

Membrane proteins that couple to cortical actin show temperature-independent diffusion. The loss of this coupling and perturbation of cortical actomyosin dynamics render the diffusion temperature dependent. The findings suggest that active fluctuations arising from dynamic actin filaments at the cortex can drive diffusion on the cell membrane.
Abstract
Molecular diffusion at the surface of living cells is believed to be predominantly driven by thermal kicks. However, there is growing evidence that certain cell surface molecules are driven by the fluctuating dynamics of cortical cytoskeleton. Using fluorescence correlation spectroscopy, we measure the diffusion coefficient of a variety of cell surface molecules over a temperature range of 24–37°C. Exogenously incorporated fluorescent lipids with short acyl chains exhibit the expected increase of diffusion coefficient over this temperature range. In contrast, we find that GPI-anchored proteins exhibit temperature-independent diffusion over this range and revert to temperature-dependent diffusion on cell membrane blebs, in cells depleted of cholesterol, and upon acute perturbation of actin dynamics and myosin activity. A model transmembrane protein with a cytosolic actin-binding domain also exhibits the temperature-independent behavior, directly implicating the role of cortical actin. We show that diffusion of GPI-anchored proteins also becomes temperature dependent when the filamentous dynamic actin nucleator formin is inhibited. However, changes in cortical actin mesh size or perturbation of branched actin nucleator Arp2/3 do not affect this behavior. Thus cell surface diffusion of GPI-anchored proteins and transmembrane proteins that associate with actin is driven by active fluctuations of dynamic cortical actin filaments in addition to thermal fluctuations, consistent with expectations from an “active actin-membrane composite” cell surface.
INTRODUCTION
The spatial organization of many cell surface molecules is scale dependent, dynamic, and influenced by interaction with the actin cortex (Mayor and Rao, 2004; Hancock, 2006; Goswami et al., 2008; Lingwood and Simons, 2010; Gowrishankar et al., 2012), the thin layer of actin cytoskeleton that is juxtaposed to the bilayer. Although the cortical actin cytoskeleton is poorly defined, there is growing evidence that it is composed simultaneously of dynamic filaments (Gowrishankar et al., 2012) and an extensively branched meshwork (Morone et al., 2006). The coupling of the membrane to these two types of actin architectures is expected to affect the dynamics and organization of membrane components.
The cell surface is compartmentalized into domains of distinct composition (Simons and Ikonen, 1997; Edidin, 2003). Whether this “compartmentalization” comes about by equilibrium or nonequilibrium mechanisms is a subject of debate. It is clear, however, that simple equilibrium explanations based on the liquid-disordered (ld)–liquid-ordered (lo) phase coexistence underpinning the original “membrane raft” proposal are likely to be inadequate (Rao and Mayor, 2014). This is because at physiological temperatures (∼37°C), large-scale phase segregation is not observed in live cell membranes, membrane blebs, or giant plasma membrane vesicles derived from the cell surface (Baumgart et al., 2007; Levental et al., 2009; Johnson et al., 2010), although the last two membrane preparations exhibit bona fide phase segregation behavior at lower temperatures (∼15–18°C).
Studies of the spatial and temporal organization of glycosylphosphatidylinositol (GPI)-anchored proteins have provided insights into the organization of a membrane component that associates with lo domains in artificial membranes. These studies have shown that GPI-anchored protein organization exhibits unusual properties not easily explained by equilibrium mechanisms (Goswami et al., 2008). These unusual features are 1) the existence of cholesterol-sensitive, GPI-anchored protein clusters and monomers in a fixed, concentration-independent ratio (Varma and Mayor, 1998; Sharma et al., 2004), 2) a heterogeneous, nonrandom spatial distribution, 3) spatial distribution and dynamics regulated by cortical actin and myosin activity, and 4) remodeling of nanoclusters (fragmentation-aggregation) with rates that are insensitive to temperature in the range 24–37°C with an abrupt crossover below 24°C, coinciding with a sharp reduction in actomyosin contractility/activity (Sheetz et al., 1984; Goswami et al., 2008). Indeed, diverse quantitative high-resolution microscopy techniques, such as fluorescence resonance energy transfer (Varma and Mayor, 1998; Sharma et al., 2004; Goswami et al., 2008), electron microscopy (Plowman et al., 2005), and near-field scanning optical microscopy (van Zanten et al., 2009), have demonstrated that in addition to GPI-anchored proteins, a variety of cell surface molecules, such as Ras (Prior et al., 2003; Plowman et al., 2005) and glycolipids (Fujita et al., 2007), form cholesterol-sensitive, actin-dependent dynamic nanoclusters.
A consistent explanation of the unusual features exhibited by GPI-anchored proteins was provided by invoking the physics of active systems (Marchetti et al., 2013) in understanding the interaction of actin filaments and the ATP-consuming myosin with the cell membrane (Gowrishankar et al., 2012). We proposed that specific cell surface molecules are actively driven by their interaction with a dynamic actomyosin system and can be classified accordingly into three types (Gowrishankar et al., 2012): inert (e.g., lipids with short acyl chains or proteins with no possibility of interaction with actin filaments, such as exogenously incorporated fluorescent, short acyl chain–containing lipids, like C5-BODIPY FL-SM), passive (molecules that exhibit an interaction with actin filaments; e.g., GPI-anchored proteins and transmembrane proteins that carry actin-binding capacity), and active (molecules that interact with and also influence cortical actin; e.g., signaling receptors such as integrin receptors and T- and B-cell receptors). Recently we showed that GPI-anchored proteins couple across the bilayer with actin-binding proteins via transbilayer interactions with inner-leaflet phosphatidylserine, involving their long acyl chains (Raghupathy et al., 2015). Predictably, passive molecules (e.g., GPI-anchored proteins) also show anomalously large density fluctuations, as a consequence of being driven by actomyosin flows (Gowrishankar et al., 2012). In contrast, inert molecules (e.g., C5-BODIPY FL-SM) exhibit conventional (Brownian) density fluctuations (Gowrishankar et al., 2012). We proposed that these features can result from nonequilibrium processes based on interaction of passive molecules with dynamic cortical actin and myosin contractility (Gowrishankar et al., 2012; Rao and Mayor, 2014). The nature of these interactions has implications for how molecules may interact at the cell surface, thereby influencing the regulation of signaling mediated by membrane receptors (Chaudhuri et al., 2011; Jaqaman et al., 2011; Gowrishankar et al., 2012). Such interactions can also influence the diffusion behavior exhibited by these types of molecules.
Prior studies on the diffusion of membrane lipids and proteins clearly revealed an influence of the cortical actin meshwork that is juxtaposed to the membrane. Single-particle tracking (SPT) studies specifically showed that the diffusion of many membrane proteins and lipids are constrained and compartmentalized by the actin meshwork and result in an effective “cage-hopping” diffusion behavior (Fujiwara et al., 2002; Umemura et al., 2008). This has led to the “picket-fence” picture of membrane organization in which certain membrane components are corralled by the stable actin mesh (Kusumi et al., 2012). Recent studies also showed that the cortical actin and microtubule–based cytoskeleton can spatially organize the diffusion of oxidized low-density lipoprotein receptor CD36 and regulate its signaling ability (Jaqaman et al., 2011). Fluorescence correlation spectroscopy (FCS) measurements in cells also suggested that the mobilities of sphingolipids, ganglioside GM1, and GPI-anchored proteins show a signature of transient trapping arising from their engagement with nanoscale lipid domains (Lenne et al., 2006) and/or the underlying cortical actin meshwork (Mueller et al., 2011).
Although the foregoing studies made a strong case for the role of the cortical actin meshwork in structuring the diffusion process in the cell surface, it is unclear whether actomyosin activity can also influence the diffusion of membrane molecules. From a first- principles perspective, molecular diffusion in any milieu is a result of random kicks provided by the fluctuating environment. The cell surface is no exception, and molecules will be driven by thermal fluctuations while encountering the impediments offered by the underlying cytoskeleton. However, for passive or active types of molecules, both active fluctuations (potentially involving cortical actomyosin) and thermal fluctuations (Brownian motion) are likely to contribute to molecular diffusion. Several studies have reported active contributions to translational diffusion of tagged particles in the cytoplasm (Brangwynne et al., 2008, 2009; Fakhri et al., 2014; Guo et al., 2014), the nucleus (Hameed et al., 2012), and the bacterial nucleoid (Weber et al., 2012), although the sources of activity are different in each case. The strategy adopted in all of these studies is to focus on those features of diffusion that are strongly affected by perturbing the sources of activity.
It is not known whether the signatures of such active fluctuations can be detected at the cell surface. Here we investigate the diffusion characteristics of fluorescently tagged inert and passive cell surface molecules by FCS (Kim et al., 2007). We report on the temperature variation of diffusion of a variety of lipid probes, GPI-anchored proteins, and transmembrane proteins with (and without) actin-binding motifs and under different conditions. The results provide evidence that passive molecules, such as GPI-anchored proteins and transmembrane protein with a cytoplasmic actin-binding domain (TM-ABD), which are capable of interacting with a dynamic cortical actin pool, exhibit diffusive motion influenced by actomyosin activity. We find that motion of these molecules is characterized by the extent of temperature variation of diffusion. This in turn reflects the nature of fluctuations driving the diffusion of the tagged molecule at the cell surface. Our results are consistent with a model of membrane dynamics and organization resulting from the active actin-membrane composite (Rao and Mayor, 2014).
