

Abstract
Sympathetic and parasympathetic control of the heart is a classic example of norepinephrine (NE) and acetylcholine (ACh) triggering opposing actions. Sympathetic NE increases heart rate and contractility through activation of β receptors, whereas parasympathetic ACh slows the heart through muscarinic receptors. Sympathetic neurons can undergo a developmental transition from production of NE to ACh and we provide evidence that mouse cardiac sympathetic nerves transiently produce ACh after myocardial infarction (MI). ACh levels increased in viable heart tissue 10–14 d after MI, returning to control levels at 21 d, whereas NE levels were stable. At the same time, the genes required for ACh synthesis increased in stellate ganglia, which contain most of the sympathetic neurons projecting to the heart. Immunohistochemistry 14 d after MI revealed choline acetyltransferase (ChAT) in stellate sympathetic neurons and vesicular ACh transporter immunoreactivity in tyrosine hydroxylase-positive cardiac sympathetic fibers. Finally, selective deletion of the ChAT gene from adult sympathetic neurons prevented the infarction-induced increase in cardiac ACh. Deletion of the gp130 cytokine receptor from sympathetic neurons prevented the induction of cholinergic genes after MI, suggesting that inflammatory cytokines induce the transient acquisition of a cholinergic phenotype in cardiac sympathetic neurons. Ex vivo experiments examining the effect of NE and ACh on rabbit cardiac action potential duration revealed that ACh blunted both the NE-stimulated decrease in cardiac action potential duration and increase in myocyte calcium transients. This raises the possibility that sympathetic co-release of ACh and NE may impair adaptation to high heart rates and increase arrhythmia susceptibility.
SIGNIFICANCE STATEMENT Sympathetic neurons normally make norepinephrine (NE), which increases heart rate and the contractility of cardiac myocytes. We found that, after myocardial infarction, the sympathetic neurons innervating the heart begin to make acetylcholine (ACh), which slows heart rate and decreases contractility. Several lines of evidence confirmed that the source of ACh was sympathetic nerves rather than parasympathetic nerves that are the normal source of ACh in the heart. Global application of NE with or without ACh to ex vivo hearts showed that ACh partially reversed the NE-stimulated decrease in cardiac action potential duration and increase in myocyte calcium transients. That suggests that sympathetic co-release of ACh and NE may impair adaptation to high heart rates and increase arrhythmia susceptibility.
Keywords: cholinergic, cytokine, myocardial infarction, sympathetic, transdifferentiation
Introduction
Autonomic control of the heart is one of the best characterized examples of sympathetic and parasympathetic nerves triggering opposing actions in a target organ. Cardiac sympathetic neurons release norepinephrine (NE) to increase heart rate and force of contraction via activation of β1 adrenergic receptors (β1AR) on sino-atrial node cells and cardiac myocytes. In contrast, parasympathetic nerves release acetylcholine (ACh) to slow the heart and decrease contractility via activation of M2 muscarinic ACh receptors (mAChR). The first demonstration of neural plasticity used these opposing actions of NE and ACh on myocytes to identify a transition from noradrenergic to cholinergic transmission in sympathetic neurons cultured with cardiac myocytes (Furshpan et al., 1976). Follow-up studies using biochemical methods confirmed that sympathetic neurons could be either noradrenergic or cholinergic (Reichardt and Patterson, 1977) and that leukemia inhibitory factor (LIF) was the “cholinergic differentiation factor” released from myocytes that triggered the suppression of NE synthesis and induction of ACh production in cultured sympathetic neurons (Yamamori et al., 1989). A similar transition in neurotransmitter phenotype was identified in vivo in sympathetic neurons innervating developing sweat glands (Schotzinger and Landis, 1988, 1990) and periosteum (Asmus et al., 2000).
Several classes of growth factors have now been shown to stimulate the induction of cholinergic properties in sympathetic neurons, including cytokines such as LIF and cardiotrophin-1 (CT-1), which act through the gp130 receptor (Yamamori et al., 1989; Habecker et al., 1995; Stanke et al., 2006), as well as Neurotrophin-3 (NT-3) and Glial Cell Line-Derived Neurotrophic Factor (GDNF) (Brodski et al., 2000; Brodski et al., 2002; Hiltunen and Airaksinen, 2004; Apostolova et al., 2007). Many studies implicate target-derived factors in stimulating cholinergic sympathetic differentiation (Schotzinger and Landis, 1988, 1990; Asmus et al., 2000; Hiltunen and Airaksinen, 2004; Stanke et al., 2006), but other data suggest that sympathetic neurons can acquire a cholinergic phenotype during development in the absence of target interactions (Schäfer et al., 1997; Furlan et al., 2013). In contrast to developmental studies, however, the genes and proteins involved in cholinergic transmission have not been detected in sympathetic neuron cell bodies after axotomy-induced nerve injury (Boeshore et al., 2004; Brumovsky et al., 2011; Wojtkiewicz et al., 2013). Instead, inflammatory cytokines such as LIF (Rao et al., 1993; Sun and Zigmond, 1996) suppress noradrenergic properties and induce production of neuropeptides thought to be important for regeneration (Hyatt-Sachs et al., 1996; Klimaschewski et al., 1996; Zigmond and Sun, 1997; Brumovsky et al., 2011; Hesp et al., 2012; Wojtkiewicz et al., 2013).
These data and others led to the conclusion that sympathetic acquisition of cholinergic properties was confined to development. Factors such as LIF that suppressed NE and induced ACh in immature sympathetic neurons were thought to suppress NE but only induce neuropeptides in mature sympathetic neurons. However, a recent study examining cardiac sympathetic neuron phenotype in failing hearts—in which LIF and CT-1 are increased chronically—revealed gp130-dependent expression of cholinergic markers in tyrosine hydroxylase (TH)-immunoreactive stellate ganglion neurons (Kanazawa et al., 2010). Most of the sympathetic neurons in the stellate ganglia project to the heart, so this suggests that sustained retrograde signaling by cardiac-derived cytokines induces cholinergic function in mature sympathetic neurons. In contrast to the extended time course of inflammation during heart failure, reperfusion of a blocked coronary artery during myocardial infarction (MI) leads to a large acute inflammatory response (Aoyama et al., 2000; Frangogiannis, 2012). Patients who survive an MI have a high risk of developing arrhythmias, especially during the first 30 d after MI (Solomon et al., 2005). Given the large inflammatory response after reperfusion and the opposing actions of ACh and NE on ion channels in cardiac myocytes, we investigated whether cardiac sympathetic nerves produced ACh after MI. Here, we provide evidence that adult cardiac sympathetic nerves transiently produce ACh after acute MI and that the induction of cholinergic genes requires gp130 expression in sympathetic neurons.
Materials and Methods
Mice.
WT C57BL/6J were obtained from Jackson Laboratories. gp130DBH-Cre/lox mice and inducible dopamine β hydroxylase (DBH-CreERT2) driver mice were generated as described previously (Stanke et al., 2006; Stubbusch et al., 2011) and were obtained from Dr. Hermann Rohrer (Max Planck Institute). Choline acetyltransferase (ChAT) is the enzyme that synthesizes ACh. To generate mice with adult sympathetic neurons that lack ChAT, we crossed DBH-CreERT2 mice with ChATlox/lox mice (Buffelli et al., 2003) that were obtained from Dr. Josh Sanes (Harvard University). Offspring that were homozygous for ChATlox/lox and expressed at least one copy of DBH-CreERT2 (ChATDBH-CreERT2/lox) were treated with tamoxifen (2 mg/d, i.p.) for 7 d to stimulate expression of Cre recombinase and to generate mice with noradrenergic neurons that lacked ChAT (iChAT KO). Treatment of mice for <7 d did not produce complete deletion of the ChAT gene in cardiac sympathetic neurons.
All mice were kept on a 12 h:12 h light/dark cycle with ad libitum access to food and water. Male and female mice 12–18 weeks old were used for all experiments. Animals from differing genotypes were age and sex matched for each experiment. All procedures were approved by either the Oregon Health and Science University (OHSU) or the University of California–Davis Institutional Animal Care and Use Committee and complied with the Guide for the Care and Use of Laboratory Animals published by the National Academies Press (Ed 8). The experimental groups used were sham-operated animals and animals that underwent ischemia–reperfusion surgery, with tissue collection for both the sham and MI groups occurring at identical times after surgery. A minimum of four animals was assigned to each group for each experiment and tissue was processed together for each type of analysis.
