

Abstract
The role of synaptic activity during early formation of neural circuits is a topic of some debate; genetic ablation of neurotransmitter release by deletion of the Munc18-1 gene provides an excellent model to answer the question of whether such activity is required for early circuit formation. Previous analysis of Munc18-1−/− mouse mutants documented their grossly normal nervous system, but its molecular differentiation has not been assessed. Munc18-1 deletion in mice also results in widespread neurodegeneration that remains poorly characterized. In this study, we demonstrate that the early stages of spinal motor circuit formation, including motor neuron specification, axon growth and pathfinding, and mRNA expression, are unaffected in Munc18-1−/− mice, demonstrating that synaptic activity is dispensable for early nervous system development. Furthermore, we show that the neurodegeneration caused by Munc18-1 loss is cell autonomous, consistent with apparently normal expression of several neurotrophic factors and normal GDNF signaling. Consistent with cell-autonomous degeneration, we demonstrate defects in the trafficking of the synaptic proteins Syntaxin1a and PSD-95 and the TrkB and DCC receptors in Munc18-1−/− neurons; these defects do not appear to cause ER stress, suggesting other mechanisms for degeneration. Finally, we demonstrate pathological similarities to Alzheimer's disease, such as altered Tau phosphorylation, neurofibrillary tangles, and accumulation of insoluble protein plaques. Together, our results shed new light upon the neurodegeneration observed in Munc18-1−/− mice and argue that this phenomenon shares parallels with neurodegenerative diseases.
SIGNIFICANCE STATEMENT In this work, we demonstrate the absence of a requirement for regulated neurotransmitter release in the assembly of early neuronal circuits by assaying transcriptional identity, axon growth and guidance, and mRNA expression in Munc18-1-null mice. Furthermore, we characterize the neurodegeneration observed in Munc18-1 mutants and demonstrate that this cell-autonomous process does not appear to be a result of defects in growth factor signaling or ER stress caused by protein trafficking defects. However, we find the presence of various pathological hallmarks of Alzheimer's disease that suggest parallels between the degeneration in these mutants and neurodegenerative conditions.
Keywords: developmental, early neuronal activity, motor neurons, Munc18-1, neurodegeneration, neurosecretion
Introduction
Early neuronal activity has been proposed to specify the fundamental properties of developing neurons, including neurotransmitter identity, connectivity, and function (Borodinsky et al., 2004; Demarque and Spitzer, 2010; Kastanenka and Landmesser, 2010; Marek et al., 2010; Crisp et al., 2011); however, this topic remains under debate (Myers et al., 2005; Varoqueaux et al., 2005; Benjumeda et al., 2013). Munc18-1-null (Munc18-1−/−) mice lack the SNARE-associated protein Munc18-1 that is essential for regulated fusion of neurotransmitter-containing vesicles (Toonen and Verhage, 2007); these mice thus lack synaptic activity (Verhage et al., 2000), providing a genetic model with which to examine the influence of such activity upon early circuit assembly. In addition, the neurodegeneration observed in Munc18-1−/− mice, but not in other models of neuronal activity loss (Myers et al., 2005; Varoqueaux et al., 2005), raises the question of the specific role of Munc18-1 in neuronal survival.
Spinal neurons of mice and chicks are electrically active soon after becoming postmitotic (Milner and Landmesser, 1999; Law et al., 2014); neurons initially fire randomly, but this firing synchronizes over time, driven by a combination of both cholinergic and GABA-ergic neurotransmission and gap-junction coupling (Milner and Landmesser, 1999; Hanson and Landmesser, 2003; Warp et al., 2012; Czarnecki et al., 2014; Law et al., 2014; Hinckley et al., 2015). Pharmacological alteration of activity patterns has been reported to disrupt the pathfinding of limb-innervating motor neurons located in the chick lateral motor column (LMC; Hanson and Landmesser, 2003; Kastanenka and Landmesser, 2010). In addition, pharmacological inhibition of activity in Xenopus laevis embryos can result in a change in the neurotransmitter identity of spinal neurons, a process mediated by the transcription factor c-jun (Borodinsky et al., 2004; Marek et al., 2010). However, blocking neuronal activity by overexpression of the Kir2.1 inwardly rectifying K+ channel in chick motor neurons fails to mimic defects in axon guidance and neuronal specification (Benjumeda et al., 2013). One explanation for this disparity is the cell-autonomous nature of Kir2.1 channel expression contrasting global pharmacological effects. In Munc18-1−/− mice, regulated neurotransmitter release is ablated in the entire nervous system, offering an unambiguous genetic model with which to examine the influence of synaptic activity upon early circuit formation in mice, which has not been assayed in the context of molecularly defined neuronal circuits.
Munc18-1−/− mice have been shown to develop normally before undergoing widespread neurodegeneration in the brainstem and forebrain (Verhage et al., 2000; Bouwman et al., 2004), but the mechanisms underlying this neurodegeneration are unclear. Developmentally programmed apoptosis is tightly regulated in spinal motor neurons, in part by limiting concentrations of motor neuron survival factors in the periphery (Pittman and Oppenheim, 1978, 1979; Oppenheim, 1996), activity-dependent axonal branching (Heeroma et al., 2003; Myers et al., 2005), and proapoptotic signals (Deppmann et al., 2008; Taylor et al., 2012). In addition, specific mechanisms cause pathological neurodegeneration, including oxidative stress, aberrant trafficking, excitotoxicity, neuroinflammation, and neurofibrillary tangle formation (for review, see Glass et al., 2010; Garden and La Spada, 2012; Huang and Mucke, 2012; Neefjes and Kant, 2014). In Munc18-1−/− animals, excessive neurodegeneration may result from acceleration of a normal program of apoptosis (Yamamoto and Henderson, 1999; Prasad et al., 2008), decreased growth factor secretion (Schulze et al., 1994; Voets et al., 2001), or some other mechanism. In particular, Munc18-1 is a known activator of CDK5 (Shetty et al., 1995), which supresses c-Jun-induced neurodegeneration (Li et al., 2002). In addition, the interaction between Munc18-1 and CDK5 (Bhaskar et al., 2004) may be of relevance to Alzheimer's disease because CDK5 influences phosphorylation of Tau and the proteolytic cleavage of APP, a process with which Munc18-1 has been linked (Sakurai et al., 2008).
Here, we demonstrate that evoked neurotransmitter release is dispensable for the normal birth, patterning, gene expression, and axon outgrowth of neurons of the spinal motor circuit in vivo by characterizing the development of Munc18-1−/− spinal cords at the molecular level. Furthermore, we characterize the degeneration that occurs in Munc18-1−/− spinal cords and demonstrate the presence of several pathological features of Alzheimer's disease.
Materials and Methods
Animals.
Embryos used in this study were a mixture of male and female (data not shown). Munc18-1−/− mice were described previously (Verhage et al., 2000). No differences were observed between Munc18-1+/− mice and Munc18-1+/+ mice; when “control” is used to indicate genotype, this represents a mixture of Munc18-1+/+ and Munc18-1+/− embryos. To generate Olig2+/cre;Munc18-1flox/flox mice, Olig2cre/+ mice (Dessaud et al., 2007) were crossed to Munc18-1flox/flox mice (Dudok et al., 2011) to generate Olig2cre/+;Munc18-1flox/+ mice, which were then crossed to generate litters with mixed genotypes. All constitutive mutant animals were housed in the animal care facilities of the Institut de Recherches Cliniques de Montréal; all conditional animals were housed in the animal care facilities of the Sloan-Kettering Institute. Animals were kept on a 12 h light/dark cycle, with food and water provided ad libitum. All experimental procedures were approved either by the Animal Care and Use Committees at McGill University and Institut de Recherches Cliniques de Montréal, in accordance with the regulations of the Canadian Council on Animal Care, or by the Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center. The morning that a vaginal plug was discovered was defined as embryonic day 0.5 (E0.5).
Calcium imaging in motor neurons.
Embryos were removed from the uterine pouch at either E11.5 or E12.5 and placed immediately into cold, oxygenated Tyrode's solution containing the following (in mm): 140 NaCl, 3 KCl, 17 NaHCO3, 12 glucose, 2 CaCl2, and 1 MgCl2. Calcium Green Dextran (Life Technologies) was injected into the brachial peripheral nerve plexus and the embryos were maintained in a circulating bath of oxygenated Tyrode's solution for >120 min to allow uptake of the dye into motor neurons. The spinal cord was then dissected into an open-book preparation and the embryos placed ventral side down in the imaging chamber, which was perfused with oxygenated Tyrode's at a rate of 2 ml/min at room temperature. Spinal cords were imaged at 1 Hz using a Zeiss Axio Observer Z1 microscope with a CSU-X1M dual cam 5000 spinning disk. Analysis of calcium activity was performed using a combination of FIJI and custom MATLAB (The MathWorks) scripts (Law et al., 2014).