RESULTS
Diffusion of GPI-anchored proteins on cell surface shows temperature independence
We used confocal-FCS to measure the diffusion coefficient D of inert lipid probes (which do not couple to dynamics of cortical actin) in the range 20–37°C (see also Lee et al., 2015). The probes—the short-chain sphingomyelin (C5-BODIPY FL-SM, B-SM) and phosphatidylcholine (C5-BODIPY FL-HPC, B-PC)—were exogenously incorporated in the outer leaflet (Figure 1, A and B, and Supplemental Figure S1). We used maximum entropy method (MEM) analysis to determine the right fitting model in an unbiased way (detailed in Materials and Methods), since the MEM algorithm fits the data to a distribution of possible correlation time scales (Sengupta et al., 2003) rather a predetermined single or multicomponent FCS model. MEM analysis of the intensity autocorrelation plots of these outer-leaflet probes showed that they could be fit well to a single diffusion time scale. The measurements, acquired at a confocal spot area (ω2 = 3 × 104 nm2), yield correlation time scales of ∼3–5 ms and a corresponding typical diffusion coefficient of ∼1–2.5 μm2/s in the range 20–37°C, reflecting a steady increase in value with increasing temperature. These values are consistent with other FCS studies on the cell surface (Lenne et al., 2006). High-resolution SPT studies show two distinct diffusion coefficients—an early, fast and a later, slow diffusion, with the latter roughly coinciding with the diffusion coefficient determined by FCS (Fujiwara et al., 2002). The origin of this discrepancy might lie in the lower spatial resolution of a confocal FCS spot compared with the nanometer-scale precision of localization of SPT (Kusumi et al., 2012).
FIGURE 1:

Membrane diffusion of GPI-anchored proteins exhibit temperature insensitivity. (A) Plasma membrane locations of the different probes used in study: GPI-anchored protein (1) and exogenously incorporated short-chain lipids, C5-BODIPY FL-SM (B-SM; 2), and C5-BODIPY FL-HPC (B-PC; 3), used for the FCS measurements on the basal membrane of the cell. (B) Confocal fluorescence images show the cell surface distribution of these fluorescent probes (1–3 are EGFP-GPI, B-SM, and B-PC, respectively) at the basal surface of CHO cells. (C) Typical diffusion coefficient, D, extracted from FCS measurements made on short-chain lipids (B-SM and B-PC) and GPI-anchored proteins, folate receptor (FR-GPI), and EGFP-CD52-GPI (CD52) in CHO cells at a confocal spot size 3 × 104 nm2 across the temperature range 20–37°C. The inert lipid probes (B-SM and B-PC) show distinct temperature dependence, whereas the GPI probes (FR-GPI and CD52) show temperature insensitivity. (D) Temperature coefficient of diffusion, Q10, is significantly higher for the lipid probes than with FR-GPI, indicating clear differences in the diffusion behavior of inert and passive membrane molecules. Scale bar, 10 μm. Data collected from 12−15 cells at each temperature for the different probes from two experiments. Error bars are SEs; ***p < 0.001; ns, nonsignificant (t test compared with B-SM).
We next studied the diffusion of two GPI-anchored proteins: 1) folate receptor (FR-GPI), labeled with a fluorescent folate analogue (PLB; Goswami et al., 2008), and 2) enhanced GFP (EGFP)–CD52-GPI (CD52; EGFP fused to the GPI signal from CD52), expressed either stably (FR-GPI) or transiently (CD52) in CHO cells. FCS measurements were made across the same temperature range (20–37°C) and confocal spot size for these probes. The values of diffusion coefficient we report for the GPI-anchored protein probes (∼0.5–2 μm2/s) are consistent with prior FCS and fluorescence recovery after photobleaching (FRAP)–based studies (Lenne et al., 2006; Nohe et al., 2006; Day and Kenworthy, 2012). However, the diffusion of GPI-anchored proteins was temperature independent (or weakly dependent) over the same range (Figure 1C and Supplemental Table S1).
One quantitative measure of temperature sensitivity for membrane diffusion is Q10 (Wey et al., 1981), the rate of change of a biochemical system on increase in temperature of 10°C (detailed in Materials and Methods). A Q10 close to 1 implies temperature independence, whereas Q10 significantly larger than 1 implies temperature dependence. Both fluorescent lipid probes showed a Q10 that is appreciably greater than 1, in sharp contrast to the GPI probes, with Q10 in the range ∼1–1.1 (Figure 1D). In Materials and Methods, we discuss other measures of temperature dependence, which also confirm that these probes show a definite and smooth increase with temperature, whereas GPI-anchored proteins remain temperature independent.
Temperature sensitivity of GPI-anchored protein diffusion is scale dependent
Prior studies on GPI-anchored proteins using FCS and FRAP (Kenworthy et al., 2004; Nohe et al., 2006; Day and Kenworthy, 2012; Bag et al., 2014) reported a temperature dependence of diffusion. Whereas our observations (Figure 1, C and D) may be at odds with these studies, a careful examination of the diffusion data on GPI-anchored proteins indicated that most measurements were acquired at a considerably larger confocal spot sizes (≥5 × 104 nm2) than those reported in Figure 1, where the confocal spot area was ω2 = 3 × 104 nm2. Hence we asked whether the temperature independence of GPI-anchored proteins could be a scale-dependent phenomenon. We tested this by measuring the diffusion of EGFP-GPI (EGFP tagged to GPI-signal of FR-GPI) expressed in CHO cells as a function of temperature at three distinct and gradually increasing confocal spot sizes (ω2 = 3 × 104 < 4.5 × 104 < 6 ×104 nm2). Although we again recover the characteristic temperature independence for EGFP-GPI at the smallest spot size of 3 × 104 nm2, at larger confocal spot areas, ω2 = 4.5 × 104 and 6 × 104 nm2, EGFP-GPI shows a crossover to a temperature-dependent increase in diffusion coefficient (Figure 2A), corroborated by significant differences in Q10 (Figure 2C and Supplemental Table S2). Thus, on change of the spot size, the diffusion of GPI-anchored proteins reverts to temperature-dependent behavior. This temperature-independent diffusion behavior and its crossover to a temperature-dependent one at larger confocal spot sizes is also observed for CD52 in Jurkat T-cells (Figure 2D and Supplemental Table S1). On the other hand, lipid probes exhibit temperature-dependent diffusion at both the small (3 × 104 nm2) and large (6 × 104 nm2) confocal spot sizes (Supplemental Figure S2).
FIGURE 2:

Temperature independence of GPI-anchored protein diffusion is scale dependent. (A) Typical diffusion coefficients of EGFP-GPI in CHO cells across 20–37°C at three different spot sizes, 3 × 104, 4.5 × 104, and 6 × 104 nm2, in a spot-variation FCS approach (inset, increasing spot sizes color-coded as blue/green/red concentric circles). Note the poor temperature dependence for the GPI-anchored proteins at the smallest spot size, 3 × 104 nm2 (A, blue), and its crossover to a distinct temperature-dependent trend at the larger spot size, 6 × 104 nm2 (A, red). (B) Jurkat T-cells expressing CD52 also show similar crossover behavior, reflected in the Q10 (inset) values. (C) A Q10 analysis shows temperature-independent diffusive behavior for the spot size 3 × 104 nm2 for multiple different constructs in CHO cells. By contrast, at the larger spot sizes, 4.5 × 104 and 6 × 104 nm2, GPI-anchored protein diffusion begins to show a distinct crossover to temperature-dependent behavior, with significantly different Q10 at spot size 6 × 104 nm2. Data collected from 10−15 cells at each temperature for different constructs and confocal spot sizes from two experiments. Error bars are SEs; ** p <0.01; ns, nonsignificant (t test for B, inset, and one-way ANOVA with Tukey’s mean comparison test for C).
We will return to this point later, when we investigate the effect of varying the cortical actin mesh size on the temperature dependence of diffusion of passive molecules. A notable feature in the D versus T data, especially for EGFP-GPI, is a sharp change in diffusion coefficient between the temperatures 20 and 24°C (**p < 0.01, Kolmogorov–Smirnov [KS] test). This is presumably due to a higher degree of variability in the measured diffusion coefficients at these temperatures. In our earlier work (Goswami et al., 2008), we reported that there is also considerable heterogeneity in the actin-driven nanocluster remodeling rates at individual temperatures. These sharp features may represent the inherent spatial differences in actin dynamics of the regions sampled in our measurements and thereby account for variations in the data. This peculiarity in the diffusion behavior of GPI-anchored proteins led us to investigate whether the coupling of GPI-anchored proteins to cortical actin and its dynamics is key to the temperature-independent diffusion.
Cholesterol depletion restores temperature-dependent diffusion of GPI-anchored proteins
Studies from our group (Sharma et al., 2004; Goswami et al., 2008; Raghupathy et al., 2015) and others (van Zanten et al., 2009) have shown that cholesterol is a key molecule in the formation, spatial distribution, and dynamics of GPI-anchored protein nanoclusters. These experiments have suggested that cholesterol is required for the coupling of the GPI-anchored proteins to the cortical actin beneath. Therefore we expected that the removal of cholesterol should qualitatively alter the dynamics of GPI-anchored proteins. Our FCS measurements showed that, in general, cholesterol depletion leads to a significant reduction in D for both inert molecules (C5-BODIPY FL-SM; Supplemental Figure S3, A and B) and passive molecules (GPI-anchored proteins; Figure 3, B and C), consistent with earlier reports (Lenne et al., 2006). This can be attributed to an overall increase in membrane viscosity due to the loss of a major component of the membrane, as well as the reorganization of the underlying actin cytoskeleton (Kwik et al., 2003). More significantly, however, we find that upon cholesterol depletion, the diffusion coefficient of both EGFP-GPI (Figure 3, Ai and B) and FR-GPI (Figure 3, Aii and C) showed a systematic increase with temperature, corroborated by a significant increase in the values of temperature coefficient Q10 (Figure 3, B and C, insets, and Supplemental Table S2). Of course, the diffusion coefficient of the inert molecule C5-BODIPY FL-SM remained temperature dependent upon similar cholesterol perturbations (Supplemental Figure S3, A and B). In summary, these results indicate that adequate cholesterol levels in the plasma membrane are necessary for the temperature-independent diffusion of GPI-proteins.
