

Abstract
Polymorphisms within Plasmodium falciparum vaccine candidate antigens have the potential to compromise vaccine efficacy. Understanding the allele frequencies of polymorphisms in critical binding regions of antigens can help in the designing of strain-transcendent vaccines. Here, we adopt a pooled deep-sequencing approach, originally designed to study P. falciparum drug resistance mutations, to study the diversity of two leading transmission-blocking vaccine candidates, Pfs25 and Pfs48/45. We sequenced 329 P. falciparum field isolates from six different geographic regions. Pfs25 showed little diversity, with only one known polymorphism identified in the region associated with binding of transmission-blocking antibodies among our isolates. However, we identified four new mutations among eight non-synonymous mutations within the presumed antibody-binding region of Pfs48/45. Pooled deep sequencing provides a scalable and cost-effective approach for the targeted study of allele frequencies of P. falciparum candidate vaccine antigens.
Malaria remains an important global public health threat with approximately 3.3 billion people living in regions with ongoing transmission. In 2013, estimates suggest that 198 million cases and 584,000 deaths were attributed to malaria.1 The development of an effective vaccine for Plasmodium falciparum, the parasite species responsible for the greatest burden of disease, is a high priority to achieve malaria elimination from endemic countries. The most advanced malaria vaccine targets the preerythrocytic stage of infection and recently completed phase III trials with modest success.2 Vaccines targeting the erythrocytic stage have generally been disappointing with very low efficacy.3 Transmission-blocking vaccines (TBVs) target antigens expressed by the gametocytes or those expressed on the parasite in the mosquito vector and aim to block transmission to the vertebrate host.4,5 Preclinical studies have identified several candidate P. falciparum antigens, including Pfs25 and Pfs48/45, against which antibodies demonstrate transmission-blocking activity.4,5
A key limitation in the development of malaria vaccines to preerythrocytic and erythrocytic stages of malaria has been antigen diversity, an issue not extensively considered for TBVs.6 This is because it has been suggested that TBV antigens generally receive less pressure from the human immune system, leading to limited diversification. Pfs25 is expressed solely during macrogametogenesis within the mosquito midgut, thus never encountering human immune pressure. As a result, this antigen may have limited diversity within parasite populations. In contrast, Pfs48/45 is expressed on gametocytes within the human host and is a target of naturally developed antibodies during infection, which may maintain greater antigenic diversity within parasite populations.7,8
Understanding the diversity of these antigens can aid in rational vaccine design, especially for the design of strain-transcendent vaccines.9 Previously, we have used pooled amplicon deep sequencing to assess diversity of drug resistance alleles in parasite populations.10,11 This approach proved to be rapid (allowing assessment of over 1,000 samples in less than 2 months) and cost effective. Here, we have modified this approach to study the diversity of two vaccine candidate antigens, Pfs25 and Pfs48/45, from six globally diverse populations of parasites. A similar approach was used to look at diversity of VAR2CSA, a candidate antigen for a malaria in pregnancy vaccine.12 In each case, we targeted the antigenic region to which neutralizing antibodies have been predicted to bind.13,14 Our deep-sequencing data, as well as previously published whole-genome sequencing data, show that these antigens are more conserved than preerythrocytic- and erythrocytic-stage antigens, but can still show significant antigenic diversity.15
In this study, DNA was extracted from 329 dried blood spots of P. falciparum field isolates collected in previous studies across six countries, four in Africa and two in Asia (Supplemental Table 1). All participants provided informed consent as approved by national institutional review boards (IRBs). Our molecular investigation of de-identified samples was approved by the University of North Carolina IRB. DNA was extracted as previously described.10 Six pools were created using 2 μL of 100 μL extracted DNA from each sample. As individual samples may contain more than one parasite variant, the diversity could be higher than the number of samples included in the pool.
