumors change during malignant progression, which also affects tumor cell functions. Thus, in addition to redundant signaling pathways, using integrin-blocking antibodies to alter how tumor cells perceived their microenvironments was shown to modulate the efficiency of cytotoxic agents (Weaver et al., 2002). Use of an in vitro approach that compared responses of a number of HER2-targeted therapeutics (including lapatinib) in multiple HER2-amplified cell lines revealed that some cells were more sensitive to lapatinib in three-dimensional (3D) Matrigel than with two-dimensional (2D) cultures using tissue culture plastic (TCP; Weigelt et al., 2010). Even in the relatively simplified 3D Matrigel cultures, there are multiple chemical and physical properties that contribute to those microenvironment-dependent drug responses. Accumulating evidence suggests that microenvironment rigidity can promote tumor progression and survival through activation of growth factor signaling pathways by enhancing integrin clustering and focal adhesion assembly (Paszek et al., 2014; Rubashkin et al., 2014) or through modulation of microRNA expression (Mouw et al., 2014). Tissue rigidity also affects cytotoxic chemotherapeutic responses in hepatocarcinoma cells because increasing matrix stiffness promotes cellular proliferation and resistance to chemotherapeutic agents (Schrader et al., 2011). Whereas this scenario is quite sensible in the context of cytotoxic chemotherapeutics, the effect of matrix rigidity on a HER2-targeted therapeutic such as lapatinib is less obvious.
Here we examined whether matrix rigidity affected lapatinib responses in HER2-amplified breast cancer cells, using polyacrylamide (PA) hydrogel–based culture substrates that enabled control over Young’s elastic modulus. The Hippo pathway mechanotransducers, transcriptional coactivator with PDZ-binding motif (TAZ) and Yes-associated protein (YAP) (Halder et al., 2012), which are also oncogenes (Wang et al., 2012), were required for modulus-dependent responses in vitro. Down-regulation of YAP in vivo slowed HER2-amplified tumor growth and improved sensitivity to lapatinib. YAP and TAZ did not mediate resistance by redundant activation of other HER-family receptors. Our results suggest that rigid microenvironments can modulate lapatinib resistance in HER2-amplified breast cancer cells via a YAP/TAZ-dependent mechanism.
RESULTS
Substrate elastic modulus is a modifier of lapatinib responses in HER2-amplified breast cancer cells
To facilitate further investigation of microenvironment-directed drug responses, we identified a breast cancer cell line and pathway-targeted drug combination that offered a potentially wide dynamic range of response. Previous work demonstrated that the use of 2D TCP versus 3D Matrigel culture microenvironments modulated the antiproliferative responses of four different HER2-targeted therapeutics that were used to treat four different HER2-amplified breast cancer cell lines. The combination of HCC1569 cells, a basal A subtype cell line (Neve et al., 2006), and lapatinib demonstrated the optimal differential response between TCP and 3D Matrigel (Weigelt et al., 2010). We first validated our analysis methodology by showing that HER2-amplified HCC1569 breast cancer cells conformed to previous findings, that is, that they are more sensitive to the antiproliferative effect of lapatinib in 3D Matrigel than with cells on 2D TCP (Weigelt et al., 2010). After plating on type 1 collagen–coated 2D TCP or in 5% “on-top” 3D Matrigel, cells were treated with dimethyl sulfoxide (DMSO) or 1.5 μM lapatinib, a dose that was comparable to the average concentration in patient blood serum (Burris et al., 2005). The magnitude of the antiproliferative effect of lapatinib was determined by measuring 5-ethynyl-2′-deoxyuridine (EdU) incorporation into nuclear DNA as a proxy for cell proliferation. Cells in 3D Matrigel were more sensitive to lapatinib than cells on TCP, with 21 ± 2.6 and 69 9.7% EdU incorporation, respectively (Figure 1, A and B). Proliferation of the HER2-negative cell line BT549 was not affected by lapatinib treatment (Figure 1, A and B). The differential antiproliferative response between TCP and 3D Matrigel is partly explained by the distinct molecular compositions of the two culture microenvironments; indeed, it is known that the increased sensitivity in three dimensions is due partly to β1 integrin–mediated extracellular matrix adhesion (Weigelt et al., 2010). However, there are other potential microenvironment characteristics that bear scrutiny in this drug response context.
FIGURE 1:

Substrate elastic modulus is a modifier of lapatinib responses in HER2-amplified cancer cells. Bar graphs show the relative incorporation of EdU expressed as a percentage of DMSO-treated cells in (A) HCC1569 and BT549 cells cultured on 2D TCP dishes or 3D in Matrigel for 48 h and then treated with lapatinib or DMSO for 48 h (n = 3; 500 cells/condition per experiment, *p < 0.05). (B) Representative images of HCC1569 and BT549 cells cultured on 2D TCP or in 3D Matrigel. EdU is pseudocolored red, and nuclear DNA is blue. (C) Bar graphs showing relative incorporation of EdU expressed as a percentage of DMSO-treated cells. HCC1569 and BT549 cells cultured on 2D TCP or 400-Pa PA gels for 48 h, followed by lapatinib (1.5 μM) or DMSO for 48 h (n = 3; 500 cells/condition per experiment, *p < 0.05). (D) Representative images of HCC1569 and BT549 cells cultured on 2D TCP or 400-Pa PA gels. EdU is pseudocolored red, and nuclear DNA is blue. Scale bars, 20 μm. (E) Dose-response curves used to calculate IC50 of lapatinib in HCC1569 cells cultured on 400-Pa PA gel (0.94 μM) vs. 2D TCP (2.66 μM). n = 3; 500 cells/condition per experiment, *p < 0.05.
