

Abstract
The early years of life are important for immune development and influences health in adulthood. While it has been established that the gut bacterial microbiome is rapidly acquired after birth, less is known about the viral microbiome (or, virome), consisting of bacteriophages and eukaryotic RNA and DNA viruses, during the first years of life. Here, we characterized the gut virome and bacterial microbiome in a longitudinal cohort of healthy infant twins. The virome and bacterial microbiome are more similar between co-twins than between non-related infants. From birth to two years of age, the eukaryotic virome and the bacterial microbiome expanded, but this was accompanied by a contraction of and shift in the bacteriophage virome composition. The bacteriophage-bacteria relationship begins from birth with a high predator-low prey dynamic, consistent with the Lotka-Volterra predator-prey model. Thus, in contrast to the stable microbiome observed in adults, the infant microbiome is highly dynamic and associated with early life changes in the composition of bacteria, viruses and bacteriophage with age.
The intestinal microbiome includes bacteria, eukaryotic viruses, bacterial viruses (bacteriophages), fungi, and archaea. It has been established that some of these microorganisms interact with the immune system and impact their host’s health1,2. Alterations in the intestinal bacterial microbiome have been implicated in a wide range of human diseases such as cirrhosis, diabetes and inflammatory bowel disease3–5. Most therapeutic strategies targeting the microbiome, such as probiotics, prebiotics and fecal microbial transplantation, aim to modulate the bacterial microbial community6,7. The bacterial microbiome starts is established soon after birth and its composition changes over the next several years towards a stereotypical ‘adult-like’ bacterial community structure8–11. This process can be influenced by multiple interacting factors such as nutrition, delivery route, antibiotic use and geographical setting8–15. Studies of twins demonstrate that infants share a more similar bacterial microbiome to their co-twin than compared to unrelated individuals14,16–18.
Much less is known about the viral microbiome (virome)19, a diverse community consisting of eukaryotic RNA and DNA viruses and bacteriophages. Emerging evidence indicate that the virome plays a role in human health. The burden of anellovirus (a eukaryotic DNA virus) is directly correlated with the degree of host immunosuppression and organ transplant outcome, and is an indicator of pediatric febrile illness and AIDS20–23. Pathogenic simian immunodeficiency virus is associated with enteric virome expansion, many of which are eukaryotic RNA viruses24. Additionally, chronic virus infection can confer increased resistance against pathogenic challenges25 indicating that the virome may provide beneficial effects to the host1. The intestinal microbiota also contains diverse bacteriophages, which in healthy adults consist mostly of members of the order Caudovirales and family Microviridae. These bacteriophages typically maintain a stable community over time17,26–28. Shifts in the enteric bacteriophage community composition have been associated with Crohn’s disease and ulcerative colitis29. However, unlike in environmental ecosystems where changes in population dynamics of bacteriophage-bacteria interactions follow a Lotka-Volterra “predator-prey” model30–32, the predator-prey relationship between bacteriophage and bacteria has yet to be observed in the human intestinal microbiome17. Metagenomic studies of the healthy infant gut virome are limited to one study of a single infant in which the DNA virome was analyzed at a single time point using modest depth Sanger sequencing33. Targeted PCR/RT-PCR studies have determined that some eukaryotic viruses, such as picornaviruses and anelloviruses, can be frequently found in stools of healthy infants34. While metagenomic analyses of the gut virome of children with diseases such as diarrhea and acute flaccid paralysis have been described35–37, to date, there has been no longitudinal analysis of the virome of a cohort of healthy infants.
Given that the bacterial microbiome is established during early infancy and likely impacts long term health8,9,14,16,38, we examined the changes in the eukaryotic viruses and bacteriophages that accompany human development. To elucidate the degree of inter-individual and intra-individual variability in the virome, we sequenced stools of a healthy monozygotic twin pair and three healthy dizygotic twin pairs. In this study, we defined ‘healthy infants’ as having no apparent underlying genetic or chronic disorders. Naturally, the infants had episodes of acute illness (Supplementary Fig. 1a). To define the virome composition and its evolution with increasing age, we compared the intestinal virome from prospectively collected stool samples collected at six time points from birth to 2 years old. Additionally, we sequenced the bacterial 16S ribosomal RNA genes of the same stools to generate an integrated view of the developing human intestinal virome and bacterial microbiome. Our results provide an in-depth timeline reconstruction of the kinetics of the infant intestinal virome and suggest the existence of “predator-prey” bacteriophage-bacteria relationship dynamics that naturally occurs during healthy human infant development.
RESULTS
Virome of infants during early development
We performed metagenomic sequencing of fecal specimens from 8 healthy infants (4 twin pairs) residing in the greater metropolitan area of Saint Louis, Missouri, United States (Fig. 1a and Supplementary Fig. 1a). Samples analyzed in this study were collected longitudinally from day of life 1–4 (defined as month 0) and at, 3, 6, 12, 18 and 24 months of age so as to define the intestinal microbiome of infants during early development39. To comprehensively detect both DNA and RNA viruses, total nucleic acid was extracted from stool specimens and subjected to two complementary amplification methods – multiple displacement amplification (MDA) and sequence independent DNA and RNA amplification (SIA) (Fig. 1a). MDA is commonly used in virome studies17,26,29 due to the high processivity of the phi29 polymerase. However, MDA cannot detect RNA viruses and its use also leads to preferential amplification of small circular DNA viruses40. To complement MDA, we used the SIA approach that is capable of detecting both RNA as well as DNA viruses35,36, although its sensitivity for DNA virus detection is generally lower than MDA (Fig. 1b). Libraries were pooled and sequenced on the Illumina MiSeq platform. On average, we obtained 549,301 (± 207,521 standard deviation (s.d.)) reads per MDA sample library and 551,592 (± 229,210 s.d.) reads per SIA sample library (Supplementary Fig. 1b). Sequencing reads were adaptor trimmed, quality filtered, taxonomically assigned and randomly sub-sampled to 200,000 reads per sample library for the analyses (details in Online Methods). To define the global virome composition, we merged the profiles of virus families identified by MDA and SIA into a presence-absence heatmap (Fig. 1c). Consistent with other PCR-based studies34, we identified eukaryotic RNA viruses such as caliciviruses, astroviruses and picornaviruses in the infant fecal specimens. Additionally, bacteriophage families were more frequently detected than eukaryotic RNA and eukaryotic DNA viruses. Only one archaeal virus family (Lipothrixviridae) was identified.