Rabbits.
Male New Zealand White rabbits were obtained from the Western Oregon Rabbit Company and kept on a 12 h:12 h light/dark cycle with ad libitum access to food and water.
Genotyping.
A two-step PCR was used to genotype both the DBH-CreERT2 and ChATlox/lox mice. For DBH-CreERT2 samples, forward (GCGTCAGAGATTTGTTGGAGGAC) and reverse (CACAGCATTGGAGTCAGAAGGG) primers were used with a 56°C annealing temperature, generating an 865 bp product. For ChATlox/lox samples, a common forward primer (GCCCTGGTCAACTCTA) was used in combination with two different reverse primers. To identify the WT allele, the reverse primer GAAATCCTGACAGATTCCAACA was used and the product was 525 bp. To identify the mutant allele, the reverse primer TTTCCGCCTCAGGACTCTTC was used and the product was 400 bp. The annealing temperature was set at 60°C for both WT and mutant samples.
Heart rate analysis.
Age-matched female WT and iChAT KO mice were intubated and anesthetized with 2% isoflurane. Heart rate was continuously monitored using subcutaneous electrodes in lead II configuration and body temperature was maintained at 37°C. Heart rate was allowed to stabilize for 10 min once body temperature reached 37°C to determine the baseline rate. Mice were then given an intraperitoneal injection of 1 mg/kg atropine (Sigma-Aldrich). Heart rate was again allowed to stabilize and 10 min of additional heart rate was recorded. Five minutes of heart rate data were averaged to determine baseline and atropine-induced heart rates.
Myocardial ischemia–reperfusion.
Ischemia–reperfusion was performed as described previously (Parrish et al., 2010; Gardner and Habecker, 2013). Adult mice were placed in an induction chamber and anesthetized with 4% isoflurane. Mice were intubated, mechanically ventilated, and maintained with 1–2% isoflurane mixed with 100% oxygen. Core body temperature was maintained at ∼37°C and a two-lead ECG was monitored throughout the surgery using a PowerLab data acquisition system (AD Instruments). A left lateral thoracotomy was performed in the fourth intercostal space and the pericardium was opened. The left anterior descending coronary artery (LAD) was reversibly ligated with an 8-0 suture for 45 min and then reperfused by release of the ligature. Occlusion was confirmed with ST segment elevation, regional cyanosis, and wall motion abnormalities. Reperfusion was confirmed by the return of color to the myocardium distal to the ligation and disappearance of ST elevation. The suture remained within the wound for the identification of the ligature site and the chest and skin were closed in layers. After surgery, animals were returned to individual cages and given regular food and water until euthanasia and tissue harvest. Buprenorphine (0.1 mg/kg) was administered as needed to ensure that animals were comfortable after surgery. All surgical procedures were performed under aseptic conditions. Sham animals underwent the procedure described above except for the LAD ligation.
HPLC analysis of NE and mass spectrometry analysis of ACh.
NE levels were measured by HPLC with electrochemical detection as described previously (Parrish et al., 2010) and ACh was quantified by mass spectrometry in the OHSU Bioanalytical Core facility as described previously (Hasan et al., 2012). Hearts were excised and cut in 2 mm transverse cross sections and the tissue below the site of LAD occlusion was separated into left and right ventricles (LV, RV). The LV was further dissected under a microscope into scar and viable (peri-infarct) tissue before freezing and storage at −80°C. Sham tissue underwent similar processing so that any ACh degradation by acetylcholinesterase during the dissection was comparable. Tissue samples from each heart were homogenized and neurotransmitters extracted at room temperature with 300 μl of perchloric acid (0.1 m) containing a 1.0 μm concentration of the internal standard dihydroxybenzylamine to correct for NE sample recovery. Catecholamines were purified from 100 μl of the supernatant by alumina extraction before analysis by HPLC. Detection limits were ∼0.05 pmol with recoveries from the alumina extraction >60%. ACh was quantified in a second aliquot of 100 μl that was filtered at 4°C before analysis on the mass spectrometer.
Immunohistochemistry.
Hearts were fixed for 1 h and stellate ganglia for 15 min in 4% paraformaldehyde. Tissue was rinsed in PBS (5 × 5 min), cryoprotected in 30% sucrose overnight, and 10 μm transverse sections were thaw mounted onto charged slides. To reduce fixative-induced autofluorescence, heart sections were rinsed in 10 mg/ml sodium borohydride in PBS 3 × 10 min and then rinsed in PBS for 10 min. Sections were then blocked in 1% BSA/0.3% Triton X-100 in PBS for 1.5 h, incubated with rabbit anti-TH (1:300; Millipore), sheep anti-TH (1:300; Serotec), rabbit anti-ChAT (1:150; Proteintech), or goat anti- vesicular ACh transporter (anti-VAChT, 1:50; Promega) overnight, rinsed 3 × 10 min in PBS, and incubated 1.5 h with donkey anti-rabbit, donkey anti-sheep, or donkey anti-goat Alexa Fluor secondary antibodies (1:500; Invitrogen). Sections were rinsed 3 × 10 min in PBS. To reduce lipofuscin-induced autofluorescence, heart sections were treated with 10 mm CuSO4 in 50 mm NH4C2H3O2 for 30 min. Slides were then dipped briefly in mqH2O and placed in PBS, coverslipped, and visualized by fluorescence microscopy. Sections from sham and MI animals were stained and photographed side by side to minimize variation between the groups due to the immunohistochemistry procedure.
Real-time PCR.
Stellate ganglia, which contain the majority of the sympathetic neurons that project to the heart, were harvested 7, 10, 14, or 21 d after sham or ischemia–reperfusion surgery and stored immediately in RNAlater. RNA was isolated from individual stellate ganglia using the Ambion RNAqueous micro kit. Total RNA was quantified by OD260 and then 200 ng of total RNA was reverse transcribed and diluted for use. Real-time PCR was performed with ABI TaqMan Universal PCR master mix in the ABI 7500. Samples were assayed using ABI prevalidated TaqMan gene expression assays for mouse genes encoding the nuclear protein Satb2, with GAPDH as a normalization control, and the proteins required for cholinergic transmission: ChAT, the enzyme that synthesizes ACh; the high-affinity choline transporter (CHT), which transports choline into the cell and is the rate-limiting step in ACh synthesis, and VAChT, which packages ACh into vesicles for release. For the PCR amplification, 2–4 μl of RT reactions (representing 5 ng of RNA template) were used in a total volume of 20 μl and each sample was assayed in duplicate. Standard curves were generated with known amounts of RNA from brain, ranging from 0.8 to 100 ng. Values for ChAT, VAChT, CHT, and Satb2 were normalized to GAPDH from the same sample. Sham controls from different time points were identical and were combined into a single sham group.
Dual optical mapping of transmembrane potential and intracellular Ca2+.
Langendorff perfusion of isolated rabbit hearts and optical mapping of transmembrane potential (Vm) and Ca2+ was performed as described previously (Myles et al., 2012). Briefly, male New Zealand White rabbits (n = 3) were anesthetized with an intravenous injection of pentobarbital sodium (50 mg/kg) containing 1000 IU of heparin. After a midsternal incision, hearts were rapidly excised and perfused at 37°C with oxygenated (95% O2, 5% CO2) modified Tyrode's solution containing the following (in mmol/L): NaCl 128.2, CaCl2 1.3, KCl 4.7, MgCl2 1.05, NaH2PO4 1.19, NaHCO3 20, and glucose 11.1, pH 7.4 ± 0.05. The flow rate (25–35 ml/min) was adjusted to maintain a perfusion pressure of 60–70 mmHg. Two Ag/AgCl disc electrodes were positioned in the bath to record an ECG analogous to a lead I configuration. Bipolar pacing electrodes were positioned on the base of the LV epicardium for pacing, which was performed at a cycle length (CL), or pacing interval, of 300 ms using a 2 ms pulse at twice the diastolic threshold. Blebbistatin (Tocris Bioscience; 10–20 μm) was added to the perfusate to eliminate motion artifact during optical recordings. Hearts were loaded with the acetoxymethyl ester form of the fluorescent intracellular Ca2+ indicator Rhod-2 (Rhod-2 AM; Invitrogen; 0.5 ml of 1 mg/ml in DMSO containing 10% pluronic acid) and subsequently stained with the voltage-sensitive dye RH237 (Invitrogen; 50 μl of 1 mg/ml in DMSO) via the coronary perfusion. The anterior epicardial surface was excited using LED light sources centered at 531 nm and band-pass filtered from 511 to 551 nm (LEX-2; SciMedia). The emitted fluorescence was collected through a THT-macroscope (SciMedia) and split with a dichroic mirror at 630 nm. The longer wavelength moiety, containing the Vm signal, was long-pass filtered at 700 nm and the shorter wavelength moiety, containing the Ca2+ signal, was band-pass filtered with a 32 nm filter centered at 590 nm. The emitted fluorescence signals were then recorded using two CMOS cameras (MiCam Ultima-L; SciMedia) with a sampling rate of 1 kHz and 100 × 100 pixels with a 31 × 31 mm field of view.