One of the major differences between TCP and 3D Matrigel is the rigidity of the culture substrate. Thus we examined whether rigidity is a modulator of responses to lapatinib in HER2-amplified breast cancer cells. Young’s elastic modulus of Matrigel has been estimated at 400 Pa (Soofi et al., 2009), on a par with that for normal breast tissue (Paszek et al., 2005). In contrast, the elastic modulus of TCP is >2 GPa (Saruwatari et al., 2005; Levental et al., 2007), which is well outside the physiological range (Kolahi et al., 2012). To examine the role played by matrix rigidity in lapatinib responses, cell culture substrates were fabricated from PA gels tuned to 400 ± 160 Pa and coated with a type 1 collagen to support cell adhesion. HCC1569 cells were more sensitive to lapatinib on 400-Pa PA gels than on TCP coated with type 1 collagen, with 50 ± 4.5 and 69 ± 4.5% EdU incorporation, respectively (Figure 1, C and D); BT549 cells were not affected by lapatinib or changes in rigidity (Figure 1, C and D). The half-maximal inhibitory concentration (IC50) of lapatinib was threefold lower on 400-Pa PA gels than with TCP, 0.9 and 2.7 μM, respectively (Figure 1E). Thus HCC1569 cells responded to lapatinib in an elastic modulus–dependent manner, showing greater resistance to the antiproliferative effect of lapatinib on rigid matrices.
YAP and TAZ are required for the modulus-dependent lapatinib responses
YAP and TAZ are Hippo pathway transcriptional coactivators that interact with the Rho/Rock pathway (Halder et al., 2012) and play an important role in transducing information about substrate rigidity from the plasma membrane into the nucleus, where a transcriptional response is generated (Dupont et al., 2011). Consistent with their role in mechanotransduction, YAP and TAZ relocated from the cytoplasm into the nucleus as substrate stiffness increased (Figure 2, A and B). We assessed the effect of YAP and TAZ knockdown by small interfering RNA (siRNA) on modulus-dependent responses to lapatinib. Both YAP and TAZ knockdown (Supplemental Figure S1A) eliminated modulus-dependent lapatinib resistance on TCP (Figure 2C). Disruption of the TEAD-YAP interaction with 2 μg/ml inhibitor verteporfin (Liu-Chittenden et al., 2012) phenocopied the effect of YAP knockdown (Figure 2D). Indeed, increasing concentrations of verteporfin diminished the effect of modulus-dependent lapatinib resistance in a synergistic manner with lapatinib (Supplemental Figure S2). YAP and TAZ were thus shown to be necessary for generating modulus-dependent lapatinib resistance.
FIGURE 2:

YAP and TAZ are required for modulus-dependent lapatinib responses. (A) Representative images of HCC1569 after 48 h culture on substrates of increasing stiffness: immunofluorescence stains represent YAP (green), TAZ (red), and nucleus (blue). Scale bars, 20 μm. (B) Bar graphs showing the proportions of single cells in which YAP and TAZ were located in the nucleus or cytoplasm or evenly distributed in both compartments as a function of stiffness (n = 3; 100 cells/condition per experiment, *p < 0.05). (C) Bar graphs showing the relative incorporation of EdU in HCC1569 cells cultured on 2D TCP and 400-Pa PA gel with YAP or TAZ knockdown by siRNA for 72 h and then treated with lapatinib (1.5 μM) or DMSO for 48 h. Results are expressed as a percentage of cells treated with DMSO and NSC siRNA-treated cells (n = 3; 500 cells/condition per experiment, *p < 0.05). (D) Bar graphs showing relative incorporation of EdU in HCC1569 cells cultured on 400-Pa and 40-kPa PA gels for 48 h and then treated with lapatinib (1.5 μM) together with verteporfin (2 μg/ml) or DMSO for 48 h. Results expressed as percentage of DMSO-treated controls (n = 3; 500/condition per experiment, *p < 0.05).
YAP knockdown in vivo increased sensitivity to lapatinib treatment
To test whether YAP similarly played a role in lapatinib responses in vivo, we used isopropyl β-d-1-thiogalactopyranoside (IPTG)–induced short hairpin RNA (shRNA) to knock down YAP in HCC1569 cells implanted in mice. Tumor volume was measured during the course of lapatinib treatment (Figure 3). Mice that received neither IPTG nor lapatinib (group A) had the maximum tumor volume (mean of volume, 1280 mm3) by day 23. Mice treated with IPTG (group B) had significantly decreased (p < 0.05) tumor volume (mean of volume, 770 mm3) compared with group A. Lapatinib treatment groups either with (group D) or without IPTG treatment (group C) had much smaller tumor volumes compared with groups A and B. Group D, which received lapatinib and had reduced YAP levels, had the smallest tumor volumes (mean of volume, 192 mm3), even compared with group C (mean of volume, 269 mm3); however, that difference was not statistically significant. These data demonstrate that YAP knockdown was sufficient to reduce growth of HER2-amplified cell lines in vivo, and they suggest that YAP knockdown and lapatinib together may have some synergistic benefit. More comprehensive animal studies are required, however, to clarify the independent versus synergistic effects.