Figure 1.

Study design and metagenomic analysis of the infant gut virome. (a) Sequencing strategy to characterize the microbiome of 8 healthy infants (4 twin pairs). (b) Heatmap of reads assigned to virus families show that the profile is influenced by the sequencing method. Comparison of representative specimens is shown: fecal specimen from infant A2 at 24 months (A2-24), infant B1 at 6 months (B1-6) and 18 months (B1-18), infant B2 at 6 months (B2-6) and 18 months (B2-18), infant C1 at 6 months (C1-6) and infant C2 at 6 months (C2-6). SIA, sequence independent DNA and RNA amplification; MDA, multiple displacement amplification. (c) Presence-absence heatmap shows the viruses identified by subject (infants A1, A2, B1, B2, C1, C2, D1 and D2) and time point (0, 3, 6, 12, 18 and 24 months) during the first two years of life.
To compare the virome biodiversity between individuals, we measured beta diversity at the virus genus level using the unweighted Bray-Curtis distance. Principal coordinate analysis of the virome (eukaryotic viruses and bacteriophages) suggested that co-twin relatedness and age contributed to the variation in virome biodiversity of these infants (Supplementary Fig. 2a,b). Agglomerative hierarchical clustering supported the hypothesis that the virome community composition was typically more similar between co-twins than between non-related infants after controlling for age (Fig. 2).
Figure 2.
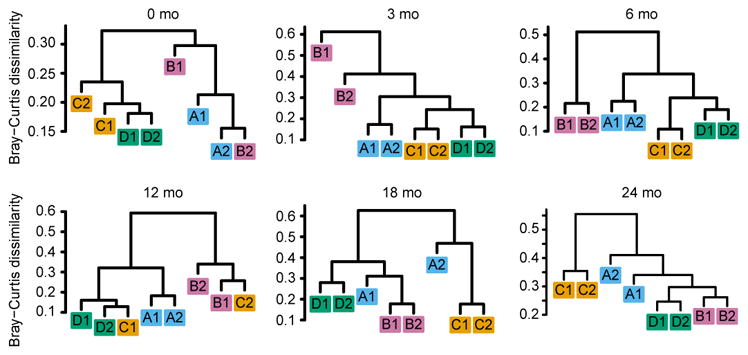
Analysis of virome beta-diversity. Agglomerative hierarchical clustering of Bray-Curtis dissimilarity of virome communities (eukaryotic viruses and bacteriophages; genera) at indicated ages.
The eukaryotic virome is acquired after birth
We next focused on the assembly of the eukaryotic virome. Eukaryotic viral population richness was low in the earliest-in-life specimens and increased thereafter (Fig. 3a, Wilcoxon test P < 0.05), suggesting that the eukaryotic virome is primarily established through environmental exposures. Of the eukaryotic RNA viruses identified, the most commonly detected viral genera were enterovirus, parechovirus, tombamovirus and sapovirus (Fig. 3b). The relative sparsity of eukaryotic viruses precluded accurate assessment of common ecological parameters (including diversity measurements and rarefaction). To determine whether co-twins harbored the same virus strains at the same time point, we analyzed sequences of parechoviruses, one of the most prevalent RNA viruses detected in our study, by assembling the reads and mapping the resulting contigs to the human parechovirus genome (Supplementary Fig. 3a). Phylogenetic analyses demonstrated that infants within a twin-pair shared nearly identical strains of parechovirus (>99.9% nucleotide (nt) identity), but different twin pairs harbored distinct strains (Fig. 3c). Consistent with this finding, strain-identical infection in co-twins was also observed for enterovirus (>99.6% nt identity), the other highly prevalent eukaryotic RNA virus (Supplementary Fig. 3b). While these findings indicate that infants within a twin-pair are frequently infected with the same virus, we also observed instances where the virus was detected in only one infant, but not in the other twin. For example, human parechovirus reads were detected in infant A1 (3 months) but not in co-twin A2. To determine whether the observed discordance might arise from sensitivity limitations of the sequencing or from differences in the viral load, we screened all the samples with a quantitative RT-PCR (qRT-PCR) assay and measured the number of human parechovirus viral copies. All five sequencing-positive samples were also positive by the qRT-PCR assay (Supplementary Fig. 3c). The three additional ‘sequencing-negative’ samples that were positive for human parechovirus by qRT-PCR had very low viral loads (18–730 viral copies/15 mg stool) (Supplementary Fig. 3c). Finally, RT-PCR and amplicon sequencing verified that ‘discordant’ infant A2 harbored the same human parechovirus isolate as its co-twin A1 (Fig. 3c). Thus, the qRT-PCR results independently validated the presence of the viruses identified by deep sequencing and further support that co-twins shared similar viromes.
Figure 3.

Alterations in the eukaryotic RNA and DNA viruses with age and evidence of shared viromes between co-twins. (a) Richness (number of observed taxa) of eukaryotic DNA and RNA virus genera (n = 8 infants). Linear regression, R2 value and 95% confidence intervals are shown. (b) Number of specimens harboring indicated eukaryotic RNA virus genera. (c) Maximum likelihood phylogeny of parechovirus sequences. Bootstrap values are indicated on the branches. (d) Number of specimens harboring indicated eukaryotic DNA virus genera. (e) Phylogenetic relationships of 61 anellovirus contigs and 12 reference strains inferred from the ORF1 amino acid alignment, generated by the maximum likelihood method. Genera are highlighted in indicated colors. (f) Presence-absence heatmap sequencing reads mapped to the anellovirus contigs. Contigs are colored by their genera phylogenetic assignment from (e). (g) Richness of anelloviruses species at indicated age (n = 8 infants) is shown. Statistical significance was assessed by Wilcoxon test (paired, non-parametric); *P = 0.01–0.05, ** P <0.01. (h) Comparison of the proportion of shared anellovirus taxa (genome contigs) acquired during the first two years of life between co-twins (n = 4 co-twin comparisons) and unrelated infants (n = 24 comparison between unrelated infants). Statistical significance was assessed by Student’s t-test; *P < 0.05.