Baseline electrophysiological parameters were determined during epicardial pacing with a 300 ms CL (equivalent to 200 bpm heart rate). To measure how the action potential duration (APD) and intracellular Ca2+ release adapt to sudden changes in heart rate, an APD restitution curve and S2/S1 ratio of Ca2+ transient (CaT) amplitude were constructed by a standard S1–S2 pacing protocol in which 12 pacing stimuli were delivered at a 300 ms interval (S1) followed by a premature S2 stimulus. The S2 interval was gradually shortened until the effective refractory period was reached and the S2 stimulus failed to propagate an action potential (AP). Adrenergic effects on APD restitution and the refractoriness of Ca2+ release were investigated using 1.5 μm NE in the perfusate. Combined adrenergic and cholinergic effects were investigated by adding 1.5 μm NE and 1.5 μm ACh to the perfusate. The sino-atrial node was removed to allow for pacing. Data analysis was performed using a commercially available analysis program as described previously (Myles et al., 2012). AP activation time was determined as the time at 50% between peak and baseline amplitude. APD at 90% repolarization (APD90) was calculated as repolarization time − activation time from data collected during pacing at various S2s. APD restitution curves were constructed by plotting APD90 versus S2 coupling length. The ratio of S2 and S1 CaT amplitude was used to assess the refractoriness of Ca2+ release.
Statistics.
Student's t test was used for a single comparison between two groups. One-way ANOVA with a Dunnett's multiple-comparison post test was used to compare surgical groups with the sham control group from the same genotype. Two-way ANOVA with a Bonferroni post test was used to compare across two genotypes and treatment groups. The slopes of APD restitution curves were compared by linear curve fit and CaT curves were analyzed using nonlinear curve fit of the S2/S1 ratio using one-phase association. Statistical analyses were performed using Prism version 5.0 (GraphPad Software).
Results
To determine whether acute MI induced cholinergic function in cardiac sympathetic neurons, we first quantified NE and ACh content in the heart 10, 14, and 21 d after sham surgery or MI. Our previous studies revealed a sustained loss of nerves in the cardiac scar 10 d after MI, with normal innervation density and NE content in the viable left ventricle surrounding the scar (Gardner and Habecker, 2013). Therefore, we dissected scar tissue away from viable “peri-infarct” tissue and quantified NE and ACh in both regions from each heart. Levels of NE and ACh were identical among shams obtained 10, 14, and 21 d after surgery, so the shams were combined into a single group. NE content was decreased in the scar compared with sham (Fig. 1A), consistent with the loss of sympathetic nerve fibers, whereas ACh content remained unchanged (Fig. 1D). The innervated tissue outside of the scar exhibited NE levels similar to shams (Fig. 1B), whereas ACh levels were increased significantly 10 and 14 d after MI, returning to sham levels 21 d after MI.
Figure 1.

Differential changes in cardiac NE and ACh content after MI. NE and ACh were quantified in scar tissue (A, C) and viable peri-infarct myocardium (B, D). NE content in scar tissue (A) decreased compared with sham tissue 10, 14, and 21 d after MI, whereas NE in viable peri-infarct myocardium did not (B). ACh content in the same scar tissue (C) did not change after MI, but ACh content in viable peri-infarct tissue increased significantly 10 and 14 d after MI, returning to sham levels 21 d post-MI. Data are shown as means ± SEM; n = 4–6 except sham n = 9; **p < 0.01, ***p < 0.001 compared with sham.
The rise in ACh within the left ventricle is consistent with the induction of cholinergic function in sympathetic neurons, but could also be due to changes in the rare cholinergic parasympathetic nerves in the ventricle. To determine whether the increase in cardiac ACh was due to sympathetic nerves, we quantified expression of the genes required for cholinergic function in the stellate ganglia, which contain most of the sympathetic neurons projecting to the heart. Ganglia were collected 10, 14, and 21 d after MI from the same mice used for cardiac NE and ACh analysis in Figure 1. To gain additional insight into the time course of ACh induction, we also examined stellate ganglia that had been collected 7 d after MI for another study. The genes required for cholinergic transmission, the synthetic enzyme ChAT, the rate-limiting CHT, and VAChT, were present a similar levels in shams from each time point, so the shams were combined into a single group. ChAT and CHT mRNA levels were increased significantly 7, 10, and 14 d after MI (Fig. 2A,B), whereas VAChT mRNA was increased significantly 10 and 14 d after MI (Fig. 2C).
Figure 2.
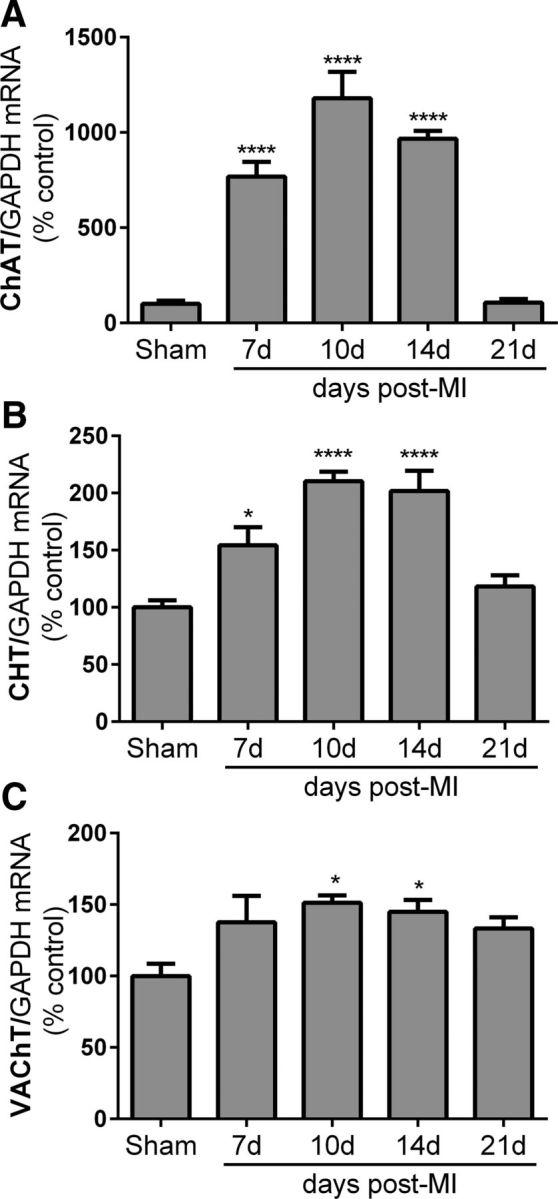
MI induces expression of cholinergic genes in cardiac sympathetic neurons. Cholinergic genes were quantified in stellate ganglia 7, 10, 14, and 21 d after MI and normalized to the associated sham controls. mRNA encoding ChAT (A), CHT (B), and VAChT (C) were increased significantly after MI. Data are shown as means ± SEM; n = 4–5 except sham n = 6; *p < 0.05, ****p < 0.0001 compared with sham.
To confirm that the increased expression of cholinergic genes in sympathetic neurons led to production of the associated proteins, immunohistochemistry was performed in stellate ganglia and heart sections 14 d after MI, when cholinergic genes were elevated and ACh levels were high in the heart. Double-label immunohistochemistry confirmed coexpression of the ACh synthetic enzyme ChAT with TH, the rate-limiting enzyme in NE synthesis, in sympathetic neuron cell bodies (Fig. 3). In contrast to the TH+/ChAT+ neurons identified 14 d after MI, sham ganglia contained predominantly TH+/ChAT− sympathetic neurons surrounded by TH−/ChAT+ preganglionic fibers. Heart sections were double labeled for TH and VAChT and, consistent with the presence of ChAT in sympathetic neuron cell bodies, TH+/VAChT+ fibers were identified in heart sections 14 d after MI. In contrast, sham hearts contained only TH+/VAChT− sympathetic fibers and rare TH−/VAChT+ parasympathetic fibers.