FIGURE 3:
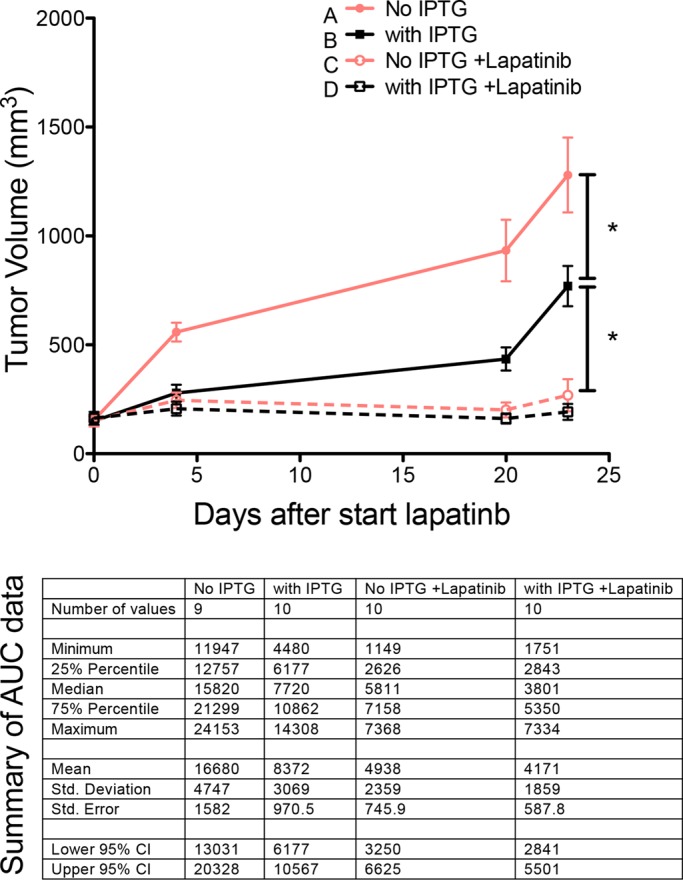
YAP knockdown has a synergistic trend of inhibition with lapatinib in vivo. Tumor volume curves as a function of time and the summary table of area-under-the-curve data for different treatment groups. The tumor volume was measured during the course of lapatinib treatment on mice that did not receive IPTG or lapatinib (group A), mice treated with IPTG only (group B), mice treated with lapatinib only (group C), and mice treated with lapatinib together with IPTG (group D).
Modulus-dependent lapatinib responses are driven by multiple factors
We sought to delineate other components of the molecular circuitry that enabled YAP to mediate the modulus-dependent response to lapatinib. Analysis of breast cancer data from the Cancer Genome Atlas (Koboldt et al., 2012) showed that YAP mRNA expression correlated positively with expression of two known YAP targets, CTGF and amphiregulin (AREG; Zhang et al., 2009), which is an EGFR ligand that has been attributed with multiple roles related to tumor invasion and drug resistance (Hurbin et al., 2002; Higginbotham et al., 2011; Supplemental Figure S3). HER2 (encoded by the ERBB2 gene) mRNA expression positively correlated with AREG and inversely correlated with EGFR and ERBB3 mRNA levels. Taken together, the results indicate that higher AREG expression correlated with YAP and HER2 expression in breast cancers.
We further examined AREG, which has been shown to mediate EGFR-HER3 heterodimer formation and activate the extracellular signal–regulated kinase (ERK)-Akt signaling pathway (Yotsumoto et al., 2010). In addition, HER3-mediated PI3K/Akt activity was correlated with lapatinib resistance in HER2-amplified breast cancer cells (Garrett et al., 2011). Paracrine AREG signaling in colorectal cancer cells also was shown to sustain ERK signaling and confer resistance to EGFR inhibitors (Hobor et al., 2014).
AREG mRNA levels in HCC1569 cells showed a modestly increasing trend as the culture substrate rigidity was increased (Figure 4A), but cell membrane surface AREG protein level increased only ∼4% (Figure 4B). To mimic the presence of a paracrine source of AREG, we added exogenous recombinant AREG (5 ng/ml) to cells, which caused increased nuclear YAP localization, even on compliant 400-Pa surfaces (Figure 4C), and on compliant surfaces conferred lapatinib resistance to the cells (Figure 4D). Simultaneous addition of exogenous AREG and the EGFR receptor inhibitor, erlotinib, reduced the resistance phenotype, demonstrating that exogenous AREG was exerting its effect partly through EGFR (Figure 4D). Knockdown of AREG by siRNA (siAREG) did not affect other ligands of EGFR, such as EGF or transforming growth factor-α, nor did it affect the receptors EGFR and HER2 (Supplemental Figure S1B). YAP knockdown with siRNA decreased AREG expression 25%, suggesting that YAP modulates AREG (Figure 4E). These data together suggest that AREG is putatively involved in the modulus-dependent lapatinib responses. However, direct targeting of AREG via siAREG showed no significant effect on modulus-dependent lapatinib responses (Figure 4F). Because AREG was reported to cause activation of HER3 (Yotsumoto et al., 2010), we examined modulus-dependent changes in HER3 phosphorylation at 1 and 48 h after attachment, as well as from 49 other receptor tyrosine kinases. Without lapatinib treatment, HER2 showed a higher phosphorylation on compliant substrates (400 Pa) within the first hour and increased phosphorylation after 48 h. However, consistent with the notion that AREG did not play a significant role in modulus-dependent responses, HER3 was unchanged within the first hour and was decreased by 48 h (Supplemental Figure S4). With lapatinib treatment, both HER2 and HER3 showed decreased phosphorylation within 1 h, and then HER3 showed a subtle increase in phosphorylation by 48 h on stiffer substrates (40 kPa). The results taken together show that modulus-dependent lapatinib resistance cannot be explained from a single YAP-AREG circuit.