Part of the pfs25 gene (nucleotides 109–474 [amino acids 37–158] of PF3D7_1031000) was amplified using barcoded primers in duplicate as previously described in a study.16 The reaction consisted of 10X Roche FastStart Hi-Fidelity Buffer (Roche, Indianapolis, IN), 400 nM forward primer (XXXXXXXXXCAGATGAGTGGTCATTTGGAA [Xs represent the barcode]), 400 nM reverse primer (TGAGCATTTGGTTTCTCCATC), 10 nM deoxynucleotide triphosphates (dNTPs), 0.5 μL Roche FastStart Hi-Fidelity Enzyme, and 5 μL DNA in a 50-μL volume. Cycling conditions were 95°C for 15 minutes, followed by 35 cycles at 94°C for 1 minute, 58°C for 1 minute, and 72°C for 2 minutes, with an extension at 72°C for 5 minutes. Library preparation, sequencing, and analysis of Pfs25 haplotypes was done as previously described (Supplemental Material).16 Part of the pfs48/45 gene (nucleotides 706–1301 [amino acids 236–433] of PF3D7_1346700) was amplified using non-barcoded primers, and each pool was indexed and library prepared individually as previously described in a study.11 The reaction consisted of 10X Roche FastStart Hi-Fidelity Buffer, 400 nM forward primer (CAAGAAGGAAAAGAAAAAGCCTTA), 400 nM reverse primer (GCCAAAAATCCATAATATGCTGA), 10 nM dNTPs, 0.5 μL Roche FastStart Hi-Fidelity Enzyme, and 5 μL DNA in a 50-μL volume. Cycling conditions were identical to those noted above. In addition to the pools, 3D7 genomic DNA was amplified and sequenced to control for sequencing error as previously described in a study.11 Analysis of sequencing was carried out as previously described using the same quality filters and a minimum 1% allele frequency to call a polymorphism.11 All sequencing was done using an Ion Torrent PGM (Life Technologies, Carlsbad, CA).
Pfs25 sequencing resulted in 414,695 reads, of which 37,038 were full length. Among the full-length reads, 35,759 were of high quality and were used in the final sample clustering to predict haplotypes. The average number of reads used to predict haplotypes in a population was 4,168 [range: 430–6,835]. We detected two haplotypes among the six pools—African populations contained the 3D7 allele exclusively, while the Asian populations were predominantly comprised of an allele with the G131A mutation (Table 1). Sequencing of control 3D7 DNA (8,632 reads) identified only the anticipated haplotype with no false alleles detected.
Table 1.
Haplotype frequency of Pfs25 in six Plasmodium falciparum populations
| Haplotype (AA polymorphism relative to PF3D7_1031000) | Haplotype frequency (%) | |||||
|---|---|---|---|---|---|---|
| Madagascar | Cambodia | DRC | India | Malawi | Tanzania | |
| No variants | 100 | 15.93 | 100 | 10.55 | 100 | 100 |
| G131A | ND | 84.07 | ND | 89.45 | ND | ND |
DRC = Democratic Republic of the Congo; ND = not detected (mutation is either not present in population or occurs at a frequency of less than 2%).
Pfs48/45 sequencing resulted in 68,292,930 bases of data. As the amplicon was sheared before sequencing, we could only determine allele frequency, rather than haplotype as with Pfs25.11 We achieved > 500-fold coverage on > 99% of positions analyzed. At the filter settings used, we saw no false-positive alleles detected from amplification and sequencing error among the 3D7 control sequencing.
We identified eight non-synonymous polymorphisms and two synonymous polymorphisms among these six populations (Table 2). Four of the nonsynonymous polymorphisms have not been described previously in genome data (gray in Table 2).15 Parasite populations harbored between 0 and 5 polymorphisms, ranging from 1% to 86% frequency. The number of polymorphisms did not correlate with the number of parasites in a pool (correlation coefficient = 0.28). The alleles were nearly equally distributed between private or shared, with six private alleles and four alleles shared between two and four populations.
Table 2.
Allele frequencies of Pfs48/45 polymorphisms in six Plasmodium falciparum populations
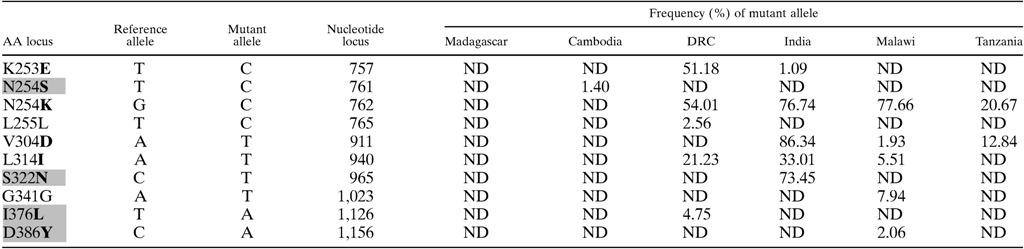
DRC = Democratic Republic of the Congo; ND = not detected (mutation is either not present in population or occurs at a frequency of less than 1.0%. Bold letters represent non-synonymous polymorphisms. Gray shading represents the novel polymorphisms described in this study.