Immunostaining and histochemistry.
Embryos were fixed in 4% PFA (Sigma-Aldrich) in PBS before cryoprotection with 30% sucrose in PBS, embedding in OCT (Sakura Finetek), and storage at − 80°C. Next, 12 μm sections were cut using a Leica cryostat microtome. Tissue was rinsed in PBS, incubated in blocking solution containing 1% heat-inactivated horse serum plus 0.05% Triton X-100 (Sigma-Aldrich) in PBS for 20 min, incubated overnight at 4°C with primary antibodies (Table 1), and diluted in blocking solution. After incubation with primary antibodies, sections were rinsed 3× in blocking solution, and then the appropriate secondary antibodies from Life Technologies were applied in the same solution at room temperature for 90 min. Sections were then rinsed three more times in PBS and coverslips applied with Mowiol. Antigen retrieval was necessary for pTau staining, and consisted of 10 min incubation in citrate buffer solution containing 10 mm citrate, 0.05% Tween 20 in H2O, pH 6.0, at 95°C. For Congo Red staining, 0.3 g of Congo Red (Sigma-Aldrich) and 0.3 g of NaCl (Sigma-Aldrich) was dissolved in an 80% solution of ethanol. A 1% volume of 1% NaOH was then added to this solution before filtering. 12-μm-thick cryosections of tissue were then submerged in this filtered solution for 30 min at room temperature and then differentiated by dipping 5–6 times into a 1% volume of 1% NaOH in 50% ethanol before rinsing and mounting. Cell counts were performed in spinal lamina IV-X of Pax2- and CCAT-stained sections by means of a custom ImageJ macro.
Table 1.
Antibodies used
| Antigen | Source | Cat. no. | Dilution (IF) | Dilution (WB) |
|---|---|---|---|---|
| Foxp1 | Gift of Bennett Novitch | — | 1/1000 | — |
| Rabbit anti-Isl1 | Gift of Tom Jessell | — | 1/1000 | — |
| Rabbit anti-Lim1 | Gift of Tom Jessell | — | 1/1000 | — |
| Rabbit anti-Munc18-1 | Gift of Matthijs Verhage | — | 1/500 | — |
| Olig2 | Gift of Bennett Novitch | — | 1/1000 | — |
| CCAT | Gift of Ricardo Dolmetsch | — | 1/100 | — |
| Guinea pig anti-Pea3 | Gift of Silvia Arber | — | 1/1000 | — |
| Iba-1 | Wako | 019-19741 | 1/100 | — |
| TrkB | Millipore | 07-225 | 1/1000 | 1/500 |
| VAChT | Synaptic Systems | 139103 | 1/500 | — |
| Neurofilament | DSHB | 2H3 | 1/100 | — |
| Alpha-synuclein | Cell Signalling Technology | 4179 | 1/100 | — |
| Nkx6.1 | DSHB | 55A10 | 1/100 | — |
| Nkx2.2 | DSHB | 5A5 | 1/100 | — |
| TrkA | Millipore | 6574 | 1/1000 | — |
| Cleaved-caspase 3 | Cell Signalling Technology | 9661-S | 1/1000 | — |
| ß-Actin | Sigma | A5441 | — | 1/10000 |
| GRP78 | AbCam | ab21685 | — | 1/500 |
| Ubiquitin | Millipore | MAB1510 | 1/500 | — |
| AT8 | Thermo Scientific | MN1020 | 1/500 | 1/1000 |
| Tau | Millipore | MAB3420 | 1/1000 | |
| PDS95 | NeuroMab | P78352 | 1/100 | — |
| Syntaxin1a | Sigma | s-0664 | 1/500 | 1/1000 |
| Neurofilament - L | Santa Cruz | sc-12980 | — | 1/500 |
| DCC | Santa Cruz | sc-6535 | 1/500 | 1/500 |
| EphA4 | Santa Cruz | sc-921 | 1/500 | — |
| c-Ret | Santa Cruz | sc167 | 1/500 | — |
In situ mRNA detection. In situ hybridizations were performed as described previously (Kao and Kania, 2011). cRNA probes were generated as follows: target sequence amplification primers were designed using Primer3 version 0.4.0 software (Untergasser et al., 2012) with a probe size set at 600–800 bp. One-step RT-PCR was performed (QIAGEN) using the appropriate primers containing T7 polymerase promoters (Invitrogen) to make and amplify cDNA template from mouse E11.5 pooled brain RNA. The PCR product was purified by gel electrophoresis in 1% agarose gel and gel extraction using QIAquick gel extraction kit (QIAGEN). The purified DNA was then reamplified by PCR. The yield of DNA was estimated using the low DNA mass ladder (Invitrogen) after gel electrophoresis. DIG-labeled RNA probes were synthesized by in vitro transcription with T7 RNA polymerase using DIG RNA labeling kit (Roche). All probes were verified by sequencing. Then, 12 μm tissue sections were fixed in 4% PFA, rinsed 3× in PBS, treated with 1 μg/ml proteinase K (in 6.25 mm EDTA, pH 8.0, Invitrogen + 50 mm Tris, pH 7.5; Fisher Scientific) for 15 min, postfixed in 4% PFA, rinsed in PBS,and then acetylated for 10 min in a mixture of 6 ml of triethanolamine (Sigma-Aldrich), 500 ml of distilled H2O, and 1.3 ml of acetic acid (Sigma-Aldrich). Samples were then rinsed and incubated in hybridization buffer containing 50% formamide (Sigma-Aldrich), 5× SSC (20× SSC is 3 m NaCl, 0.3 m NaAc), 5× Denhardt's (Sigma-Aldrich), and 500 μg/ml salmon sperm DNA (Roche) in H2O for 2 h, followed by overnight hybridization at 72°C with DIG-labeled probes at a concentration of 2–5 ng/μl. After hybridization, samples were immersed in 5×SSC at 72°C, followed by 2 45 min washes in 0.2× SSC at 72°C and 1 5 min wash in 0.2×SSC at room temperature. Tissues were then rinsed with B1 buffer containing 0.1 m Tris, pH 7.5, and 0.15 m NaCl (Fisher Scientific) for 5 min, blocked with B2 buffer (10% heat inactivated horse serum in B1) for 1 h at room temperature, and incubated with anti-DIG antibody (1:5000 in B2; Roche) overnight at 4°C. Samples were then rinsed with B1 and equilibrated with B3 buffer containing 0.1 m Tris, pH 9.5, 0.1 m NaCl, and 0.05 m MgCl2 (Fisher Scientific). To detect bound anti-DIG antibodies, samples were incubated with B4 buffer containing [100 mg/ml NBT, 50 mg/ml BCIP (Roche), and 400 mm levamisol (Sigma-Aldrich) in B3 in the dark. The reaction was stopped by immersion in H2O.
mRNA expression microarray analysis.
Lumbar spinal cords were isolated from E12.5 mice dissected in cold PBS and stored in RNAlater at − 80°C until RNA extraction using a QIAGEN RNAeasy kit; these samples were then sent to the Centre for Applied Genomics at the Hospital for Sick Children (Toronto, Ontario, Canada) for further analysis. RNA quality was tested using an Agilent Bioanalyzer before performing standard cDNA synthesis and in vitro transcription (Affymetrix WT PLUS kit). Then, 400 μg of RNA was used for in vitro transcription and 5.5 μg of cDNA was used for hybridization to the Mouse Gene 2.0 ST chip. Scanning was performed using the Affymetrix GeneChip Scanner 3000 and analysis done using Affymetrix Expression Console to obtain signal-log ratios of the control to the sample. Hybridizations of four biological replicates for both WT and Munc18−/− genotypes were performed. Robust multiarray (RMA) preprocessing and expression comparison was performed upon signal-log ratios of probe hybridization intensity using R software (version 3.1.1; limma and oligo packages); p-values were adjusted for multiple comparisons using the false discovery rate method (Benjamini and Hochberg, 1995; Verhage et al., 2000).
Analysis of ER stress.