Anelloviruses were the most prevalent DNA eukaryotic virus family detected (Fig. 3d). Almost all anelloviruses were previously unknown and highly divergent from previously described anelloviruses (Fig. 3e and Supplementary Fig. 3d). As anellovirus load has previously been associated with changes in host immune status20–22, we hypothesized that changes in infant immunity (for instance, waning of maternal antibodies) may be reflected by changes in anellovirus prevalence or abundance. Because of the large number of novel anellovirus reads detected, we curated a set of unique anellovirus contigs (shared < 95% nt identity) to functionally serve as “reference genomes”. We then mapped sequencing reads from each specimen to the anellovirus reference contigs to determine the prevalence of each contig (Fig. 3f and Supplementary Fig. 3e). This approach yielded 98.6% concordance with PCR assays designed to detect three specific anellovirus contigs (Supplementary Fig. 3f). Anelloviruses were rarely detected earlier than 3 months of age, but soon increased significantly (P < 0.05), peaking at 6–12 months of age (Fig. 3g). Notably, one infant (C1) harbored at least 47 anellovirus species at 12 months of age. Moreover, co-twins shared a higher proportion of anelloviruses than did non-related infants (Fig. 3h). Further, in some instances, the same anelloviruses could be detected from stools from the same infant that were collected up to 12 months apart (Fig. 3f), suggesting either persistence or a stable source of recurrent infection.
Bacteriophage community contracts and shifts in composition with age
While DNA bacteriophages were detected in all samples, analysis of the SIA-generated data did not yield any RNA phages. In the absence of RNA phage, we chose to focus the subsequent bacteriophage analyses on the MDA-generated data so that our findings could be comparable to other bacteriophage virome studies which have used MDA17,26,29. In contrast to the eukaryotic virome, bacteriophage richness was greatest in the earliest in life specimens at 0 months and decreased with age (Fig. 4a, Wilcoxon test P < 0.01). Richness rarefaction curves demonstrated that the rate of bacteriophage species accumulation indeed decreased with age, suggesting that the decrease in richness was unlikely to be attributed to sampling bias (Fig. 4b). Likewise, bacteriophage diversity decreased with age (Fig. 4c, Wilcoxon test P < 0.01). The interpersonal variation in the bacteriophage virome was lower (that is, it was more similar) between co-twins than between unrelated infants (Fig. 4d). The most abundant bacteriophages were from the Caudovirales order (Siphoviridae, Inoviridae, Myoviridae and Podoviridae families) and Microviridae family, consistent with other studies26,29,33. However, there was a marked shift in the community composition towards an increased relative abundance of Microviridae bacteriophages by 24 months of age (Fig. 4e and Supplementary Fig. 4a). This shift in Microviridae abundance was also seen in the SIA data indicating that it was not an artifact of method bias (Supplementary Fig. 4b). An increase in Microviridae species richness was observed indicating that this expansion was not driven by a particular species (Supplementary Fig. 4c,d). The relative abundance of Caudovirales was inversely correlated with Microviridae (Fig. 4f). crAssphage, a globally ubiquitous bacteriophage41, was detected in only one specimen (infant A2, 24 month; 50,355 reads), suggesting that crAssphage is not acquired early in life. Thus, early infant development was marked by a contraction of bacteriophage community richness and diversity, and accompanied by a shift towards a predominantly Microviridae composition.
Figure 4.

Decrease in bacteriophage richness and diversity with age coincides with a shift in bacteriophage composition. (a) Richness of bacteriophage species is shown (n = 8 infants). Linear regression, R2 value and 95% confidence intervals are shown. (b) Rarefaction curves show the acquisition of bacteriophage species richness (500 permutations). Curves from samples at the same age are indicated in colors. (c) Alpha diversity (Shannon index) of bacteriophage species is plotted (n = 8 infants). Linear regression, R2 value and 95% confidence intervals. (d) Bray-Curtis distance of the bacteriophage virome at the genus level within twin pairs (white) (n = 4 co-twin comparisons) compared to unrelated infants (shaded) (n = 24 comparison between unrelated infants). Statistical significance was assessed by Student’s t-test; *P = 0.01–0.05, **P < 0.01. (e) Relative abundance of bacteriophage families. (f) Plot shows the relationship of the Microviridae family abundance compared to Caudovirales order abundance (n = 48 sampling time points). Linear regression and 95% confidence intervals are shown, and the Spearman correlation coefficient is indicated.
Bacterial microbiome changes in infants
The ecological signatures of a healthy intestinal bacterial microbiota during early infancy have been characterized by increasing richness and diversity as bacterial populations mature into a more stable ‘adult-like’ population by 2–3 years old8,9,11. Given the dramatic changes we observed in the bacteriophage virome, we sought to understand if these changes correlated with changes in the bacterial microbiome. Therefore, we performed bacterial 16S rRNA bacterial gene sequencing to generate an average of 67,569 reads per sample (s.d. ± 39,862 reads). Quality-filtered sequence reads were clustered into operational taxonomic units (OTUs) at a 97% identity threshold. Principal coordinate analyses of unweighted UniFrac distance matrices indicated that variation in the bacterial community was associated with age (Supplementary Fig. 5a,b). Consistent with other studies8–10, bacterial richness (Fig. 5a, Wilcoxon test P < 0.001 and Supplementary Fig. 5c) and diversity (Fig. 5b, Wilcoxon test P < 0.001) also increased with age. Overall, we identified increasingly abundant Clostridia (Firmicutes) (Fig. 5c and Supplementary Fig. 5d). This was preceded by the predominance of Bacilli (Firmicutes) OTUs at 0 months, an increase in Gammaproteobacteria (Proteobacteria) and Actinobacteria (Actinobacteria) abundance at 3 and 6 months, and an increase in Bacteroidia (Bacteroidetes) abundance at 12, 18 and 24 months. The inter-individual variation between co-twins was less than between unrelated infants (Fig. 5d). Hence, the bacterial microbiota of infants in this study was consistent with the expected trajectory of changes previously observed8–10.