FIGURE 3:

GPI-anchored protein diffusion becomes temperature dependent upon cholesterol depletion. CHO cells expressing EGFP-GPI (Ai) or FR-GPI (Aii) were treated with 10 mM MβCD (for 45 min) to reduce the levels of membrane cholesterol, and FCS measurements were made from the basal membrane at the smallest spot size (ω2 = 3 × 104 nm2). Compared to untreated cells (controls same as in Figure 2), FCS measurements on cholesterol-depleted (MβCD) cells expressing EGFP-GPI (B) or FR-GPI (C) show a marked reduction in their diffusion coefficients, as well as a systematic dependence on temperature, also seen in the significant increase in Q10 in both cases. Scale bar, 10 μm; at least 10–12 cells at each temperature from two experiments. Error bars are SEs; ***p < 0.001, **p < 0.01 (t test).
Diffusion of GPI-anchored proteins on blebs is temperature dependent
We next explored the effect of detaching the actin cytoskeleton on the diffusion behavior of passive molecules, such as GPI-anchored proteins. Giant membrane blebs or cell-attached giant plasma membrane vesicles (Baumgart et al., 2007) induced by brief exposure to ethanol (5–10% [vol/vol]) represent a good system with which to study lipid diffusion in the absence of cortical actin (Figure 4Ai). With regard to passive molecules, we previously showed that the GPI-anchored protein organization is extremely sensitive to the loss of dynamic actin; on blebs, we find a complete abrogation of nanoclustering of the GPI-anchored proteins (Goswami et al., 2008). Here we find that the diffusion of FR-GPI on blebs shows a clear temperature dependence, with D > 4 μm2/s (Figure 4B). Predictably, we see an appreciable increase in the D of lipids on these blebs due to a combination of effects that include a local loss in hydrodynamic friction, a smoothening of short-wavelength membrane folds, and a reduction in steric effects arising from the cortical meshwork. Diffusion on membrane blebs has also been reported by multiple techniques, including SPT (Murase et al., 2004), FCS (Golebiewska et al., 2008), and FRAP (Tank et al., 1982), to be much faster than on plasma membrane with attached actin cortex. Consistent with this, we find that the inert molecule C5-BODIPY FL-SM also shows enhanced but still temperature-dependent diffusion on blebs (Supplemental Figure S6C). In addition, we see a distinct temperature-dependent diffusion of GPI-APs on blebs reflected in the significant increase of Q10 values (Figure 4C and Supplemental Table S2).
FIGURE 4:

GPI-anchored protein diffusion on membrane blebs devoid of cortical actin shows temperature dependence upon actomyosin perturbations. (A) CHO cells expressing FR-GPI (labeled with PLB) were acutely treated with 10% ethanol (vol/vol) for 15 min to induce the formation of large and stable blebs (Ai; note bleb in inset) or with inhibitor of actin polymerization LatA (Aii) and imaged on a confocal microscope. We carried out FCS measurements on the topmost confocal plane for blebs (+ in inset of Ai) and the basal membrane for the other perturbations (+ in Aii), with spot size ω2 = 3 × 104 nm2. (B) Typical diffusion coefficients of FR-GPIs determined by FCS measurements on blebs or by perturbations of cortical actin polymerization by LatA or myosin contractility by a cocktail of ML7 and Y27632 (ML7+Y27). These treatments make the FR-GPI diffusion temperature dependent compared with the untreated cell (control, dimethyl sulfoxide [DMSO] vehicle treatment). (C) The extent of temperature dependence is measured by the significant increase in the value of Q10 compared with the control case. Scale bar, 10 μm. Data collected from 10−12 cells at each temperature for all of the perturbations from two experiments. Error bars, SEs; ***p < 0.001, **p < 0.01, and *p < 0.05 (one-way ANOVA, Tukey’s mean comparison test).
Perturbation of actin and myosin renders GPI-anchored protein diffusion temperature dependent
We then asked whether perturbations of cortical actin and its activity affect the diffusion of GPI-anchored proteins and their temperature variation. To do this, we first inhibited F-actin polymerization by treating cells with titrated amounts (∼2 μM) of latrunculin A, a G-actin–sequestering agent (Figure 4ii). In earlier work, we observed that at this concentration, there was a loss of dynamic actin filaments (Gowrishankar et al., 2012) and nanocluster remodeling activity (Goswami et al., 2008). On latrunculin A treatment, our FCS data showed that the diffusion coefficient of GPI-anchored proteins became temperature dependent (Figure 4, B and C), reflected by a small (but statistically significant) change in Q10.
At first glance, our observations appear at odds with earlier FCS (Lenne et al., 2006) and FRAP studies (Day and Kenworthy, 2012), which concluded that GPI-anchored protein diffusion is unaffected upon actin perturbations. Indeed, at 37°C, the diffusion coefficient of GPI probes does not show significant differences between control and latrunculin A–perturbed cells (Lenne et al., 2006), in agreement with our experiments (Figure 4B). However, at lower temperatures (20–28°C), the diffusion coefficient of GPI APs in latrunculin A–treated cells is much lower than the control, resulting in an overall temperature dependence.
To perturb actomyosin activity, we modulated the activity of myosin regulatory light chain (MRLC) phosphorylation, a key modulator of nonmuscle myosin II activity (Vicente-Manzanares et al., 2009). Here we used a cocktail comprising inhibitors against the two predominant MRLC kinases, myosin light chain kinase (MLCK; inhibited by ML-7) and Rho-associated protein kinase (ROCK; inhibited by Y-27632), to reduce global myosin activity (Totsukawa et al., 2000; Katoh et al., 2001). On such a perturbation, FCS measurements on FR-GPI showed that D increases smoothly with temperature, exhibiting a modest (yet statistically significant) increase in Q10 (Figure 4, B and C, and Supplemental Table S2).
Although the temperature dependence of the diffusion of inert molecules is retained upon these perturbations, the extent of variation as measured by Q10 is reduced (Supplemental Figure S3E). Quite generally, we find that the value of the diffusion coefficient of both inert and passive molecules is lower than the control (Supplemental Figure S3, D and E, and Figure 4B). This may reflect the fact that perturbations of cortical actin and its activity affect some global property, such as membrane tension, which could in turn alter molecular diffusion on the membrane (by changing both the local environment of the tagged particle and the excess area stored in the short- wavelength folds of the membrane).
To summarize, our observations show that perturbing both cortical actin and the linkage of the GPI-anchored proteins to the actin (by the removal of cholesterol) and its actomyosin activity makes the diffusion of GPI-anchored proteins thermally driven.
Diffusion of a transmembrane actin-binding probe is also temperature independent
To test directly whether it is the molecular association with cortical actin filaments that gives rise to the temperature-independent diffusion, we measured the diffusion coefficient of a model transmembrane protein fused to an actin-binding domain (ABD) from ezrin at its cytoplasmic domain (TM-ABD; Figure 5Aii), which can interact directly with the cortical actin (Gowrishankar et al., 2012). To generate a negative control, we also constructed an actin-binding mutant (R579A mutation in the ezrin ABD) counterpart, TM-ABD* (Figure 5Aiii), which eliminates its binding to actin. TM-ABD was previously shown to form nanoclusters that depend on its ability to bind actin filaments, whereas the TM-ABD* protein is unable to form nanoclusters (Gowrishankar et al., 2012). FCS measurements (at the confocal spot size of 3 × 104 nm2) on TM-ABD across the temperature range 20–37°C (Figure 5, B and C) shows an overall temperature-independent diffusion. In contrast, the diffusion of TM-ABD* exhibits temperature- dependent diffusion in the same temperature range. This is also reflected in a significant difference in the values of Q10 between TM-ABD and TM-ABD*. These results provide further evidence suggesting that membrane molecules that bind, either directly or indirectly, to actin filaments exhibit a diffusion coefficient that is independent of temperature.
FIGURE 5:

TM-ABD protein diffusion on the cell membrane is temperature independent, whereas the mutant is temperature dependent. (A) Schematic (Ai) shows the membrane location of TM-ABD, a chimeric construct with an N-terminal reporter domain, the folate receptor (FBP), and then a transmembrane domain followed by a C-terminal actin-binding domain from ezrin (ABD) or its R579A mutant (ABD/*), which cannot bind actin. CHO cells expressing a transmembrane actin-binding probe, TM-ABD (Aii), or its nonbinding counterpart (Aiii) are labeled with PLB and imaged on a confocal microscope. We made FCS measurements on the flat membrane regions of both cells across different temperatures. (B) The TM-ABD exhibits temperature-independent diffusion, whereas TM-ABD* does not. (C) Measurement of Q10 shows that whereas TM-ABD has a value close to 1, TM-ABD* has a value that is significantly higher. Scale bar, 10 μm. Data from 10–12 cells at each temperature for both constructs from two experiments. Error bars are SEs; **p < 0.01(t test).