Mouse embryonic fibroblasts were isolated from WT E11.5 embryos by dissection of limbs and body walls in cold PBS, followed by mincing with a razor blade. Minced tissue was then incubated with 25% trypsin (Invitrogen) at 37°C for 10 min before the addition of cold Dulbecco's modified Eagle's medium (DMEM; Invitrogen) containing 10% fetal bovine serum (FBS; Invitrogen) and 1% penicillin/streptomycin (P/S; Invitrogen), and trituration through a fire-polished glass pasteur pipette. Fibroblasts were pelleted by centrifugation, resuspended in DMEM + 10% FBS + 1% P/S, then grown to 95% confluence in standard culture medium. ER stress was induced by the addition of medium containing 2 μm tunicamycin for 12 h. Cells were then pelleted and RNA isolated as described above. RNA from the spinal cords of E13.5 mice was also harvested as described above. RT-PCR was performed using the following primers.
Western blotting.
Whole spinal cords were dissected from E13.5 embryos in cold PBS and snap frozen in a dry ice/ethanol bath. Spinal cords were lysed in RIPA buffer containing 120 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS in 50 mm Tris, pH 8.0, for 15 min on ice with occasional vortexing. Lysates were centrifuged at 4°C to remove the insoluble pellet and protein concentration determined by Bradford assay. Lysates were run on a 7.5% polyacrylamide gel (20 μg of protein per lane) and then transferred to a nitrocellulose membrane (Millipore). Membranes were blocked for 2 h in TBS-T (150 mm NaCl, 10 mm Tris, 0.05% Tween 20, pH 8.0) containing either 1% bovine serum albumin (BSA; Invitrogen, for phospho-proteins) or 10% skimmed milk powder (for all other proteins). Primary antibodies were diluted in TBS-T containing either 0.1% or 1% BSA (Table 2) and membranes were stained overnight at 4°C. After 3 rinses in TBS-T,the appropriate HRP-conjugated secondary antibodies (Jackson Laboratories) were applied to membranes for 90 min. After secondary labeling, membranes were rinsed twice in TBS-T and then treated with ECL reagent (GE Healthcare) and exposed to film, which was then developed. Quantifications were performed by analyzing relative densities of exposed film using ImageJ.
Table 2.
LMC death during development
| Age and spinal level | Genotype | N | Mean no. of caspase+ cells per LMC | SEM | p-value |
|---|---|---|---|---|---|
| E11.5 brachial | +/+ | 3 | 0.24 | 0.12 | 0.264 |
| E11.5 brachial | −/− | 4 | 0.09 | 0.05 | |
| E12.5 lumbar | +/+ | 6 | 0.68 | 0.11 | 0.00265 |
| E12.5 lumbar | −/− | 4 | 2.37 | 0.47 | |
| E12.5 brachial | +/+ | 4 | 1.16 | 0.54 | 0.01364 |
| E12.5 brachial | −/− | 3 | 4.78 | 0.21 | |
| E13.5 lumbar | +/+ | 3 | 1.59 | 0.53 | 0.0007 |
| E13.5 lumbar | −/− | 3 | 6.91 | 0.20 | |
| E13.5 brachial | +/+ | 4 | 2.40 | 0.10 | 0.0061 |
| E13.5 brachial | −/− | 5 | 16.01 | 2.57 |
Statistical comparisons.
Unless otherwise noted, statistical comparisons were performed using two-tailed Student's t tests.
Results
Early expression of Munc18-1 and spontaneous spinal neuron activity
To ascertain the influence of synaptic activity upon early neuronal circuit formation, we characterized the development of Munc18-1−/− mice, which lack evoked neurotransmitter release (Verhage et al., 2000). Although Munc18-1 expression is restricted to the nervous system and adrenal medulla in the adult (Pevsner et al., 1994a,b; Voets et al., 2001), the other two mouse Munc18 family genes, Munc18–2 and Munc18–3, are ubiquitously expressed (Hata and Südhof, 1995; Tellam et al., 1995), although their expression does not compensate for the loss of synaptic activity caused by Munc18-1 mutation (Verhage et al., 2000). To clarify the expression of Munc18-1 at early developmental stages, we monitored its mRNA expression in the spinal cords of E11.5 and E13.5 embryos and found it overlapping with NeuN, a marker of postmitotic neurons (Fig. 1A–C).
Figure 1.

Early expression of Munc18-1 and loss of spontaneous activity in Munc18-1−/− mice. At E11.5, Munc18-1 mRNA in the spinal cord (A) is restricted to a region containing NeuN postmitotic neurons (B). At E13.5, Munc18-1 expression is restricted to the nervous system (C). Calcium imaging of motor neurons at E11.5 reveals large, synchronous calcium transients that persist for several seconds in control animals (D; frequency = 0.7 ± 0.046 events/min, event length = 6.8 ± 0.17 s, n = 64 cells, n = 7 embryos). Munc18-1−/− mice also display calcium transients at E11.5 that are more frequent and greater in length than those in control animals (D; frequency = 0.91 ± 0.060 events/min, event length = 14.3 ± 0.96 s, n = 19 cells, n = 4 embryos; p(frequency) = 0.025, p(length) < 0.0001). In control animals at E12.5, large calcium transients occur synchronously in multiple neurons (E; frequency = 1.17 ± 0.047 events/min, event length = 5.78 ± 0.27 s, n = 93 cells, n = 4 embryos); in contrast, Munc18-1−/− neurons display very little activity, with only asynchronous transients occurring in single neurons (E; frequency = 0.08 ± 0.032 events/min, event length = 4.4 ± 0.14 s, n = 153 cells, n = 3 embryos; p(frequency) < 0.0001, p(length) < 0.0001).
Mouse spinal neurons are spontaneously active at E12 (Milner and Landmesser, 1999), but the earliest time point at which loss of synaptic activity was demonstrated in Munc18-1−/− mice was at E18.5 (Verhage et al., 2000). To determine whether Munc18-1 is critical for spontaneous activity at earlier developmental stages, we monitored calcium fluxes in spinal cords from control and Munc18-1−/− littermates at E11.5 (Fig. 1D) by retrograde labeling of motor neurons with calcium green dextran. Motor neurons in control animals (n = 7) displayed large calcium transients persisting for several seconds and occurring in multiple neurons simultaneously (Fig. 1D). Calcium transients were observed in Munc18-1−/− animals (n = 4) at this age (Fig. 1D); these transients were more frequent (p < 0.0001) and longer (p = 0.0254) than those in control embryos. These data demonstrate that neuronal activity at this age is present and not dependent upon evoked neurotransmitter release, possibly being driven through gap-junction-dependent mechanisms (Hanson and Landmesser, 2003; Bittman et al., 2004; Czarnecki et al., 2014). At E12.5, calcium imaging of control animals (n = 4) revealed transients that occurred in multiple motor neurons simultaneously (Fig. 1E), as described previously in chick (Wang et al., 2009; Law et al., 2014); in contrast, motor neurons from Munc18-1−/− animals (n = 3) were largely inactive compared with controls (p < 0.0001) and the transients displayed by active motor neurons were shorter (p < 0.0001) and occurred only in single neurons (Fig. 1E). Together, these data show that, at E11.5, neuronal activity is present, possibly mediated by gap junctions, but that by E12.5 neuronal activity requires Munc18-1-dependent neurotransmitter release, as has been seen in previous studies (Hanson and Landmesser, 2003; Czarnecki et al., 2014).
Molecular characterization of spinal neuron development in the absence of neurotransmitter release
Although nervous system assembly appears morphologically normal in Munc18-1−/− animals (Verhage et al., 2000), the extent of molecular differentiation of neuronal circuits in such mutants has not been examined in detail. To do this, we focused on the spinal cord, the development of which is very well characterized at the molecular level and some aspects of which have been studied previously in the context of electrical activity (Milner and Landmesser, 1999; Hanson and Landmesser, 2003, 2004; Benjumeda et al., 2013; Law et al., 2014).
Motor neurons and many of their associated interneurons arise from ventral spinal cord progenitors expressing the transcription factors Nkx2.2, Nkx6.2, and Olig2 (Novitch et al., 2001; Vallstedt et al., 2001). Similar numbers of these progenitors were found in WT and Munc18-1−/− E11.5 embryos (Fig. 2A–F; Olig2, p = 0.64; Nkx2.2, p = 0.88). The distribution of spinal interneurons expressing the transcription factors Chx10, Evx1, En1, and the C-terminal fragment of the ion channel CaV1.2 (CCAT), which may influence gene transcription under the influence of synaptic activity, was also similar in WT and mutant embryos between E11.5 and E13.5 (Fig. 2G–N, E12.5; data not shown). To assess motor neuron differentiation, we examined the expression of the postmitotic motor neuron markers FoxP1, Lim1, and Isl1 (Tsuchida et al., 1994; Dasen et al., 2008; Rousso et al., 2008) at the lumbar level of E13.5 mice after the specification and migration of LMC neurons had been completed (Hollyday and Hamburger, 1977; Novitch et al., 2001; Palmesino et al., 2010). We found no differences between WT and Munc18-1−/− littermates, as determined by the expression of LMC transcription factors FoxP1, Lim1, and Isl1 (Fig. 2O–W; Isl1, p = 0.67; Lim1, p = 0.58; Foxp1, p = 0.16). The lateral and medial subtypes of LMC neurons defined by, respectively, Lim1+ and Isl1+ expression in Foxp1+ LMC neurons were also generated normally (Fig. 2X,Y; Isl1+/Foxp1+, p = 0.126; Lim1+/Foxp1+, p = 0.91).