FIGURE 4:

YAP-dependent amphiregulin protein regulation is involved in modulus-dependent lapatinib responses. Bar graphs showing (A) AREG mRNA expression level measured by qRT-PCR and (B) cell surface AREG measured by FACS in HCC1569 cultured on TCP or 400-Pa PA gels for 96 h (n = 3; 10,000 cells/condition per experiment, *p < 0.05). (C) Bar graphs showing the proportions of single cells in which YAP and TAZ were located in the nucleus or cytoplasm or evenly distributed in both compartments as a function of stiffness in HCC1569 cells cultured on TCP or 400-Pa PA gel for 48 h and then treated with AREG (5 ng/ml) for 48 h (n = 3; 100 cells/condition per experiment, *p < 0.05). (D) Bar graphs showing the relative incorporation of EdU, expressed as a percentage of DMSO-treated cells in HCC1569 cultured on TCP or 400-Pa PA gels for 48 h and then treated with lapatinib (1.5 mM), AREG (5 ng/ml), and erlotinib (1.5 mM) for 48 h (n = 3; 500 cells/condition per experiment, *p < 0.05). (E) Bar graphs showing intracellular AREG protein levels measured by enzyme-linked immunosorbent assay in HCC1569 with NSC siRNA or YAP knockdown by siRNA for 72 h (n = 3, *p < 0.05). (F) Bar graphs show the relative incorporation of EdU, expressed as a percentage of DMSO- and NSC siRNA–treated cells in HCC1569 cultured on 400-Pa and 40-kPa PA gels with AREG knockdown by siRNA for 72 h and then treated with lapatinib (1.5 μM) or DMSO for 48 h (n = 3; 500 cells/condition per experiment, *p < 0.05).
DISCUSSION
Here we demonstrate that the mechanical property of microenvironments may influence resistance to and efficacy of the HER2 pathway–targeted therapeutic lapatinib in HER2-amplified breast cancer cells. Although engineered culture substrates necessarily oversimplify the tumor microenvironment compared with in vivo, they can reveal important mechanistic elements of cellular responses by winnowing down the possible candidate pathways involved in a given response. We specifically probed the property of elastic modulus on cells at low density on PA gels to minimize the confounding effects of cell–cell contact. YAP activation is regulated also by cell–cell contact (Zhao et al., 2007); high cell density inhibits YAP activation by inducing YAP phosphorylation. In our engineered system, YAP and TAZ activation correlated with resistance to lapatinib, and when YAP was knocked out in orthotopically implanted tumors grown in mice, tumor growth slowed and they became more sensitive to lapatinib. The resistance phenotype is not exclusively modulus dependent, but by isolating and studying that one physical property of the matrix, we showed that the Hippo pathway is likely an important component of resistance in HER2-targeted kinase inhibitors.
YAP has been attributed dual roles in tumor genesis in breast cancer. Studies in vitro show that exogenous expression of YAP in cells can promote cell growth, suggesting that YAP has a role as a tumor promoter (Wang et al., 2012). Others studies reported potential tumor-suppressive roles for YAP. Loss of heterozygosity at the YAP locus in a number of luminal breast cancers and shRNA knockdown of YAP in some breast cancer cell lines suppress anoikis and promotes tumor growth in vivo (Yuan et al., 2008). Moreover, YAP expression was reduced in some invasive carcinoma samples compared with normal breast tissues (Tufail et al., 2012). The multiple roles of YAP in tumorigenesis may depend on the stage of progression of a given cell; for example, active YAP enhances tumor growth when expressed in mammary carcinomas but not when expressed in a nonmalignant mammary epithelial cell line (Lamar et al., 2012). YAP expression in breast cancers may be subtype dependent (Kim et al., 2014), and YAP is notably present in stromal cells (Calvo et al., 2013) and not only epithelial cells, which further complicates the in vivo situation. In addition, YAP and TAZ exhibit distinct functions; for example, YAP-knockout mice are embryonic lethal (Morin-Kensicki et al., 2006), but TAZ- knockout mice can be viable, although the animals have kidney disease (Hossain et al., 2007). Here we showed that either YAP or TAZ knockdown in vitro can eliminate modulus-dependent lapatinib responses and confer more sensitivity to lapatinib, suggesting redundant roles in the context of modulus-dependent response to lapatinib.
The photosensitizer verteporfin (Visudyne; Novartis, Basel, Switzerland) is used as photodynamic therapy for neovascular lesions in the eye (Michels and Schmidt-Erfurth, 2001). In the absence of photoactivation, verteporfin can disrupt TEAD-YAP association and inhibit YAP-induced liver overgrowth (Liu-Chittenden et al., 2012), suggesting a pharmacological strategy for regulating the transcriptional activities of YAP, which requires TEAD-family proteins to bind to DNA. Our data show that verteporfin has a synergistic effect with lapatinib in vitro, indicating that there is a potential benefit to testing verteporfin in the context of lapatinib resistance.