In the literature, there have been eight non-synonymous polymorphisms previously reported within the region of pfs25 that we amplified.15 In this study, we identified two pfs25 variants—the 3D7 allele and an allele with the G131A polymorphism. Consistent with past reports, African pools were comprised exclusively of the 3D7 allele, while the Asian pools were composed primarily of the G131A allele (> 80%).15 In fact, our allele frequencies were nearly identical to those determined from genome sequencing databases—G131A has a reported allele frequency of 0.807 in south Asia and 0.840 in eastern southeast Asia.15
A higher number of non-synonymous polymorphisms have been reported for Pfs48/45, with 14 occurring within the amplified region. Pfs48/45 mutant allele frequencies tend to be higher, and high frequencies occur in a more diverse geographic range than with Pfs25.15 Notable differences between our results and previous results include 1) a lack of V304D in Cambodia where previous allele frequencies in eastern southeast Asia were 0.805, 2) the presence of S322N in India, and 3) the lack of mutant alleles at amino acid 253 in Malawi and Tanzania (K253E previously reported at 0.524). Of note, we also described several new polymorphisms in this region including N254S, S322N, I376L, and D386Y (Table 2). In addition, there was a predominance of non-synonymous mutations detected in our samples, suggestive of diversifying selection occurring on this region of Pfs48/45 (a high dN/dS ratio).
Pooled deep sequencing provides a cost-effective and scalable approach to studying allele frequencies in P. falciparum populations.10 Here we add to the available information about variation in P. falciparum transmission blocking vaccine candidates. Limitations in our study include not assessing the complete length of each gene, which means some diversity was likely missed. However, we did target regions predicted to be important for antibody binding.13,14 Second, we used a set of samples collected previously for other purposes, representing different ages, clinical symptoms, and years collected. This may introduce biases for which we cannot account. Finally, several of our populations had less than 50 samples available for sequencing, but were included to provide a diverse geographical range for this survey.
Although genomic databases are extremely useful, our data suggest that they may have limitations for determining allele frequencies in important regions of candidate vaccine antigens. This is in part due to the limitation in the number of samples within these databases and the expenses associated with whole-genome sequences. Our pooled deep-sequencing approach has previously been shown to produce high-quality estimates of allele frequencies within samples, is scalable and cost effective with more than 50 samples being evaluated.10 Using this approach, we identified new alleles in populations and provide novel insights into the diversity of TBV candidate antigens. Future studies could use this approach to supplement genome databases to provide more robust data concerning allele frequencies of targeted critical antigens involved in malaria vaccine development.
Supplementary Material
Footnotes
Financial support: This project was funded by the National Institute for Allergy and Infectious Diseases (NIAID) grant R01AI089819 (JJJ), R21AI101427 (NK), R21 AI103466 (NK), and R01AI107949 (SRM). Christian M. Parobek was supported by the National Institute of General Medicine Sciences T32GM008719, T32GM007092, and F30AI109979. Nicholas F. Brazeau was funded by a grant from the IDSA Medical Scholars Program. David L. Saunders and Chanthap Lon were supported by the U.S. Department of Defense Global Emerging Surveillance Program. Billy Ngasala and Andreas Mårtensson were supported by The Swedish Development Cooperation Agency (SIDA) (Bil-Tz 16/9875007059). Andreas Mårtensson was also supported by The Swedish Medical Research Council (2013-6594). Sample collection in Sainte Marie was financially supported by Institut Pasteur de Madagascar.