Perturbation of actin filament dynamics but not the actin mesh architecture leads to temperature dependence of GPI-anchored protein diffusion
Thus far, we have seen that the temperature independence of the diffusion coefficient of passive molecules on the cell membrane is due to their coupling with actin at the cortex. We now ask what configuration or architecture of actin is responsible for this “non-Brownian” behavior. We recall that the two configurations of actin, namely the branched meshwork and the dynamic actin pool, could be produced by different nucleators (Pollard, 2007). For instance, the branched meshwork is predominantly nucleated by Arp2/3 (Svitkina and Borisy, 1999; Wu et al., 2012). Pharmacological perturbation of Arp2/3 using CK-666 (Nolen et al., 2009) results in the reduction of cytoskeletal branching and lamellar extension (Wu et al., 2012). In contrast, perturbation of formins by treating cells with the inhibitor SMIFH2 (Rizvi et al., 2009) has little effect on the visible architecture of Arp2/3-dependent lamellipodia.
On the other hand, there is increasing evidence that the formins, major nucleators of filamentous actin in cells (Goode and Eck, 2007), are responsible for nucleating a different configuration of actin at the cortex (Pruyne et al., 2002). FCS and SMP tracking of EGFP-tagged actin-binding domain of utrophin (UTR-ABD) provided evidence for the presence of dynamic short actin filaments at the cortex of length l ≈ 200 nm (associated with a diffusion time scale of ∼10 ms; Gowrishankar et al., 2012). We find a marked reduction in the levels of this dynamic actin pool in SMIFH2-treated (25 μM for 2 h) cells but not in CK-666 treated (50 μM for 2 h) cells (Figure 6, B and C), suggesting the role of formins as a specific nucleator of this dynamic filament pool at the cortex.
FIGURE 6:

Perturbation of formin activity leads to loss of dynamic actin filaments and temperature dependence of GPI-anchored protein diffusion. (A) Schematic shows the EGFP-tagged actin-binding domain of utrophin (UTR-ABD) used here to study the dynamics of actin filaments by FCS. Confocal images of CHO cells expressing this probe (control, Ai) show distinct labeling of actin stress fibers interspersed with diffuse regions corresponding to the cortical meshwork (Ai) where FCS measurements were made. In contrast, cells (Aii) treated with formin inhibitor SMIFH2 for 2 h showed a marked reduction of stress fibers. Note changes in the shape of the treated cells, with a pronounced increase in the lamellipodial extensions (Ai, Di). (B) Autocorrelation decays, fits (B, black line), and residuals (B, bottom), together with the corresponding distributions of diffusion time scales (C) obtained from maximum entropy method (MEM) analysis of the FCS data for the probe UTR-ABD in control (blue) and SMIFH2-treated (red) and CK-666–treated (green) cells. The slow–diffusion time scale component (∼10 ms; C, blue line) corresponds to the diffusion of the short actin pool, which disappears in SMIFH2-treated cells (C, green line). (D, E) We carried out FCS measurements on the flat membrane regions of cells expressing EGFP-FR-GPI and treated with SMIFH2 and CK-666 (confocal image in Di and Dii, respectively) across different temperatures. (E) EGFP-GPI exhibits temperature-dependent diffusion upon perturbation of formins with SMIFH2, whereas CK-666 treated cells show a characteristic temperature independence as in the control (DMSO vehicle treated). (F) Comparative analysis of Q10 shows a higher extent of the temperature dependence of GPI-anchored protein diffusion in SMIFH2-treated cells. Scale bar, 5 μm; data collected from 10−12 cells at each temperature for both the constructs from two experiments. Error bars represent SEs; **p < 0.01; ns, nonsignificant (t test compared with control).
The loss of dynamic actin filaments upon formin perturbation affects the diffusion characteristics of GPI-anchored proteins. FCS measurements were made at different temperatures on CHO cells stably expressing EGFP-GPI (Figure 6, Di and E) pretreated with SMIFH2 as described. These studies showed that the diffusion coefficient becomes temperature dependent upon formin perturbations (Figure 6E), with Q10 ≈ 1.4 (Figure 6 F). Consistent with the result that Arp2/3 activity is not the nucleator of short dynamic filaments, we find that the temperature-independent diffusion of EGFP-GPI is maintained upon Arp2/3 perturbations, with Q10 ≈ 1 (Figure 6, E and F).
The absence of any discernible effect of the branched actin nucleator (Arp2/3) inhibitor CK-666 on the diffusion characteristics of GPI-anchored proteins prompted us to examine whether altering the nature of the static actin mesh in any other way had an effect on GPI-anchored protein diffusion. As highlighted in previous work (Morone et al., 2006), we explored this issue on cells with widely different mesh sizes. Our studies thus far had been conducted on CHO cells, which, from SPT (Murase et al., 2004; Umemura et al., 2008) measurements, display mesh sizes in the range of 40–100 nm. We now measured the diffusion characteristics of GPI-anchored proteins in normal rat kidney epithelial (NRK) cells (Figure 7A), which have a much larger mesh size (∼250 nm) than CHO cells (Murase et al., 2004; Morone et al., 2006). Our FCS measurements in NRK cells on GPI-anchored CD52 at the smallest confocal spot size (ω2 = 3 × 104 nm2) show that the diffusion coefficient shows a sharp jump from 24 to 28°C (*p < 0.05, KS test), after which it is temperature independent in the range 28–37°C (Figure 7B), with Q10 ≈ 1.05–1.1 (Figure 7C), similar to CHO cells (Figure 2, A and C). The value of the diffusion coefficient is also roughly the same in the two cell types. In addition, similar to CHO and Jurkat cells, diffusion in NRK cells also shows a distinct crossover to a temperature-dependent behavior at the larger confocal spot area (ω2 = 6 × 104 nm2; Figure 7B), although the diffusion coefficients at the larger spot size are much reduced compared with those at the smaller spot size. Taken together, our results suggest that the coupling of passive molecules to juxtamembrane dynamic actin filaments renders their diffusion nonthermal.
FIGURE 7:

Temperature-independent diffusion of GPI-anchored protein is not affected by actin mesh architecture. (A) Confocal image of NRK cell expressing EGFP-CD52-GPI (CD52). (B) Typical diffusion coefficients of CD52 measured across different temperatures at two confocal spot sizes, ω2 = 3 × 104 and 6 × 104 nm2. At the larger spot size, although we see a marked reduction of Dtypical values, there is a distinct crossover from a weaker temperature dependence at the smaller spot size (blue) to a distinct temperature dependence at the larger spot size (red), which is also reflected in the Q10 values at these two spot sizes (C). Scale bar, 10 μm. Data from at least 10−12 cells at each temperature from two experiments. Error bars are SEs; *p < 0.05 (t test).
DISCUSSION
The diffusion of inert cell surface molecules, which do not interact with cortical actin, such as short-chain lipids or model transmembrane proteins that do not have actin-binding properties, was shown in our experiments to increase smoothly with increasing temperature in the range 20–37°C. Inert molecules experience thermal fluctuations alone and hence must have thermal diffusion coefficient D = μkBT, where kB is the Boltzmann constant, and the mobility μ is inversely proportional to the membrane viscosity, η. The local membrane viscosity could in general depend both on temperature and local organization of the membrane components. By contrast, our results highlight that passive cell surface molecules that interact with actin filaments (see later discussion), such as GPI-anchored proteins or transmembrane proteins that couple directly to actin (TM-ABD), exhibit a different diffusion behavior. Their diffusion coefficient is independent of (or very weakly dependent on) temperature in the same range.
Previous studies from our laboratory showed that passive molecules that couple to the actin cytoskeleton are organized into nanoclusters by the active driving of the underlying actin filaments, resulting in relatively immobile nanoclusters. This observation (Goswami et al., 2008; Gowrishankar et al., 2012) and its explanation (Rao and Mayor, 2014) imply that there is a connection with membrane components and underlying dynamic actin filaments. For transmembrane proteins that have actin-binding motifs in their cytoplasmic tails or can recruit such actin couplers (like ezrin; Doherty and McMahon, 2008), the connection to the dynamic filaments is obvious. Recent experiments from our laboratory also showed that the coupling of GPI-anchored proteins across the bilayer to the underlying cortical actin is mediated by cholesterol-dependent transbilayer interactions of saturated long acyl chain present in the GPI-anchor coupling with saturated long–acyl chain phosphatidylserine at the inner leaflet (Raghupathy et al., 2015). This transient stabilization of local lo compositional fluctuations of the membrane at the inner leaflet in association with actin filaments provides a mechanism that allows outer-leaflet GPI-anchored proteins to be driven by dynamic actin filaments. We suggest that the active fluctuations arising from these interactions can in turn influence the diffusive motion of passive molecules on the cell surface.
