

Abstract
Generalized spike-wave seizures involving abnormal synchronization of cortical and underlying thalamic circuitry represent a major category of childhood epilepsy. Inborn errors of Cacna1a, the P/Q-type voltage-gated calcium channel α subunit gene, expressed throughout the brain destabilize corticothalamic rhythmicity and produce this phenotype. To determine the minimal cellular lesion required for this network disturbance, we used neurotensin receptor 1 (Ntsr1) cre-driver mice to ablate floxed Cacna1a in layer VI pyramidal neurons, which supply the sole descending cortical synaptic input to thalamocortical relay cells and reticular interneurons and activate intrathalamic circuits. Targeted Cacna1a ablation in layer VI cells resulted in mice that display a robust spontaneous spike-wave absence seizure phenotype accompanied by behavioral arrest and inhibited by ethosuximide. To verify the selectivity of the molecular lesion, we determined that P/Q subunit proteins were reduced in corticothalamic relay neuron terminal zones, and confirmed that P/Q-mediated glutamate release was reduced at these synapses. Spike-triggered exocytosis was preserved by N-type calcium channel rescue, demonstrating that evoked release at layer VI terminals relies on both P/Q and N-type channels. Whereas intrinsic excitability of the P/Q channel depleted layer VI neurons was unaltered, T-type calcium currents in the postsynaptic thalamic relay and reticular cells were dramatically elevated, favoring rebound bursting and seizure generation. We find that an early P/Q-type release defect, limited to synapses of a single cell-type within the thalamocortical circuit, is sufficient to remodel synchronized firing behavior and produce a stable generalized epilepsy phenotype.
SIGNIFICANCE STATEMENT This study dissects a critical component of the corticothalamic circuit in spike-wave epilepsy and identifies the developmental importance of P/Q-type calcium channel-mediated presynaptic glutamate release at layer VI pyramidal neuron terminals. Genetic ablation of Cacna1a in layer VI neurons produced synchronous spike-wave discharges in the cortex and thalamus that were inhibited by ethosuximide. These mice also displayed N-type calcium channel compensation at descending thalamic synapses, and consistent with other spike-wave models increased low-threshold T-type calcium currents within postsynaptic thalamic relay and reticular neurons. These results demonstrate, for the first time, that preventing the developmental homeostatic switch from loose to tightly coupled synaptic release at a single class of deep layer cortical excitatory output neurons results in generalized spike-wave epilepsy.
Keywords: Cacna1a, mouse, plasticity, spike-wave, T-type, tottering
Introduction
Since the first description of cortical spike-wave seizures in a child with staring spells (Berger, 1933), identifying the essential neuronal circuitry underlying this pattern generation has been a central challenge in epilepsy research. Studies in inbred rat models (van Luijtelaar and Zobeiri, 2014; Depaulis et al., 2015) and >20 defined monogenic mouse models (Maheshwari and Noebels, 2014) uniformly point to defects in the reciprocal interplay of long-range corticothalamic and thalamocortical circuitry during the stereotyped synchronous discharge, the electroencephalographic signature of absence epilepsy and related clinical syndromes. The canonical circuit defined in these models involves the near simultaneous activation of deep layer neurons in a low-threshold region of somatosensory cortex (Meeren et al., 2002; Polack et al., 2007) maintained by intrinsic rebound bursting of thalamic cells, whereas intracortical and intrathalamic GABAergic synaptic inhibition modulates both nodes of the loop (Huguenard and McCormick, 2007). However, in models where the dysfunctional gene is known, the defective protein is widely distributed throughout cells within and outside this network and the minimal elements necessary for the pathological discharge development are unknown.
The voltage-gated P/Q-type calcium channel was the first to be linked to the spike-wave phenotype and remains an index gene for the disorder. Loss of function mutations within the P/Q channel complex in the α pore-forming subunit encoded by Cacna1a, along with accessory β4 and α2δ-2 subunits, result in a phenotypic triad of spike-wave epilepsy, episodic dystonia, and ataxia (Maheshwari and Noebels, 2014). Discovered in the tottering mouse, which contains a point mutation in Cacna1a that reduces current in single P/Q-type channels (Noebels and Sidman, 1979; Fletcher et al., 1996; Wakamori et al., 1998); the absence phenotype appears in many other allelic, partial knockdown, and homozygous knock-out models of Cacna1a in mouse (Jun et al., 1999; Saito et al., 2009; Noebels, 2012), rat (Tokuda et al., 2007), and human (Jouvenceau et al., 2001; Imbrici et al., 2004). The P/Q channel is widely expressed at presynaptic terminals where, along with less effective N- and R-type channels, it regulates neurotransmitter release (Neher and Sakaba, 2008; Catterall et al., 2013). P/Q channel loss does not eliminate synaptic transmission, which can be rescued in part by N- and R-type-mediated exocytosis at central synapses (Qian and Noebels, 2000, 2001; Inchauspe et al., 2007), however, deficient P/Q function disrupts normal patterns of spontaneous and evoked release. Loss of P/Q increases jitter, depressing synaptic transmission fidelity (Depetris et al., 2008), and targeted mutation of the IQ motif impairs Ca2+ calmodulin kinase modulation of short-term synaptic facilitation (Yan et al., 2014). An age- and synapse-specific switch from pure N-type to mixed N-P/Q-type dominant release occurs during early postnatal development, and most terminals in adult brain display roughly equally shared control of quantal release by these subtypes (Iwasaki et al., 2000).
In addition to altered transmission, mice bearing genomic P/Q subunit loss of function mutations display upregulation of low voltage-activated (LVA) T-type calcium currents in thalamic relay neurons, an important element of downstream circuit plasticity favoring spike-wave epilepsy generation (Zhang et al., 2002; Ernst et al., 2009; Cain and Snutch, 2013). This mechanism remains undefined, because there is no evidence for upregulaton or alternative splicing of Cacna1G, the gene responsible for LVA calcium current in these cells (Ernst and Noebels, 2009). It is also unclear whether thalamic remodeling depends upon intrinsic loss of P/Q calcium current or abnormal synaptic input. Recently, a knock-in Cacna1a mouse with the YFP tagged channel flanked by loxP sites was created, allowing selective removal of Cacna1a from cre-expressing cells to investigate its role in microcircuits (Mark et al., 2011). To further elucidate the role of the P/Q channel within the thalamocortical spike-wave circuit and its ability to regulate lasting thalamic excitability changes, we generated mice with the channel removed in layer VI corticothalamic pyramidal neurons and investigated the impact of this single mutated projection on the generation of spike-wave epilepsy.
Materials and Methods
Mouse strains
Mice of both sexes were maintained in the Baylor Developmental Neurogenetics breeding colony. The floxed Cacna1aCitrine (C57BL/6129/SvJae) strain was obtained from Drs Melanie Mark and Stefan Herlitze at Ruhr University of Bochem. These mice have been characterized and show no change in channel function due to the construct (Mark et al., 2011). Tg(Ntsr1-cre)GN220Gsat/Mmucd mice generated by the GENSAT project were obtained from the Mutant Mouse Regional Resource Center (www.mmrrc.org/catalog/sds.php?mmrrc_id = 17266). These mice show highly specific cre-driven reporter expression confined to large cortical layer VI pyramidal projection neurons. Cacna1aCitrine mice were crossed with Ntsr-1 Cre mice to ablate Cacna1a in these neurons (Cacna1aNtsr(−/−)). To verify Cre expression in layer VI neurons, Ntsr1 cre mice were crossed with the reporter mouse B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J from The Jackson Laboratory. For physiology experiments, to identify layer VI cells with tdTomato expression, Ntsr1 cre mice, Cacna1aCitrine mice were crossed with the tdTomato reporter mice to generate Cacna1aNtsr(+/+)/tdTomato and Cacna1aNtsr(−/−)/tdTomato. Mouse breeding and all experiments were performed under IACUC approved protocols at Baylor College of Medicine. Genotyping primers were as follows: Cacna1a citrine forward: TATATCATGGCCGACAAGCA; Cacna1a citrine reverse: TTCGGTCTTCACAAGGAACC; Cacna1a wild-type forward: GGGGTCTGACTTCTGATGGA; Cacna1a wild-type reverse: AAGTTGCACACAGGGCTTCT; Ntsr-cre forward: GACGGCACGCCCCCCTTA; Ntsr-cre reverse: CGGCAAACGGACAGAAGCATT; tdTomato mutant forward: GGCATTAAAGCAGCGTATCC; tdTomato mutant reverse: CTGTTCCTGTACGGCATGG; tdTomato wild-type forward: AAGGGAGCTGCAGTGGAGTA; tdTomato wild-type reverse: CTGTTCCTGTACGGCATGG.
Immunohistochemistry
After deep anesthesia, mice of either sex were perfused intracardially with ice-cold PBS, and then with 4% paraformaldehyde in PBS. Five to six mouse brains per genotype at 8–10 weeks of age were removed and cryoprotected overnight in 30% sucrose in PBS. Brains were embedded in OCT and frozen. Coronal cryostat sections (20 μm thickness) were collected onto glass slides, air dried for 30 min, and washed 3 times in PBS (Invitrogen). For tdTomato mice, sections were permeabilized and blocked in a solution consisting of 5% BSA, 0.3% Triton in PBS for 30 min. Slides were incubated with mouse anti-parvalbumin (1:1000, Sigma-Aldrich) overnight in blocking solution. After washing three times with PBS, slides were incubated with goat anti-mouse FITC-conjugated secondary antibody (Invitrogen, 1:500) for 1 h at room temperature. Slides were washed three times and cured with Prolong gold with DAPI (Invitrogen) and imaged using wide-field fluorescent microscopy. For Cacna1a staining, slides were stained as described previously with specific antibodies against mouse anti-parvalbumin and rabbit anti-P/Q-type calcium channel (Synaptic Systems; 1:500). TRITC-conjugated goat anti-rabbit was also used for this staining (Invitrogen). For densitometry analysis, the thalamic reticular nucleus was identified by parvalbumin-positive staining and measurements were made from the thalamic reticular nucleus; layer VI of the cortex; and the ventral posterolateral (VPL), ventral posteromedial (VPM), vental anterolateral (VAL), and posterior nucleus (PO) regions of the thalamus. Staining intensity of the hilar mossy fiber region of the dentate gyrus was used as an internal control for comparison across sections. Densitometric analysis was performed on wild-type and mutant sections using ImageJ by averaging the intensity of FITC-staining imaged from three separate regions (mean intensity within individual sections of 6480 square pixels) and three sections from five individual mice for Cacna1aCitrine and Cacna1aNtsr(−/−) genotypes and 6 Cacna1aNtsr(+/+) mice.