Authors' addresses: Jonathan J. Juliano, Nicholas F. Brazeau, and Irving Hoffman, Department of Medicine, Division of Infectious Diseases, University of North Carolina at Chapel Hill, Chapel Hill, NC, E-mails: jonathan_juliano@med.unc.edu, nbrazeau1@gmail.com, and irving_hoffman@med.unc.edu. Christian M. Parobek, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC, E-mail: christian_parobek@med.unc.edu. Billy Ngasala, Department of Parasitology, Muhimbili University of Health and Allied Sciences, Dar el Salam, United Republic of Tanzania, E-mail: bngasala70@yahoo.co.uk. Milijaona Randrianarivelojosia, Department of Malariology, Institut Pasteur de Madagascar, Antananarivo, Madagascar, E-mail: milijaon@pasteur.mg. Chanthap Lon and David L. Saunders, Department of Immunology and Medicine, Armed Forces Research Institute of Medical Sciences, Bangkok, Thailand, E-mails: chanthapl.ca@afrims.org and david.saunders@afrims.org. Kashamuka Mwandagalirwa and Antoinette Tshefu, School of Public Health, University of Kinshasa, Kinshasa, The Democratic Republic of the Congo, E-mails: mkashamuka@yahoo.com and antotshe@yahoo.com. Ravi Dhar, Sushant Lok, Gurgaon, India, E-mail: rdhar_in@yahoo.com. Bidyut K. Das, Department of Medicine, SCB Medical College, Odisha, India, E-mail: bidyutdas@hotmail.com. Francis Martinson, University of North Carolina Project, Lilongwe, Malawi, E-mail: francis_martinson@med.unc.edu. Andreas Mårtensson, Infectious Diseases Unit, Department of Medicine, Karolinska Institutet, Stockholm, Sweden, E-mail: andreas.martensson@ki.se. Nirbhay Kumar, Department of Tropical Medicine, School of Public Health and Tropical Medicine, Tulane University, New Orleans, LA, E-mail: nkumar@tulane.edu. Steven R. Meshnick, Department of Epidemiology, University of North Carolina at Chapel Hill, Chapel Hill, NC, E-mail: meshnick@email.unc.edu.
References
- 1.WHO . World Malaria Report 2014. Geneva, Switzerland: World Health Organization; 2014. [Google Scholar]
- 2.RTS,S Clinical Trials Partnership Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet. 2015;386:31–45. doi: 10.1016/S0140-6736(15)60721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thera MA, Doumbo OK, Coulibaly D, Diallo DA, Kone AK, Guindo AB, Traore K, Dicko A, Sagara I, Sissoko MS, Baby M, Sissoko M, Diarra I, Niangaly A, Dolo A, Daou M, Diawara SI, Heppner DG, Stewart VA, Angov E, Bergmann-Leitner ES, Lanar DE, Dutta S, Soisson L, Diggs CL, Leach A, Owusu A, Dubois MC, Cohen J, Nixon JN, Gregson A, Takala SL, Lyke KE, Plowe CV. Safety and immunogenicity of an AMA-1 malaria vaccine in Malian adults: results of a phase 1 randomized controlled trial. PLoS One. 2008;3:e1465. doi: 10.1371/journal.pone.0001465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter R. Transmission blocking malaria vaccines. Vaccine. 2001;19:2309–2314. doi: 10.1016/s0264-410x(00)00521-1. [DOI] [PubMed] [Google Scholar]
- 5.Carter R, Mendis KN, Miller LH, Molineaux L, Saul A. Malaria transmission-blocking vaccines—how can their development be supported? Nat Med. 2000;6:241–244. doi: 10.1038/73062. [DOI] [PubMed] [Google Scholar]
- 6.Takala SL, Coulibaly D, Thera MA, Batchelor AH, Cummings MP, Escalante AA, Ouattara A, Traore K, Niangaly A, Djimde AA, Doumbo OK, Plowe CV. Extreme polymorphism in a vaccine antigen and risk of clinical malaria: implications for vaccine development. Sci Transl Med. 2009;1:2ra5. doi: 10.1126/scitranslmed.3000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bousema JT, Drakeley CJ, Kihonda J, Hendriks JC, Akim NI, Roeffen W, Sauerwein RW. A longitudinal study of immune responses to Plasmodium falciparum sexual stage antigens in Tanzanian adults. Parasite Immunol. 2007;29:309–317. doi: 10.1111/j.1365-3024.2007.00948.x. [DOI] [PubMed] [Google Scholar]