Many studies into resistance to HER2-targeted cancer therapeutics have focused on intrinsic cellular mechanisms, such as compensatory pathway activation, but the tumor microenvironment will likely play an important role as well. Indeed, greater matrix rigidity leads to increased resistance to the antiproliferative effects of lapatinib in HER2-amplified breast cancer cells on culture substrates engineered to mimic different levels of matrix rigidity. Several studies have shown in various cancer contexts that increasing matrix stiffness can both promote chemotherapeutic resistance (Schrader et al., 2011; Sharma et al., 2014; Zustiak et al., 2014) and decrease sensitivity to Raf kinase inhibitors (Nguyen et al., 2014); however, the mechanisms underlying these elastic modulus–dependent effects are not well defined.
Lapatinib resistance also has been linked to compensatory activation of HER3 (Amin et al., 2010), but we did not find that HER3 was strongly activated in the context of increased matrix rigidity. To mimic levels found in patient serum, we used 1.5 μM lapatinib, a concentration considered high (Amin et al., 2010), and measured HER3 phosphorylation 48 h after lapatinib treatment, whereas compensatory activation of HER3 may occur after 72 h and at a lower lapatinib concentration. Taken together, and experimental differences notwithstanding, these reports suggest that resistance to HER2-targeted kinase inhibitors is likely a multifaceted challenge still to be overcome.
That most failures in drug development are due to a lack of efficacy suggests that our preclinical development toolbox does a poor job of predicting compound activity in vivo (Baker and Chen, 2012). Multiwell TCP plates are still the substrate of choice for much of modern drug screening, which ignores an obvious lack of context (Labarge et al., 2014). Some high throughput (HT)–compatible 3D culture systems are being developed to overcome this problem, but retooling of HT systems and improvements in image analysis algorithms remain significant barriers to wide-scale adoption. Adaptation of 2D hydrogels that are controlled for tissue-like elastic moduli and are conjugated with tissue-like molecular milieus to HT systems might present an intermediate step that can both take advantage of existing HT systems and recapitulate some key elements of in vivo microenvironments that are crucial for determinants of drug responses.
Our data show that microenvironment rigidity influenced lapatinib responses in HER2-amplified breast cancer cell lines and that YAP and TAZ are important in this context. Our findings underscore the importance of microenvironmental effects in drug development and suggest potential therapeutic benefits of verteporfin in HER2-targeted treatment.
MATERIALS AND METHODS
Cell culture and drug treatment
HCC1569 (American Type Culture Collection, Manassas, VA) and BT549 breast cancer cell lines (a gift from Joe W. Gray, Oregon Health and Science University, Portland, OR) were maintained in RPMI 1640 (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (FBS; Gemini Bio-Products, West Sacramento, CA), and 1% penicillin/streptomycin/glutamine (Invitrogen). Genotypes of the cell lines were authenticated by STR profiling (DDC Medical, Fairfield, OH). For drug treatment in 2D cultures, cells were cultured in 24-well plates with RPMI 1640 with 1% FBS and 1% penicillin/streptomycin/glutamine for 48 h after initial adhesion and then treated with lapatinib (1.5 μM; LC Laboratories, Woburn, MA) for an additional 48 h. For drug treatment in 3D cultures, cells were cultured in 24-well plates coated with Matrigel (BD Biosciences, San Jose, CA) following the so-called “on-top” protocol adapted from Lee et al. (2007), using a 5% Matrigel drip, and then drugs or control were added on day 4 after cell plating for an additional 48 h. Other pharmaceutical and recombinant protein modulators were added concurrently with lapatinib: verteporfin (Sigma-Aldrich, St. Louis, MO) was added at 0.2, 2, and 10 μg/ml, recombinant human amphiregulin (Sigma-Aldrich) at 5 ng/ml, and erlotinib at 1.5 μM.
Tunable elastic modulus cell culture substrate fabrication
Polyacrylamide gels were polymerized on 12-mm-diameter coverslips etched with 0.1 M NaOH, adapted from Tse and Engler (2010). Acrylamide 3% and bis-acrylamide 0.06% were used to generate 400-Pa PA gels. Sulfo-SANPAH (0.5 mM; ProteoChem, Loves Park, IL) was added on PA gels and activated by ultraviolet light exposure for 10 min. PA gels were washed with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer and then incubated with type 1 collagen at room temperature for 2 h (0.1 mg/ml in 50 mM HEPES from calf skin; Sigma-Aldrich). Gels were rinsed with copious amounts of phosphate-buffered saline (PBS) before placing them in 24-well plates treated with polyHEMA (0.133 ml at 12 mg/ml in 95% EtOH; Sigma-Aldrich) for cell culture.
Proliferation assay
EdU incorporation and staining were performed according to the manufacturer’s protocol (Invitrogen). Nuclei were stained with Hoechst 33342. Images were acquired by a Zeiss 710 LSM (Carl Zeiss, Jena, Germany) confocal microscope, and images were analyzed with ImageJ (National Institutes of Health, Bethesda, MD). Drug response values are expressed as a percentage of DMSO-treated cells.
Transfection
Cells were transfected with YAP, WWTR1 (TAZ), AREG, or nonsilencing control siRNA (NSC; SMARTpool: ON-TARGET plus, GE Dharmacon, Lafayette, CO) with a fluorescein isothiocyanate label (siGLO Green Transfection Indicator; GE Dharmacon), using DharmaFECT 2 Transfection Reagent (GE Dharmacon) according to the manufacturer’s protocol 72 h before assay performance.