Figure 5.

Bacterial community expansion with age. (a) Richness (number of observed bacterial OTUs) of bacterial OTUs(n = 8 infants). Linear regression, R2 value and 95% confidence intervals are shown. (b) Bacterial alpha diversity (Faith’s phylogenetic diversity)(n = 8 infants). Linear regression, R2 value and 95% confidence intervals are shown. (c) Relative abundance of bacterial families based on 16S rRNA gene sequences. (d) Unifrac distance of the bacterial community compared within twin pairs (white) (n = 4 co-twin comparisons) and between unrelated infants (shaded) (n = 24 comparison between unrelated infants). Statistical significance was assessed by Student’s t-test; **P < 0.01.
“Predator-prey”-like bacteriophage and bacteria relationships
Although bacteriophage-bacteria relationships in oceans display predator-prey dynamics30,31, both bacteriophage and bacteria populations are relatively stable in the adult intestine17,26. However, this relationship has yet to be defined in infants. In our cohort, bacteriophage diversity was inversely correlated with bacterial diversity (Fig. 6a). By examining temporal trends, we found that the microbiome shifted from a high bacteriophage-low bacterial diversity community at 0 months towards a low bacteriophage-high bacterial diversity community by 24 months of age. Consistent with this finding, bacteriophage and bacteria richness were inversely correlated in an age-dependent manner (Fig. 6b). Further reflecting these ecological trends, correlations between specific genera of bacteriophage and bacteria, calculated using a linear mixed model, were dominated by negative correlations. (Supplementary Fig. 6). Thus, the infant virome and bacterial microbiome evolves in a dynamic trajectory during the early years of life.
Figure 6.
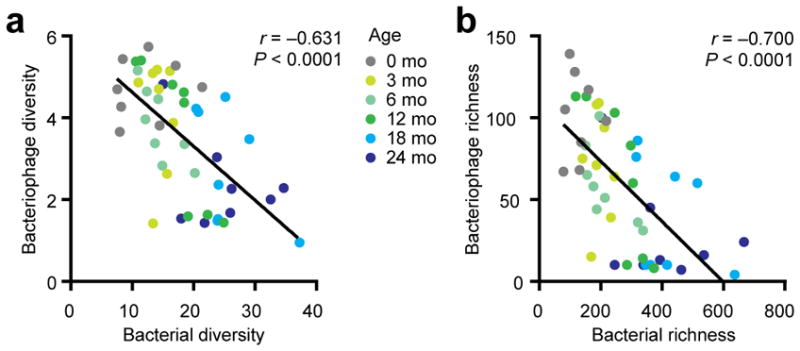
Inverse relationships between bacteriophage and bacteria. (a) Correlation between bacteriophage diversity and bacterial diversity (n = 48 sampling time points). Line indicates linear regression, and the Spearman correlation coefficient is shown. Color spectrum indicates age progression from 0–24 months. (b) Correlation plot between bacteriophage richness and bacterial richness(n = 48 sampling time points). Line indicates linear regression, Spearman correlation coefficient is shown. Color spectrum indicates age progression from 0–24 months.
DISCUSSION
Interactions between the intestinal microbiota impact host physiology, development, and immunity. Assembly of the infant intestinal bacterial microbiome likely ordains an adult microbiome that will have long term implications on host phenotype such as obesity, inflammatory bowel disease, and food allergies4,14. However, much less is known about how the virome develops in the infant, its impact on the bacterial microbiome, and its role in human health. Here, we longitudinally defined the complete virome and bacterial communities of 8 infants (4 co-twin pairs) and uncover the microbial milestones of healthy infant virome development. The twin study design enabled us to determine that the infant microbiome (virome and bacterial community) is more similar between co-twins than between unrelated infants. This result contrasts to a study of adult co-twins in which the DNA virome was unique to each individual and twins were not more similar to each other than unrelated individuals17. One possible interpretation is that while “twin-ness” matters during infancy, this may reflect that environmental exposures are the primary drivers of virome composition, as the infant twins generally still share a common environment. This was evident by the detection in co-twins of near-identical strains of eukaryotic viruses when sampled at the same time points.
Anelloviruses have been proposed to serve as biomarkers of functional immunocompetence because changes in anellovirus load in the sera have been associated with immunosuppression levels in transplant recipients20,21. Additionally, anelloviruses have been frequently detected by PCR in the serum of infants42. We observed an expansion of anellovirus richness in the gut at 6 and 12 months old; most of these anelloviruses were highly divergent from known anelloviruses, underscoring the value of using unbiased deep-sequencing approaches to systematically define the virome, as they may be difficult to detect using PCR assays. We speculate that this expansion could be the result of lowered immune state as it coincides with the nadir of human IgG, as maternal antibodies wane.
To date, no evidence indicates that “kill-the-winner”43 dynamics occur in the human intestinal microbiota, whereby a peak in the bacterial (prey) population precedes the increase in bacteriophages (predator), which subsequently decreases bacterial (prey) populations. While this is the most commonly recognized aspect of the classical Lotka-Volterra “predator-prey” model, the model also describes the reciprocal relationship whereby limited prey diversity controls predator abundance (that is, predator peaks precedes prey peaks, also referred to as a reversed predator-prey cycle)32,44. Our study (Fig. 6) suggests bacteriophage-bacteria interactions in early infant development begin with the latter dynamics of the Lotka-Volterra model32. We posit that at birth, bacteriophage diversity is high (Fig. 4, 0 month) but the bacteriophage population is unsustainable because of low bacterial colonization density (Fig. 5, 0 month). This leads to a contraction in the bacteriophage virome (Fig. 4), thereby relieving the predatory pressure on the bacterial community, allowing it to establish and colonize the gut (Fig. 5). In turn, this drives a shift in the bacteriophage composition (including increased Microviridae abundance) that has been selected for in the newly-established bacterial community. In a 2.5 year longitudinal study of a single adult, Microviridae were the predominant bacteriophage taxon present26, raising a question about how and when the dynamic microbial state during infancy transitions into the stable community that has been reported in adults9,17,26. Additionally, our study is unable to address the source of the early bacteriophage diversity, but our data raise the possibility of vertical or prepartum transmission (the median day of first sampling was day 2.6 of life). Nonetheless, our data will serve as a reference for healthy infant microbiota development. While bacteriophage contraction associated with healthy development at birth might be a generalizable phenotype, it is possible that the specific founder bacteriophage composition (identities) might differ by external factors (e.g. geography and diet9,28,36).