In situ hybridization
Hybridizing probes for Cacna1 g mRNA transcripts were designed as described by Ernst et al. (2009). Sagittal sections of a representative Cacna1aNtsr(+/+) and Cacna1aNtsr(−/−) adult female brain were probed and analyzed by the Baylor College of Medicine RNA In Situ Hybridization core, following standard procedures (Carson et al., 2005; Yaylaoglu et al., 2005; Lein et al., 2007). Representative sections from each genotype were analyzed by scanning densitometry and the images pseudocolored for intensity of cellular Cacna1g mRNA expression.
Quantitative real-time PCR
Quantitative real-time PCR was used to assess regional mRNA transcript levels from Cacna1aNtsr(+/+), Cacna1aCitrine, and Cacna1aNtsr(−/−) mice (N = 3 each) by dissection of the cortex, thalamus, and cerebellum. In brief, total RNA was extracted with TRIzol reagent (Invitrogen), quantified, and treated with DNase (Ambion). The Phusion kit from Ambion was used to reverse transcribe mRNA into cDNA. Using SyberGreen Advantage qPCR mix (Clontech Laboratories), probes for Cacna1 g transcripts included forward: GCCTTCGACGACTTCATCTT and reverse: GTGTCTCCCAGGTAACATTTCT. For GAPDH control, primers included forward: AACAGCAACTCCCACTCTTC and reverse: CCTGTTGCTGTAGCCGTATT. Real-time qPCR was performed on an Applied Biosystems 7500 Real-Time PCR System. The average threshold cycle (CT), which indicates the fractional cycle number of amplified PCR product that reaches a fixed threshold value, was determined for each sample by running three technical replicates. To report quantitative ratios between mice, we used the comparative threshold method (2−ΔΔCT) which uses an internal control (GAPDH) and compared both Cacna1aCitrine mice and Cacna1aNtsr(−/−) to Cacna1aNtsr(+/+) mice (Schmittgen and Livak, 2008; Ernst et al., 2009).
EEG recordings
Silver wire electrodes (0.005 inch diameter) soldered to a microminiature connector were implanted bilaterally into the subdural space over frontal and parietal cortex of mice under Avertin anesthesia (250 mg/kg) 2 weeks before recording. Simultaneous EEG and behavioral monitoring was performed using a digital video-electroencephalograph (Harmonie 6.1, Stellate Systems) upon adult mice of either sex aged 8 weeks while moving freely in the test cage. EEG activity from adult wild-type (Cacna1aNtsr(+/+), N = 3 and Cacna1aCitrine, N = 4) and mutant (Cacna1aNtsr(−/−), N = 4) mouse genotypes was assayed during three separate recording sessions of at least 4 h each. To assess spike-wave discharge activity, a 12 h sample of EEG recording was analyzed in each mouse where spike-wave discharges (>1 s) were counted and reported as spike-wave discharge/h. To study the drug sensitivity of spike-wave seizures in Cacna1aNtsr(−/−) mice, the T-type calcium channel antagonist, ethosuximide, was injected intraperitoneally at a concentration of 200 mg/kg following a 10 min baseline recording. After a 5 min postinjection recovery period, spike-wave discharges were counted for 10 min. In one Cacna1aNtsr(−/−) mouse, bilateral electrodes were also positioned stereotaxically in ventrobasal thalamus (coordinates from bregma, 1.4 mm posterior, 1.5 mm lateral, and 3 mm depth) and electrode coordinates were verified visually following the recording session.
Slice electrophysiology
Evoked EPSCs in thalamic neurons.
Coronal slices of a hemisphere from mice of either sex were cut at a 300 μm thickness and at an angle of 55° with the longitudinal axis to include intact corticothalamic circuitry (Agmon and Connors, 1991). The brain slices were first incubated at a temperature of 35°C for 45 min then incubated in ACSF at room temperature for 1 h before recording. The ACSF contained the following (in mm): 125 NaCl, 3 KCl, 1.25 Na2PO4, 26 NaHCO3, 2 MgCl2, 2 CaCl2, and 10 glucose.
To record evoked EPSCs, the patch pipette solution contained the following (in mm): 135 CsCl, 10 HEPES, 10 EGTA, 5 QX-314, 4 MgATP, with the pH maintained at 7.3 with CsOH. Whole-cell voltage-clamp recordings were made at room temperature (∼23°C). The input resistance of the recording pipette was 3–4 MΩ and the holding membrane potential was set at −70 mv. A bipolar tungsten wire stimulation electrode was positioned in the juxtathalamic white matter ∼400–600 μm away from the recorded cell. The stimulation pulse duration was 0.1 ms. The stimulation intensity was determined by finding the minimal stimulation magnitude required to reliably evoke EPSCs and increasing the current pulse four to five times in magnitude. GABA transmission was blocked with bicuculline (30 μm) and NMDA receptors were blocked with D-APV (20 μm) added to the bath. A pair of pulses separated by an interval of 50 ms was used to evoke synaptic transmission at the corticothalamic synapses. The effect of 1 μm N-type Ca2+ channel toxin ω-CTX GVIA (Bachem AG) on neurotransmitter release was quantified by comparing the peak amplitude of EPSCs before and after bath perfusion of the channel blocker.
Low-voltage activated calcium currents and firing behavior in thalamic and reticular neurons.
Horizontal slices (300 μm thick) were prepared from 4- to 6-week-old adult mice of either sex. Slices were perfused with ACSF at 2 mm/min at 30°C. Neurons in the ventrobasal complex (thalamic relay neurons) and nucleus reticular thalamus (reticular interneurons) were visually identified using differential interference contrast optics at 4× and 40× magnification (BX-51W microscope, Olympus) based on their anatomical location. Whole-cell configuration was obtained with patch pipettes (2–4MΩ) containing cesium gluconate solution (in mm: 110 cesium gluconate, 10 HEPES, 8 NaCl, 1 MgCl2, 10 BAPTA, 2 ATP, 0.3 GTP, 5 QX-314, 280 mOsm, pH 7.2). All neurons were voltage-clamped at −70 mV. Access resistances were <25 MΩ and were compensated by 70%. Data were acquired using a MultiClamp 700B amplifier, digitized by Digidata 1332 and analyzed with pClamp 9.2 software (Molecular Devices). After establishment of stable whole-cell configuration, LVA currents were evoked by an initial step to −50 mV from prior hyperpolarization. Steady-state inactivation (SSI) of the current was analyzed using 2 s hyperpolarizing prepulses at varying holding membrane potentials from −110 to −40 mV. Data were fitted with Boltzmann's equation using Prism software. The time dependency of de-inactivation was tested by changing the duration (0–2 s) of the −110 mV prepulses.
Characterization of intrinsic excitability in layer VI cortical neurons.
For identification of layer VI cortical neurons, Cacna1aNtsr(+/+) and Cacna1aNtsr(−/−) mice of either sex were generated by additional mouse crosses to the tdTomato reporter mice, B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J. Coronal corticothalamic slices (300 μm) were prepared from Cacna1aNtsr(−/−)/tdTomato and age-matched Cacna1aNtsr(+/+)/tdTomato mice of either sex. Slices were superfused with ACSF (in mm: 126 NaCl, 10 glucose, 1 MgSO4, 3 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, and 0.4 ascorbic acid (equilibrated with 95% O2/5% CO2) at 2 ml/min.
The Cre-expressing layer VI cortical neurons were identified by their td-Tomato epi-fluorescence (excitation 560 ± 20 nm bandpass, emission 585 nm long-pass) and thalamic relay and reticular neurons were identified as described above. Patch pipettes were filled with an internal solution (in mm: 130 potassium gluconate, 10 HEPES, 1 MgCl2, 8 NaCl, 0.05 EGTA, 2 Na-ATP, 0.3 Na-GTP, pH adjusted to 7.2 with KOH) and had a 2–3 MΩ tip resistance. After establishment of stable whole-cell configuration, membrane excitability was tested in current-clamp mode. Membrane potential was held at −60 mV and a series of command currents (0.5 s, −200 to +200 pA) were injected. Signals were acquired at 20 kHz, amplified with a Multiclamp 200B amplifier, digitized by Digidata1322A and analyzed with pClamp9.2 software (Molecular Devices). Series resistances >20 MΩ were excluded form analysis. Bridge balance was adjusted manually.
Behavioral analysis
Adult mice of either sex were trained over 3 d of trials on an automated rotating rod (Ugo Basile). Mice were placed carefully on the rotating rod and the latency to fall (seconds) was measured for each animal while the speed increased from 4 to 40 rpm over a 3 min interval. On the third day, with experimenters blinded to genotype, mice were serially tested three times and the average latency to fall is reported.
For gait analysis, with examiners blinded to the genotype, adult mice of either sex were placed on a flat surface to observe spontaneous gait. Animals were scored on a graded ataxia scale of 0–3 (Guyenet et al., 2010), where normal movement included body weight supported on all limbs with abdomen off the ground scored a 0 for ataxia. Such mice could not be discerned from wild-type littermates. Slight limping or tremor scored a 1. A severe tremor, severe limp, flattened pelvis, or hind limbs angled away from the body scored a 2. If the mouse moved with more severe difficulty, it would score a 3.
For the ledge test, mice of either sex were lifted and placed onto the lip of an empty plastic mouse cage. The mouse was observed to walk along the ledge and lower itself into the cage. Easy movement along the ledge and proper, graceful lowering into the cage scored a 0. Loss of footing, but mostly coordinated mice scored a 1. Ineffective hind leg movement or improper lowering into the cage scored a 2. Mice that fell or refused to move even with gentle nudging scored a 3 (Guyenet et al., 2010).