An explanation for our data may be found in the recently proposed active composite model (Gowrishankar et al., 2012; Rao and Mayor, 2014) of the cell surface. This framework considers the cell surface as a composite of the multicomponent plasma membrane and the cortical actin configuration on which it rests. The cortical actin is composed of two species; one is a relatively static branched and cross-linked meshwork (Morone et al., 2006), and the other comprises short and dynamic polar actin filaments (Gowrishankar et al., 2012). The local composition and dynamics of the membrane coupled with the cortical actin are thereby affected by active currents and stresses generated by the dynamic of actin and myosin at the cortex (Gowrishankar 2012). According to the active composite model (see Figure 8), the passive molecule can couple to locally correlated flows of dynamic cortical actin filaments (of scale ∼200 nm; Gowrishankar et al., 2012). This yields a mean duty ratio for the passive molecules that depends on both the local concentration of oriented dynamic filaments (aster zone) and the binding/unbinding rate. When the passive particles are bound to these filaments, their diffusion is driven by actomyosin activity, whereas in the unbound state, the passive particles diffuse freely due to thermal fluctuations. Indeed, prior studies showed that active processes involving actomyosin activity, such as ATP-dependent myosin motility on actin filaments in vitro (Sheetz et al., 1984) and the dynamic remodeling of GPI-anchored protein nanoclusters (Goswami et al., 2008), show temperature insensitivity above 24°C. We argue here that the observed temperature independence of passive molecules reflects such active processes. This also explains why perturbations of cholesterol (which couples the outer leaflet components to the actin), actin polymerization (latrunculin A), myosin activity (ML7+Y27 cocktail), and dynamic filament nucleator formin (SMIFH2) all affect the temperature independence of GPI-anchored protein diffusion.
FIGURE 8:
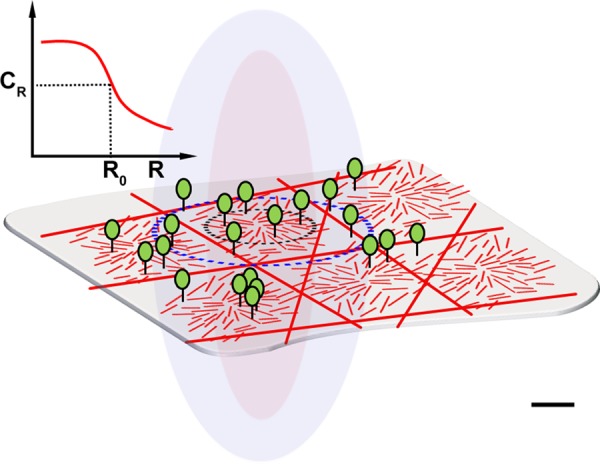
Schematic of FCS measurements on an active composite cell surface. The active composite model of the cell membrane explains how the diffusion of passive molecules can be driven by active fluctuations imparted by the cortical actin filaments. We study the tagged GPI-anchored proteins (green) on the outer leaflet of the plasma membrane (gray translucent sheet) at different confocal spot sizes, represented by the black and the blue dashed circles. According to the active composite cell surface model, the GPI-anchored proteins are driven by the fluctuating dynamics of the underlying cortical actin filaments (short red lines) in the presence of stable and slowly remodeling branched actin meshwork (long red rods) and exhibit active diffusion at long time scales (see Discussion and the Supplemental Material). The mean concentration of oriented polar actin filaments, CR (where R is the confocal spot size), is roughly constant over a scale (R0) and then decreases (inset, graph). This will result in crossover from temperature-independent active diffusion to thermal diffusion at larger spot sizes, which we observe. Suggestive scale bar, 100 nm.
There is a crossover from temperature-independent diffusion at small confocal spot sizes to temperature-dependent diffusion at larger spot sizes. How does our model of active driving by dynamic actin filaments account for this? From the discussion thus far, passive molecules will be subject to both active and thermal fluctuations. The relative contribution from the former is large 1) in regions where the concentration of polar and oriented dynamic filaments is high, that is, in the aster zone, and 2) over the scale of the filament that is advected, convolved with the binding and unbinding rates of the filaments with the passive molecules (Figure 8). This is the case when the FCS experiments are done using small confocal spot sizes, R. As R increases beyond a scale R0 (the aster zone), the contribution of the active fluctuations from dynamic cortical actin falls appreciably (Figure 8), and the relative contribution from thermal fluctuations starts to increase, leading to a crossover from “active” diffusion (temperature independent) to thermal diffusion (temperature dependent). Moreover, at larger spot sizes, passive molecules can undergo higher chances of cage hopping (Kusumi et al., 2012) that arise from the temperature-dependent fluctuations of the branched meshwork. Thus the effective diffusion of the molecules would also reflect overall temperature dependence at the larger spot size.
Along the same lines, prior FRAP (Kenworthy et al., 2004) and spatial-imaging FCS (Bag et al., 2014) studies on GPI-anchored proteins and other diffusions showed a clear temperature dependence. This is contrary to what we see at the smallest confocal spot size. However, FRAP is often acquired by looking at the recovery of a bleached spot whose size ranges from 1 to 5 μm (Jacobson et al., 1984; Kenworthy et al., 2004; Day and Kenworthy, 2012), whereas spatial FCS looks at spatial scales of ≥0.5 μm2 (Bag et al., 2014). Clearly, both techniques monitor diffusion and dynamics over a much larger spatial scale (compared with the sub–200 nm regime for the smallest spot size of FCS measurement. Moreover, FRAP recovery data indicates the extent of turnover, which includes both diffusion (via active driving and cage hopping) and binding/unbinding or transient trapping events, as well as thermally driven motion. Whereas active driving would confer temperature independence, the latter processes are thermally driven. Hence monitoring kinetics over larger length scales (as in FRAP/imaging-FCS) can outweigh the contributions from active driving. Moreover, the diffusion coefficients monitored by spatial assays like FRAP and imaging-FCS are often lower in magnitude than those obtained from FCS.
In the context of the active actin membrane composite model, FRAP and imaging-FCS measurements would map dynamics over the scale R ≫ R0, and hence, as expected, the effective diffusion will have stronger contributions from thermal kicks, binding-unbinding to actin corrals, or partitioning into nanodomains, all of which are obviously temperature driven, in turn rendering the diffusion behavior temperature dependent.
The configuration of cortical actin that is responsible for this temperature-independent diffusion of GPI-anchored proteins is likely to be the dynamic short actin filaments juxtaposed to the inner membrane leaflet and driven by myosin motors (see the Supplemental Material for further justification). The absence of any palpable effect upon inhibiting the branched actin nucleator Arp2/3 or by changing the actin mesh size leads us to conclude that the branched cortical actin mesh may not be responsible for imparting the active fluctuations fueling the non-Brownian diffusion of GPI-anchored proteins at small observation spot sizes. Instead, the branched actin-meshwork density and architecture might regulate the effective diffusion coefficient we obtain at the higher spot sizes (>200 nm). This can explain why we observe lower values of the diffusion coefficient of GPI-anchored proteins in NRK cells at the larger spot size (6 × 104 nm2). One plausible reason could be the intrinsic differences in the organization of actin meshwork/corrals between the CHO (or Jurkat cells) and the NRK cells. Although CHO (Murase et al., 2004) and Jurkat (Dustin and Davis, 2014) cells have been reported to exhibit smaller mesh sizes (<100 nm, considerably smaller than the FCS spot size), NRKs exhibit a much more complex nested meshwork, with compartments of ∼ 250 nm nested within larger, 750-nm actin structures (Fujiwara et al., 2002; Ritchie et al., 2003). At a spot size of 6 × 104 nm2, the possibilities of encountering the nested branched actin network in NRK cells increase. The presence of these nested corrals can impede membrane diffusion in NRK cells to a larger magnitude than with CHO cells, leading to a markedly lower diffusion coefficient.
Consistent with this, inhibition of the activity of myosin and actin polymerization renders the diffusion of passive molecules temperature dependent. This treatment also affects the value of the diffusion coefficient of inert molecules. This must reflect the fact that perturbations of cortical actin and its activity must affect some global property, such as membrane tension, which could in turn alter molecular diffusion on the membrane by changing the local environment of the tagged particle or the excess area stored in the short-wavelength folds of the membrane.
The active composite cell membrane allows for both trapping and cage hopping arising from interactions of membrane components with the long-lived branched actin mesh (reminiscent of the picket-fence model) and active driving of passive molecules by the dynamic actin filaments. The SPT and stimulated emission depletion–FCS measurements (Fujiwara et al., 2002; Murase et al., 2004; Umemura et al., 2008; Mueller et al., 2011) were perfectly suited for detailed investigations of the former (also discussed in detail in the Supplemental Material). On the other hand, by identifying a distinctive qualitative trend of passive molecular diffusion, namely its temperature independence, our FCS studies have revealed the latter.
In conclusion, we see that both the nanocluster remodeling (Goswami et al., 2008; Gowrishankar et al., 2012) and the diffusion coefficient of passive molecules (this work), such as GPI-anchored proteins and TM-ABD, are independent of temperature in the physiological range 24–37°C. These properties have important implications for chemical reactions and hence signaling at the cell surface (Chaudhuri et al., 2011). This is particularly significant because several transmembrane signaling receptors (T-cell receptor, integrin receptors, epidermal growth factor receptor) either have direct links with actin or associate with protein domains (e.g., ezrin/radixin/moesin proteins) that recruit filamentous actin (Doherty and McMahon, 2008; Jaqaman and Grinstein, 2012). Signaling reactions involving these receptors are likely to be buffered from temperature-dependent diffusion by engagement with the dynamic cortical actin activity. This is guaranteed by having an active mechanism for molecular organization at the cell surface.
MATERIALS AND METHODS
Plasmids, cell culture, and labeling
The following constructs were used (with references):
Human FR-GPI, described in detail earlier (Varma and Mayor, 1998).
EGFP-GPI, a chimeric model GPI-AP, where the EGFP was fused to the GPI signal from FR-GPI (Sharma et al., 2004).
EGFP-CD52-GPI(CD52), a chimeric model GPI-AP, where the EGFP was fused to the GPI signal from CD52 (Greene et al., 2014).