Figure 3.
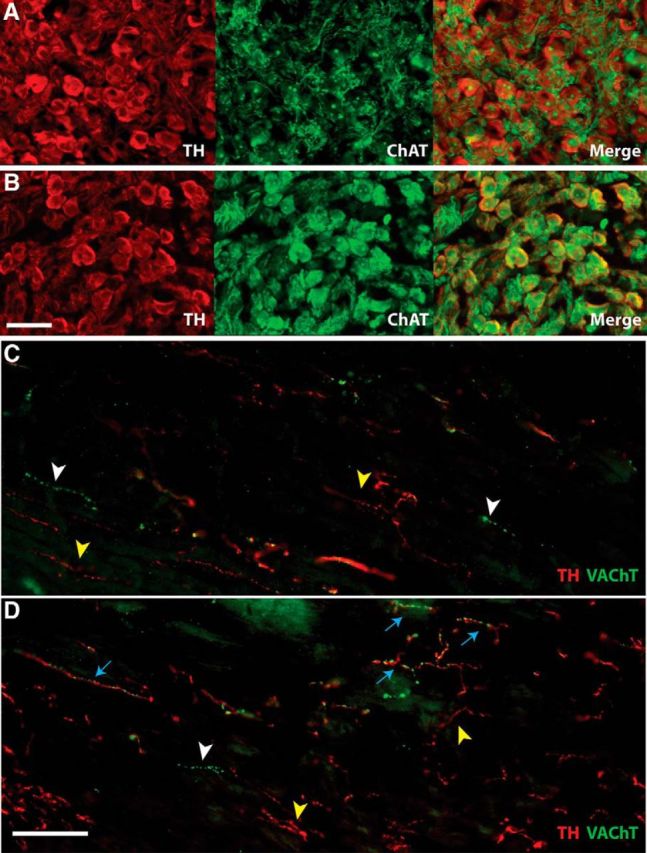
A, B, Stellate ganglion sections 14 d after sham (A) or MI (B) stained for TH (red) to identify noradrenergic neurons and ChAT (green) to identify cholinergic neurons. Scale bar, 50 μm. In sham ganglia, only preganglionic fibers are positive for ChAT, whereas sympathetic cell bodies are TH+. After MI, however, TH+ sympathetic neurons also express ChAT. C, D, Heart sections after sham (C) or MI (D) stained for TH (red) and VAChT (green). Scale bar, 100 μm. Yellow arrowheads identify TH+/VAChT− sympathetic fibers; white arrowheads identify TH−/VAChT+ parasympathetic fibers; blue arrows identify fibers positive for both noradrenergic and cholinergic markers, which were observed only after MI. Note that parasympathetic fibers are sparse in the left ventricle whereas sympathetic fibers are abundant.
Finally, to confirm that the rise in cardiac ACh observed after MI was due solely to increased ACh production in sympathetic neurons and not to changes in parasympathetic transmission, we generated mice in which the ChAT gene was deleted in adult sympathetic neurons. Adult mice homozygous for a ChATlox/lox allelle (Buffelli et al., 2003) and expressing an inducible Cre recombinase driven by the dopamine β hydroxylase promoter (DBHCre-ERT2) (Stubbusch et al., 2011) were treated for 7 d with tamoxifen to drive deletion of the ChAT gene in noradrenergic neurons (iChAT KO). ChAT mRNA was near the limit of detection in WT sham ganglia, making it difficult to confirm deletion of the ChAT gene in stellates from unoperated iChAT KO mice. Therefore, we performed MI surgery to induce cholinergic genes in tamoxifen-treated mice and collected hearts and stellate ganglia 14 d after MI. Cholinergic gene expression and neurotransmitter levels were quantified and compared with the values obtained previously for WT mice 14 d after MI (Figs. 1, 2). Real-time PCR confirmed deletion of the ChAT gene in cardiac sympathetic neurons. ChAT mRNA levels in post-MI iChAT KO mice treated with tamoxifen were similar to the low levels present in WT sham mice, and significantly lower than ChAT mRNA in WT mice 14 d after MI, denoted by the dashed line in Figure 4A. In contrast, the other cholinergic genes were expressed at high levels similar to those observed in WT post-MI mice rather than sham mice. Once deletion of ChAT mRNA was confirmed in stellate ganglia, neurotransmitter content was quantified in viable peri-infarct myocardium from the same mice. Deletion of ChAT from sympathetic neurons abolished the post-MI increase in cardiac ACh content, resulting in ACh levels significantly lower than WT post-MI hearts (Fig. 4B) and similar to those in sham hearts (sham 0.127 ± 0.03 pmol/mg, n = 9; iChAT KO 0.065 ± 0.02 pmol/mg, n = 4). In contrast, NE levels, which should not be affected by deletion of ChAT in sympathetic neurons, were identical to values obtained earlier from WT mice 14 d after MI (Fig. 4B). This suggests that the increased ACh observed in the left ventricle after MI was due to production of ACh by sympathetic nerves rather than other sources.
Figure 4.
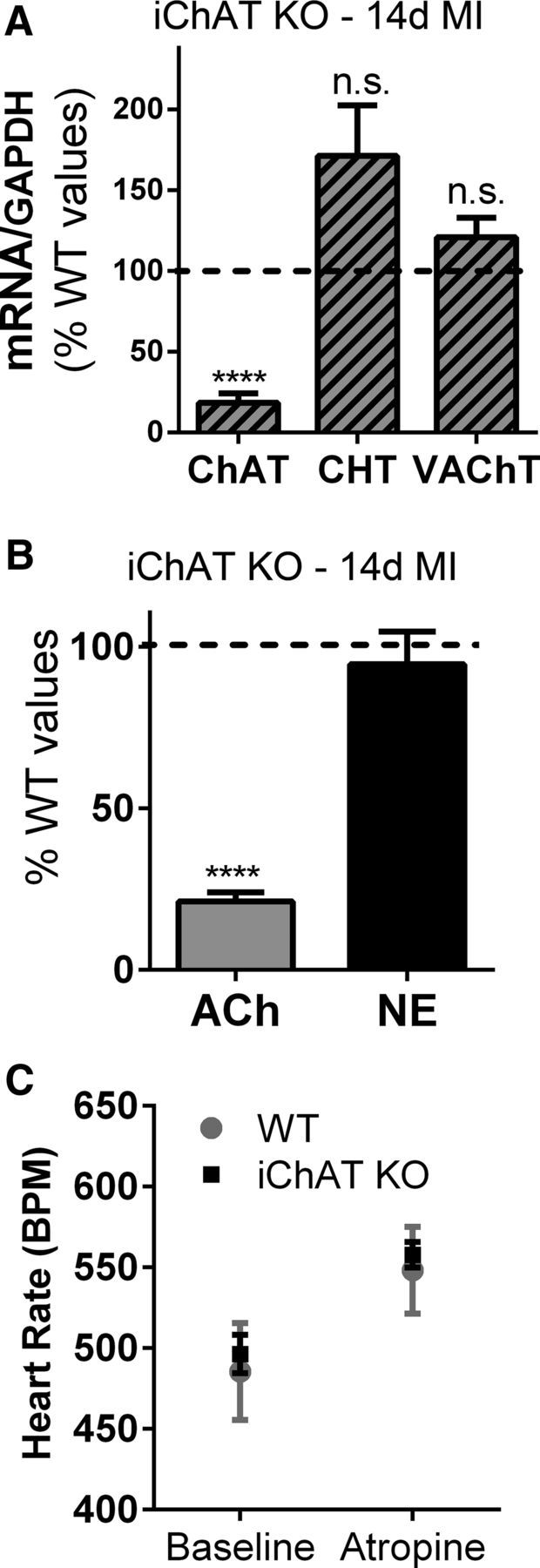
Deletion of ChAT in adult sympathetic neurons prevents the increase in left ventricle ACh after MI. A, Expression of cholinergic genes. ChAT, CHT, and VAChT mRNA were quantified 14 d after MI in ChATDBHCreERT2/lox mice that had been treated with tamoxifen for 7 d (iChAT KO) and normalized to GAPDH mRNA in the same samples. Data are graphed as a percentage of control using the levels of each gene previously observed in WT mice 14 d post-MI as the control values (dotted black line). ChAT mRNA was present at levels similar to sham controls and was significantly lower than WT post-MI mice, whereas CHT and VAChT mRNA levels were not significantly different from WT mice after MI. B, Ventricular ACh and NE. The loss of the ChAT gene in sympathetic neurons resulted in ACh levels 14 d after MI that were similar to sham hearts and significantly lower than the post-MI levels previously identified in WT mice (dotted black line). NE levels were unchanged compared with WT mice. Data are shown as means ± SEM; n = 4; ****p < 0.0001 compared with WT 14 d post-MI. C, Parasympathetic control of heart rate. Heart rate was quantified in unoperated WT and iChAT KO mice before and after atropine injection (mean ± SEM, n = 3).