Statistical analysis
Unless otherwise noted, the data presented represents the mean ± SEM. Statistical tests were conducted as mentioned in the text using either Student's t tests, Kruskal–Wallis, or one-way ANOVAs as appropriate using GraphPad Prism software. In some cases, where variances between the data populations were not equal, the Welch's correction was used on a t test and this is noted in the text. For quantitative real-time PCR analysis, p values were generated by a one-way ANOVA within each brain region comparing the three different genotypes (Cacna1aNtsr(+/+), Cacna1aCitrine, Cacna1aNtsr(−/−)).
Results
Selective knockdown of P/Q-type calcium channels in layer VI corticothalamic projection neurons
To selectively delete P/Q-type calcium channels from layer VI pyramidal neurons, we crossed Cacna1aCitrine (Mark et al., 2011) mice with TgNtsr1-Cre. The Cacna1aCitrine mice consist of a conditional knock-in of the Citrine gene in the first exon of Cacna1a surrounded by loxP sites to permit Cre recombinase-mediated removal of the P/Q-type calcium channel exon 1 (Mark et al., 2011). TgNtsr1-Cre mice, express Cre recombinase under the control of the neurotensin-1 (Ntsr1) promotor that selectively labels a consistent subpopulation of layer VI neurons innervating thalamic relay neurons (Olsen et al., 2012; Bortone et al., 2014), as well as collateral innervation of parvalbumin-positive interneurons residing in the thalamic reticular nucleus (Bourassa and Deschenes, 1995). To verify selective expression of Cre recombinase in layer VI projection neurons in these mice, we crossed male TgNtsr1-Cre mice with female tdTomato reporter mice (B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J). In coronal sections of adult brain, tdTomato expression was clearly detected and confined to layer VI pyramidal neurons (Fig. 1A,B), axonal projections within the subcortical white matter (Fig 1A, 1C), and terminals in the ventrobasal thalamus and thalamic reticular nucleus (Fig. 1A,C). Within the deep layers of neocortex, the tdTomato-labeled cellular expression did not overlap with parvalbumin-positive cells, indicating that the interneurons within layer VI were spared (Fig. 1B). No cell-specific tdTomato staining was observed in other brain regions except for rare scattered cells detected in the dentate gyrus of the hippocampal formation (Fig. 1A,D) and no tdTomato-positive neurons were detected elsewhere in the forebrain or cerebellum.
Figure 1.

The Ntsr-1 cre drives expression of tdTomato in layer VI projection neurons to the thalamic reticular nucleus and ventrobasal thalamus. A, Stitched 10× images of the projections from layer VI through the internal capsule to the nRT and thalamus. Cell nuclei are marked with DAPI in blue, TdTomato staining in red indicates Ntsr Cre+ cells, and FITC staining in green marks parvalbumin interneurons throughout the brain. Scale bar, 500 μm. B, Image (10×) of layer VI cortical projection neurons. Scale bar, 200 μm. C, Image (10×) of the nRT and thalamus shows no cre-driven cellular staining, but diffuse label due to axonal projections from layer VI neurons. Scale bar, 200 μm. D, Image (10×) of hippocampal dentate gyrus shows scattered cells expressing tdTomato. Scale bar, 200 μm.
To determine whether the expression of Cre recombinase in Cacna1aCitrine mice decreased the P/Q channel population in the terminal fields of layer VI projection neurons, immunohistochemistry was performed using a specific antibody to the P/Q channel (Fig. 2A). We used the ratio of the thalamic terminal field staining intensity over the intensity of the axon terminal-rich hilar mossy fiber region of the dentate gyrus as an internal standard to control for staining variability between sections. We found significantly decreased P/Q staining intensity in the VAL, VPM, and VPL nuclei of the thalamus in Cacna1aNtsr(−/−) (0.83 ± 0.02, ANOVA, p = 0.0001, n = 5) compared with wild-type mice (Cacna1aNtsr(+/+) 1.00 ± 0.03, n = 6; Cacna1aCitrine 1.03 ± 0.01, n = 5; Fig. 2B). Because these thalamic nuclei also receive thalamopetal axon terminals originating from nRT and other brain regions, there is substantial residual P/Q staining due to the overlapping presence of nonablated terminals. However, despite their innervation by layer VI axon collaterals, no measurable decreases of P/Q antibody labeling were detected in the nRT (ANOVA, p = 0.11; Fig. 2C) or deep cortical layer (ANOVA, p = 0.50; Fig. 2D) compared with the mossy fiber region of the dentate gyrus, presumably because these subregions comingle with an even larger population of nonablated axon terminals. These data confirm the specificity of Ntsr1 Cre ablation of P/Q calcium channels at layer VI terminals in the ventrobasal thalamus.
Figure 2.
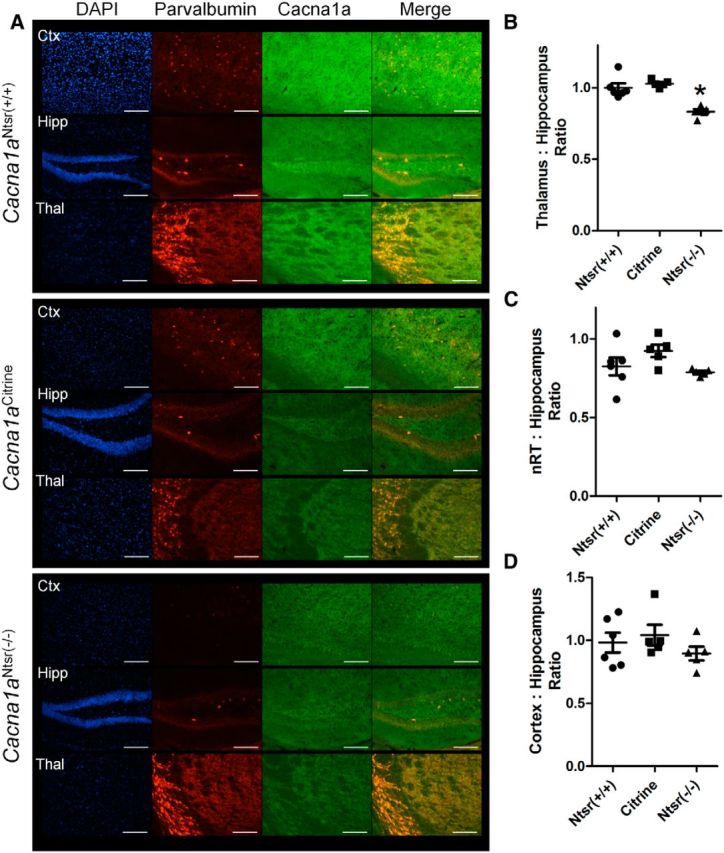
Immunostaining reveals specific knockdown of Cacna1a in ventrobasal regions of the thalamus. A, Representative staining in (top) wild-type (Cacna1aNtsr(+/+), (middle) Cacna1aCitrine, and (lower) Cacna1aNtsr(−/−)mice of cortex, hippocampus, and thalamus. TRITC-conjugated parvalbumin staining was used to identify the nRT. FITC staining marks P/Q calcium channels and DAPI marks cell nuclei. Scale bars, 100 μm. B, Densitometric ratio of the ventrobasal thalamus compared with the mossy fiber region of the hippocampus within slices of wild-type (Cacna1aNtsr(+/+), n = 6, 1.00 ± 0.03; Cacna1aCitrine, n = 5, 1.03 ± 0.01; Cacna1aNtsr(−/−), n = 5, 0.83 ± 0.02) reveal decreased P/Q immunostaining (ANOVA, *p = 0.0001). C, Densitometric ratio of the nRT compared with the mossy fiber region of the hippocampus within slices of wild-type (Cacna1aNtsr(+/+), n = 6, 0.82 ± 0.06; Cacna1aCitrine, n = 5, 0.92 ± 0.04; Cacna1aNtsr(−/−), n = 5, 0.79 ± 0.09) show no decreased P/Q immunostaining (ANOVA, p = 0.11). D, Densitometric ratio of the layer V/VI cortex compared with the mossy fiber region of the hippocampus within slices of wild-type (Cacna1aNtsr(+/+), n = 6, 0.98 ± 0.08; Cacna1aCitrine, n = 5, 1.04 ± 0.08; Cacna1aNtsr(−/−), n = 5, 0.89 ± 0.05) show no decreased P/Q immunostaining (ANOVA, p = 0.50).
Functional loss of presynaptic P/Q-type channels at the corticothalamic relay synapse in Cacna1aNtsr(−/−) mice
To ascertain whether the channel ablation was reflected in functional knockdown of P/Q channel-mediated release at the layer VI-thalamic neuron terminals, we investigated synaptic transmission at the corticothalamic terminal synapses in the ventrobasal thalamus using an N-type channel-specific toxin to dissect the contribution of these two major presynaptic calcium channel subtypes. Mutation of presynaptic P/Q-type channels in tottering mice result in a shift of channel reliance of neurotransmitter release from P/Q-type to N-type at both corticohippocampal and mossy fiber hippocampal synapses (Qian and Noebels, 2000, 2001). As shown in the sample traces in Figure 3A, EPSCs evoked by paired-pulse stimulation of corticothalamic axons exhibit large amplitude paired pulse facilitation (PPF) of cortical-thalamic synaptic transmission in both Cacna1aNtsr(+/+) and Cacna1aNtsr(−/−) animals (Fig. 3B; t test, p = 0.51). In Cacna1aNtsr(+/+) control brain slices, the mean PPF ratio was 3.4 ± 0.6 (n = 12), which does not significantly differ from the Cacna1aNtsr(−/−) group (PPF ratio 3.2 ± 0.5, n = 12). Application of 1 μm ω-CTX GVIA, a N-type calcium channel blocker, revealed that neurotransmitter release at the synapse in Cacna1aNtsr(−/−) slices was more sensitive to blockade of N-type channels than Cacna1aNtsr(+/+) slices (Fig. 3C; t test, p = 0.0001). On average, the N-type calcium channel toxin reduced the evoked EPSC amplitude by 71 ± 11% (n = 12) in Cacna1aNtsr(−/−) as compared with 50 ± 12% (n = 12) in the Cacna1aNtsr(+/+) group. This evidence demonstrates a severe functional loss of presynaptic P/Q-type channels in layer VI presynaptic thalamic terminals in Cacna1aNtsr(−/−) mice.
Figure 3.