Immunofluorescence staining
Cells were fixed in 4% paraformaldehyde at room temperature for 10 min, blocked with PBS, 5% normal goat serum, and 0.1% Triton X-100 at room temperature for 30 min, and then incubated with primary antibodies anti-YAP (1:100; Santa Cruz Biotechnology, Dallas, TX) and anti-TAZ (1:200; Cell Signaling Technology, Beverly, MA) overnight at 4°C. Primary antibodies were visualized with fluorescent secondary antibodies raised in goats (1:500; Invitrogen) together with Hoechst 33342 (1:200; Sigma-Aldrich) incubated at room temperature for 2 h. Images were acquired with a Zeiss 710 LSM confocal microscope. Cell segmentation and single-cell fluorescence intensities were analyzed with Matlab script adapted from Pelissier et al., 2014. For quantification of YAP/TAZ localization, the ratios of mean fluorescence intensity in the cytoplasmic (C) and nuclear (N) compartments of segmented cells were used. The cutoffs of log2 ratios were used to establish three classes: C > N (X < −0.074), N = C (−0.074 < X < 0.074), and N > C (X > 0.074).
Real-Time PCR
Total RNA was extracted with TRIzol (Invitrogen) and purified by RNeasy prep (Qiagen, Valencia, CA). cDNA was synthesized with SuperScript III RT (Invitrogen). Transcripts levels were measured by quantitative real-time PCR (qRT-PCR) with iTaq SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA) and Light Cycler480 (Roche, Indianapolis, IN). Primer sequences were as follows: YAP, 5′-AGCCAGTTGCAGTTTTCAGG-3′ and 5′-AGCAGCAATGGACAAGGAAG-3′; TAZ (WWTR1), 5′-GGAGAAAACGCAGGACAAAC-3′ and 5′-TCATTGAAGAGGGGGATCAG-3′; AREG, 5′-GTGGTGCTGTCGCTCTTGATA-3′ and 5′-ACTCACAGGGGAAATCTCACT-3′; and glyceraldehyde-3-phosphate dehydrogenase, 5′-AAGGTGAAGGTCGGAGTCAAC-3′ and 5′-GGGGTCATTGATGGCAACAATA-3′.
Flow cytometry
Cells were collected via EDTA-PBS (0.4% EDTA) treatment without trypsin on ice. After washing with PBS, cells were blocked with PBS containing 2% bovine serum albumin, 5% normal goat serum, and 5 mM EDTA on ice for 30 min. Cells were incubated with the primary antibody anti-AREG (1:100; R&D Systems, Minneapolis, MN) on ice for 30 min, washed with PBS, and then treated with the secondary antibody on ice for 15 min. After two PBS washes, the level of AREG bound on cell membrane was measured with a FACSCalibur (Becton-Dickinson, San Jose, CA).
AREG enzyme-linked immunosorbent assay
The intracellular AREG protein level was measured according to the manufacturer’s protocol (Abcam, Cambridge, MA), after 72 h in HCC1569 cells cultured on 2D TCP and 400-Pa PA gel with YAP knockdown by siRNA or nonsilencing control.
Animal experiments
Six-week-old female nu−/− mice were obtained from Taconic (Germantown, NY) and housed five per cage with chow and water ad libitum in a controlled animal barrier. After 1 wk, the animals were injected subcutaneously into the upper flank with (3.5–5) × 106 shRNA YAP HCC1569 cells. On day 13 after tumor injection, when the average tumor volume was 150–200 mm3, IPTG (Sigma-Aldrich) and lapatinib were administered for 2 wk. IPTG was mixed into the drinking water at 10 mM/1% glucose in light-protected bottles and changed every 2–3 d. Lapatinib was administered at 75 mg/kg/day body weight divided into twice-daily dosing by oral gavage. Tumor dimensions (width, height, and depth) were measured biweekly. At the time of killing, tumors were harvested and either immediately snap frozen or fixed in Formalin. Animals were monitored for toxicity by measuring weight, assessing overall activity, and performing necropsy. All experimental procedures were followed according to the University of California, San Francisco, Animal Welfare Committee’s approved policies and guidelines.
Human phospho–receptor tyrosine kinase array
HCC1569 cells were cultured on 400-Pa and 40-kPa PA gels for 48 h, treated with lapatinib (1.5 μM) or DMSO, and then harvested at 1 or 48 h after lapatinib treatment. The phosphorylations of 49 different receptor tyrosine kinases were measured according to the manufacturer’s protocol (ARY001B, lot 1323072; R&D Systems).
Statistics
Significance was considered p < 0.05 or better using t tests and Pearson correlations. Those tests and area-under-the-curve calculations were performed with Prism (GraphPad, La Jolla, CA). *p < 0.05, **p < 0.01, and ***p < 0.001.
Supplementary Material
Acknowledgments
M.L. is supported by National Institutes of Health Grants NIA R00AG033176 and R01AG040081, the U.S. Department of Energy (DE-AC02-05CH11231), a Congressionally Directed Medical Research Programs/Breast Cancer Research Program Era of Hope Scholar Award, and the California Breast Cancer Research Program and Anita Tarr Turk Fund for Breast Cancer Research (20IB-0109).
Abbreviations used:
- AREG
amphiregulin
- EGFR
epidermal growth factor receptor
- HER2/3
human epidermal growth factor receptor 2/3
- HGF
hepatocyte growth factor
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- MET
hepatocyte growth factor receptor
- PA
polyacrylamide
- TCP
tissue culture plastic.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-07-0456) on September 2, 2015.