It has been well described that diet, antibiotics, and mode of delivery influence the bacterial microbiome8,11–15,28. While our cohort included individuals that varied in terms of zygosity, breastfeeding status, and mode of delivery, the small cohort size precluded assessment of the role of these factors on the microbiome. Regardless, our current study provides detailed insight into the dynamic interactions between viruses and bacteria in the gut during early development.
Online methods
Samples
This study was approved by the Human Research Protection Office of Washington University School of Medicine in St. Louis. We obtained consent from the mothers of twin infants to collect fecal specimens from their children monthly through age two years39. Fecal specimens were couriered to the laboratory in insulated envelopes containing frozen packs and stored at −80°C until analysis, as described39. Data collected included mode of delivery, medications given to infants, feeding content, and episodes of illness (fever/vomiting/diarrhea) by regular interviews of parents or reviews of medical records from the physicians of the twins (Supplementary Fig. 1a). The 4 twin pairs (8 infants) in this study were chosen representative of healthy infants that varied in terms of zygosity, breastfeeding status, sex (6 males and 2 females) and mode of delivery. In this study, we defined these infants as ‘healthy’ as they had no apparent underlying genetic or chronic disorders. There were no other exclusion criteria. The infants had episodes of acute illness (Supplementary Fig. 1a). The nomenclature used to label specimens in the study begins with the infant twin pair designation followed by the age (e.g. A2-24 refers to a specimen from the second infant in twin pair “A” collected at 24 month age). For the purpose of clarity in the manuscript, the age of life was defined as 0 months (avg 2.6 days, s.d. ±1.1 days), 3 months (avg 98.0 days, s.d. ±2.7 days), 6 months (avg 192.5 days, s.d. ±4.6 days), 12 months (avg 365.8 days, s.d. ±10.2 days), 18 months (avg 545.0 days, s.d. ±17.4 days) and 24 months (avg 718.5 days, s.d. ±11.6 days).
Virome sequencing
Fecal specimens (approximately 200 mg) were diluted in phosphate-buffered saline (PBS) in a 1:6 ratio and filtered through a 0.45-μm-pore-size membrane. Total nucleic acid was extracted from the filtrate on the COBAS Ampliprep instrument (Roche) according to the manufacturer’s recommendation. Samples were randomized using a random number generator (to define grouping of samples per sequencing run) and then subjected to the following amplification methods. The sequence independent DNA and RNA amplification (SIA) was performed on the total nucleic acid with primers consisting of a base-balanced 16 nt specific sequence upstream of a random 15-mer (15 Ns) for random priming as previously described35, and used for Nextera DNA library construction (Illumina). For multiple displacement amplification (MDA), total nucleic acid was amplified with Phi29 polymerase (GenomiPhi V2 kit, GE Healthcare) according to the manufacturer’s instructions and used for Nextera DNA library construction (Illumina). Libraries were purified and size-selected using Agencourt Ampure XP beads (Beckman-Coulter), followed by quantification using a 2100 Bioanalyzer (Agilent Technologies). Multiplexed SIA libraries were pooled and sequenced separately from multiplexed MDA libraries. One SIA sample (C2-0) failed library construction and was not sequenced. Additionally, to evaluate the level of specimen cross-contamination that might occur after library construction (e.g. mixed clusters that lead to index misidentification45, bioinformatic demultiplexing error, etc.), a uniquely-indexed library of cDNA derived from the nematode Orsay virus RNA1 segment46 was included in the pool for each sequencing run. Hence, 24 libraries and 1 Orsay virus control library were pooled at equimolar per sequencing run and sequenced on the Illumina MiSeq platform at the Center for Genome Sciences & Systems Biology at Washington University (total of 4 MiSeq sequencing runs, 2x250 paired-end reads, MiSeq v2 reagent kit).
Virome sequence processing
Investigators were blinded to the group allocation (i.e. age, twin pair, time point) during processing of virome sequences up to taxonomic assignment. Sequencing reads were demultiplexed and adapter sequences were trimmed. Overlapping reads were joined using fastq-join in the ea-utils package47. Low quality nucleotides were trimmed and discarded at a quality filter of Q30 Phred quality score. Candidate viral reads were identified by querying against a customized virus database. The customized virus database is comprised of all sequences with the “Viruses” superkingdom taxonomic classification from the publicly available NCBI NT and NR database (downloaded on Novemeber 7, 2013). CD-HIT48 was used to minimize sequence redundancy (98% identity over 98% of the sequence length), resulting in a customized viral NT database of 449,469 sequences and a viral NR database of 621,095 sequences. The customized viral databases for this study can be downloaded at the following: (viral NT) http://pathology.wustl.edu/virusseeker/data/VirusDBNT_20131107_ID98.tgz; (viral NR) http://pathology.wustl.edu/virusseeker/data/VirusDBNR_20131107_ID98.tgz. Sequencing reads were queried against the customized viral database sequentially using BLASTn (e-value cutoff 1E−10), followed by BLASTx (e-value cutoff 1E−3). False positive viral sequences were filtered by sequentially querying the candidate viral reads against the NCBI NT database using MegaBLAST (e-value cutoff 1E−10), BLASTn (e-value cutoff 1E−10), and the NCBI NR database using BLASTx (e-value cutoff 1E−3) to remove sequences that have a top BLAST hit corresponding to a non-viral sequence (e.g. human, fungal, etc.) as previously described49. The taxonomic assignment for sequencing reads was determined by the taxonomy ID of the top BLAST result. Family and genus taxonomic assignments were parsed from the BLAST output using in-house perl and python scripts. Bacteriophage species taxonomic assignment was determined using the lowest-common ancestor algorithm implemented in Megan (v5.8.6)50 with the following parameters: Min Support: 1, Min Score: 40.0, Max Expected: 0.01, Top Percent: 10.0, Min-Complexity filter: 0.44. Due to the presence of low complexity/repetitive regions in the reads, the following false-positive virus family taxonomic assignments were omitted from the analyses: Herpesviridae (3 reads), Mimiviridae (1 read) and Phycodnaviridae (2 reads). To assess for specimen cross-contamination, we evaluated each demultiplexed dataset for the presence of reads that map to the control Orsay virus that was pooled into each MiSeq sequencing run as described above. The average number of Orsay virus sequencing reads obtained from the control library itself was 463,315 reads (s.d. ± 106,337 reads). None of the 48 infant specimens yielded any Orsay virus sequencing reads.