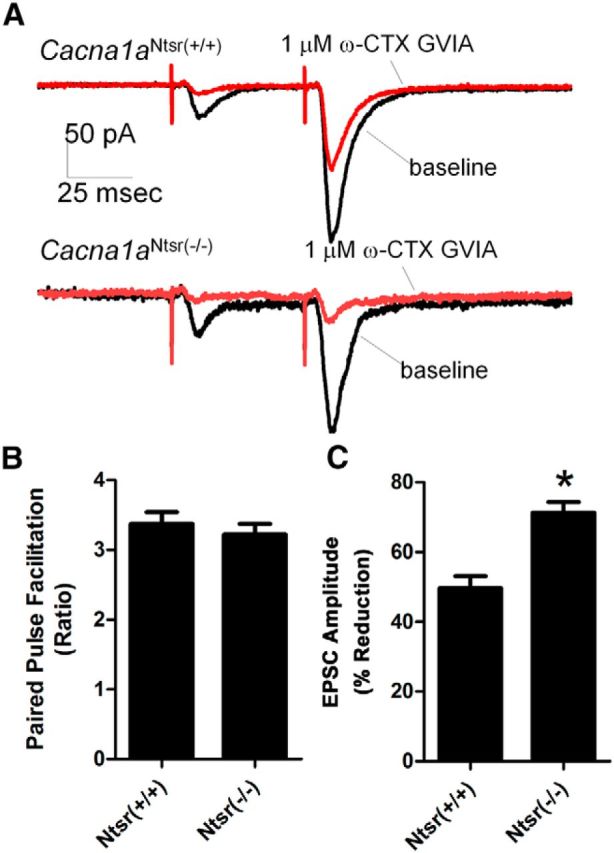
Electrophysiological recordings from the cortical thalamic synapse reveal functional loss of P/Q calcium channels. A, Example traces of PPF within the ventrobasal thalamus when stimulated in the internal capsule before and after the application of N-type calcium channel blocker, ω-CTX GVIA. Scale bar, 50 ms. B, Baseline PPF is not significantly different (t test, p = 0.51) between Cacna1aNtsr(+/+) (PPF ratio 3.4 ± 0.6, n = 12) and Cacna1aNtsr(−/−) mice (PPF ratio 3.2 ± 0.5, n = 12). C, Application of 1 μm ω-CTX GVIA resulted in a significant EPSC decrease (t test, *p = 0.0001) of 50 ± 12% in Cacna1aNtsr(+/+) slices (n = 12) and 71 ± 11% in Cacna1aNtsr(−/−) slices (n = 12).
Layer VI cell intrinsic properties are unchanged in Cacna1aNtsr(−/−) mice
To further investigate the extent of Cacna1a ablation on the excitability of the parent layer VI pyramidal neurons, which might impact levels of excitatory drive in descending corticothalamic axons to the thalamus, whole-cell recordings of these cells in Cacna1aNtsr(+/+) and Cacna1aNtsr(−/−) mice labeled with the tdTomato reporter were performed (Fig. 4A,B, representative trace; n = 9 cells each). There was no significant difference, at any injected voltage, in the number of action potentials generated in cells from Cacna1aNtsr(+/+)/tdTomato and Cacna1aNtsr(−/−)/tdTomato mice (Fig 4C; ANOVA interaction, p = 0.9118). Additionally, no difference was seen in the resting membrane potential (Fig. 4D; t test, p = 0.30), membrane resistance (Fig. 4E; t test, p = 0.10), cell capacitance (Fig. 4F; t test, p = 0.85), or frequency of spontaneous EPSCs (Fig. 4G; t test with Welch's correction, p = 0.33) of Cacna1aNtsr(+/+)/tdTomato or Cacna1aNtsr(−/−)/tdTomato cells. This indicates that selective knock-out of Cacna1a in layer VI neurons does not significantly alter their intrinsic membrane firing properties, and results primarily in a specific impairment of neurotransmitter release.
Figure 4.

Whole-cell patch-clamp electrophysiology of layer VI cells. A, Representative tdTomato expression in layer VI for identification to patch-clamp. Scale bar, 200 μm. B, Representative voltage step protocol and current response by a Cacna1aNtsr(+/+) cell. C, Number of action potentials elicited by injected positive current for Cacna1aNtsr(+/+) cells (n = 9) and Cacna1aNtsr(−/−) cells (n = 9). No significant difference was found at any current step. (ANOVA, p = 0.88 and 0.91 for column factor or interaction). D, No difference in resting membrane potential of layer VI projection neurons. The mean resting membrane potential for Cacna1aNtsr(+/+) cells was 69.5 ± 1.9 mV and for Cacna1aNtsr(−/−) cells 72.0 ± 1.4 mV (n = 9 each, t test, p = 0.30). E, No difference in membrane resistance of layer VI projection neurons. The mean membrane resistance for Cacna1aNtsr(+/+) cells was 174.0 ± 9.7 MΩ and for Cacna1aNtsr(−/−) cells 153.5 ± 7.0 MΩ (n = 9 each, t test, p = 0.11). F, No difference in membrane capacitance of layer VI projection neurons. The mean membrane capacitance for Cacna1aNtsr(+/+) cells was 53.5 ± 6.9 pF and for Cacna1aNtsr(−/−) cells was 55.2 ± 6.1 pF (n = 9 each, t test, p = 0.85). G, No difference in the frequency of spontaneous EPSCs of layer VI projection neurons. Frequency of spontaneous EPSCs for Cacna1aNtsr(+/+) cells was 1.59 ± 0.36 Hz and for Cacna1aNtsr(−/−) cells 2.61 ± 0.96 Hz (n = 9 each, t test with Welch's correction, p = 0.34).
Functional knock-out of P/Q calcium channels in layer VI projection neurons results in spike-wave epilepsy
To assess whether reduction of P/Q calcium channels in layer VI projection neurons is sufficient to produce spike-wave epilepsy, video-EEG experiments were performed on wild-type and Cacna1aNtsr(−/−) mice (Fig. 5A). During extensive monitoring, Cacna1aNtsr(−/−) mice (n = 4) displayed frequent seizures (33.2 ± 5.8 spike-wave discharges/h, Kruskal–Wallis, p < 0.0001, range: 0–156 spike-wave discharges/h), consisting of bilaterally synchronous, 5–7 spikes/s discharges that always coincided with behavioral arrest of the mice. Depth electrodes placed in the ventrobasal thalamus, as well as over frontal cortex detected concomitant spike-wave activity in both regions of one Cacna1aNtsr(−/−) mouse, confirming the involvement of both structures in the aberrant discharges (Fig. 5C). In contrast, no EEG abnormalities were found in Cacna1aNtsr(+/+) (n = 3) or Cacna1aCitrine (n = 4) mice (Fig. 5B).
Figure 5.

Cacna1aNtsr(−/−) mice have spike-wave epilepsy. A, Representative 12 s traces of wild-type (Cacna1aNtsr(+/+), Cacna1aCitrine) and Cacna1aNtsr(−/−) mice electroencephalographic recordings from bilateral electrodes over frontal cortex. Cacna1aNtsr(−/−) mice display stereotyped 5–7 spike/s spike-wave discharges. Scale bar, 500 μV and 500 ms. B, Spike-wave discharge rates from Cacna1aNtsr(+/+), N = 3, 12 h/mouse; Cacna1aCitrine, N = 4, 12 h/mouse; and Cacna1aNtsr(−/−) mice, N = 4, 12 h/mouse. Spike-wave discharges were only seen in Cacna1aNtsr(−/−) mice. C, Representative bilateral EEG recording from the frontal cortex with depth electrodes placed in the ventrobasal thalamus show spike-wave discharges in both regions of the brain. Scale bar, 500 μV and 500 ms. D, Representative EEG recording from a Cacna1aNtsr(−/−) frontal cortex before and after intraperitoneal injection of 200 mg/kg ethosuximide shows suppression of spike-wave discharges after drug exposure. Scale bar, 500 μV and 500 ms.
To further characterize the spike-wave seizures, we investigated whether they were sensitive to ethosuximide, a drug with T-type calcium channel blocking activity, which effectively suppresses seizures in tottering and all other reported SWD mouse models. Figure 5D shows a sample of baseline EEG traces recorded before and after 200 mg/kg intraperitoneal ethosuximide injection, demonstrating the similar rapid suppression of spike-wave seizures in this model. In three Cacna1aNtsr(−/−) mice, we saw spike-wave discharges decrease from 2.4 ± 0.7 spike-wave seizures/min to 0 ± 0 spike-wave seizures/min (data not shown).
Selective knock-out of P/Q calcium channels in cortical layer VI output neurons increases T-type calcium currents in thalamic relay and reticular neurons
LVA (T-type) calcium currents in thalamic relay neurons are required for spike-wave seizures (Kim et al., 2001), and elevation of this current in thalamic relay neurons is a common, sufficient, but not essential, feature of mutant mice with spike-wave seizures (Zhang et al., 2002; Song et al., 2004; Ernst et al., 2009). Thus, we examined whether similar membrane current remodeling of ventrobasal thalamic relay neurons occurred in Cacna1aNtsr(−/−) mutant mice. Whole-cell LVA currents in response to membrane depolarization were recorded from thalamic relay neurons in brain slices maintained in vitro. LVA current was reliably evoked in both Cacna1aNtsr(+/+) and Cacna1aNtsr(−/−) slices with typical fast kinetics (Fig. 6A; 10–90% decay) of 44.1 ± 11.7 ms (Cacna1aNtsr(+/+), n = 11) and 39.5 ± 4.7 ms (Cacna1aNtsr(−/−), n = 10) and strong dependency on prior membrane hyperpolarization. Peak LVA current density was significantly elevated 1.7-fold in ventrobasal thalamic neurons of Cacna1aNtsr(−/−) (12.38 ± 1.15 pA/pF, n = 10) slices compared with wild-type (7.36 ± 1.05 pA/pF, n = 11) neurons (t test, p = 0.0044, data not shown). In addition, SSI, defined by half-maximum voltage of the prior hyperpolarization voltage, showed a significant depolarizing shift in the mutant neurons (Fig. 6B; −82.2 ± 2.1 mV Cacna1aNtsr(+/+), n = 11; −75.3 ± 1.2 mV Cacna1aNtsr(−/−), n = 10; t test, p = 0.0109) indicating that LVA currents can be evoked by a smaller hyperpolarizing input in the mutant neurons. In contrast, the dependency on duration of the hyperpolarizing prepulse was not significantly altered (data not shown). We also examined rebound burst firing in ventrobasal thalamus and found no difference between Cacna1aNtsr(+/+) (1.14 ± 0.14 bursts, n = 7) and Cacna1aNtsr(−/−) (1.29 ± 0.18 bursts, n = 7) thalamic relay neurons (representative trace Fig. 6C; t test, p = 0.55). Additionally, we observed no significant difference in resting membrane potential (Fig. 6D; t test, p = 0.80), membrane resistance (Fig. 6E; t test, p = 0.18), latency to burst (Fig. 6F,G, representative trace; t test, p = 0.41), or number of action potentials per burst (Fig. 6H; t test, p = 0.99).