Figure 2.

Neuronal specification in Munc18-1−/− mice. At E11.5, Olig2 (A, B, E) and Nkx2.2 (C, D, F) progenitors are present in numbers similar to WT: Olig2: 47.49 ± 2.65 (+/+, n = 3), 49.16 ± 1.863 (−/−, n = 3, p = 0.64; Nkx2.2: 130.2 ± 2.48 (+/+, n = 2), 127.9 ± 1.57 (−/−, n = 2), p = 0.88. The number and distribution of Nkx6.1 (A, B) progenitors appears similar in both genotypes. The numbers and distributions of Chx10+ (G, H), En1+ (I, J), Evx+ (K, L), and Cav1.2+ (M, N) neurons also appear similar between genotypes at E12.5. In the lumbar spinal cord of E13.5 mice, numbers of Isl1+ (O, P, U; 46.65 ± 6.33, +/+, n = 4, 42.68 ± 6.07 (−/−, n = 5, p = 0.67), Lim1+ (Q, R, V; 27.59 ± 4.58, +/+, n = 4, 32.08 ± 6.44 (−/−, n = 3, p = 0.58), and Foxp1+ (S, T, W; 76.64 ± 7.37, +/+, n = 4, 87.66 ± 2.66 (−/−, n = 5, p = 0.17) motor neurons are similar between genotypes. Proportions of Isl1+ (X; 56.88 ± 1.59, +/+, n = 4, 49.03 ± 6.54, −/−, n = 5, p = 0.3) and Lim1+ (Y; 41.35 ± 3.56, +/+, n = 4, 42.26 ± 7.17 −/−, n = 3, p = 0.91) LMC motor neurons are also similar.
Electrical activity has previously been reported to influence the growth and guidance of axons (Nishiyama et al., 2003; Hanson and Landmesser, 2004), but this was not assessed in Munc18-1−/− mutants (Verhage et al., 2000), prompting us to examine the guidance of LMC motor axons in the limb, a process that is well characterized at the molecular level (Kao et al., 2012). At E12.5, both mutant and WT motor axons extended into the limb to a similar extent and segregated into ventral and dorsal limb nerves (Fig. 3A,B), with EphA4 expression appropriately labeling LMC axons in the dorsal limb nerve (Fig. 3C,D; Helmbacher et al., 2000) and DCC labeling in both branches of motor axons (Fig. 3E,F; Bai et al., 2011). At E13.5, both WT and mutant motor axons reach their distal limb muscle targets with similar frequency, as assessed by expression of neurofilament and the motor axon marker vesicular acetylcholine transferase (VAChT; Fig. 3G,H), demonstrating that deletion of Munc18-1 does not impair axon outgrowth.
Figure 3.
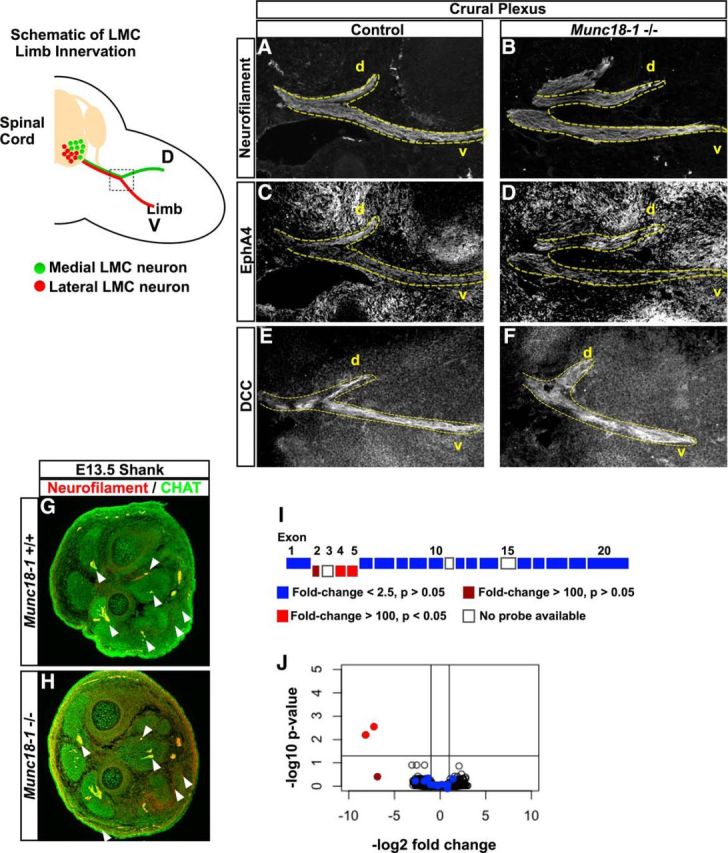
Motor axon growth and mRNA expression in Munc18-1−/− mice. A schematic of limb innervation by LMC neurons shows the dorsoventral (D-V) divergence of axons from neurons of the medial (red) and lateral (green) LMC at the base of the limb. In the E12.5 crural plexus, axons diverge normally at the base of the limb as determined by neurofilament expression (A, B). These axons also express EphA4 in the dorsal nerve branches (C, D) and DCC in both branches (E, F). In the distal hindlimbs of E13.5 mice (G, H), motor axons defined by coexpression of neurofilament (red) and ChAT (green) are present in both WT and Munc18-1−/− mice. Arrowheads point to innervation found in both genotypes. I, Schematic of the Munc18-1 allele. Exons deleted in the Munc18-1-null allele are aligned below the remaining exons; color indicates the expression level of exons in Munc18-1−/− mice compared with WT as determined by microarray. J, Volcano plot of mRNA expression at the probe level in Munc18-1−/− spinal cords versus WT. Only the exons deleted in the Munc18-1-null allele are downregulated (red dots); exons retained in the-null allele (blue dots) are not downregulated.
Many studies of synaptic activity during neuronal differentiation argue for its role in regulating gene expression (Borodinsky et al., 2004; Demarque and Spitzer, 2010; Marek et al., 2010). To determine the extent of gene expression changes in Munc18-1−/− spinal cords during the early stages of neural circuit formation, we isolated mRNA from the lumbar spinal cords of E12.5 WT and Munc18-1−/− embryos, which was then labeled and hybridized to Affymetrix Mouse Gene 2.0 arrays and probe signal intensities were compared using RMA processing. Single-probe-level quantification using p-values adjusted by the false discovery rate method (Benjamini and Hochberg, 1995) revealed a significant loss of expression of the exons deleted in the Munc18-1-null allele (>100-fold, Fig. 3I,J; Verhage et al., 2000). Surprisingly, no other significant gene expression differences between WT and Munc18-1−/− animals were detected, demonstrating that the absence of synaptic activity during the early stage of spinal cord development does not lead to any detectable changes in gene expression, even at the level of differential splicing. Together, these data demonstrate that, even in the absence of synaptic activity, the molecular specification of the murine spinal motor circuits proceeds normally without obvious changes in neuronal identity, axon extension, or mRNA expression.
Early onset and increased neuronal apoptosis in Munc18-1−/− embryos
Previous studies of Munc18-1−/− mice reveal widespread cell death throughout the CNS (Verhage et al., 2000); however, the mechanisms underlying it remain largely unexplored. Motor neurons are normally generated in excess and undergo a period of apoptotic death resulting in motor neuron numbers commensurate with their target muscle identity (Hamburger, 1975; Lance-Jones, 1982; Gould and Oppenheim, 2011). Given that silencing of neuronal activity can reduce apoptosis in motor neurons (Hory-Lee and Frank, 1995; Myers et al., 2005; Varoqueaux et al., 2005), we aimed to determine how this reduction in activity might influence the neurodegeneration observed in Munc18-1−/− mice during this developmental window. We thus examined the number of apoptotic motor neurons, in WT and Munc18-1−/− mutants before and during the period of naturally occurring cell death (Yamamoto and Henderson, 1999). Specifically, we examined lumbar and cervical spinal motor neurons between E11.5, before the onset of LMC apoptosis, and E13.5, during the peak of LMC apoptosis, by immunostaining for the apoptotic marker Cleaved Caspase-3 (CC3; Tewari et al., 1995) and the LMC marker Foxp1 (Fig. 4A–H). At E11.5, we did not detect any difference in the number of CC3+ LMC neurons between mutant and WT embryos (Fig. 4A,B,K, Table 2), but at E12.5, there was a significant increase in their numbers in Munc18-1−/− mice compared with their WT littermates (Fig. 4C,D,K, Table 2). This disparity increased over time (Fig. 4E–I,K, Table 2) such that, in the E13.5 brachial spinal cord, there were >8-fold more CC3+ LMC cells in mutants compared with controls (Fig. 4I–K, Table 2). This increased cell death was not restricted to the LMC, being apparent throughout the spinal cord, indicating the loss of both motor neurons and interneurons (Fig. 4H,J, arrowheads). By E16.5, compared with WT and Munc18-1 heterozygotes, very few Isl1+ cells remained in the Munc18-1−/− spinal cord (Fig. 4L,M), whereas neurons of the dorsal root ganglia (DRG) were spared (Fig. 4L,M, arrowheads).