REFERENCES
- Amin DN, Sergina N, Ahuja D, McMahon M, Blair JA, Wang D, Hann B, Koch KM, Shokat KM, Moasser MM. Resiliency and vulnerability in the HER2-HER3 tumorigenic driver. Sci Transl Med. 2010;2:16ra17. doi: 10.1126/scitranslmed.3000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM, Chen CS. Deconstructing the third dimension—how 3D culture microenvironments alter cellular cues. J Cell Sci. 2012;125:3015–3024. doi: 10.1242/jcs.079509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris HA, 3rd, Hurwitz HI, Dees EC, Dowlati A, Blackwell KL, O’Neil B, Marcom PK, Ellis MJ, Overmoyer B, Jones SF, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol. 2005;23:5305–5313. doi: 10.1200/JCO.2005.16.584. [DOI] [PubMed] [Google Scholar]
- Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637–646. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CT, Kim H, Liska D, Gao S, Christensen JG, Weiser MR. MET activation mediates resistance to lapatinib inhibition of HER2-amplified gastric cancer cells. Mol Cancer Ther. 2012;11:660–669. doi: 10.1158/1535-7163.MCT-11-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sanchez V, Chakrabarty A, Dave B, Cook RS, Pao W, McKinely E, et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci USA. 2011;108:5021–5026. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev. Mol Cell Biol. 2012;13:591–600. doi: 10.1038/nrm3416. [DOI] [PubMed] [Google Scholar]
- Higginbotham JN, Demory Beckler M, Gephart JD, Franklin JL, Bogatcheva G, Kremers GJ, Piston DW, Ayers GD, McConnell RE, Tyska MJ, Coffey RJ. Amphiregulin exosomes increase cancer cell invasion. Curr Biol. 2011;21:779–786. doi: 10.1016/j.cub.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobor S, Van Emburgh BO, Crowley E, Misale S, Di Nicolantonio F, Bardelli A. TGF-alpha and amphiregulin paracrine network promotes resistance to EGFR blockade in colorectal cancer cells. Clin Cancer Res. 2014;20:6429–6438. doi: 10.1158/1078-0432.CCR-14-0774. [DOI] [PubMed] [Google Scholar]
- Hossain Z, Ali SM, Ko HL, Xu J, Ng CP, Guo K, Qi Z, Ponniah S, Hong W, Hunziker W. Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc Natl Acad Sci USA. 2007;104:1631–1636. doi: 10.1073/pnas.0605266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurbin A, Dubrez L, Coll JL, Favrot MC. Inhibition of apoptosis by amphiregulin via an insulin-like growth factor-1 receptor-dependent pathway in non-small cell lung cancer cell lines. J Biol Chem. 2002;277:49127–49133. doi: 10.1074/jbc.M207584200. [DOI] [PubMed] [Google Scholar]
- Kim M, Agarwal S, Tripathy D. Updates on the treatment of human epidermal growth factor receptor type 2-positive breast cancer. Curr Opin Obstet Gynecol. 2013;26:27–33. doi: 10.1097/GCO.0000000000000043. [DOI] [PubMed] [Google Scholar]
- Kim SK, Jung WH, Koo JS. Yes-associated protein (YAP) is differentially expressed in tumor and stroma according to the molecular subtype of breast cancer. Int J Clin Exp Pathol. 2014;7:3224–3234. [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolahi KS, Donjacour A, Liu X, Lin W, Simbulan RK, Bloise E, Maltepe E, Rinaudo P. Effect of substrate stiffness on early mouse embryo development. PLoS One. 2012;7:e41717. doi: 10.1371/journal.pone.0041717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kun Y, How LC, Hoon TP, Bajic VB, Lam TS, Aggarwal A, Sze HG, Bok WS, Yin WC, Tan P. Classifying the estrogen receptor status of breast cancers by expression profiles reveals a poor prognosis subpopulation exhibiting high expression of the ERBB2 receptor. Hum Mol Genet. 2003;12:3245–3258. doi: 10.1093/hmg/ddg347. [DOI] [PubMed] [Google Scholar]
- Labarge MA, Parvin B, Lorens JB. Molecular deconstruction, detection, and computational prediction of microenvironment-modulated cellular responses to cancer therapeutics. Adv Drug Deliv Rev. 2014;70:123–131. doi: 10.1016/j.addr.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci USA. 2012;109:E2441–2450. doi: 10.1073/pnas.1212021109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GY, Kenny PA, Lee EH, Bissell MJ. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods. 2007;4:359–365. doi: 10.1038/nmeth1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental I, Georges PC, Janmey PA. Soft biological materials and their impact on cell function. Soft Matter. 2007;3:299–306. doi: 10.1039/b610522j. [DOI] [PubMed] [Google Scholar]
- Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin AM, Gilmer TM. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69:6871–6878. doi: 10.1158/0008-5472.CAN-08-4490. [DOI] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina PJ, Goodin S. Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther. 2008;30:1426–1447. doi: 10.1016/j.clinthera.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Michels S, Schmidt-Erfurth U. Photodynamic therapy with verteporfin: a new treatment in ophthalmology. Semin Ophthalmol. 2001;16:201–206. doi: 10.1076/soph.16.4.201.10298. [DOI] [PubMed] [Google Scholar]