Figure 6.

T-type calcium channel currents are increased in Cacna1aNtsr(−/−) thalamic relay neurons. A, Sample traces of low voltage-activated calcium currents from ventrobasal thalamic relay neurons in Cacna1aNtsr(+/+) and Cacna1aNtsr(−/−) cells with prior hyperpolarization steps to activate the currents. Currents were elevated 1.7-fold in Cacna1aNtsr(−/−) cells. B, SSI (half-maximum voltage of the prior hyperpolarization voltage), showed a significant depolarizing shift in Cacna1aNtsr(−/−) ventrobasal thalamic neurons (−75.3 ± 1.2 mV, n = 10) from Cacna1aNtsr(+/+) (−82.2 ± 2.1 mV; t test, p = 0.011). C, Sample traces of rebound bursts from ventrobasal thalamic relay neurons in Cacna1aNtsr(+/+) and Cacna1aNtsr(−/−). No difference in rebound bursts was observed. D, Resting membrane potential of Cacna1aNtsr(+/+) (65.1 ± 1.2 mV, n = 7) and Cacna1aNtsr(−/−) (64.5 ± 1.4 mV, n = 7; t test, p = 0.80) cells. E, Membrane resistance of Cacna1aNtsr(+/+) (97.7 ± 13.7 MΩ, n = 7) and Cacna1aNtsr(−/−) (101.4 ± 7.6 MΩ, n = 7; t test, p = 0.18) cells. F, Representative trace of first rebound burst following hyperpolarization. Scale bar, 25 mV/50 ms. G, Latency to burst of Cacna1aNtsr(+/+) (32.9 ± 4.8 ms, n = 7) and Cacna1aNtsr(−/−) (26.0 ± 3.4 ms; n = 7; t test, p = 0.41) cells. H, Number (N) of action potentials in burst for Cacna1aNtsr(+/+) (7.4 ± 0.9 AP, n = 7) and Cacna1aNtsr(−/−) (8.7 ± 0.9 AP; n = 7; t test, p = 0.99) cells.
We also investigated intrinsic excitability and LVA currents in the thalamic reticular nucleus (nRT). Similar to thalamic relay neurons, peak LVA current was significantly elevated by 1.86-fold (t test, p = 0.001) in Cacna1aNtsr(−/−) nRT neurons (17.67 ± 2.00 pA/pF, n = 10) compared with Cacna1aNtsr(+/+) neurons (9.48 ± 0.57 pA/pF, n = 10; Fig. 7A). In contrast to ventrobasal thalamus, the SSI voltage was not altered in nRT neurons (Fig. 7B; −81.6 ± 2.5 mV Cacna1aNtsr(+/+), n = 10; −78.1 ± 1.0 mV Cacna1aNtsr(−/−), n = 10; t test with Welch's correction, p = 0.23). Correlated with this shift, nRT neurons showed increased rebound bursting following a brief hyperpolarizing pulse (Cacna1aNtsr(−/−); 10.57 ± 2.22 bursts, n = 7) compared with Cacna1aNtsr(+/+) neurons (2.12 ± 0.79 bursts, n = 8, t test with Welch's correction, p = 0.009) Figure 7C. Additionally, we observed no significant difference in resting membrane potential (Fig. 7D; t test, p = 0.79), membrane resistance (Fig. 7E; t test, p = 0.18), latency to burst (representative trace Figs. 7F, 6G; t test, p = 0.73), or number of action potentials per burst (Fig. 7H; t test, p = 0.25). Although both nRT and thalamic relay neurons show increased T-type currents, the divergent effects on rebound bursting is likely due to the additional complex interaction of HCN channels, SK channels, and other intrinsic burst control properties that differ between thalamic cell types (Budde et al., 2005; Zhang et al., 2009; Dreyfus et al., 2010; Cain et al., 2015). Although untested in this study, these additional channels may contribute to rebounding bursting and might also be affected by ablation of Cacna1a in layer IV.
Figure 7.

T-type calcium channel currents are increased in Cacna1aNtsr(−/−) reticular thalamic interneurons. A, Sample traces of low-voltage activated calcium currents from nRT neurons in Cacna1aNtsr(+/+) and Cacna1aNtsr(−/−) cells with prior hyperpolarization steps to activate the currents. B, SSI was not changed between in Cacna1aNtsr(+/+) (−81.6 ± 2.5 mV, n = 10) and Cacna1aNtsr(−/−) nRT neurons (−78.1 ± 1.0 mV, n = 10, t test with Welch's correction, p = 0.23). C, Sample traces of rebound bursts show a significant difference from nRT neurons in Cacna1aNtsr(+/+) (2.12 ± 0.79 bursts, n = 10) and Cacna1aNtsr(−/−) (10.57 ± 2.22 bursts, n = 10, t test, p = 0.009). D, Resting membrane potential of Cacna1aNtsr(+/+) (69.7 ± 4.1 mV, n = 8) and Cacna1aNtsr(−/−) (71.1 ± 2.2 mV, n = 7; t test, p = 0.10) cells. E, Membrane resistance of Cacna1aNtsr(+/+) (195.7 ± 19.1 MΩ, n = 8) and Cacna1aNtsr(−/−) (193.1 ± 18.2 MΩ, n = 7; t test, p = 0.79) cells. F, Representative trace of first rebound burst following hyperpolarization. Scale bar, 25 mV/50 ms. G, Latency to burst of Cacna1aNtsr(+/+) (67.8 ± 10.9 ms, n = 7) and Cacna1aNtsr(−/−) (52.5 ± 9.4 ms; n = 7; t test, p = 0.73) cells. H, Number of action potentials in burst for Cacna1aNtsr(+/+) (5.0 ± 0.9 AP, n = 7) and Cacna1aNtsr(−/−) (5.6 ± 0.6 AP; n = 7; t test, p = 0.41) cells.
We next examined whether the enhanced T-type calcium current could be explained by elevated transcription of Cacna1 g, the sole gene responsible for LVA current in mouse thalamic relay neurons. Figure 8A–D are representative cell-based densitometric in situ hybridization images from a littermate pair of Cacna1aNtsr(+/+) and Cacna1aNtsr(−/−) mice showing no detectable difference in the expression pattern or level of Cacna1g mRNA transcripts. The mRNA transcript levels were also analyzed by quantitative real-time PCR on dissected regions of Cacna1aNtsr(+/+), Cacna1aCitrine, and Cacna1aNtsr(−/−) brain (n = 3 per genotype) using the comparative threshold method (see Materials and Methods), Cacna1aNtsr(+/+) mice were used as a control for Cacna1aNtsr(−/−) and Cacna1aCitrine mice. The cerebellum, cortex, and thalamus were dissected from each mouse and revealed no significant difference of Cacna1 g mRNA transcript levels (Fig. 8E). Within the thalamus, mRNA levels of Cacna1aNtsr(−/−) were 1.15 ± 0.14-fold of Cacna1aNtsr(+/+) levels and Cacna1aCitrine transcript levels were 1.26 ± 0.05-fold (one-way ANOVA, p = 0.16). Similarly, there was not a significant difference found in cortex mRNA levels (Cacna1aNtsr(−/−) 1.01 ± 0.11-fold; Cacna1aCitrine 1.09 ± 0.12-fold, one-way ANOVA, p = 0.76) or in cerebellum (Cacna1aNtsr(−/−) 0.92 ± 0.0.10-fold; Cacna1aCitrine 0.92 ± 0.09-fold, one-way ANOVA, p = 0.73). The finding of comparable mRNA levels between the wild-type and mutant is consistent with the lack of significant detectable transcriptional changes in other related spike-wave mouse models, including stg, tg, and lh (Zhang et al., 2002; Song et al., 2004), and suggest that the measured current alterations are more likely explained by alternative post-translational modifications that modulate T-type currents (Chemin et al., 2014; Todorovic and Jevtovic-Todorovic, 2014).
Figure 8.

Cacna1g mRNA transcript levels are not altered in Cacna1aNtsr(−/−) mice. A, RNA in situ hybridization of Cacna1g mRNA in Cacna1aNtsr(+/+) brain. B, RNA in situ hybridization of Cacna1g in Cacna1aNtsr(−/−) brain. C, Pseudocolored cell-based densitometry of in situ hybridization image of Cacna1g mRNA in Cacna1aNtsr(+/+) brain. D, Pseudocolored RNA in situ hybridization image of Cacna1g in Cacna1aNtsr(−/−) brain. E, Normalized ratio of Cacna1g mRNA transcripts revealed no difference in mRNA expression within either the cerebellum, the cortex, or the thalamus of Cacna1aNtsr(−/−) mice (n = 3) compared with Cacna1aNtsr(+/+) mice (black square, n = 3) or Cacna1aCitrine mice (red circle, n = 3).