- 8.Bousema T, Roeffen W, Meijerink H, Mwerinde H, Mwakalinga S, van Gemert GJ, van de Vegte-Bolmer M, Mosha F, Targett G, Riley EM, Sauerwein R, Drakeley C. The dynamics of naturally acquired immune responses to Plasmodium falciparum sexual stage antigens Pfs230 and Pfs48/45 in a low endemic area in Tanzania. PLoS One. 2010;5:e14114. doi: 10.1371/journal.pone.0014114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barry AE, Arnott A. Strategies for designing and monitoring malaria vaccines targeting diverse antigens. Front Immunol. 2014;5:359. doi: 10.3389/fimmu.2014.00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor SM, Parobek CM, Aragam N, Ngasala BE, Martensson A, Meshnick SR, Juliano JJ. Pooled deep sequencing of Plasmodium falciparum isolates: an efficient and scalable tool to quantify prevailing malaria drug-resistance genotypes. J Infect Dis. 2013;208:1998–2006. doi: 10.1093/infdis/jit392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor SM, Parobek CM, DeConti DK, Kayentao K, Coulibaly SO, Greenwood BM, Tagbor H, Williams J, Bojang K, Njie F, Desai M, Kariuki S, Gutman J, Mathanga DP, Martensson A, Ngasala B, Conrad MD, Rosenthal PJ, Tshefu AK, Moormann AM, Vulule JM, Doumbo OK, Ter Kuile FO, Meshnick SR, Bailey JA, Juliano JJ. Absence of putative artemisinin resistance mutations among Plasmodium falciparum in sub-Saharan Africa: a molecular epidemiologic study. J Infect Dis. 2015;211:680–688. doi: 10.1093/infdis/jiu467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bordbar B, Tuikue Ndam N, Renard E, Jafari-Guemouri S, Tavul L, Jennison C, Gnidehou S, Tahar R, Gamboa D, Bendezu J, Menard D, Barry AE, Deloron P, Sabbagh A. Genetic diversity of VAR2CSA ID1-DBL2Xb in worldwide Plasmodium falciparum populations: impact on vaccine design for placental malaria. Infect Genet Evol. 2014;25:81–92. doi: 10.1016/j.meegid.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 13.Outchkourov N, Vermunt A, Jansen J, Kaan A, Roeffen W, Teelen K, Lasonder E, Braks A, van de Vegte-Bolmer M, Qiu LY, Sauerwein R, Stunnenberg HG. Epitope analysis of the malaria surface antigen pfs48/45 identifies a subdomain that elicits transmission blocking antibodies. J Biol Chem. 2007;282:17148–17156. doi: 10.1074/jbc.M700948200. [DOI] [PubMed] [Google Scholar]
- 14.Sharma B. Structure and mechanism of a transmission blocking vaccine candidate protein Pfs25 from P. falciparum: a molecular modeling and docking study. In Silico Biol. 2008;8:193–206. [PubMed] [Google Scholar]
- 15.Venkatesan M, Gadalla NB, Stepniewska K, Dahal P, Nsanzabana C, Moriera C, Price RN, Martensson A, Rosenthal PJ, Dorsey G, Sutherland CJ, Guerin P, Davis TM, Menard D, Adam I, Ademowo G, Arze C, Baliraine FN, Berens-Riha N, Bjorkman A, Borrmann S, Checchi F, Desai M, Dhorda M, Djimde AA, El-Sayed BB, Eshetu T, Eyase F, Falade C, Faucher JF, Froberg G, Grivoyannis A, Hamour S, Houze S, Johnson J, Kamugisha E, Kariuki S, Kiechel JR, Kironde F, Kofoed PE, LeBras J, Malmberg M, Mwai L, Ngasala B, Nosten F, Nsobya SL, Nzila A, Oguike M, Otienoburu SD, Ogutu B, Ouedraogo JB, Piola P, Rombo L, Schramm B, Some AF, Thwing J, Ursing J, Wong RP, Zeynudin A, Zongo I, Plowe CV, Sibley CH, WWARN AL. ASAQ Molecular Marker Study Group Polymorphisms in Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes: parasite risk factors that affect treatment outcomes for P. falciparum malaria after artemether-lumefantrine and artesunate-amodiaquine. Am J Trop Med Hyg. 2014;91:833–843. doi: 10.4269/ajtmh.14-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin JT, Hathaway NJ, Saunders DL, Lon C, Balasubramanian S, Kharabora O, Gosi P, Sriwichai S, Kartchner L, Chuor CM, Satharath P, Lanteri C, Bailey JA, Juliano JJ. Using amplicon deep sequencing to detect genetic signatures of Plasmodium vivax relapse. J Infect Dis. 2015;212:999–1008. doi: 10.1093/infdis/jiv142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.