Figure 4.

Cell death in the spinal cord of Munc18-1−/− mice. Staining of Foxp1 (red) and cleaved caspase-3 (green) demonstrates the increased extent of cell death in the Munc18-1−/− spinal cord from the E11.5 brachial spinal cord (A, B, K; Table 2) to the E13.5 brachial spinal cord (I–K; Table 2; C–H show the interim stages of development) in both the LMC and adjacent interneurons (white arrowheads). By E16.5, Isl1 staining reveals the degeneration of the majority of the spinal cord (L, M) in Munc18-1−/− animals, although the DRG (arrowheads) remain intact.
Cell-autonomous and non-cell-autonomous motor neuron death in Munc18-1 mutants
Munc18-1 regulates neurotrophin-containing large dense-core vesicle fusion in adrenochromaffin cells (Voets et al., 2001; Dieni et al., 2012), suggesting the possibility that neuronal apoptosis in Munc18-1−/− mutants might be caused by a deficiency in neurotrophin secretion. A corollary of this would be that WT neurons might rescue Munc18-1−/− neurons from apoptosis by providing secreted neurotrophins, as suggested by in vitro manipulations (Heeroma et al., 2004). To test the rescue of Munc18-1−/− neurons by control neurons in vivo, we used a conditional floxed allele of Munc18-1 (Munc18-1flox; Dudok et al., 2011) in combination with Cre recombinase expressed under the control of the Olig2 motor-neuron-specific promoter (Olig2cre; Dessaud et al., 2007). Excision of Munc18-1 in Olig2-derived motor neurons was confirmed by the loss of Munc18-1 protein expression in the spinal ventral roots that exclusively carry motor axons (Fig. 5A,B, arrowheads). Motor neuron apoptosis was assayed in Olig2cre/+;Munc18-1flox/flox (conditional) embryos by detecting CC3 in the E13.5 brachial spinal cord, where the greatest difference exists between levels of apoptosis in Munc18-1−/− and WT embryos. Surprisingly, Olig2cre/+;Munc18-1flox/flox mice exhibited similar numbers of apoptotic LMC neurons to Olig2+/+;Munc18-1flox/flox controls (Fig. 5C–E, p = 0.12), many fewer than in Munc18-1−/− embryos (Fig. 5E, p = 0.005). To determine whether this rescue of motor neuron death in conditional knock-outs was permanent, we examined the expression of the Isl1 motor neuron transcription factor at E16.5. In Olig2cre/+;Munc18-1flox/flox spinal cords, we observed that, compared with controls, only ∼10% of motor neurons remained (Fig. 5H; p = 0.007), whereas other spinal neurons, such as those expressing CCAT or Pax2, appeared to be unaffected by the elimination of Munc18-1 in motor neurons (Fig. 5I–N; CCAT p = 0.242; Pax2 p = 0.687). These experiments show that conditional deletion of Munc18-1 in motor neurons leads to a temporary reprieve from apoptosis, although cell-autonomous motor neuron degeneration still occurs.
Figure 5.
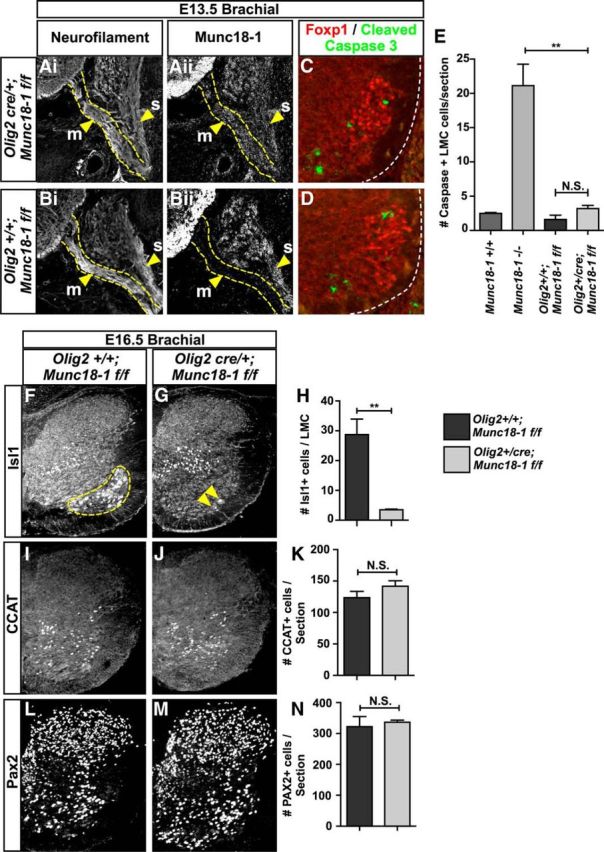
Cell death in Munc18-1 conditional mutants. Staining of neurofilament (Ai, Bi) and Munc18-1 (Aii, Bii) in Olig2+/cre;Munc18-1flox/flox mice reveals ablation of Munc18-1 expression in motor axons (m, arrowheads), but not sensory axons (s), demonstrating the loss of Munc18-1 specifically in motor neurons of Olig2+/cre;Munc18-1flox/flox mice (B) compared with Olig2+/+;Munc18-1flox/flox mice (A). Staining with Foxp1 (red) and cleaved caspase-3 (green) reveals the extent of cell death in the E13.5 brachial spinal cord of Olig2+/+;Munc18-1flox/flox mice (C, E; 1.61 ± 0.62, n = 4) compared with Olig2+/cre;Munc18-1flox/flox mice (D, E; 3.19 ± 0.48, n = 3, p = 0.12). Comparison between Olig2+/cre;Munc18-1flox/flox and Munc18-1−/− mice reveals a significant decrease in the frequency of caspase+ LMC neurons (E; 16.01 ± 2.57, Munc18-1−/−, n = 5, vs 3.19 ± 0.48, n = 3, p = 0.007) in Olig2+/cre;Munc18-1flox/flox mice. At E16.5, Isl1 staining reveals the absence of motor neurons in the ventral spinal cord of Olig2+/cre;Munc18-1flox/flox mice compared with Olig2+/+;Munc18-1flox/flox littermates (F–H; 28.7 ± 5.2, Olig2+/+;Munc18-1flox/flox, n = 2, 3.47 ± 0.26 (Olig2+/cre;Munc18-1flox/flox, n = 3, p = 0.0074). CCAT+ (I–K) and Pax2+ (L–N) neuron counts reveal no differences between Olig2+/cre;Munc18-1flox/flox (CCAT = 141.7 ± 8.69, n = 3; Pax2 = 336.7 ± 6.489, n = 3) and Olig2+/+;Munc18-1flox/flox mice (CCAT = 123.7 ± 9.838, n = 3, p = 0.242; Pax2 = 322.2 ± 32.67, n = 3, p = 0.687).
Neurotrophic signaling in Munc18-1 mutant spinal cords
Neurotrophic support promoting LMC neuron survival may originate from the periphery and from within the CNS (Oppenheim, 1996; Gould and Oppenheim, 2011). To assay peripheral LMC survival factor signaling, we took advantage of the observation that the expression of the transcription factor Pea3 is induced in motor neurons by a peripheral source of the neurotrophin GDNF (Haase et al., 2002). In E13.5 Munc18-1−/− spinal cords, Pea3 and the GDNF receptor c-Ret were expressed as in controls (Fig. 6A–F), which, combined with the fact that Munc18-1 is not prominently expressed outside of the nervous system (Fig. 1C), suggests that a loss of peripheral neurotrophic support is unlikely to be responsible for Munc18-1−/− LMC neuron apoptosis.
Figure 6.