Constitutive DBH-Cre driver lines can cause recombination in cardiac parasympathetic neurons due to transient expression of DBH in neural crest precursor cells (Parrish et al., 2009). Although this should not occur in the DBH-CreERT2 mice used for this study (Stubbusch et al., 2011), we confirmed retention of ChAT expression and ACh production in parasympathetic neurons by examining parasympathetic control of heart rate in unoperated WT and iChAT KO mice. Basal heart rate was the same in both genotypes and blockade of cholinergic transmission with atropine generated an identical increase in heart rate (Fig. 4C), confirming intact parasympathetic transmission in iChAT KO hearts.
Several different classes of growth factors can induce cholinergic function in sympathetic neurons, including cytokines that act via gp130 signaling, NT-3, and GDNF (Yamamori et al., 1989; Brodski et al., 2000; Brodski et al., 2002; Stanke et al., 2006). NT-3 expression is unchanged in the heart after ischemia–reperfusion (Hiltunen et al., 2001), whereas GDNF levels are low in adult heart (Miwa et al., 2010) and have not been examined after injury. In contrast, gp130 cytokines such as LIF and CT-1 are elevated in the left ventricle after MI (Aoyama et al., 2000; Gritman et al., 2006), where they affect the remodeling of cardiac myocytes (Haghikia et al., 2011; Oba et al., 2012) and trigger the local loss of TH from sympathetic nerves via proteasomal degradation (Parrish et al., 2010; Shi and Habecker, 2012). We hypothesized that cytokine activation of gp130 was responsible for the induction of cholinergic function in cardiac sympathetic neurons after MI. To test that hypothesis, we quantified expression of the genes associated with cholinergic transmission in stellate ganglia from WT mice and mice with noradrenergic neurons that lack gp130 (gp130 KO). Cytokine stimulation of cholinergic function in sympathetic neurons requires the nuclear matrix protein Satb2, which is not involved in the induction of cholinergic genes by other growth factors (Apostolova et al., 2010). Therefore, we also quantified Satb2 mRNA in WT and gp130 KO ganglia. Three days after MI, cholinergic genes were not increased compared with shams (data not shown), but 7 d after MI, the mRNAs encoding ChAT, CHT, VAChT, and Satb2 were all increased significantly in WT neurons, but not in neurons lacking gp130 (Fig. 5). These data suggest that inflammatory cytokines stimulate ACh production in cardiac sympathetic neurons after acute MI, just as they do in nonischemic heart failure (Kanazawa et al., 2010). In addition, basal levels of CHT and ChAT mRNA were significantly lower in the gp130 KO stellates than in the WT stellates.
Figure 5.

Neuronal gp130 is required for cholinergic gene expression after MI. ChAT (A), CHT (B), VAChT (C), and Satb2 (D) mRNA were quantified in WT and neuronal gp130 KO (hashed bars) stellate ganglia 7 d after sham surgery or MI. Data are shown as means ± SEM; n = 4 except WT 7 d MI n = 3; *p < 0.05, **p < 0.01, ***p < 0.001 compared with sham of the same genotype; #p < 0.05 compared with sham WT (two-way ANOVA).
To understand the functional consequences of cholinergic sympathetic transmission in the heart, we first investigated the effect of exogenous neurotransmitters in a normal heart. The transmural (epicardial to endocardial) gradient in APD is critical for normal activation and repolarization of the left ventricle (Costantini et al., 2005) and the gradient in APD is matched by a transmural gradient of sympathetic innervation. Disrupting the innervation gradient in a normal heart is arrhythmogenic (Ieda et al., 2007; Lorentz et al., 2010), highlighting the importance of neuronal regulation of cardiac repolarization. To elucidate the impact of NE and ACh, we focused on regulation of APD and calcium release using a standard S1–S2 pacing protocol. Control hearts were paced under baseline conditions and then adrenergic effects on APD restitution and CaTs were investigated using 1.5 μm NE in the absence or presence of 1.5 μm ACh. Global application of NE decreased APD across all pacing intervals, consistent with allowing the ventricle to adapt to the higher heart rates present during sympathetic stimulation. The addition of ACh together with NE resulted in APDs that were somewhat longer (Fig. 6A,C), which would likely make it harder for the heart to adapt to higher heart rates and may increase the likelihood of arrhythmia. Application of NE also increased CaT amplitude after a premature stimulus (Fig. 6B,D), whereas coapplication of ACh with NE reduced the CaT amplitude back toward baseline levels, suggesting that cardiac contractility would be blunted with co-release of ACh and NE.
Figure 6.

Functional effects of NE and ACh co-release. A, C, Application of NE (1.5 μm) alone dramatically shortens APD90 at all premature (S2) pacing intervals to allow the heart to quickly adapt to high heart rates during sympathetic activity. Coapplication of ACh (1.5 μm) does not allow the APD90 to shorten to the same extent, which may lead to arrhythmia. B, D, Application of NE alone also increased the size of CaTs after premature stimuli (increased S2/S1 ratio), whereas coapplication of ACh reduced the effect of NE (B, D). The S2/S1 ratio was calculated as the S2 CaT amplitude divided by S1 CaT amplitude (D). Data are shown as means ± SEM; n = 3.
Discussion
Our data indicate that cardiac sympathetic neurons transiently produce ACh together with NE in the weeks after MI. Several lines of evidence suggest that the increase in ACh content observed in viable heart tissue after MI is due to the induction of ACh in sympathetic nerves rather than increased production by parasympathetic nerves. First, there are few parasympathetic fibers detectable in the mouse left ventricle, in contrast to robust innervation to the SA node and cardiac conduction system (Mabe et al., 2006), and functional studies indicate that stimulating cardiac parasympathetic nerves has little impact on contractility of the left ventricle (Matsuura et al., 1997; Takahashi et al., 2003; Brack et al., 2010). Second, the transient increase in cardiac ACh content coincided with increased expression of the genes required for ACh production in the stellate ganglion and the appearance of ChAT and VAChT protein in TH+ cardiac sympathetic neurons. Third, selective deletion of the ChAT gene in adult sympathetic neurons prevented the increase in cardiac ACh content after MI. These data are consistent with ACh production in cardiac sympathetic nerves after MI, which is interesting because cholinergic properties have also been observed in adult sympathetic neurons during heart failure (Kanazawa et al., 2010), but have not been seen in other injury paradigms, including axotomy (Boeshore et al., 2004; Wojtkiewicz et al., 2013) and inflammatory colitis (Skobowiat et al., 2010).
The large fraction of neurons within the stellate ganglion projecting to the heart facilitate use of this model to track changes in neurotransmitter phenotype. Approximately 92% of rat stellate neurons project to the heart (Pardini et al., 1989), so mRNA isolated from rodent stellate ganglia is primarily from cardiac neurons. Therefore, it is not surprising that we were able to detect increases in cholinergic genes within the stellate after MI and widespread coexpression of ChAT protein in TH+ neurons. Most of these cholinergic sympathetic neurons project to the heart, consistent with the increase in ACh content in the heart and identification of VAChT in TH+ axons within the heart. However, some of the noncardiac neurons in stellate ganglia project to sweat glands in the front paws, which also have a TH+/ChAT+ phenotype in the mouse (Guidry and Landis, 1995, 1998). These neurons were relatively rare and were also observed in sham ganglia (data not shown).