Neurological phenotype of Cacna1aNtsr(−/−) mice
Genomic mutations reducing Cacna1a function produce mice with various degrees of ataxia; however, the functional contribution of descending corticothalamic projections to abnormal motor control has never been assessed. To ascertain whether the layer VI P/Q channel ablation in Cacna1aNtsr(−/−) mice lead to a motor coordination phenotype, we performed three assays for balance, agility, and gait. Using a rotating rod, we found no significant differences between the wild-type groups (Cacna1aNtsr(+/+), n = 9; Cacna1aCitrine, n = 6) and Cacna1aNtsr(−/−) (n = 9) adult mice (Fig. 9A; ANOVA, p = 0.17). Similarly, no significant difference in gait was detected between wild-type (Cacna1aNtsr(+/+), n = 9; Cacna1aCitrine, n = 6) and Cacna1aNtsr(−/−) (n = 9) mice (Fig. 9B; ANOVA, p = 0.17). A small but significant difference (0.85 ± 0.19 compared with 0.27 ± 0.14 and 0.18 ± 0.12, ANOVA, p = 0.017), in performance score was found between Cacna1aNtsr(−/−) mice (n = 13) and their wild-type (Cacna1aNtsr(+/+), Cacna1aCitrine) littermates (n = 11) using a ledge test (Guyenet et al., 2010) to assay motor coordination (Fig. 9C). Unlike genomic ataxic mutant models of Cacna1a, it was not possible to visually detect adult Cacna1aNtsr(−/−) mice with certainty by their gait or spontaneous motor behavior, indicating that the severity of these motor phenotypes is accounted for by P/Q deficits outside of the corticothalamic pathway. We also did not detect any episodes of repetitive limb dyskinesia like those observed in the tg mutant.
Figure 9.
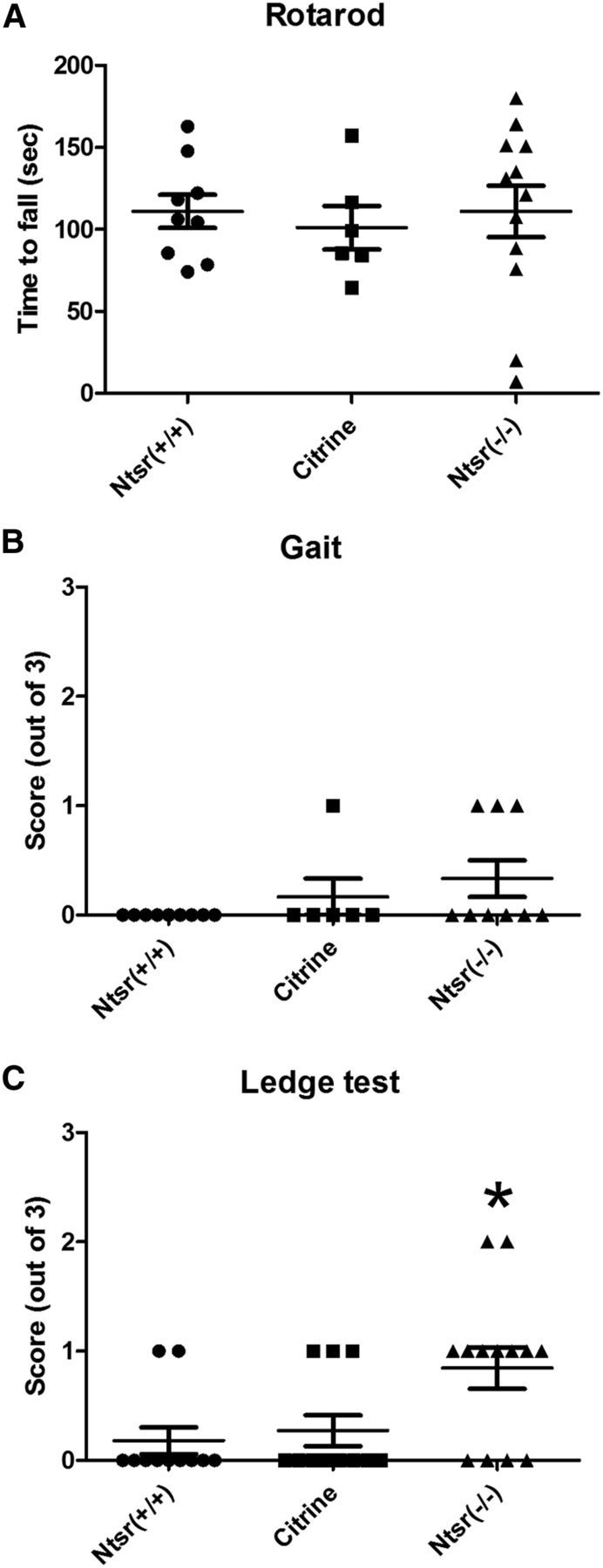
Minimal motor deficit in Cacna1aNtsr(−/−) mutants. A, Rotarod: average latency to fall over three trials in wild-type (Cacna1aNtsr(+/+), n = 9, 111 ± 10 s; Cacna1aCitrine, n = 6, 101 ± 13 s; and Cacna1aNtsr(−/−), n = 9, 111 ± 16 s). No significant difference was observed (ANOVA, p = 0.89). B, Gait test: functional score (range 0–3) of wild-type (Cacna1aNtsr(+/+), n = 9, 0 ± 0; Cacna1aCitrine, n = 6, 0.17 ± 0.17; and Cacna1aNtsr(−/−), n = 9, 0.33 ± 0.17). No significant difference was observed (ANOVA, p = 0.17). C, Ledge test: functional score (range 0–3) of wild-type (Cacna1aNtsr(+/+), n = 11, 0.18 ± 0.12; Cacna1aCitrine, n = 11, 0.27 ± 0.14; and Cacna1aNtsr(−/−), n = 13, 0.85 ± 0.19, ANOVA, *p = 0.017), which was a small, but significant, deficit in motor coordination.
Discussion
Together with its rich monogenic landscape, absence epilepsy is an especially complex synchronization disorder to physiologically dissect, since the identity of critical disease microcircuitry underlying the phenotype in each model is obscured by widespread and largely functionally redundant expression of the disease-causing mutant protein. In the case of P/Q-type calcium channelopathy, none of the models expressing genomic P/Q channel defects reveal which limb, if any, of the spike-wave circuit is inherently sufficient to generate the distinctive discharge, because P/Q channels contribute to synaptic release at each of its nodes as well as the remainder of the nervous system.
Here we used conditional cre-driven mutagenesis of Cacna1a within a highly targeted subpopulation of layer VI neocortical excitatory pyramidal cells to pinpoint synapses originating from a solitary cell type within the canonical thalamocortical network, and determined that impaired synaptic release at their terminals is sufficient for the genesis of generalized, spike-wave absence epilepsy. Seizures generated by ablating P/Q channels from layer VI pyramidal neuron terminals were phenotypically identical to those present in genomic Cacna1a mutations, including spike burst frequency (5–7 spikes/s), concomitant behavioral arrest, and ethosuximide sensitivity. We also determined that impairment of layer VI projections to the thalamus was not sufficient to account for the ataxia or episodic dystonia characteristic of genomic Cacna1a deficiency. This conditional mutant model reduces the network complexity required to solve a mechanism for the aberrant spike-wave phenotype to a single, defined, cortical synaptic output and may simplify the analysis and modeling of critical dynamic firing patterns and downstream molecular plasticity that contribute to a stable spike-wave synchronization phenotype.
Layer VI neurons: isolated dynamic release defect at the distant corticothalamic projection synapse remodels thalamic neuron excitability
The “top down” suppressant feedback of layer VI output following thalamic afferent input to a cortical column has been confirmed in visual (Olsen et al., 2012) and somatosensory (Crandall et al., 2015) cortex, where it acts as an inhibitory gain control of ascending sensory-evoked responses. Electrical stimulation of corticothalamic fibers also alters intrinsic oscillation frequency in this circuit (Bal et al., 2000; Blumenfeld and McCormick, 2000). Thus, although a complete silencing of this synapse might be expected to impede rhythmicity, we found that P/Q channel-deficient layer VI synaptic output modified thalamic T-currents, rebound burst firing, and released aberrant network oscillatory activity.
At the layer VI–thalamic relay synapse, we found evidence for shared P/Q- and N-type calcium channel control of evoked neurotransmitter release, with partial compensation by non-P/Q presynaptic calcium channel subtypes as described at other descending cortical and intrahippocampal central excitatory synapses (Qian and Noebels, 2000, 2001). Despite preservation of unitary evoked release events, the dynamic properties of P/Q deficient synapses are altered because of unequal coupling of N-type channel subunits to vesicular release nanodomains; tightly coupled P/Q channels trigger fast, synchronous exocytosis, whereas N-type channels mediate less synchronous release (Neher and Sakaba, 2008; Catterall et al., 2013). Thus P/Q-deficient synapses, when analyzed at motor nerve terminals or the presynaptic Calyx of Held, show an increase in synaptic release delay (jitter; Depetris et al., 2008) and impaired short-term facilitation (Inchauspe et al., 2007) that, depending on the degree of rescue by N- and R-type calcium channels, may significantly alter network synaptic synchrony. This strongly implies a role for P/Q-mediated short-term plasticity at corticothalamic synapses in preserving normal patterns of thalamic oscillations.
Along with functional release defects that impact dynamic circuit behavior, we also detected a persistent postsynaptic excitability change in relay cells, similar in magnitude to that found previously in genomic spike-wave mutant models, which was due solely to the isolated corticofugal synaptic defect (Tsakiridou et al., 1995; Zhang et al., 2002, 2004; Broicher et al., 2008; Ernst et al., 2009). Importantly, in our conditional model, the transsynaptic enhancement of thalamic LVA current occurred in the presence of wild-type P/Q-type channel expression in the relay cells, demonstrating that T-type current remodeling is presynaptically driven, and independent of the native P/Q-type channel status in the postsynaptic cells. Transsynaptic plasticity in thalamocortical pathways due to abnormal deprivation of afferent sensory input during early brain development and later in adulthood is well documented in somatosensory circuits (Yu et al., 2012; Li et al., 2013). Our findings clearly indicate that altered release from descending cortical inputs is also sufficient to persistently modify intrinsic thalamic membrane properties and have clear implications for neurodevelopment. Because we found that a similar magnitude of LVA calcium current enhancement (∼50%) due to overexpression of a BAC transgene for Cacna1g T-type channels is sufficient to produce the spike-wave phenotype (Ernst et al., 2009), and the majority of native Cacna1g expression is within thalamic relay cells, our results add to the evidence that thalamic T-type remodeling is a sufficient, although not necessarily exclusive, molecular basis for the spike-wave phenotype.