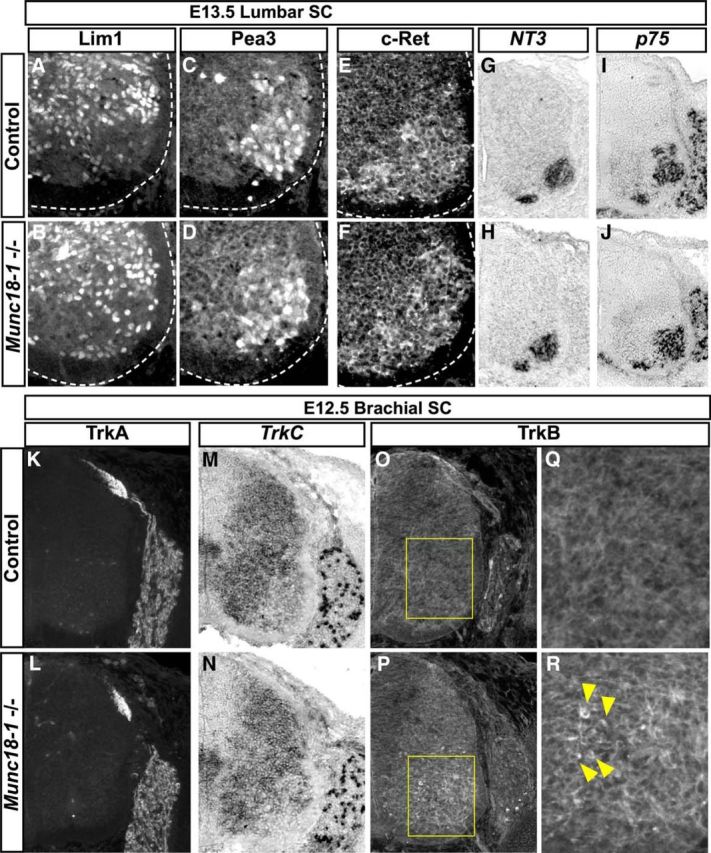
Expression and transduction of growth factor signaling in Munc18-1−/− mice. Normal transduction of GDNF signals from the limb upregulates Pea3 in a population of spinal motor neurons in both control (A, C) and Munc18-1−/− animals (B, D). Consistent with this, the GDNF receptor c-Ret is expressed normally in Munc18-1−/− motor neurons (E, F). Expression of NT3 (G, H) and p75 mRNAs (I, J) are also normal in Munc18-1−/− lumbar spinal cords at E13.5. Expression levels and distribution of TrkA protein (K, L) and TrkC mRNA (M, N) is comparable between control and Munc18-1−/− animals at E12.5. Immunostaining of TrkB reveals accumulations of signal in neurons within the ventral spinal cord of Munc18-1−/− animals that are not present in WT (O–R).
In the CNS, our microarray data demonstrate that the expression of mRNAs of neurotrophic factors and their receptors was normal in Munc18-1−/− spinal cords (Fig. 3I,J). Because whole spinal cord mRNA expression profiling does not allow the determination of which cells express a given mRNA and due to the possibility of apoptosis induction by local changes in expression of neurotrophin receptors and their ligands (Bamji et al., 1998; Béchade et al., 2002; Nikoletopoulou et al., 2010), we examined p75, TrkA, TrkC, and NT3 mRNAs and protein localization in E13.5 Munc18-1−/− spinal cord, where we found them to be indistinguishable from controls (Fig. 6G–N).We also examined the expression of the TrkB receptor, which, in addition to providing neurotrophic support (Klein et al., 1993), is also important for activity-dependent synaptic refinement (Wang and Poo, 1997). In WT mice, TrkB is expressed in DRG neurons and in a filamentous pattern throughout the spinal cord, consistent with its reported dendritic localization (Fig. 6O,Q; Yan et al., 1997). However, in Munc18-1−/− embryos, TrkB protein appears to accumulate in cells adjacent to the LMC at E12.5 (Fig. 6P,R) and E13.5 (Fig. 7C) despite normal mRNA expression (Fig. 3I,J), suggesting that trafficking of the TrkB receptor is compromised in Munc18-1−/− mice, which may reduce the transduction of survival-promoting BDNF signaling.
Figure 7.
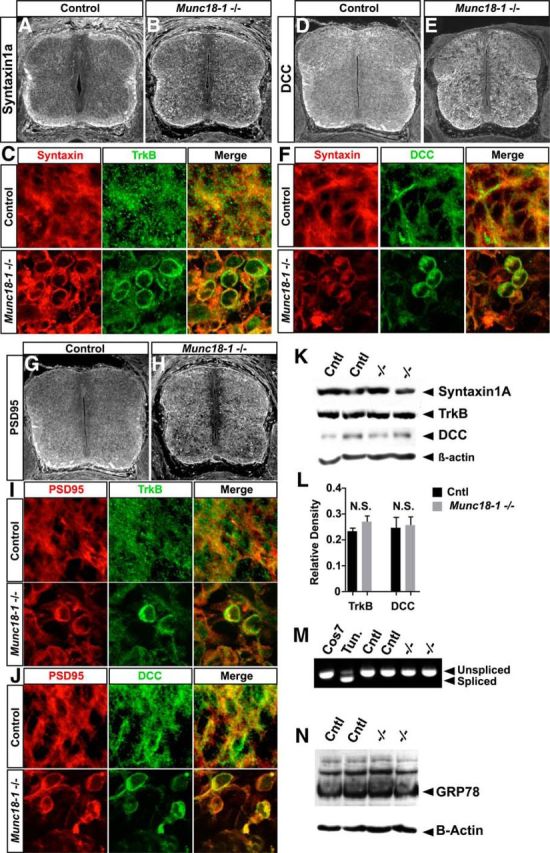
Protein mistrafficking in Munc18-1−/− mice. Syntaxin1a is expressed throughout the spinal cord, particularly in fiber tracts (A, B). In Munc18-1−/− spinal cords, accumulation of Syntaxin1a can be seen in the soma of neurons (B, C). TrkB and Syntaxin1a accumulate in the same neurons in Munc18-1−/− spinal cords (C). Similar to Syntaxin1a, DCC accumulates in Munc18-1−/− spinal neurons (D, E). DCC and Syntaxin can accumulate in the same neurons (F). The postsynaptic protein PSD-95 is expressed in the spinal cord and accumulates similarly to Syntaxin1a (G, H). Cells in which PSD-95 accumulates also display accumulation of TrkB (I) or DCC (J). Western blotting reveals that Syntaxin1a, DCC, and TrkB are found at the same molecular weight in control and Munc18-1−/− animals (K). In addition, expression levels of TrkB and DCC are unaffected in Munc18-1−/− spinal cords (L; TrkB, p = 0.212 n = 6; DCC, p = 0.855, n = 4). RT-PCR directed against XBP-1 reveals that tunicamycin treatment induces XBP-1 splicing in mouse embryonic fibroblasts, but that this splicing is not present in control or Munc18-1−/− spinal cords at E13.5 (M). Western blotting for GRP78 reveals no differences in expression levels between control and Munc18-1−/− animals at E13.5 (N).
Intracellular protein trafficking defects in Munc18-1−/− neurons
The mislocalization of TrkB in Munc18-1−/− neurons (Fig. 6P–R) and the observation that, in C. elegans, UNC-18 is necessary for the correct trafficking of the homolog of mouse Syntaxin-1a (McEwen and Kaplan, 2008) suggests that Munc18-1 may influence the trafficking of other proteins. In addition, in Munc18-1−/− E18 mouse cortex, Syntaxin1a protein levels are reduced, suggesting that Munc18-1 stabilizes Syntaxin1a, perhaps acting as a chaperone (Toonen et al., 2005). We thus considered whether the aberrant localization of TrkB receptors (Fig. 6M–O) might be a result of more general protein trafficking defects in Munc18-1−/− mutants. At E13.5, similar levels of the Syntaxin1a protein were present in mutant and control spinal cords as determined by Western blot (Fig. 7K); however, its localization appeared abnormal in Munc18-1−/− mutants, in which essentially all spinal neurons exhibit somatic accumulations of Syntaxin1a (Fig. 7A–C). Moreover, at the level of single cells, Syntaxin1a and TrkB protein colocalize extensively (Fig. 7C). We next examined the cellular distribution of the axonal protein DCC, the distribution of which can be influenced by activity (Bouchard et al., 2008), and found that, at E13.5, its spinal cord expression appeared to be increased in Munc18-1−/− mutants (Fig. 7D,E) and coincided with Syntaxin1a's somatic accumulation (Fig. 7F), although its levels in peripheral motor axons were comparable between genotypes at E12.5 (Fig. 3E,F). We also examined the localization of the postsynaptic protein PSD-95, which normally interacts with the Munc18-1-binding partner CDK5 (Morabito et al., 2004); at E13.5, in the absence of Munc18-1, the localization of PDS95 appeared disrupted, with multiple neurons showing strong accumulation of PSD-95 and overlap with mislocalised TrkB and DCC (Fig. 7I,J).