The acquisition of cholinergic function required expression of the gp130 cytokine receptor in sympathetic neurons, suggesting that retrograde signaling by target-derived cytokines induced the acquisition of a cholinergic neurotransmitter phenotype in adult sympathetic neurons. This was further supported by the HPLC analysis Satb2 mRNA within the stellate ganglia because Satb2 mediates cytokine stimulation of cholinergic genes in sympathetic neurons, but is not involved in the induction of cholinergic genes by other factors (Apostolova et al., 2010). Previous studies have shown that removal of gp130 from sympathetic neurons also prevents the local suppression of noradrenergic transmission in the left ventricle after acute MI (Parrish et al., 2010) and induction of cholinergic transmission in heart failure (Kanazawa et al., 2010). Together, these data suggest that retrograde signaling by gp130 in adult neurons can alter sympathetic neurotransmitter phenotype in a manner similar to the gp130-dependent noradrenergic to cholinergic conversion of sympathetic neurons innervating developing sweat glands (Stanke et al., 2006).
A technical advance of our study was the development of a method to quantify cholinergic and noradrenergic properties simultaneously by measuring NE and ACh within the same heart tissue. Sensitive HPLC assays for NE and other catecholamines have been available for many years, but it was not possible to quantify ACh directly until the recent development of a sensitive mass spectrometry assay. Therefore, cholinergic function in the past was assayed indirectly by measuring ChAT activity in tissue homogenates (Schotzinger and Landis, 1988) or by the lack of a potassium permanganate precipitate in EM analysis of vesicles (Landis, 1976; Kanazawa et al., 2010). The advent of a sensitive assay for ACh has allowed us to quantify both NE and ACh in the same tissue and thus track relative changes over time. Our earlier studies of the post-MI ventricle-quantified NE content in the entire left ventricle (Li et al., 2004; Parrish et al., 2010; Lorentz et al., 2011), but newer data indicate that the scar remains denervated for an extended period of time after ischemia–reperfusion (Gardner and Habecker, 2013). Therefore, we dissected scar tissue away from the viable innervated left ventricle before analysis of NE and ACh for these studies. One consequence was a delay in freezing the tissue, which resulted in a small loss of ACh due to metabolism by acetylcholinesterase. This was not the case for NE, which was more stable. To control for the degradation of ACh, tissue from sham hearts was processed in the same way as infarcted hearts so that any loss of ACh would be comparable across all groups. Therefore, we are confident that the relative changes in ACh content reported here are accurate, but the exact levels of ACh reported may underestimate the amount present in vivo.
The functional consequences of sympathetic cholinergic transmission in the heart are not yet understood. Many functional studies have been performed in mice with noradrenergic neurons that lack gp130 (Parrish et al., 2008; Parrish et al., 2010; Kanazawa et al., 2010), but removal of gp130 from all DBH-expressing cells does not simply prevent the acquisition of cholinergic function in sympathetic neurons innervating the heart. Constitutive noradrenergic gp130 knock-out mice have autonomic imbalance due to disrupted central autonomic control (Parrish et al., 2009), elevated ACh content in cardiac parasympathetic nerves (Hasan et al., 2012), altered neuropeptide expression after MI (Alston et al., 2011), and impaired axon regeneration (Pellegrino and Habecker, 2013). Importantly, the promiscuous expression of DBH in neural crest cells results in the deletion of target genes in cardiac parasympathetic neurons as well as sympathetic neurons (Parrish et al., 2009), necessitating the change to the inducible DBH-CreERT2 that does not generate recombination in parasympathetic neurons (Stubbusch et al., 2011). Therefore, elucidating the role of sympathetic cholinergic transmission after MI will require detailed functional studies in iChAT KO mice, which have not yet been characterized.
As a first test to elucidate the impact of combined noradrenergic and cholinergic transmission in the ventricle, we examined the effect of global NE and ACh on cardiac APD and calcium transients. Activating cardiac β receptors with exogenous NE decreased APD and flattened the slope of the APD restitution curve, consistent with previous studies (Hartzell, 1988). The shallower slope of the APD restitution curve at short cycle lengths is suggestive of decreased arrhythmia propensity (Nash et al., 2006; Selvaraj et al., 2007) and allows the heart to function properly at high heart rates. Likewise, NE increased the size of myocyte CaTs after a premature stimuli, which normally produce small transients due to refractory ryanodine receptors and low calcium stores within the sarcoplasmic reticulum (SR). NE activation of β receptors stimulates faster recovery of ryanodine receptors and enhanced filling of SR calcium stores by the SR calcium transport ATPase, resulting in larger CaTs and increased cardiac contractility even at high heart rates. Coadministration of ACh with NE blunted the NE-stimulated increase in CaT and increased APD, suggesting decreased contractility and impaired adaptation to fast heart rates when ACh is present together with NE. This contrasts with the normal situation of ACh release only from parasympathetic nerves, which strongly decreases heart rate and has limited direct effect on ventricular APD (Matsuura et al., 1997; Brack et al., 2010).
Together, our data indicate that cardiac sympathetic nerves produce ACh in addition to NE after MI. The production of ACh is transient and corresponds to a period of cardiac remodeling that is characterized by high levels of inflammatory cytokines and macrophages within the heart and includes a period of particularly high arrhythmia risk. The induction of cholinergic genes requires expression of the gp130 cytokine receptor in sympathetic neurons, suggesting that retrograde signaling by heart-derived cytokines stimulates the acquisition of the cholinergic phenotype. The functional consequences of cholinergic sympathetic transmission remain unclear, but our data suggest that release of ACh together with NE from sympathetic nerves may impair adaptation to fast heart rates and increase arrhythmia propensity.
Footnotes
This work was supported by the National Institutes of Health (Grant HL068231 to B.A.H., Grant HL111600 to C.M.R., and Grant T32HL094294 to R.T.G.). We thank Joshua Yuan, Michelle Stamm, Cassandra Dunbar, Richard Bayles, and the OHSU Bioanalytical Shared Resource for technical assistance; and Drs. Hermann Rohrer and Joshua Sanes for transgenic mice.
The authors declare no competing financial interests.