We previously identified spike-wave discharges following conditional ablation of Cacna1a in cerebellar circuits in purky (Mark et al., 2011) and quirky mutants (Maejima et al., 2013). Although thalamic T-type currents were not measured, both models disturb developmental patterns of thalamic signaling, suggesting a similar mechanism. However, these cre-promoter lines also showed sparse forebrain cellular expression, whereas in the case of the NTSR-cre driver studied here, reporter expression was confined to the cortical layer VI neurons, and we detected limited expression only among a small number of hippocampal cells unrelated to thalamocortical circuitry.
Layer VI neurons: local excitatory and inhibitory control of the spike-wave circuit
Intracortical circuitry plays a major role in spike-wave seizure generation, beginning with the terminals of regular spiking deep pyramidal neurons of layer VI whose output we targeted in this study. In vivo microelectrode analysis in somatosensory cortex of an inbred rat model of spike-wave epilepsy revealed that layer VI cells show the strongest coupling to the cortical EEG seizure compared with neurons in layer IV and their activation systematically preceded firing in more superficial layers (Polack et al., 2007). In wild-type mice, dual whole-cell recording in disinhibited cortical slices demonstrate that layer VI intracortical terminals are essential for horizontal synchrony during epileptiform bursting (Golshani and Jones, 1999).
Recent details on the functional connectivity of the layer VI pyramidal cell projectome reveal an elaborate intracortical synaptic microcircuitry activated by these axon collaterals, and how they differ between motor (Yamawaki and Shepherd, 2015) and sensory cortex (Thomson, 2010). Layer VI neurons activate layer V cells (Kim et al., 2014), and this projection is stronger in sensory than motor cortex. Thalamic afferents contact layer VI as well as layer IV neurons (Constantinople and Bruno, 2013), but have diverse regional and laminar patterns (Clascá et al., 2012). Layer IV projects back to layer VI (Tanaka et al., 2011), and this local excitatory loop is faster and stronger in the neocortical microcircuit than that driven from the thalamic projection.
Along with these excitatory connections, there is a strong feedforward input onto a diverse and translaminar group of inhibitory neurons (Bortone et al., 2014). In rat neocortex, fast spiking interneurons in layer V ramify around pyramidal cell somata and show P/Q-mediated synaptic depression, whereas burst firing interneurons synapsing on pyramidal cell dendrites show N-type-mediated facilitation (Ali and Nelson, 2006). Adding to this anatomical complexity, it is not yet known whether P/Q-type release coupling predominates at these intracortical synapses, and how mutation affects their development. In cerebellar cortex, the P/Q channel is the predominant adult form at excitatory synapses (Baur et al., 2015), however in neocortex, thalamocortical-evoked EPSPs remain N-type dominant (Iwasaki et al., 2000). There are distinct and still unexplained differences in P/Q channel-mediated release at excitatory versus inhibitory synapses (Ermolyuk et al., 2013). Analysis of a gain-of-function mutation in Cacna1a showed enhanced transmission at intracortical excitatory synapses, while sparing release at fast-spiking interneurons, suggesting that presynaptic calcium channels are differentially sensitive to modulation in different cell types (Vecchia et al., 2014).
If a synaptic release defect is present in layer VI intracortical axon collaterals similar to that found at the distant thalamic synapse, the isolated P/Q deficiency in this model creates an imbalance of network excitation and inhibition at both the intracortical and thalamic nodes. We found no intrinsic excitability difference in the P/Q channel-ablated layer VI neurons themselves, and therefore little evidence that altered firing properties per se lead to seizure generation. Additional evidence favoring the complex disruption of finely tuned signaling between intracortical excitatory and inhibitory neurons during generalized seizures was found following conditional P/Q ablation in mice using cre-drivers for parvalbumin+ (Nkx2.1baccre), somatostatin (SSTcre), and pyramidal (Emx1cre) neurons. P/Q-deficient synaptic impairment of these interneuron subtypes only produced spike-wave seizure patterns when simultaneously removed from both inhibitory and excitatory cortical neurons (Rossignol et al., 2013). Because the degree of excitation and inhibition at each synapse in this loop varies depending on the pattern of activity (Crandall et al., 2015), detailed modeling of this circuitry will be essential to visualize the dynamic balance of synaptic strength underlying the aberrant oscillating activity.
Footnotes
This work was supported by an Epilepsy Foundation Postdoctoral fellowship grant (V.C.B.), NINDS NS029709 (J.L.N.), and the Blue Bird Circle Foundation. We thank the RNA In Situ Hybridization Core facility at Baylor College of Medicine, which is supported by a Shared Instrumentation grant from the NIH (1S10 OD016167) and the NIH IDDRC Grant (1U54 HD083092) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
The authors declare no competing financial interests.
References
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-J. [DOI] [PubMed] [Google Scholar]
- Ali AB, Nelson C. Distinct Ca2+ channels mediate transmitter release at excitatory synapses displaying different dynamic properties in rat neocortex. Cereb Cortex. 2006;16:386–393. doi: 10.1093/cercor/bhi117. [DOI] [PubMed] [Google Scholar]
- Bal T, Debay D, Destexhe A. Cortical feedback controls the frequency and synchrony of oscillations in the visual thalamus. J Neurosci. 2000;20:7478–7488. doi: 10.1523/JNEUROSCI.20-19-07478.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur D, Bornschein G, Althof D, Watanabe M, Kulik A, Eilers J, Schmidt H. Developmental tightening of cerebellar cortical synaptic influx-release coupling. J Neurosci. 2015;35:1858–1871. doi: 10.1523/JNEUROSCI.2900-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger H. Über das elektrenkephalogramm des menschen. Archiv Psychiatri Nervenkrankheit. 1933;98:231–254. doi: 10.1007/BF01814645. [DOI] [Google Scholar]
- Blumenfeld H, McCormick DA. Corticothalamic inputs control the pattern of activity generated in thalamocortical networks. J Neurosci. 2000;20:5153–5162. doi: 10.1523/JNEUROSCI.20-13-05153.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortone DS, Olsen SR, Scanziani M. Translaminar inhibitory cells recruited by layer 6 corticothalamic neurons suppress visual cortex. Neuron. 2014;82:474–485. doi: 10.1016/j.neuron.2014.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourassa J, Deschênes M. Corticothalamic projections from the primary visual cortex in rats: a single fiber study using biocytin as an anterograde tracer. Neuroscience. 1995;66:253–263. doi: 10.1016/0306-4522(95)00009-8. [DOI] [PubMed] [Google Scholar]
- Broicher T, Kanyshkova T, Meuth P, Pape HC, Budde T. Correlation of T-channel coding gene expression, IT, and the low threshold Ca2+ spike in the thalamus of a rat model of absence epilepsy. Mol Cell Neurosci. 2008;39:384–399. doi: 10.1016/j.mcn.2008.07.012. [DOI] [PubMed] [Google Scholar]
- Budde T, Caputi L, Kanyshkova T, Staak R, Abrahamczik C, Munsch T, Pape HC. Impaired regulation of thalamic pacemaker channels through an imbalance of subunit expression in absence epilepsy. J Neurosci. 2005;25:9871–9882. doi: 10.1523/JNEUROSCI.2590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain SM, Snutch TP. T-type calcium channels in burst-firing, network synchrony, and epilepsy. Biochim Biophys Acta. 2013;1828:1572–1578. doi: 10.1016/j.bbamem.2012.07.028. [DOI] [PubMed] [Google Scholar]
- Cain SM, Tyson JR, Jones KL, Snutch TP. Thalamocortical neurons display suppressed burst-firing due to an enhanced Ih current in a genetic model of absence epilepsy. Pflugers Arch. 2015;467:1367–1382. doi: 10.1007/s00424-014-1549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson JP, Eichele G, Chiu W. A method for automated detection of gene expression required for the establishment of a digital transcriptome-wide gene expression atlas. J Microsc. 2005;217:275–281. doi: 10.1111/j.1365-2818.2005.01450.x. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Leal K, Nanou E. Calcium channels and short-term synaptic plasticity. J Biol Chem. 2013;288:10742–10749. doi: 10.1074/jbc.R112.411645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Cazade M, Lory P. Modulation of T-type calcium channels by bioactive lipids. Pflugers Arch. 2014;466:689–700. doi: 10.1007/s00424-014-1467-5. [DOI] [PubMed] [Google Scholar]
- Clascá F, Rubio-Garrido P, Jabaudon D. Unveiling the diversity of thalamocortical neuron subtypes. Eur J Neurosci. 2012;35:1524–1532. doi: 10.1111/j.1460-9568.2012.08033.x. [DOI] [PubMed] [Google Scholar]
- Constantinople CM, Bruno RM. Deep cortical layers are activated directly by thalamus. Science. 2013;340:1591–1594. doi: 10.1126/science.1236425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall SR, Cruikshank SJ, Connors BW. A corticothalamic switch: controlling the thalamus with dynamic synapses. Neuron. 2015;86:768–782. doi: 10.1016/j.neuron.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaulis A, David O, Charpier S. The genetic absence epilepsy rat from Strasbourg as a model to decipher the neuronal and network mechanisms of generalized idiopathic epilepsies. J Neurosci Methods. 2015 doi: 10.1016/j.jneumeth.2015.05.022. Advance online publication. Retrieved June 9, 2015. [DOI] [PubMed] [Google Scholar]
- Depetris RS, Nudler SI, Uchitel OD, Urbano FJ. Altered synaptic synchrony in motor nerve terminals lacking P/Q-calcium channels. Synapse. 2008;62:466–471. doi: 10.1002/syn.20516. [DOI] [PubMed] [Google Scholar]