Given that both TrkB and DCC are glycosylated proteins (Reale et al., 1994; Haniu et al., 1995) and that aberrant glycosylation may result in somatic accumulation of misfolded proteins (Raposo et al., 1995), we examined the extent of DCC and TrkB glycosylation in spinal cords of E13.5 mice by Western blot and quantified these to verify that Munc18-1 ablation did not lead to overexpression of these proteins. In the absence of Munc18-1, both DCC and TrkB migrated at the same molecular weight as in controls (Fig. 7K), suggesting that a lack of glycosylation is not responsible for the mislocalization of TrkB and DCC in Munc18-1−/− mutants. Quantification revealed no significant differences in levels of expression between control and Munc18-1−/− spinal cords (TrkB: p = 0.26, n = 6; DCC: p = 0.71, n = 4; Fig. 7L), demonstrating that the aggregation of TrkB and DCC was not due to their overexpression.
Protein mislocalization to the endoplasmic reticulum resulting in ER stress has been proposed as a pathogenic mechanism in some neurodegenerative disorders (Saxena et al., 2009). We thus monitored the presence of spliced transcripts of the XBP-1 gene, a known indicator of ER stress (Yoshida et al., 2001), in spinal cords of Munc18-1−/− mutants and controls at E13.5, when many neurons are apoptotic (Fig. 4) and/or display accumulations of Syntaxin1a, DCC, TrkB, and/or PSD-95 (Fig. 7). As a positive control, we used tunicamycin-treated mouse embryonic fibroblasts producing an ER-stress-specific spliced isoform of XBP-1 mRNA, detectable by PCR (Fig. 7M). In contrast, we did not detect this isoform in Munc18-1−/− spinal cords (Fig. 7M). In addition, the expression of GRP78, another marker of ER-stress responses (Haze et al., 1999), was unchanged in E13.5 mutants (Fig. 7N), demonstrating that, despite the mislocalization of Syntaxin1a, PSD-95, TrkB, and DCC, the cellular pathways that respond to ER stress are unlikely to be activated in Munc18-1−/− mutants.
Neurodegenerative markers in Munc18-1−/− neurons
To gain further insight into neuronal degeneration in Munc18-1−/− mutants, we next examined hallmarks of neurodegenerative diseases such as activated inflammatory response (Glass et al., 2010), α-synuclein expression (Bendor et al., 2013), phospho-Tau localization (Lee et al., 2001), Congo Red reactivity (Hardy and Selkoe, 2002; Ross and Poirier, 2004), and ubiquitination (Atkin and Paulson, 2014). At E13.5, the time of intense neurodegeneration in Munc18-1−/− mutants, the microglial markers Iba1 and Cd11b, which are upregulated in some neurodegenerative conditions (Glass et al., 2010), appears unchanged in Munc18-1−/− mutants (Fig. 8A–D). In addition, we did not detect changes in the expression of α-synuclein in the spinal cords of Munc18-1−/− embryos (Fig. 8E,F).
Figure 8.
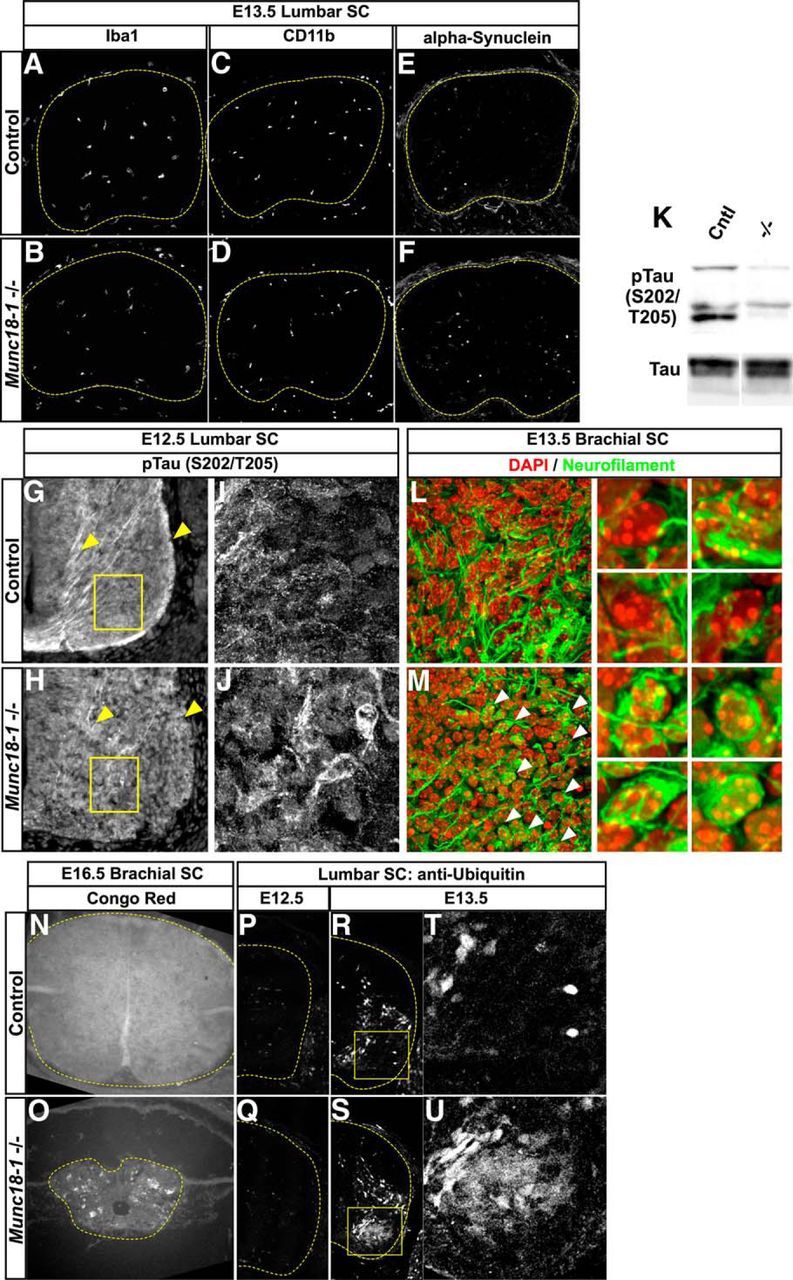
Neurodegenerative markers in Munc18-1−/− mice. Immunostaining for the microglial markers Iba1 (A, B) and CD11b (C, D) reveal no differences between control and Munc18-1−/− mice. Alpha synuclein expression also appears similar between control and Munc18-1−/− mice (E, F). Immunostaining for pTau expression in Munc18-1−/− mice at E12.5 reveals accumulations within neurons in the spinal cord that are not present in WT (G–J) and a reduction of expression in axons (G, H, arrowheads). Western blotting reveals a reduction in pTau, but not total Tau, levels in lysates from E13.5 Munc18-1−/− spinal cords (K). Immunostaining at E13.5 reveals multiple aggregations of neurofilament in Munc18-1−/− animals (M, arrowheads) that are not present in controls (L). Congo Red staining reveals UV-emitting accumulations of signal in Munc18-1−/− degenerating spinal cords at E16.5 that are not present in controls (N, O). Immunostaining for ubiquitinated proteins at E12.5 reveals no differences between control and Munc18-1−/− animals (P, Q); however, at E13.5, there is an increase in ubiquitin staining in motor neurons of Munc18-1−/− mice (R–U).
Munc18-1 is known to interact with Cdk5, a kinase that phosphorylates Tau (Bhaskar et al., 2004), and aberrant phosphorylation of Tau is a known component of some neurodegenerative diseases including ALS and Alzheimer's (Buée et al., 2000; Lee et al., 2001; Rudrabhatla, 2014). We monitored Tau phosphorylation at Ser202 and Thr205 (pTau) by immunostaining of E12.5 spinal cords with the AT8 antibody (Oddo et al., 2003; Ballatore et al., 2007) and found that, in E12.5 Munc18-1−/− spinal cords, there was an abnormal reduction in pTau levels (Fig. 8G–J, arrowheads), confirmed by Western blotting (Fig. 8K) and accompanied by accumulation of pTau within neuronal cell bodies in the ventral spinal cord (Fig. 8G–I), demonstrating that Munc18-1−/−-associated neurodegeneration exhibits a tauopathy hallmark (Varoqueaux et al., 2002; Myers et al., 2005; Ballatore et al., 2007; Benjumeda et al., 2013; Puchtler et al., 1961). Neurofilament immunostaining at E13.5 revealed neurofibrillary tangles appearing in many Munc18-1−/− neurons (Fig. 8L,M). The perinuclear accumulation of neurofilament immunoreactivity was similar to that of PSD-95 and Syntaxin1a (Fig. 8L,M, arrowheads, insets; cf. Fig. 7B,H).