Virome analysis
Sequencing reads generated by the SIA and MDA methods were rarefied (subsampling without replacement) to 200,000 reads per sample method (5 iterations) based on the sequencing depth (Supplementary Fig. 1b). The number of reads detected for a given viral taxon was plotted as a heatmap. Consistent results were obtained in the analyses across all iterations. Thus, results obtained from a representative iteration are shown. To define the global virome composition, we merged the rarefied SIA and MDA data and plotted it as an unweighted (presence/absence) heatmap. This merged data was used in analyses of the global virome (eukaryotic viruses and bacteriophages). The prevalence of eukaryotic viruses was inadequate to accurately assess most common ecological measurements (e.g. diversity measurements, rarefaction) in a standalone analysis of the eukaryotic virome community. To analyze the bacteriophage virome community, we used the MDA data as it had a better representation of DNA viruses (Fig. 1b), and so as to allow our findings to be comparable to other bacteriophage virome studies which historically have used MDA17,26,29. Ecological analyses including richness and diversity measurements (Shannon index, Bray-Curtis dissimilarity), agglomerative hierarchical clustering and rarefaction curve analyses were performed using the vegan R package51. Rarefaction curves were performed using 500 permutations. Principal coordinates analyses (PCoA) was performed using Emperor52.
To examine inter-individual virus isolates, sequencing reads were mapped to reference genomes (human parechovirus 1 (FM178558), human enterovirus B (NC_001472) and crAssphage (JQ995537)) using bowtie 2 and geneious53,54. Maximum likelihood (ML) phylogenetic trees were constructed with PhyML (version 3.00)55 using appropriate evolution models as assessed by jModelTest2 (version 2.16) and ProtTest (version 2.4) accordingly56,57. Support for ML trees was assessed by 1000 nonparametric bootstraps, and analyses were performed at least twice.
Human parechovirus analysis
A previously described pan-parechovirus TaqMan RT-PCR assay targeting conserved sequences in the 5′UTR region of the genome was used to screen all samples for parechovirus58. The following primers were used: AN345F (5′-GTAACASWWGCCTCTGGGSCCAAAAG -3′) and AN344R (5′-GGCCCCWGRTCAGATCCAYAGT -3′), with probe AN257 (5′/6-FAM/CCTRYGGGTACCTYCWGGGCATCCTTC/TAMRA/ -3′). The qRT-PCR was performed using the TaqMan Fast Virus 1-Step Master Mix (Applied Biosystems). The 20 μL reaction included 5 μL of extracted sample, 10 pmol of each primer, and 5 pmol of probe. The following cycling conditions were used: 50°C for 5 min, 95°C for 20 sec, 40 cycles of 95°C for 3 sec and 58°C for 30 sec. To generate a standard curve for this assay, in vitro transcribed RNA was generated from a plasmid containing the region of interest using MEGAscript (Ambion) per the manufacturer’s protocol. Serial dilutions of the in vitro transcribed RNA from 5 X 106 to 5 copies were used to generate a standard curve and a limit of detection of 5 copies was defined. Samples were tested in a 96-well plate format with 5 water-only negative controls. All 5 negative controls were negative for parechovirus. We sought to obtain the orthologous 3D region from parechovirus-positive samples in order to perform a phylogenetic comparison. 3′ RACE was performed with ThermoScript reverse transcriptase (Life Technologies) using an oligo (dT)20 primer, and subsequently PCR amplified with a primer specific for human parechovirus (5′-CCAGGTTAACAATGAACTATGGCAG -3′). Although human parechovirus was detected in specimens D1-12 and D2-12 by the Taqman assay (at low viral copies), we were unable to amplify the human parechovirus from these specimens by 3′RACE and RT-PCR.
Anellovirus analysis
Many of the anellovirus sequences shared limited identity to known anelloviruses, suggesting that they were highly divergent. Therefore, we first sought to curate the anellovirus genome sequences. Contigs were assembled de novo from all QC-filtered reads using Newbler (version 2.8)59 and queried against the above customized viral database using BLASTx to identify anellovirus contigs. Anellovirus contigs greater than 500 nt were aligned to reference anellovius genomes: alphatorquevirus TTV1 (AB008394), TTV-P1C1 (AF298585), TTV SIA109 (FJ426280), TTV8 genotype 22 (AB054647); betatorquevirus TTmV1 TLMV-CBD279 (AB026931), TTV-like TLMV-CLC062 (AB038625), TTV-like TTMV_LY2 (JX134045), TTmV5 TGP96 (AB041962), TTV-like LIL-y1 (EF538880), TTV-like LIL-y2 (EF538881), TTmV3 (NC_014088); gammatorquevirus TTmV1 MD1-073 (AB290918), TTmV MDJHem8-2 (AB303557), TTmV MDJN1 (AB303558). Contigs that shared > 95% nucleotide identity were combined by taking the consensus. Additionally, to determine the phylogenetic relationships of the anelloviruses, only contigs that encoded ORF1 were used for further analyses. ML phylogenetic trees were constructed from the ORF1 amino acid alignment that included the above anellovirus reference genomes. Support for ML trees (LG + I + G + F) was assessed by 1000 nonparametric bootstraps. Analyses were performed at least twice.