- Dreyfus FM, Tscherter A, Errington AC, Renger JJ, Shin HS, Uebele VN, Crunelli V, Lambert RC, Leresche N. Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of ITwindow. J Neurosci. 2010;30:99–109. doi: 10.1523/JNEUROSCI.4305-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolyuk YS, Alder FG, Surges R, Pavlov IY, Timofeeva Y, Kullmann DM, Volynski KE. Differential triggering of spontaneous glutamate release by P/Q-, N- and R-type Ca2+ channels. Nat Neurosci. 2013;16:1754–1763. doi: 10.1038/nn.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst WL, Noebels JL. Expanded alternative splice isoform profiling of the mouse Cav3.1/alpha1G T-type calcium channel. BMC Mol Biol. 2009;10:53. doi: 10.1186/1471-2199-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst WL, Zhang Y, Yoo JW, Ernst SJ, Noebels JL. Genetic enhancement of thalamocortical network activity by elevating alpha 1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. J Neurosci. 2009;29:1615–1625. doi: 10.1523/JNEUROSCI.2081-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy JD, Jr, Hawkes R, Frankel WN, Copeland NG, Jenkins NA. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/S0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- Golshani P, Jones EG. Synchronized paroxysmal activity in the developing thalamocortical network mediated by corticothalamic projections and “silent” synapses. J Neurosci. 1999;19:2865–2875. doi: 10.1523/JNEUROSCI.19-08-02865.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp. 2010;39:e1787. doi: 10.3791/1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 2007;30:350–356. doi: 10.1016/j.tins.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Imbrici P, Jaffe SL, Eunson LH, Davies NP, Herd C, Robertson R, Kullmann DM, Hanna MG. Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain. 2004;127:2682–2692. doi: 10.1093/brain/awh301. [DOI] [PubMed] [Google Scholar]
- Inchauspe CG, Forsythe ID, Uchitel OD. Changes in synaptic transmission properties due to the expression of N-type calcium channels at the calyx of Held synapse of mice lacking P/Q-type calcium channels. J Physiol. 2007;584:835–851. doi: 10.1113/jphysiol.2007.139683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Momiyama A, Uchitel OD, Takahashi T. Developmental changes in calcium channel types mediating central synaptic transmission. J Neurosci. 2000;20:59–65. doi: 10.1523/JNEUROSCI.20-01-00059.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenceau A, Eunson LH, Spauschus A, Ramesh V, Zuberi SM, Kullmann DM, Hanna MG. Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet. 2001;358:801–807. doi: 10.1016/S0140-6736(01)05971-2. [DOI] [PubMed] [Google Scholar]
- Jun K, Piedras-Rentería ES, Smith SM, Wheeler DB, Lee SB, Lee TG, Chin H, Adams ME, Scheller RH, Tsien RW, Shin HS. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc Natl Acad Sci U S A. 1999;96:15245–15250. doi: 10.1073/pnas.96.26.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW, Shin HS. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron. 2001;31:35–45. doi: 10.1016/S0896-6273(01)00343-9. [DOI] [PubMed] [Google Scholar]
- Kim J, Matney CJ, Blankenship A, Hestrin S, Brown SP. Layer 6 corticothalamic neurons activate a cortical output layer, layer 5a. J Neurosci. 2014;34:9656–9664. doi: 10.1523/JNEUROSCI.1325-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Li H, Fertuzinhos S, Mohns E, Hnasko TS, Verhage M, Edwards R, Sestan N, Crair MC. Laminar and columnar development of barrel cortex relies on thalamocortical neurotransmission. Neuron. 2013;79:970–986. doi: 10.1016/j.neuron.2013.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Wollenweber P, Teusner LU, Noebels JL, Herlitze S, Mark MD. Postnatal loss of P/Q-type channels confined to rhombic-lip-derived neurons alters synaptic transmission at the parallel fiber to purkinje cell synapse and replicates genomic Cacna1a mutation phenotype of ataxia and seizures in mice. J Neurosci. 2013;33:5162–5174. doi: 10.1523/JNEUROSCI.5442-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshwari A, Noebels JL. Monogenic models of absence epilepsy: windows into the complex balance between inhibition and excitation in thalamocortical microcircuits. Prog Brain Res. 2014;213:223–252. doi: 10.1016/B978-0-444-63326-2.00012-0. [DOI] [PubMed] [Google Scholar]
- Mark MD, Maejima T, Kuckelsberg D, Yoo JW, Hyde RA, Shah V, Gutierrez D, Moreno RL, Kruse W, Noebels JL, Herlitze S. Delayed postnatal loss of P/Q-type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations. J Neurosci. 2011;31:4311–4326. doi: 10.1523/JNEUROSCI.5342-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM, Lopes da Silva FH. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. 2002;22:1480–1495. doi: 10.1523/JNEUROSCI.22-04-01480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Noebels JL. The voltage-gated calcium channel and absence epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's basic mechanisms of the epilepsies. Ed 4. New York: Oxford UP; 2012. [Google Scholar]
- Noebels JL, Sidman RL. Inherited epilepsy: spike-wave and focal motor seizures in the mutant mouse tottering. Science. 1979;204:1334–1336. doi: 10.1126/science.572084. [DOI] [PubMed] [Google Scholar]
- Olsen SR, Bortone DS, Adesnik H, Scanziani M. Gain control by layer six in cortical circuits of vision. Nature. 2012;483:47–52. doi: 10.1038/nature10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 2007;27:6590–6599. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Noebels JL. Presynaptic Ca(2+) influx at a mouse central synapse with Ca(2+) channel subunit mutations. J Neurosci. 2000;20:163–170. doi: 10.1523/JNEUROSCI.20-01-00163.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Noebels JL. Presynaptic Ca2+ channels and neurotransmitter release at the terminal of a mouse cortical neuron. J Neurosci. 2001;21:3721–3728. doi: 10.1523/JNEUROSCI.21-11-03721.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol E, Kruglikov I, van den Maagdenberg AM, Rudy B, Fishell G. Ca 2.1 ablation in cortical interneurons selectively impairs fast-spiking basket cells and causes generalized seizures. Ann Neurol. 2013;74:209–222. doi: 10.1002/ana.23913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H, Okada M, Miki T, Wakamori M, Futatsugi A, Mori Y, Mikoshiba K, Suzuki N. Knockdown of Cav2.1 calcium channels is sufficient to induce neurological disorders observed in natural occurring Cacna1a mutants in mice. Biochem Biophys Res Commun. 2009;390:1029–1033. doi: 10.1016/j.bbrc.2009.10.102. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Song I, Kim D, Choi S, Sun M, Kim Y, Shin HS. Role of the alpha1G T-type calcium channel in spontaneous absence seizures in mutant mice. J Neurosci. 2004;24:5249–5257. doi: 10.1523/JNEUROSCI.5546-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka YR, Tanaka YH, Konno M, Fujiyama F, Sonomura T, Okamoto-Furuta K, Kameda H, Hioki H, Furuta T, Nakamura KC, Kaneko T. Local connections of excitatory neurons to corticothalamic neurons in the rat barrel cortex. J Neurosci. 2011;31:18223–18236. doi: 10.1523/JNEUROSCI.3139-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM. Neocortical layer 6, a review. Front Neuroanat. 2010;4:13. doi: 10.3389/fnana.2010.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic SM, Jevtovic-Todorovic V. Redox regulation of neuronal voltage-gated calcium channels. Antioxid Redox Signal. 2014;21:880–891. doi: 10.1089/ars.2013.5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda S, Kuramoto T, Tanaka K, Kaneko S, Takeuchi IK, Sasa M, Serikawa T. The ataxic groggy rat has a missense mutation in the P/Q-type voltage-gated Ca2+ channel alpha1A subunit gene and exhibits absence seizures. Brain Res. 2007;1133:168–177. doi: 10.1016/j.brainres.2006.10.086. [DOI] [PubMed] [Google Scholar]
- Tsakiridou E, Bertollini L, de Curtis M, Avanzini G, Pape HC. Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J Neurosci. 1995;15:3110–3117. doi: 10.1523/JNEUROSCI.15-04-03110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Luijtelaar G, Zobeiri M. Progress and outlooks in a genetic absence epilepsy model (WAG/Rij) Curr Med Chem. 2014;21:704–721. doi: 10.2174/0929867320666131119152913. [DOI] [PubMed] [Google Scholar]
- Vecchia D, Tottene A, van den Maagdenberg AM, Pietrobon D. Mechanism underlying unaltered cortical inhibitory synaptic transmission in contrast with enhanced excitatory transmission in CaV2.1 knockin migraine mice. Neurobiol Dis. 2014;69:225–234. doi: 10.1016/j.nbd.2014.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamori M, Yamazaki K, Matsunodaira H, Teramoto T, Tanaka I, Niidome T, Sawada K, Nishizawa Y, Sekiguchi N, Mori E, Mori Y, Imoto K. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem. 1998;273:34857–34867. doi: 10.1074/jbc.273.52.34857. [DOI] [PubMed] [Google Scholar]
- Yamawaki N, Shepherd GM. Synaptic circuit organization of motor corticothalamic neurons. J Neurosci. 2015;35:2293–2307. doi: 10.1523/JNEUROSCI.4023-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Leal K, Magupalli VG, Nanou E, Martinez GQ, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by neuronal calcium sensor-1 induces short-term synaptic facilitation. Mol Cell Neurosci. 2014;63:124–131. doi: 10.1016/j.mcn.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaylaoglu MB, Titmus A, Visel A, Alvarez-Bolado G, Thaller C, Eichele G. Comprehensive expression atlas of fibroblast growth factors and their receptors generated by a novel robotic in situ hybridization platform. Dev Dyn. 2005;234:371–386. doi: 10.1002/dvdy.20441. [DOI] [PubMed] [Google Scholar]
- Yu X, Glen D, Wang S, Dodd S, Hirano Y, Saad Z, Reynolds R, Silva AC, Koretsky AP. Direct imaging of macrovascular and microvascular contributions to BOLD fMRI in layers IV–V of the rat whisker-barrel cortex. Neuroimage. 2012;59:1451–1460. doi: 10.1016/j.neuroimage.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Renaud LP, Kolaj M. Properties of a T-type Ca2+channel-activated slow afterhyperpolarization in thalamic paraventricular nucleus and other thalamic midline neurons. J Neurophysiol. 2009;101:2741–2750. doi: 10.1152/jn.91183.2008. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Mori M, Burgess DL, Noebels JL. Mutations in high-voltage-activated calcium channel genes stimulate low-voltage-activated currents in mouse thalamic relay neurons. J Neurosci. 2002;22:6362–6371. doi: 10.1523/JNEUROSCI.22-15-06362.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Vilaythong AP, Yoshor D, Noebels JL. Elevated thalamic low-voltage-activated currents precede the onset of absence epilepsy in the SNAP25-deficient mouse mutant coloboma. J Neurosci. 2004;24:5239–5248. doi: 10.1523/JNEUROSCI.0992-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]