- Morin-Kensicki EM, Boone BN, Howell M, Stonebraker JR, Teed J, Alb JG, Magnuson TR, O’Neal W, Milgram SL. Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol Cell Biol. 2006;26:77–87. doi: 10.1128/MCB.26.1.77-87.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouw JK, Yui Y, Damiano L, Bainer RO, Lakins JN, Acerbi I, Ou G, Wijekoon AC, Levental KR, Gilbert PM, et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat Med. 2014;20:360–367. doi: 10.1038/nm.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TV, Sleiman M, Moriarty T, Herrick WG, Peyton SR. Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials. 2014;35:5749–5759. doi: 10.1016/j.biomaterials.2014.03.058. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, Hudak JE, Lakins JN, Wijekoon AC, Cassereau L, et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature. 2014;511:319–325. doi: 10.1038/nature13535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pelissier FA, Garbe JC, Ananthanarayanan B, Miyano M, Lin C, Jokela T, Kumar S, Stampfer MR, Lorens JB, LaBarge MA. Age-related dysfunction in mechanotransduction impairs differentiation of human mammary epithelial progenitors. Cell Rep. 2014;7:1926–1939. doi: 10.1016/j.celrep.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012;17:1–16. doi: 10.1615/critrevoncog.v17.i1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubashkin MG, Cassereau L, Bainer R, DuFort CC, Yui Y, Ou G, Paszek MJ, Davidson MW, Chen YY, Weaver VM. Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylinositol (3,4,5)-triphosphate. Cancer Res. 2014;74:4597–4611. doi: 10.1158/0008-5472.CAN-13-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusnak DW, Alligood KJ, Mullin RJ, Spehar GM, Arenas-Elliott C, Martin AM, Degenhardt Y, Rudolph SK, Haws TF, Jr, Hudson-Curtis BL, Gilmer TM. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 2007;40:580–594. doi: 10.1111/j.1365-2184.2007.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saruwatari L, Aita H, Butz F, Nakamura HK, Ouyang J, Yang Y, Chiou WA, Ogawa T. Osteoblasts generate harder, stiffer, and more delamination-resistant mineralized tissue on titanium than on polystyrene, associated with distinct tissue micro- and ultrastructure. J Bone Miner Res. 2005;20:2002–2016. doi: 10.1359/JBMR.050703. [DOI] [PubMed] [Google Scholar]
- Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quaas A, Walsh S, Benten D, Forbes SJ, Wells RG, Iredale JP. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192–1205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Santiskulvong C, Rao J, Gimzewski JK, Dorigo O. The role of Rho GTPase in cell stiffness and cisplatin resistance in ovarian cancer cells. Integr Biol. 2014;6:611–617. doi: 10.1039/c3ib40246k. [DOI] [PubMed] [Google Scholar]
- Soofi SS, Last JA, Liliensiek SJ, Nealey PF, Murphy CJ. The elastic modulus of Matrigel as determined by atomic force microscopy. J Struct Biol. 2009;167:216–219. doi: 10.1016/j.jsb.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse JR, Engler AJ. Preparation of hydrogel substrates with tunable mechanical properties. Curr Protoc Cell Biol. 2010 doi: 10.1002/0471143030.cb1016s47. Chapter 10, Unit 10.16. [DOI] [PubMed] [Google Scholar]
- Tufail R, Jorda M, Zhao W, Reis I, Nawaz Z. Loss of Yes-associated protein (YAP) expression is associated with estrogen and progesterone receptors negativity in invasive breast carcinomas. Breast Cancer Res Treatment. 2012;131:743–750. doi: 10.1007/s10549-011-1435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su L, Ou Q. Yes-associated protein promotes tumour development in luminal epithelial derived breast cancer. Eur J Cancer. 2012;48:1227–1234. doi: 10.1016/j.ejca.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JC, Giancotti F, Werb Z, Bissell MJ. beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2:205–216. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B, Lo AT, Park CC, Gray JW, Bissell MJ. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res Treatment. 2010;122:35–43. doi: 10.1007/s10549-009-0502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Petricoin EF, 3rd, Zhao S, Liu L, Osada T, Cheng Q, Wulfkuhle JD, Gwin WR, Yang X, Gallagher RI, Bacus S, Lyerly HK, Spector NL. An heregulin-EGFR-HER3 autocrine signaling axis can mediate acquired lapatinib resistance in HER2+ breast cancer models. Breast Cancer Res. 2013;15:R85. doi: 10.1186/bcr3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yotsumoto F, Fukami T, Yagi H, Funakoshi A, Yoshizato T, Kuroki M, Miyamoto S. Amphiregulin regulates the activation of ERK and Akt through epidermal growth factor receptor and HER3 signals involved in the progression of pancreatic cancer. Cancer Sci. 2010;101:2351–2360. doi: 10.1111/j.1349-7006.2010.01671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Tomlinson V, Lara R, Holliday D, Chelala C, Harada T, Gangeswaran R, Manson-Bishop C, Smith P, Danovi SA, et al. Yes-associated protein (YAP) functions as a tumor suppressor in breast. Cell Death Differ. 2008;15:1752–1759. doi: 10.1038/cdd.2008.108. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ji JY, Yu M, Overholtzer M, Smolen GA, Wang R, Brugge JS, Dyson NJ, Haber DA. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat Cell Biol. 2009;11:1444–1450. doi: 10.1038/ncb1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zustiak S, Nossal R, Sackett DL. Multiwell stiffness assay for the study of cell responsiveness to cytotoxic drugs. Biotechnol Bioeng. 2014;111:396–403. doi: 10.1002/bit.25097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.