To determine the prevalence of the anelloviruses bioinformatically, sequencing reads from each sample were mapped to the curated anellovirus genome contigs using bowtie 2 and SAMtools53,60. We evaluated the concordance of the in silico prevalence analysis with PCR assays for three curated anelloviruses: an alphatorquevirus anellovirus Contig2355 (forward primer 5′-GTAGCCAGAATAAGAACTATGCCC -3′, reverse primer 5′-TACTGTCTAAAACCTGGAAGTTGC -3′); a betatorquevirus anellovirus Contig2737 (forward primer 5′-TCCAAGAGACTTTAAACCAGGCC -3′, reverse primer 5′-GGAACTCCTGGATTGTCCCATC -3′); a gammatorquevirus anellovirus Contig2393 (forward primer 5′-CTGATGTAGATGATGGACATGGC -3′, reverse primer 5′-CATGAGCTTTGTTGCAGAAAGTC -3′). These three anellovirus contigs were chosen based on the following criteria: 1) a representative of each genus (alpha, beta, and gamma) was selected, and 2) based on sequence data, the contigs were either present in multiple timepoints from an infant or detected across multiple infants. PCR was performed with Taq DNA polymerase (Life technologies) under the following cycling conditions: 95°C for 5 min, 40 cycles of 95°C for 30 sec, 58°C for 30 sec, 72°C for 23 sec, followed by 72°C for 10 min. Products were visualized by electrophoresis using 2% agarose gels.
Bacterial 16S rRNA gene sequencing
Nucleic acid was extracted from fecal specimens that were disrupted by bead beating as described previously29. PCR was performed using Golay-barcoded primers specific for the V4 region (F515/R806) as previously described29. Equimolar libraries were pooled and sequenced using an Illumina MiSeq sequencer (2x250 paired-end reads, MiSeq v2 reagent kit) at the Center for Genome Sciences & Systems Biology at Washington University. 4 specimens (A1-0, A2-0, D1-0, D2-0) yielded insufficient reads (< 10,000 reads). Hence, we repeated the 16S PCR for these 4 specimens at 40 PCR cycles and re-sequenced their libraries in a subsequent MiSeq sequencing run.
Bacterial 16S rRNA gene analysis
16S analysis was performed with QIIME (Quantitative Insights Into Microbial Ecology, version 1.8.0)61. Sequences were quality filtered at Q20 Phred quality score and demultiplexed. Sequences were assigned to closed reference operational taxonomic units (OTUs) at a 97% identity threshold using the Greengenes database (version 13.8)62. Investigators were blinded to the group allocation (i.e. age, twin pair, time point) during processing of 16S rRNA gene sequences up to OTU taxonomic assignment. To account for inter-sample sequencing depth variability, all samples were rarefied to 10,000 reads per sample (10 iterations), which exceed the generally accepted minimum sequencing depth previously described63. Consistent results were obtained in the 16S analyses across all iterations. Thus, results obtained from a representative iteration are shown. Alpha diversity (Faith’s phylogenetic diversity), OTU richness and UniFrac distance was calculated using QIIME. Rarefaction curves were performed using the vegan R package using 500 permutations. PCoA plots were visualized using Emperor.
Correlation network
A linear mixed model was used to investigate the relative abundance changes of bacteria and bacteriophage over time. A linear mixed model takes into account repeated measurements from the same subjects at different time points. In the model, the relative abundance of bacteria and phage were log transformed, sample collection time was designated a fixed effect and subjects were designated as random effects. P values from multiple comparisons were corrected using False Discovery Rate (q value). A q value less than 0.05 was considered statistically significant. Bacteria and phage correlation was plotted using Cytoscape. The analyses were performed in R version 3.1.2.
Statistics
Student’s t-test (two-sided) was performed with SAS. Normal distribution and equal variances between the groups were verified. Wilcoxon test (paired, non-parametric) was applied to compare the eukaryotic virus richness (Fig. 3a), bacteriophage richness (Fig. 4a), bacteriophage diversity (Fig. 4c), bacterial richness (Fig. 5a) and bacterial diversity (Fig. 5b) between matched samples at 0 month compared to 24 month. Wilcoxon test, linear regression and Spearman correlation was performed with GraphPad Prism. P values and 95% confidence intervals are shown accordingly.
Supplementary Material
Acknowledgments
We thank the families and their physicians for their participation in, and cooperation with, the study. This was work was supported in part by the Children’s Discovery Institute (MD-FR-2013-292) and the National Institutes of Health (5P30 DK052574 (Biobank, DDRCC) to P.I.T. and UH3AI083265 to P.I.T. and B.B.W.). P.I.T. and B.B.W. received funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and the Foundation for the National Institutes of Health (made possible by support from the Gerber Foundation). D.W. holds an Investigator in the Pathogenesis of Infectious Disease award from the Burroughs Wellcome Fund. E.S.L. is an Eli & Edythe Broad Fellow of the Life Sciences Research Foundation.
Footnotes
Competing financial interests:
The authors declare no competing financial interests.
Accession codes:
Sequence data have been deposited to the NCBI Sequence Read Archive under BioProject accession number SRP058399.
Author contributions:
E.S.L., L.R.H., D.W. conceived and designed the experiments. L.D., I.K.B., E.S.L. prepared samples for sequencing. E.S.L and I.K.B. performed PCR experiments. E.S.L., G.Z. and Y.Z. processed and analyzed the sequencing data. P.I.T., B.B.W., M.N. recruited the study participants and managed the metadata. E.S.L., L.R.H., D.W. and P.I.T. wrote and edited the manuscript.