TM-ABD/ABD*, a chimeric model transmembrane actin-binding/nonbinding probe, where the transmembrane form of the folate receptor is fused to a cytosolic actin-binding domain from ezrin (c-terminal ezrin ABD) or its mutant nonbinding counterpart (ABD*; Gowrishankar et al., 2012).
UTR-ABD, a EGFP fusion to utrophin actin-binding domain used for labeling of actin filaments (Burkel et al., 2007).
CHO cell lines stably expressing either GPI-anchored GFP (EGFP-GPI, GPI signal from FR-GPI), folate receptor (FR-GPI), and transmembrane (FR-TM-ABD/ABD*) constructs were maintained in Ham’s F12 medium (folic acid–free for FR-GPI; HiMedia, India) and imaged as described previously (Varma and Mayor, 1998).
Rat basophilic leukemia (RBL2H3) cells were cultured in DMEM-GlutaMAX (Life Technologies, Carlsbad, CA) supplemented with 15% fetal bovine serum (FBS; Atlanta Biologicals, Flowery Branch, GA). Jurkat T-cells were cultured in RPMI1640 medium (Life Technologies) supplemented with 1 mM sodium pyruvate (Cellgro, Manassas, VA), 100 μg/ml penicillin/streptomycin (Cellgro), and 10% FBS (Atlanta Biologicals) and were imaged as described previously (Triffo et al., 2012; Lee et al., 2015). NRK fibroblast cells were maintained in Ham’s F12 medium as described previously (Fujiwara et al., 2002).
Cells expressing GFP-tagged membrane proteins were treated with 75 μg/ml cycloheximide for 3 h at 37°C to clear Golgi-associated fluorescence before imaging (Sabharanjak et al., 2002). FR-GPI–expressing cells were labeled with fluorescent analogues of folic acid Nα-pteroyl-Nε-BodipyTMR-l-lysine (PLB) at the cell surface at saturating concentration (∼400 nM) on ice for 1 h. Transient transfections reported here (for the constructs CD52 and UTR-ABD) were carried out ∼12−14 h before their preparation for the experiments. This ensured low levels of expression required for the FCS measurements. The cells were transferred to prewarmed buffers (150 mM NaCl, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, pH 7.2–7.4) before being placed on a temperature-controlled microscope stage and imaged within 30 min. Temperature-sensitive measurements were done by using a microscope stage connected with a temperature controller (Polyscience, Niles, IL). The controller has both refrigerating and heating capacity, and the temperature of the stage was changed and maintained by circulating water at a defined temperature. The imaging dish (jacketed with a metal ring for better temperature equilibration and heat distribution) was then placed on the stage and allowed to equilibrate for 5–10 min. After this, the bath temperature was monitored by a thermocouple needle probe (Physitemp Instruments, Clifton, NJ) placed in the buffer near the cells. The temperature variation was seen around ±0.5°C. A single experimental trial (and its repeats) included measurements at all four or five temperature points across 20–37°C on the same day, with a fresh batch of cells (control or perturbation) being used for each temperature.
Fluorescent lipid analogues and cell surface labeling
The following BODIPY-FL–conjugated lipid analogues (Life Technologies, Carlsbad, CA) were used:
sphingomyelin (SM): C5-BODIPY FL-SM and C12-BODIPY FL-SM, chain-labeled; and
phosphocholine (PC): C5-BODIPY FL-HPC and C12-BODIPY FL-HPC, chain-labeled.
Fluorescently labeled lipid probes were complexed with BSA (1:1 or 1:2) in a 0.5–1 μM solution and incorporated onto the CHO cell surface by incubating for 15–20 min on ice (Goswami et al., 2008; Lee et al., 2015). Postlabeling, excess lipid–BSA complex was removed, and the cells were washed thoroughly with cold HEPES-based buffer to minimize the levels of freely diffusing probes in the medium before imaging. To avoid probe internalization, all of the FCS measurements were made within 30 min.
Treatments and pharmacological perturbations
Cholesterol depletions were carried out by treating the cells with 10 mM methyl-β-cyclodextrin (MβCD) at 37°C for 30 min, followed by exogenous labeling of fluorescent probes. Stable membrane blebs for FCS measurements were generated by incubating the cells with 10% ethanol (vol/vol) in HEPES-based buffer for 15–20 min at 37°C (Baumgart et al., 2007). Actin perturbations were effected by incubating the cells with actin-polymerization inhibitor latrunculin A (LatA) at 2 μM for 15 min before the FCS measurements. To reduce myosin activity in the cells, we pretreated cells using a cocktail of inhibitors against the two myosin light chain kinases, ROCK (Y27 at 20 μM) and MLCK (ML7 at 20 μM), for 1 h at 37°C. Perturbation of formin activity was carried out by pretreatment of cells with the formin inhibitor SMIFH2 at 25 μM for 2 h at 37°C. Arp2/3 activity was perturbed by treating cells with CK-666 at 50 μM for 2 h at 37°C. Unless otherwise mentioned, inhibitors with reversible effects were maintained in the buffer during the imaging and measurements.
FCS instrumentation and analysis
FCS measurements were made on cells (Kim et al., 2007) expressing EGFP-tagged probes or labeled with fluorescent analogues as mentioned, using either the ConfoCor 2 module on a Zeiss 510 Meta NLO (Carl Zeiss) or gallium arsenide array detectors on a Zeiss 780 (Carl Zeiss) confocal microscope. The cells were imaged with the 488-nm laser illumination using a 40×/1.2 numerical aperture water-immersion lens using the descanned PMT detectors in the confocal scan head. Manually selected regions close to the cell periphery or lamellum were moved to the center of the field. The laser beam for FCS overfills the back-focal plane of the objective to form a diffraction-limited confocal volume and was parked at the center of the field at a specified z-distance (dictated by high counts per molecule readout) from the coverslips onto the basal membrane of the cells. Pinhole alignment was always done before initiating a day’s experiment. The confocal spot size was calibrated each day of the experiment by measuring the three-dimensional diffusion coefficient of rhodamine 6G (Sigma-Aldrich, St. Louis, MO) in water, for which the diffusion coefficient is known (Billaudeau et al., 2013). For spot-variation measurements, the confocal spot size was increased by reducing the width of the laser beam at the back-focal plane of the objective and calibrating the resultant confocal spot size as described earlier. The laser power was titrated to ensure that triplet fraction stayed low (≤20%). Each measurement constitutes fluorescence intensity–time traces, typically collected over 10 s each. The intensity–time trace was autocorrelated in real time using the on-board software correlator.
The data on intensity autocorrelation function G(τ) versus time τ were fitted to the conventional formula (Schwille et al., 1999) for the distribution of diffusing species, where Ai is the contribution of the ith component to the total amplitude of the autocorrelation function with the diffusion time scale τD(i),
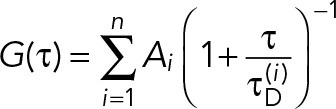 (1) (1)
|
To account for a possible triplet contribution, we use a multicomponent decay model (Banks and Fradin, 2005),
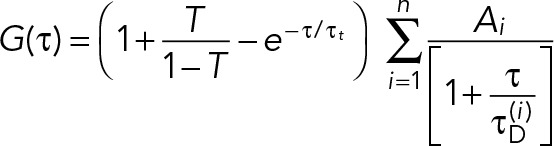 (2) (2)
|
where T is the average triplet fraction and τt is the triplet time scale. To allow for the possibility of anomalous diffusion, <δr2> ∼ tα, we use a generalization (Schwille et al., 1999; Banks and Fradin, 2005),
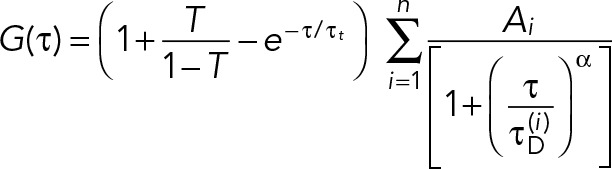 (3) (3)
|
To ensure a bias-free estimation of the number of diffusing components, we use a fitting algorithm based on the maximum entropy method (MEM-FCS; Sengupta et al., 2003).
Although we expect the probes to yield single-component diffusion at the cell surface, we often observe a fast-diffusing (t ≈ 0.3 ms) component arising from intracellular/luminal EGFP very close to the plasma membrane, especially for EGFP constructs (Supplemental Figure S4). To avoid any ambiguity, we fitted the entire data set to single-component or discrete multicomponent decay models, based on their MEM-FCS distribution profile. The goodness of the fits was judged based on the value of reduced χ2, evenly distributed residuals across the full extent of the data, and whether visual inspection of the fit accurately described the autocorrelation decay data. We extracted the diffusion time scales from all of the multiple iterations (∼6−10) and its repeats (n ≈ 10−15 cells at each temperature from two independent experiments) to calculate the diffusion coefficients. We then used this entire data set to generate a cumulative frequency distribution of the calculated diffusion coefficient. We fit the distribution to an error function (cumulative normal distribution) to obtain a most probable value (or typical value) representing the typical diffusion coefficient, Dtypical, for each temperature (Supplemental Figure S1). The error bars signify the errors in the estimation of Dtypical. We computed the derivative of the best-fit error function to obtain a Gaussian that describes the original distribution and estimated its width, σ. The error of estimation of this mean can be obtained by dividing σ by the square root of total number of data points (n) or the SE. We plotted this error along with Dtypical. This was done because fitting the cumulative frequency distribution data to an error function provides a more accurate estimate of Dtypical. We displayed the raw data for representative FCS curves under different conditions and the fits to the same with residuals (Supplemental Figure S5). The FCS diffusion law scaling–type analysis of the EGFP-GPI data at 37°C (Supplemental Figure S6) showed a positive y-intercept, as reported earlier (Lenne et al., 2006), suggesting nanoscale domain–partitioned diffusion of GPI-APs.