References
- Alston EN, Parrish DC, Hasan W, Tharp K, Pahlmeyer L, Habecker BA. Cardiac ischemia–reperfusion regulates sympathetic neuropeptide expression through gp130-dependent and independent mechanisms. Neuropeptides. 2011;45:33–42. doi: 10.1016/j.npep.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama T, Takimoto Y, Pennica D, Inoue R, Shinoda E, Hattori R, Yui Y, Sasayama S. Augmented expression of cardiotrophin-1 and its receptor component, gp130, in both left and right ventricles after myocardial infarction in the rat. J Mol Cell Cardiol. 2000;32:1821–1830. doi: 10.1006/jmcc.2000.1218. [DOI] [PubMed] [Google Scholar]
- Apostolova G, Dorn R, Ka S, Hallböök F, Lundeberg J, Liser K, Hakim V, Brodski C, Michaelidis TM, Dechant G. Neurotransmitter phenotype-specific expression changes in developing sympathetic neurons. Mol Cell Neurosci. 2007;35:397–408. doi: 10.1016/j.mcn.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Apostolova G, Loy B, Dorn R, Dechant G. The sympathetic neurotransmitter switch depends on the nuclear matrix protein Satb2. J Neurosci. 2010;30:16356–16364. doi: 10.1523/JNEUROSCI.3502-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmus SE, Parsons S, Landis SC. Developmental changes in the transmitter properties of sympathetic neurons that innervate the periosteum. J Neurosci. 2000;20:1495–1504. doi: 10.1523/JNEUROSCI.20-04-01495.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeshore KL, Schreiber RC, Vaccariello SA, Sachs HH, Salazar R, Lee J, Ratan RR, Leahy P, Zigmond RE. Novel changes in gene expression following axotomy of a sympathetic ganglion: a microarray analysis. J Neurobiol. 2004;59:216–235. doi: 10.1002/neu.10308. [DOI] [PubMed] [Google Scholar]
- Brack KE, Coote JH, Ng GA. Vagus nerve stimulation inhibits the increase in Ca2+ transient and left ventricular force caused by sympathetic nerve stimulation but has no direct effects alone–epicardial Ca2+ fluorescence studies using fura-2 AM in the isolated innervated beating rabbit heart. Exp Physiol. 2010;95:80–92. doi: 10.1113/expphysiol.2009.048215. [DOI] [PubMed] [Google Scholar]
- Brodski C, Schnürch H, Dechant G. Neurotrophin-3 promotes the cholinergic differentiation of sympathetic neurons. Proc Natl Acad Sci U S A. 2000;97:9683–9688. doi: 10.1073/pnas.160080697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodski C, Schaubmar A, Dechant G. Opposing functions of GDNF and NGF in the development of cholinergic and noradrenergic sympathetic neurons. Mol Cell Neurosci. 2002;19:528–538. doi: 10.1006/mcne.2001.1093. [DOI] [PubMed] [Google Scholar]
- Brumovsky PR, Seroogy KB, Lundgren KH, Watanabe M, Hökfelt T, Gebhart GF. Some lumbar sympathetic neurons develop a glutamatergic phenotype after peripheral axotomy with a note on VGLUT(2)-positive perineuronal baskets. Exp Neurol. 2011;230:258–272. doi: 10.1016/j.expneurol.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffelli M, Burgess RW, Feng G, Lobe CG, Lichtman JW, Sanes JR. Genetic evidence that relative synaptic efficacy biases the outcome of synaptic competition. Nature. 2003;424:430–434. doi: 10.1038/nature01844. [DOI] [PubMed] [Google Scholar]
- Costantini DL, Arruda EP, Agarwal P, Kim KH, Zhu Y, Zhu W, Lebel M, Cheng CW, Park CY, Pierce SA, Guerchicoff A, Pollevick GD, Chan TY, Kabir MG, Cheng SH, Husain M, Antzelevitch C, Srivastava D, Gross GJ, Hui CC, et al. The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell. 2005;123:347–358. doi: 10.1016/j.cell.2005.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan A, Lübke M, Adameyko I, Lallemend F, Ernfors P. The transcription factor Hmx1 and growth factor receptor activities control sympathetic neurons diversification. EMBO J. 2013;32:1613–1625. doi: 10.1038/emboj.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furshpan EJ, MacLeish PR, O'Lague PH, Potter DD. Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: evidence for cholinergic, adrenergic, and dual-function neurons. Proc Natl Acad Sci U S A. 1976;73:4225–4229. doi: 10.1073/pnas.73.11.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RT, Habecker BA. Infarct-derived chondroitin sulfate proteoglycans prevent sympathetic reinnervation after cardiac ischemia–reperfusion injury. J Neurosci. 2013;33:7175–7183. doi: 10.1523/JNEUROSCI.5866-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritman K, Van Winkle DM, Lorentz CU, Pennica D, Habecker BA. The lack of cardiotrophin-1 alters expression of interleukin-6 and leukemia inhibitory factor mRNA but does not impair cardiac injury response. Cytokine. 2006;36:9–16. doi: 10.1016/j.cyto.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidry G, Landis SC. Sympathetic axons pathfind successfully in the absence of target. J Neurosci. 1995;15:7565–7574. doi: 10.1523/JNEUROSCI.15-11-07565.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidry G, Landis SC. Target-dependent development of the vesicular acetylcholine transporter in rodent sweat gland innervation. Dev Biol. 1998;199:175–184. doi: 10.1006/dbio.1998.8929. [DOI] [PubMed] [Google Scholar]
- Habecker BA, Pennica D, Landis SC. Cardiotrophin-1 is not the sweat gland-derived differentiation factor. Neuroreport. 1995;7:41–44. doi: 10.1097/00001756-199512000-00009. [DOI] [PubMed] [Google Scholar]
- Haghikia A, Stapel B, Hoch M, Hilfiker-Kleiner D. STAT3 and cardiac remodeling. Heart Fail Rev. 2011;16:35–47. doi: 10.1007/s10741-010-9170-x. [DOI] [PubMed] [Google Scholar]
- Hartzell HC. Regulation of cardiac ion channels by catecholamines, acetylcholine and second messenger systems. Prog Biophys Mol Biol. 1988;52:165–247. doi: 10.1016/0079-6107(88)90014-4. [DOI] [PubMed] [Google Scholar]
- Hasan W, Woodward WR, Habecker BA. Altered atrial neurotransmitter release in transgenic p75 (-/-) and gp130 KO mice. Neurosci Lett. 2012;529:55–59. doi: 10.1016/j.neulet.2012.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesp ZC, Zhu Z, Morris TA, Walker RG, Isaacson LG. Sympathetic reinnervation of peripheral targets following bilateral axotomy of the adult superior cervical ganglion. Brain Res. 2012;1473:44–54. doi: 10.1016/j.brainres.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen JO, Laurikainen A, Väkevä A, Meri S, Saarma M. Nerve growth factor and brain-derived neurotrophic factor mRNAs are regulated in distinct cell populations of rat heart after ischaemia and reperfusion. J Pathol. 2001;194:247–253. doi: 10.1002/path.878. [DOI] [PubMed] [Google Scholar]
- Hiltunen PH, Airaksinen MS. Sympathetic cholinergic target innervation requires GDNF family receptor GFR[alpha]2. Mol Cell Neurosci. 2004;26:450–457. doi: 10.1016/j.mcn.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Hyatt-Sachs H, Bachoo M, Schreiber R, Vaccariello SA, Zigmond RE. Chemical sympathectomy and postganglionic nerve transection produce similar increases in galanin and VIP mRNA but differ in their effects on peptide content. J Neurobiol. 1996;30:543–555. doi: 10.1002/(SICI)1097-4695(199608)30:4<543::AID-NEU9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Ieda M, Kanazawa H, Kimura K, Hattori F, Ieda Y, Taniguchi M, Lee JK, Matsumura K, Tomita Y, Miyoshi S, Shimoda K, Makino S, Sano M, Kodama I, Ogawa S, Fukuda K. Sema3a maintains normal heart rhythm through sympathetic innervation patterning. Nat Med. 2007;13:604–612. doi: 10.1038/nm1570. [DOI] [PubMed] [Google Scholar]
- Kanazawa H, Ieda M, Kimura K, Arai T, Kawaguchi-Manabe H, Matsuhashi T, Endo J, Sano M, Kawakami T, Kimura T, Monkawa T, Hayashi M, Iwanami A, Okano H, Okada Y, Ishibashi-Ueda H, Ogawa S, Fukuda K. Heart failure causes cholinergic transdifferentiation of cardiac sympathetic nerves via gp130-signaling cytokines in rodents. J Clin Invest. 2010;120:408–421. doi: 10.1172/JCI39778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimaschewski L, Grohmann I, Heym C. Target-dependent plasticity of galanin and vasoactive intestinal peptide in the rat superior cervical ganglion after nerve lesion and re-innervation. Neuroscience. 1996;72:265–272. doi: 10.1016/0306-4522(95)00551-X. [DOI] [PubMed] [Google Scholar]
- Landis SC. Rat sympathetic neurons and cardiac myocytes developing in microcultures: correlation of the fine structure of endings with neurotransmitter function in single neurons. Proc Natl Acad Sci U S A. 1976;73:4220–4224. doi: 10.1073/pnas.73.11.4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Knowlton D, Van Winkle DM, Habecker BA. Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am J Physiol Heart Circ Physiol. 2004;286:H2229–H2236. doi: 10.1152/ajpheart.00768.2003. [DOI] [PubMed] [Google Scholar]