References
- 1.Virgin HW. The virome in mammalian physiology and disease. Cell. 2014;157:142–150. doi: 10.1016/j.cell.2014.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Norman JM, Handley SA, Virgin HW. Kingdom-agnostic metagenomics and the importance of complete characterization of enteric microbial communities. Gastroenterology. 2014;146:1459–1469. doi: 10.1053/j.gastro.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qin N, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513:59–64. doi: 10.1038/nature13568. [DOI] [PubMed] [Google Scholar]
- 4.Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. 2011;10:311–323. doi: 10.1016/j.chom.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borody TJ, Khoruts A. Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol. 2012;9:88–96. doi: 10.1038/nrgastro.2011.244. [DOI] [PubMed] [Google Scholar]
- 7.Gritz EC, Bhandari V. The human neonatal gut microbiome: a brief review. Front Pediatr. 2015;3:17. doi: 10.3389/fped.2015.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koenig JE, et al. Succession of microbial consortia in the developing infant gut microbiome. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl 1):4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yatsunenko T, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subramanian S, et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature. 2014;510:417–421. doi: 10.1038/nature13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Backhed F, et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe. 2015;17:690–703. doi: 10.1016/j.chom.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 12.Dominguez-Bello MG, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.La Rosa PS, et al. Patterned progression of bacterial populations in the premature infant gut. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:12522–12527. doi: 10.1073/pnas.1409497111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walter J, Ley R. The human gut microbiome: ecology and recent evolutionary changes. Annu Rev Microbiol. 2011;65:411–429. doi: 10.1146/annurev-micro-090110-102830. [DOI] [PubMed] [Google Scholar]
- 16.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5:e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reyes A, et al. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature. 2010;466:334–338. doi: 10.1038/nature09199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodrich JK, et al. Human genetics shape the gut microbiome. Cell. 2014;159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oh J, et al. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514:59–64. doi: 10.1038/nature13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Vlaminck I, et al. Temporal response of the human virome to immunosuppression and antiviral therapy. Cell. 2013;155:1178–1187. doi: 10.1016/j.cell.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beland K, et al. Torque Teno virus in children who underwent orthotopic liver transplantation: new insights about a common pathogen. J Infect Dis. 2014;209:247–254. doi: 10.1093/infdis/jit423. [DOI] [PubMed] [Google Scholar]
- 22.McElvania TeKippe E, et al. Increased prevalence of anellovirus in pediatric patients with fever. PLoS One. 2012;7:e50937. doi: 10.1371/journal.pone.0050937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, et al. AIDS alters the commensal plasma virome. J Virol. 2013;87:10912–10915. doi: 10.1128/JVI.01839-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Handley SA, et al. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell. 2012;151:253–266. doi: 10.1016/j.cell.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barton ES, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;447:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- 26.Minot S, et al. Rapid evolution of the human gut virome. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12450–12455. doi: 10.1073/pnas.1300833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breitbart M, et al. Metagenomic analyses of an uncultured viral community from human feces. J Bacteriol. 2003;185:6220–6223. doi: 10.1128/JB.185.20.6220-6223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minot S, et al. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res. 2011;21:1616–1625. doi: 10.1101/gr.122705.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Norman JM, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160:447–460. doi: 10.1016/j.cell.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parsons RJ, Breitbart M, Lomas MW, Carlson CA. Ocean time-series reveals recurring seasonal patterns of virioplankton dynamics in the northwestern Sargasso Sea. ISME J. 2012;6:273–284. doi: 10.1038/ismej.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hennes KP, Simon M. Significance of bacteriophages for controlling bacterioplankton growth in a mesotrophic lake. Appl Environ Microbiol. 1995;61:333–340. doi: 10.1128/aem.61.1.333-340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cortez MH, Weitz JS. Coevolution can reverse predator-prey cycles. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:7486–7491. doi: 10.1073/pnas.1317693111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breitbart M, et al. Viral diversity and dynamics in an infant gut. Research in microbiology. 2008;159:367–373. doi: 10.1016/j.resmic.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 34.Kapusinszky B, Minor P, Delwart E. Nearly constant shedding of diverse enteric viruses by two healthy infants. J Clin Microbiol. 2012;50:3427–3434. doi: 10.1128/JCM.01589-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finkbeiner SR, et al. Metagenomic analysis of human diarrhea: viral detection and discovery. PLoS Pathog. 2008;4:e1000011. doi: 10.1371/journal.ppat.1000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holtz LR, et al. Geographic variation in the eukaryotic virome of human diarrhea. Virology. 2014;468–470:556–564. doi: 10.1016/j.virol.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kapoor A, et al. A highly prevalent and genetically diversified Picornaviridae genus in South Asian children. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20482–20487. doi: 10.1073/pnas.0807979105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olszak T, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gurnee EA, et al. Gut colonization of healthy children and their mothers with pathogenic ciprofloxacin-resistant Escherichia coli. J Infect Dis. 2015 doi: 10.1093/infdis/jiv278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edwards RA, Rohwer F. Viral metagenomics. Nat Rev Microbiol. 2005;3:504–510. doi: 10.1038/nrmicro1163. [DOI] [PubMed] [Google Scholar]
- 41.Dutilh BE, et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat Commun. 2014;5:4498. doi: 10.1038/ncomms5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ninomiya M, Takahashi M, Nishizawa T, Shimosegawa T, Okamoto H. Development of PCR assays with nested primers specific for differential detection of three human anelloviruses and early acquisition of dual or triple infection during infancy. J Clin Microbiol. 2008;46:507–514. doi: 10.1128/JCM.01703-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez-Valera F, et al. Explaining microbial population genomics through phage predation. Nat Rev Microbiol. 2009;7:828–836. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- 44.Thingstad TF. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol Oceanogr. 2000;45:1320–1328. [Google Scholar]
- 45.Kircher M, Heyn P, Kelso J. Addressing challenges in the production and analysis of illumina sequencing data. BMC Genomics. 2011;12:382. doi: 10.1186/1471-2164-12-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Felix MA, et al. Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol. 2011;9:e1000586. doi: 10.1371/journal.pbio.1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aronesty E. ea-utils : Command-line tools for processing biological sequencing data. 2011 [Google Scholar]
- 48.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 49.Zhao G, et al. Identification of novel viruses using VirusHunter--an automated data analysis pipeline. PLoS One. 2013;8:e78470. doi: 10.1371/journal.pone.0078470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oksanen JF, et al. vegan: Community Ecology Package. 2013. R package version 2.0-10. [Google Scholar]
- 52.Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience. 2013;2:16. doi: 10.1186/2047-217X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kearse M, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 56.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 57.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nix WA, et al. Detection of all known parechoviruses by real-time PCR. J Clin Microbiol. 2008;46:2519–2524. doi: 10.1128/JCM.00277-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McDonald D, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caporaso JG, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.