Quantification of the extent of temperature variation
Temperature coefficient, Q10.
Because of the slight scatter in the D versus T data, we used the temperature coefficient, Q10, as a quantitative measure of the extent of temperature dependence. Q10 is a measure of the rate of change of a biological or chemical process due to increase in temperature by 10°C. This has been successfully used to monitor the temperature dependence of membrane diffusion (Wey et al., 1981). We computed Q10 using the formula
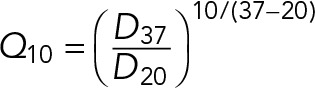 |
where D37 and D20 correspond to Dtypical at 37 and 20°C, respectively.
The errors in Q10 were propagated from the errors in Dtypical (at 20 or 37°C) in accordance with the standard rules of error propagation (Taylor, 1997). The Q10 values and their differences (between control and perturbation scenarios) were compared for statistical significance by t test (unpaired with Welch’s correction) or one-way analysis of variance (ANOVA) with Tukey’s mean comparison tests using GraphPad Prism 6 (GraphPad, La Jolla, CA). A detailed description of the statistical data is presented in Supplemental Table S2, and individual plots show the statistical significance.
Analysis of local slope.
We also analyzed the local slopes of the D versus T plot. We did this by using a piecewise cubic interpolation, SPLINE, or PCHIP routines in Matlab (MathWorks, Natick, MA), but we prefer the last, since the monotonicity property of PCHIP is more desirable than the smoothness property of SPLINE. We then plotted the local slopes as a function of T. When the slopes were consistently >0 over this temperature range, we declared it to be temperature-dependent diffusion. When the slopes take on both positive and negative values over this temperature range, that is, when they oscillate about 0 with low amplitude, we declared it to be temperature- independent diffusion. Examples of the analysis of local slopes are presented in Supplemental Figure S7.
Statistical comparison of overall linear slope.
This was done by linear regression analysis and fitting of the D versus T plot using GraphPad Prism 6. A linear fit of the data yields the overall (global) slope for any given condition. A temperature-independent measurement (or scenario) would have a slope close to 0, and the value is expected to be statistically insignificant compared with a 0 slope. For temperature- dependent cases, the slopes are expected to show statistically significant differences when compared with 0. The data from this type of analysis, presented in Supplemental Table S3, complement and corroborate the Q10 data.
Supplementary Material
Acknowledgments
We thank Akihiro Kusumi for gifting us the NRK cells. We thank Debanjan Goswami and Pragya Srivastava for help with analysis of FCS data, Sudipta Maiti for discussions, and the Central Imaging and Flow Facility at the National Centre for Biological Sciences for FCS instrumentation. M.R. acknowledges a grant from the Simons Foundation. S.M. is a J.C. Bose Fellow (Department of Science and Technology, Government of India) and acknowledges support from Human Frontier Science Program Grant RGP0027/2012. S.S. acknowledges fellowship support from the National Centre for Biological Sciences–Tata Institute of Fundamental Research Graduate program.
Glossary
Abbreviations used:
- FCS
fluorescence correlation spectroscopy
- FRAP
fluorescence recovery after photobleaching
- GPI
glycosylphosphatidylinositol
- NRK
normal rat kidney epithelial
- SPT
single-particle tracking
- TM-ABD
transmembrane protein with actin-binding domain.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-06-0397) on September 16, 2015.
REFERENCES
- Bag N, Yap DHX, Wohland T. Temperature dependence of diffusion in model and live cell membranes characterized by imaging fluorescence correlation spectroscopy. Biochim Biophys Acta. 2014;1838:802–813. [PubMed] [Google Scholar]
- Banks DS, Fradin C. Anomalous diffusion of proteins due to molecular crowding. Biophys J. 2005;89:2960–2971. doi: 10.1529/biophysj.104.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart T, Hammond AT, Sengupta P, Hess ST, Holowka DA, Baird BA, Webb WW. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc Natl Acad Sci USA. 2007;104:3165–3170. doi: 10.1073/pnas.0611357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billaudeau C, Mailfert S, Trombik T, Bertaux N, Rouger V, Hamon Y, He HT, Marguet D. Probing the plasma membrane organization in living cells by spot variation fluorescence correlation spectroscopy. Methods Enzymol. 2013;519:277–302. doi: 10.1016/B978-0-12-405539-1.00010-5. [DOI] [PubMed] [Google Scholar]
- Brangwynne CP, Koenderink GH, MacKintosh FC, Weitz DA. Cytoplasmic diffusion: molecular motors mix it up. J Cell Biol. 2008;183:583–587. doi: 10.1083/jcb.200806149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Koenderink GH, MacKintosh FC, Weitz DA. Intracellular transport by active diffusion. Trends Cell Biol. 2009;19:423–427. doi: 10.1016/j.tcb.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Burkel BM, Von Dassow G, Bement WM. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil Cytoskeleton. 2007;64:822–832. doi: 10.1002/cm.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri A, Bhattacharya B, Gowrishankar K, Mayor S, Rao M. Spatiotemporal regulation of chemical reactions by active cytoskeletal remodeling. Proc Natl Acad Sci USA. 2011;108:14825–14830. doi: 10.1073/pnas.1100007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CA, Kenworthy AK. Mechanisms underlying the confined diffusion of cholera toxin B-subunit in intact cell membranes. PLoS One. 2012;7:e34923. doi: 10.1371/journal.pone.0034923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty GJ, McMahon HT. Mediation, modulation, and consequences of membrane-cytoskeleton interactions. Annu Rev Biophys. 2008;37:65–95. doi: 10.1146/annurev.biophys.37.032807.125912. [DOI] [PubMed] [Google Scholar]
- Dustin ML, Davis SJ. TCR signaling: the barrier within. Nat Immunol. 2014;15:136–137. doi: 10.1038/ni.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edidin M. Lipids on the frontier: a century of cell-membrane bilayers. Nat Rev Mol Cell Biol. 2003;4:414–418. doi: 10.1038/nrm1102. [DOI] [PubMed] [Google Scholar]
- Fakhri N, Wessel AD, Willms C, Pasquali M, Klopfenstein DR, MacKintosh FC, Schmidt CF. High-resolution mapping of intracellular fluctuations using carbon nanotubes. Science. 2014;344:1031–1035. doi: 10.1126/science.1250170. [DOI] [PubMed] [Google Scholar]
- Fujita A, Cheng J, Hirakawa M, Furukawa K, Kusunoki S, Fujimoto T. Gangliosides GM1 and GM3 in the living cell membrane form clusters susceptible to cholesterol depletion and chilling. Mol Biol Cell. 2007;18:2112–2122. doi: 10.1091/mbc.E07-01-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Ritchie K, Murakoshi H, Jacobson K, Kusumi A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J Cell Biol. 2002;157:1071–1081. doi: 10.1083/jcb.200202050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golebiewska U, Nyako M, Woturski W, Zaitseva I, McLaughlin S. Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol Biol Cell. 2008;19:1663–1669. doi: 10.1091/mbc.E07-12-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- Goswami D, Gowrishankar K, Bilgrami S, Ghosh S, Raghupathy R, Chadda R, Vishwakarma R, Rao M, Mayor S. Nanoclusters of GPI-anchored proteins are formed by cortical actin-driven activity. Cell. 2008;135:1085–1097. doi: 10.1016/j.cell.2008.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar K, Ghosh S, Saha S, Rumamol C, Mayor S, Rao M. Active remodeling of cortical actin regulates spatiotemporal organization of cell surface molecules. Cell. 2012;149:1353–1367. doi: 10.1016/j.cell.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Greene AC, Lord SJ, Tian A, Rhodes C, Kai H, Groves JT. Spatial organization of epha2 at the cell-cell interface modulates trans-endocytosis of ephrina1. Biophys J. 2014;106:2196–2205. doi: 10.1016/j.bpj.2014.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Ehrlicher AJ, Jensen MH, Renz M, Moore JR, Goldman RD, Lippincott-Schwartz J, Mackintosh FC, Weitz D A. Probing the stochastic, motor-driven properties of the cytoplasm using force spectrum microscopy. Cell. 2014;158:822–832. doi: 10.1016/j.cell.2014.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hameed FM, Rao M, Shivashankar G V. Dynamics of passive and active particles in the cell nucleus. PLoS One. 2012;7:e45843. doi: 10.1371/journal.pone.0045843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock JF. Lipid rafts: contentious only from simplistic standpoints. Nat Rev Mol Cell Biol. 2006;7:456–462. doi: 10.1038/nrm1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K, O’Dell D, August JT. Lateral diffusion of an 80,000-dalton glycoprotein in the plasma membrane of murine fibroblasts: relationships to cell structure and function. J Cell Biol. 1984;99:1624–1633. doi: 10.1083/jcb.99.5.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Grinstein S. Regulation from within: the cytoskeleton in transmembrane signaling. Trends Cell Biol. 2012;22:515–526. doi: 10.1016/j.tcb.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Kuwata H, Touret N, Collins R, Trimble WS, Danuser G, Grinstein S. Cytoskeletal control of CD36 diffusion promotes its receptor and signaling function. Cell. 2011;146:593–606. doi: 10.1016/j.cell.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Stinson BM, Go MS, Carmona LM, Reminick JI, Fang X, Baumgart T. Temperature-dependent phase behavior and protein partitioning in giant plasma membrane vesicles. Biochim Biophys Acta. 2010;1798:1427–1435. doi: 10.1016/j.bbamem.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Kano Y, Amano M, Kaibuchi K, Fujiwara K. Stress fiber organization regulated by MLCK and Rho-kinase in cultured human fibroblasts. Am J Physiol Cell Physiol. 2001;280:C1669–C1679. doi: 10.1152/ajpcell.2001.280.6.C1669. [DOI] [PubMed] [Google Scholar]