- Lorentz CU, Alston EN, Belcik T, Lindner JR, Giraud GD, Habecker BA. Heterogeneous ventricular sympathetic innervation, altered {beta} adrenergic receptor expression, and rhythm instability in mice lacking p75 neurotrophin receptor. Am J Physiol Heart Circ Physiol. 2010;298:H1652–H1660. doi: 10.1152/ajpheart.01128.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentz CU, Woodward WR, Tharp K, Habecker BA. Altered norepinephrine content and ventricular function in p75NTR-/- mice after myocardial infarction. Auton Neurosci. 2011;164:13–19. doi: 10.1016/j.autneu.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabe AM, Hoard JL, Duffourc MM, Hoover DB. Localization of cholinergic innervation and neurturin receptors in adult mouse heart and expression of the neurturin gene. Cell Tissue Res. 2006;326:57–67. doi: 10.1007/s00441-006-0215-3. [DOI] [PubMed] [Google Scholar]
- Matsuura W, Sugimachi M, Kawada T, Sato T, Shishido T, Miyano H, Nakahara T, Ikeda Y, Alexander J, Jr, Sunagawa K. Vagal stimulation decreases left ventricular contractility mainly through negative chronotropic effect. Am J Physiol. 1997;273:H534–H539. doi: 10.1152/ajpheart.1997.273.2.H534. [DOI] [PubMed] [Google Scholar]
- Miwa K, Lee JK, Takagishi Y, Opthof T, Fu X, Kodama I. Glial cell line-derived neurotrophic factor (GDNF) enhances sympathetic neurite growth in rat hearts at early developmental stages. Biomed Res. 2010;31:353–361. doi: 10.2220/biomedres.31.353. [DOI] [PubMed] [Google Scholar]
- Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local beta-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res. 2012;110:1454–1464. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash MP, Bradley CP, Sutton PM, Clayton RH, Kallis P, Hayward MP, Paterson DJ, Taggart P. Whole heart action potential duration restitution properties in cardiac patients: a combined clinical and modelling study. Exp Physiol. 2006;91:339–354. doi: 10.1113/expphysiol.2005.031070. [DOI] [PubMed] [Google Scholar]
- Oba T, Yasukawa H, Hoshijima M, Sasaki K, Futamata N, Fukui D, Mawatari K, Nagata T, Kyogoku S, Ohshima H, Minami T, Nakamura K, Kang D, Yajima T, Knowlton KU, Imaizumi T. Cardiac-specific deletion of SOCS-3 prevents development of left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol. 2012;59:838–852. doi: 10.1016/j.jacc.2011.10.887. [DOI] [PubMed] [Google Scholar]
- Pardini BJ, Lund DD, Schmid PG. Organization of the sympathetic postganglionic innervation of the rat heart. J Auton Nerv Syst. 1989;28:193–201. doi: 10.1016/0165-1838(89)90146-X. [DOI] [PubMed] [Google Scholar]
- Parrish DC, Gritman K, Van Winkle DM, Woodward WR, Bader M, Habecker BA. Postinfarct sympathetic hyperactivity differentially stimulates expression of tyrosine hydroxylase and norepinephrine transporter. Am J Physiol Heart Circ Physiol. 2008;294:H99–H106. doi: 10.1152/ajpheart.00533.2007. [DOI] [PubMed] [Google Scholar]
- Parrish DC, Alston EN, Rohrer H, Nkadi P, Woodward WR, Schütz G, Habecker BA. Infarction-induced cytokines cause local depletion of tyrosine hydroxylase in cardiac sympathetic nerves. Exp Physiol. 2010;95:304–314. doi: 10.1113/expphysiol.2009.049965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish DC, Alston EN, Rohrer H, Hermes SM, Aicher SA, Nkadi P, Woodward WR, Stubbusch J, Gardner RT, Habecker BA. The absence of gp130 in dopamine {beta} hydroxylase-expressing neurons leads to autonomic imbalance and increased reperfusion arrhythmias. Am J Physiol Heart Circ Physiol. 2009;297:H960–H967. doi: 10.1152/ajpheart.00409.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MJ, Habecker BA. STAT3 integrates cytokine and neurotrophin signals to promote sympathetic axon regeneration. Mol Cell Neurosci. 2013;56:272–282. doi: 10.1016/j.mcn.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Sun Y, Escary JL, Perreau J, Tresser S, Patterson PH, Zigmond RE, Brulet P, Landis SC. Leukemia inhibitory factor mediates an injury response but not a target- directed developmental transmitter switch in sympathetic neurons. Neuron. 1993;11:1175–1185. doi: 10.1016/0896-6273(93)90229-K. [DOI] [PubMed] [Google Scholar]
- Reichardt LF, Patterson PH. Neurotransmitter synthesis and uptake by isolated sympathetic neurones in microcultures. Nature. 1977;270:147–151. doi: 10.1038/270147a0. [DOI] [PubMed] [Google Scholar]
- Schäfer MK, Schütz B, Weihe E, Eiden LE. Target-independent cholinergic differentiation in the rat sympathetic nervous system. Proc Natl Acad Sci U S A. 1997;94:4149–4154. doi: 10.1073/pnas.94.8.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotzinger RJ, Landis SC. Cholinergic phenotype developed by noradrenergic sympathetic neurons after innervation of a novel cholinergic target in vivo. Nature. 1988;335:637–639. doi: 10.1038/335637a0. [DOI] [PubMed] [Google Scholar]
- Schotzinger RJ, Landis SC. Acquisition of cholinergic and peptidergic properties by sympathetic innervation of rat sweat glands requires interaction with normal target. Neuron. 1990;5:91–100. doi: 10.1016/0896-6273(90)90037-G. [DOI] [PubMed] [Google Scholar]
- Selvaraj RJ, Picton P, Nanthakumar K, Chauhan VS. Steeper restitution slopes across right ventricular endocardium in patients with cardiomyopathy at high risk of ventricular arrhythmias. Am J Physiol Heart Circ Physiol. 2007;292:H1262–H1268. doi: 10.1152/ajpheart.00913.2006. [DOI] [PubMed] [Google Scholar]
- Shi X, Habecker BA. gp130 cytokines stimulate proteasomal degradation of tyrosine hydroxylase via extracellular signal regulated kinases 1 and 2. J Neurochem. 2012;120:239–247. doi: 10.1111/j.1471-4159.2011.07539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skobowiat C, Gonkowski S, Calka J. Phenotyping of sympathetic chain ganglia (SChG) neurons in porcine colitis. J Vet Med Sci. 2010;72:1269–1274. doi: 10.1292/jvms.10-0081. [DOI] [PubMed] [Google Scholar]
- Solomon SD, Zelenkofske S, McMurray JJ, Finn PV, Velazquez E, Ertl G, Harsanyi A, Rouleau JL, Maggioni A, Kober L, White H, Van de Werf F, Pieper K, Califf RM, Pfeffer MA Valsartan in Acute Myocardial Infarction Trial (VALIANT) Investigators. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N Engl J Med. 2005;352:2581–2588. doi: 10.1056/NEJMoa043938. [DOI] [PubMed] [Google Scholar]
- Stanke M, Duong CV, Pape M, Geissen M, Burbach G, Deller T, Gascan H, Otto C, Parlato R, Schütz G, Rohrer H. Target-dependent specification of the neurotransmitter phenotype: cholinergic differentiation of sympathetic neurons is mediated in vivo by gp 130 signaling. Development. 2006;133:141–150. doi: 10.1242/dev.02189. [DOI] [PubMed] [Google Scholar]
- Stubbusch J, Majdazari A, Schmidt M, Schütz G, Deller T, Rohrer H. Generation of the tamoxifen-inducible DBH-Cre transgenic mouse line DBH-CT. Genesis. 2011;49:935–941. doi: 10.1002/dvg.20773. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zigmond RE. Involvement of leukemia inhibitory factor in the increases in galanin and vasoactive intestinal peptide mRNA and the decreases in neuropeptide Y and tyrosine hydroxylase mRNA in sympathetic neurons after axotomy. J Neurochem. 1996;67:1751–1760. doi: 10.1046/j.1471-4159.1996.67041751.x. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Maehara K, Onuki N, Saito T, Maruyama Y. Decreased contractility of the left ventricle is induced by the neurotransmitter acetylcholine, but not by vagal stimulation in rats. Jpn Heart J. 2003;44:257–270. doi: 10.1536/jhj.44.257. [DOI] [PubMed] [Google Scholar]
- Wojtkiewicz J, Równiak M, Crayton R, Gonkowski S, Robak A, Zalecki M, Majewski M, Klimaschewski L. Axotomy-induced changes in the chemical coding pattern of colon projecting calbindin-positive neurons in the inferior mesenteric ganglia of the pig. J Mol Neurosci. 2013;51:99–108. doi: 10.1007/s12031-013-0007-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamori T, Fukada K, Aebersold R, Korsching S, Fann MJ, Patterson PH. The cholinergic neuronal differentiation factor from heart cells is identical to leukemia inhibitory factor. Science. 1989;246:1412–1416. doi: 10.1126/science.2512641. [DOI] [PubMed] [Google Scholar]
- Zigmond RE, Sun Y. Regulation of neuropeptide expression in sympathetic neurons: paracrine and retrograde influences. Ann NY Acad Sci. 1997;814:181–197. doi: 10.1111/j.1749-6632.1997.tb46157.x. [DOI] [PubMed] [Google Scholar]