- Kenworthy AK, Nichols BJ, Remmert CL, Hendrix GM, Kumar M, Zimmerberg J, Lippincott-Schwartz J. Dynamics of putative raft-associated proteins at the cell surface. J Cell Biol. 2004;165:735–746. doi: 10.1083/jcb.200312170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SA, Heinze KG, Schwille P. Fluorescence correlation spectroscopy in living cells. Nat Methods. 2007;4:963–973. doi: 10.1038/nmeth1104. [DOI] [PubMed] [Google Scholar]
- Kusumi A, Fujiwara TK, Chadda R, Xie M, Tsunoyama TA, Kalay Z, Kasai RS, Suzuki KGN. Dynamic organizing principles of the plasma membrane that regulate signal transduction: commemorating the fortieth anniversary of Singer and Nicolson’s fluid-mosaic model. Annu Rev Cell Dev Biol. 2012;28:215–250. doi: 10.1146/annurev-cellbio-100809-151736. [DOI] [PubMed] [Google Scholar]
- Kwik J, Boyle S, Fooksman D, Margolis L, Sheetz MP, Edidin M. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc Natl Acad Sci USA. 2003;100:13964–13969. doi: 10.1073/pnas.2336102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IH, Saha S, Polley A, Huang HH, Mayor S, Rao M, Groves JT. Live cell plasma membranes do not exhibit a miscibility phase transition over a wide range of temperatures. J Phys Chem. 2015;B 119:4450–4459. doi: 10.1021/jp512839q. [DOI] [PubMed] [Google Scholar]
- Lenne P-F, Wawrezinieck L, Conchonaud F, Wurtz O, Boned A, Guo X-J, Rigneault H, He H-T, Marguet D. Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO J. 2006;25:3245–3256. doi: 10.1038/sj.emboj.7601214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental I, Byfield FJ, Chowdhury P, Gai F, Baumgart T, Janmey PA. Cholesterol-dependent phase separation in cell-derived giant plasma-membrane vesicles. Biochem J. 2009;424:163–167. doi: 10.1042/BJ20091283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Marchetti MC, Joanny JF, Ramaswamy S, Liverpool TB, Prost J, Rao M, Simha RA. Hydrodynamics of soft active matter. Rev Mod Phys. 2013;85:1143–1189. [Google Scholar]
- Mayor S, Rao M. Rafts: scale-dependent, active lipid organization at the cell surface. Traffic. 2004;5:231–240. doi: 10.1111/j.1600-0854.2004.00172.x. [DOI] [PubMed] [Google Scholar]
- Morone N, Fujiwara T, Murase K, Kasai RS, Ike H, Yuasa S, Usukura J, Kusumi A. Three-dimensional reconstruction of the membrane skeleton at the plasma membrane interface by electron tomography. J Cell Biol. 2006;174:851–862. doi: 10.1083/jcb.200606007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller V, Ringemann C, Honigmann A, Schwarzmann G, Medda R, Leutenegger M, Polyakova S, Belov VN, Hell SW, Eggeling C. STED nanoscopy reveals molecular details of cholesterol- and cytoskeleton-modulated lipid interactions in living cells. Biophys J. 2011;101:1651–1660. doi: 10.1016/j.bpj.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase K, Fujiwara T, Umemura Y, Suzuki K, Iino R, Yamashita H, Saito M, Murakoshi H, Ritchie K, Kusumi A. Ultrafine membrane compartments for molecular diffusion as revealed by single molecule techniques. Biophys J. 2004;86:4075–4093. doi: 10.1529/biophysj.103.035717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohe A, Keating E, Fivaz M, Van Der Goot FG, Petersen NO. Dynamics of GPI-anchored proteins on the surface of living cells. Nanomedicine. 2006;2:1–7. doi: 10.1016/j.nano.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature. 2009;460:1031–1034. doi: 10.1038/nature08231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowman SJ, Muncke C, Parton RG, Hancock JF. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci USA. 2005;102:15500–15505. doi: 10.1073/pnas.0504114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu Rev Biophys Biomol Struct. 2007;36:451–477. doi: 10.1146/annurev.biophys.35.040405.101936. [DOI] [PubMed] [Google Scholar]
- Prior IA, Muncke C, Parton RG, Hancock JF. Direct visualization of ras proteins in spatially distinct cell surface microdomains. J Cell Biol. 2003;160:165–170. doi: 10.1083/jcb.200209091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruyne D, Evangelista M, Yang C, Bi E, Zigmond S, Bretscher A, Boone C. Role of formins in actin assembly: nucleation and barbed-end association. Science. 2002;297:612–615. doi: 10.1126/science.1072309. [DOI] [PubMed] [Google Scholar]
- Raghupathy R, Anilkumar AA, Polley A, Singh PP, Yadav M, Johnson C, Suryawanshi S, Saikam V, Sawant SD, Panda A, et al. Transbilayer lipid interactions mediate nanoclustering of lipid-anchored proteins. Cell. 2015;161:581–594. doi: 10.1016/j.cell.2015.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao M, Mayor S. Active organization of membrane constituents in living cells. Curr Opin Cell Biol. 2014;29:126–132. doi: 10.1016/j.ceb.2014.05.007. [DOI] [PubMed] [Google Scholar]
- Ritchie K, Iino R, Fujiwara T, Murase K, Kusumi A. The fence and picket structure of the plasma membrane of live cells as revealed by single molecule techniques (review) Mol Membr Biol. 2003;20:13–18. doi: 10.1080/0968768021000055698. [DOI] [PubMed] [Google Scholar]
- Rizvi SA, Neidt EM, Cui J, Feiger Z, Skau CT, Gardel ML, Kozmin SA, Kovar DR. Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chem Biol. 2009;16:1158–1168. doi: 10.1016/j.chembiol.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabharanjak S, Sharma P, Parton RG, Mayor S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated clathrin-independent pinocytic pathway. Dev Cell. 2002;2:411–423. doi: 10.1016/s1534-5807(02)00145-4. [DOI] [PubMed] [Google Scholar]
- Schwille P, Korlach J, Webb WW. Fluorescence correlation spectroscopy with single-molecule sensitivity on cell and model membranes. Cytometry. 1999;36:176–182. doi: 10.1002/(sici)1097-0320(19990701)36:3<176::aid-cyto5>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Sengupta P, Garai K, Balaji J, Periasamy N, Maiti S. Measuring size distribution in highly heterogeneous systems with fluorescence correlation spectroscopy. Biophys J. 2003;84:1977–1984. doi: 10.1016/S0006-3495(03)75006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Varma R, Sarasij RC, Ira, Gousset K, Krishnamoorthy G, Rao M, Mayor S. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell. 2004;116:577–589. doi: 10.1016/s0092-8674(04)00167-9. [DOI] [PubMed] [Google Scholar]
- Sheetz MP, Chasan R, Spudich JA. ATP-dependent movement of myosin in vitro: characterization of a quantitative assay. J Cell Biol. 1984;99:1867–1871. doi: 10.1083/jcb.99.5.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009–1026. doi: 10.1083/jcb.145.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank DW, Wu ES, Webb WW. Enhanced molecular diffusibility in muscle membrane blebs: release of lateral constraints. J Cell Biol. 1982;92:207–212. doi: 10.1083/jcb.92.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JR. An Introduction to Error Analysis: The Study of Uncertainties in Physical Measurements. Sausalito, CA: University Science Books; 1997. [Google Scholar]
- Totsukawa G, Yamakita Y, Yamashiro S, Hartshorne DJ, Sasaki Y, Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J Cell Biol. 2000;150:797–806. doi: 10.1083/jcb.150.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triffo SB, Huang HH, Smith AW, Chou ET, Groves JT. Monitoring lipid anchor organization in cell membranes by PIE-FCCS. J Am Chem Soc. 2012;134:10833–10842. doi: 10.1021/ja300374c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemura YM, Vrljic M, Nishimura SY, Fujiwara TK, Suzuki KGN, Kusumi A. Both MHC class II and its GPI-anchored form undergo hop diffusion as observed by single-molecule tracking. Biophys J. 2008;95:435–450. doi: 10.1529/biophysj.107.123018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zanten TS, Cambi A, Koopman M, Joosten B, Figdor CG, Garcia-Parajo MF. Hotspots of GPI-anchored proteins and integrin nanoclusters function as nucleation sites for cell adhesion. Proc Natl Acad Sci USA. 2009;106:18557–18562. doi: 10.1073/pnas.0905217106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma R, Mayor S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature. 1998;394:798–801. doi: 10.1038/29563. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber SC, Spakowitz AJ, Theriot JA. Nonthermal ATP-dependent fluctuations contribute to the in vivo motion of chromosomal loci. Proc Natl Acad Sci USA. 2012;109:7338–7343. doi: 10.1073/pnas.1119505109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wey CL, Cone RA, Edidin MA. Lateral diffusion of rhodopsin in photoreceptor cells measured by fluorescence photobleaching and recovery. Biophys J. 1981;33:225–232. doi: 10.1016/S0006-3495(81)84883-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–987. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.