Next, we performed Congo Red staining, which is a histochemical method allowing the detection of structures rich in β-sheets, including the amyloid plaques common in Alzheimer's disease (Jankowsky et al., 2001; Borodinsky et al., 2004; Demarque and Spitzer, 2010; Puchtler et al., 1961). Cortices of aged mice carrying the 3xTg-AD transgenes [Tg(APPSwe,tauP301L)1Lfa; Jankowsky et al., 2001] contain many amyloid plaques that stain strongly with Congo Red and may be visualized by fluorescent microscopy using Texas Red and ultraviolet filters (data not shown). At E16.5, intense Congo Red staining could be detected throughout Munc18-1−/− spinal cords compared with nondegenerating WT spinal cords (Fig. 8N,O). Finally, because ubiquitination of misfolded proteins is also a hallmark of several neurodegenerative diseases (Atkin and Paulson, 2014), we examined its extent in Munc18-1−/− mutants (Fig. 8P–U). Although, at E12.5, we did not detect any ubiquitin upregulation in Munc18-1−/− mutants (Fig. 8P,Q), at E13.5, mutant spinal cords contained many neurons with elevated ubiquitin levels (Fig. 8R–U), suggesting that Munc18-1 loss results in increased protein degradation. Together, these results demonstrate that, despite the absence of activated ER stress response, Munc18-1 mutants have misregulated Tau phosphorylation, neurofibrillary tangles, amyloid accumulation, and an increased ubiquitination, all common features of neurodegenerative disorders.
Discussion
Summary
In this report, by studying the Munc18-1−/− nervous system, we demonstrate that synaptic activity is dispensable for the early formation of spinal motor circuits at the levels of axon guidance, differentiation of transcriptional identity, and mRNA expression. We also show that aspects of neurotrophin signaling are normal in Munc18-1 mutants, but uncover evidence of abnormal accumulation of multiple proteins that is not a result of misglycosylation and does not lead to ER stress responses. Munc18-1−/− neurons degenerate in a cell-autonomous fashion in the absence of inflammatory responses or α-synuclein accumulation; however, Munc18-1−/− neurons display Alzheimer's disease hallmarks such as aberrant tau phosphorylation, accumulation of neurofibrillary tangles in neurons, and amyloid accumulations in degenerating spinal cords. Here, we discuss these results in the context of previous findings regarding the role of neuronal activity in development, the cellular function of Munc18-1, and neurodegeneration.
Role of synaptic activity in spinal neuron development
It has been proposed that patterned neural activity influences the early development of neuronal circuits before its function in synaptic competition and pruning (for review, see Spitzer, 2006); however, this has been controversial, with some reports demonstrating that early activity is dispensable for normal development (Varoqueaux et al., 2002; Myers et al., 2005; Benjumeda et al., 2013). In this study, we demonstrate that, in vivo, the molecular specification of motor neurons and interneurons in the murine spinal cord, the growth of their axons, and the expression of mRNA is unaffected by the loss of synaptic activity.
Experiments in which synaptic activity can be demonstrated to affect aspects of neuronal differentiation (Borodinsky et al., 2004; Demarque and Spitzer, 2010) and path finding (Ming et al., 2001; Nishiyama et al., 2003; Hanson and Landmesser, 2004; Kastanenka and Landmesser, 2010) rely on pharmacological means or overexpression of ion channels to disrupt activity. However, a study of ChAT mutant mice, in which residual glutamatergic neurotransmission results in reduced motor circuit activity, failed to reveal any differences in motor circuit formation other than an increased number of motor neurons due to lack of acetylcholine-mediated competition (Myers et al., 2005). Similarly, a study of cortical mRNAs from E14.5–E18.5 Munc18-1−/− mice revealed only a single neurotransmitter identity gene expression change at the oldest age examined (Bouwman et al., 2006). In our study, the absence of neurotransmission precludes confounding effects of compensatory neurotransmitter activity (Myers et al., 2005) and our survey of the transcriptional identity of motor neurons and interneurons provides the most complete evidence for the absence of requirement neurotransmitter release in murine neural circuit formation. Alongside other studies (Varoqueaux et al., 2002; Myers et al., 2005; Benjumeda et al., 2013), our analysis indicates that the early events in the molecular diversification of neurons do not critically depend on synaptic activity because both evoked and spontaneous neurotransmitter release are blocked in Munc18-1−/− mice (Verhage et al., 2000).
Mechanisms of neurodegeneration in Munc18-1−/− spinal cords
Motor neurons are generated in excess and undergo programmed cell death (Hamburger, 1975; Chu-Wang and Oppenheim, 1978). Although it is less well described, programmed cell death also occurs in spinal interneurons (Prasad et al., 2008). Our study shows that, in Munc18-1−/− mice, LMC neuron death begins earlier (E12.5) and is more extensive than normal. This phenotype is not observed in other mutants in which synaptic vesicle release is blocked, such as in the Munc13–1;Munc13–2 double mutant mice (Varoqueaux et al., 2002, 2005), suggesting that it is not a consequence of synaptic activity loss. As suggested by in vitro studies (Heeroma et al., 2004), the degeneration of Munc18-1−/− neurons appears to be partially suppressed by neighboring neurons with WT Munc18-1 or Munc18-1flox alleles, demonstrating that the mechanisms that lead to degeneration can be temporarily rescued by an influence from control neurons. However, such rescue is only temporary because Munc18-1−/− neurons degenerate, suggesting that the apoptotic mechanism inevitably overwhelms any prosurvival contribution from control neurons. Despite the lack of secretion in Drosophila melanogaster embryos lacking the Munc18 homolog Rop (Harrison et al., 1994) and the reduction in dense-core vesicle docking in adrenochromaffin cells lacking Munc18-1 (Voets et al., 2001), it seems unlikely that Munc18-1−/− degeneration is caused by lack of trophic factor support for the following reasons: (1) neuronal survival factor mRNA expression of in the CNS appears normal; (2) Munc18-1−/− motor neurons can transduce neurotrophic signals correctly; (3) LMC neurons in motor-neuron-specific knock-outs of Munc18-1 degenerate in the presence of presumably normal growth factor support from both the CNS and periphery; and (4) when activity-dependent competition for growth factors is reduced or eliminated, greater numbers of motor neurons survive (Pittman and Oppenheim, 1978; Myers et al., 2005; Varoqueaux et al., 2005). Despite accumulation of the TrkB receptor in a subpopulation of neurons (Figs. 6O–R, 7B,C), which may result in lower cellular response to BDNF signals, the size of that subpopulation of neurons is too small to account for the near-complete degeneration of the spinal cord observed in Munc18-1−/− mice. Together, these results suggest a neurotrophic support-independent mechanism for Munc18-1−/− neurodegeneration.
Similarities between Munc18-1−/− and neurodegenerative disease
Our results suggest that the mistrafficking of proteins such as DCC and TrkB in Munc18-1−/− spinal cords is not caused by aberrant glycosylation and does not result in ER stress, so how do Munc18-1 mutant neurons degenerate? Our molecular characterization of Munc18-1−/− mutants reveals the presence of striking pathological neurodegenerative hallmarks. Munc18-1 associates with membrane microdomains rich in APP and miRNA knock-down of Munc18-1 can lead to a shift in APP distribution to domains containing BACE1, promoting the formation of toxic αβ fragments of APP (Sakurai et al., 2008). These αβ fragments are toxic before their aggregation into fibrils visible with light microscopy (Harmeier et al., 2009).
In Munc18-1−/− mice, similar to WT, there is a correlation between the onset of degeneration and the time since cell cycle exit (Gould et al., 1999). Given that the expression of Munc18-1 and APP appears restricted to postmitotic neurons (Visel et al., 2004) and that the accumulation of toxic forms of APP can lead to neurodegeneration (Harmeier et al., 2009), we propose that, among other effects, the loss of Munc18-1 causes misprocessing of APP in postmitotic neurons, leading to degeneration of these neurons within a few days of their exit from the cell cycle.
The reduction of Tau phosphorylation observed by Western blotting is most likely due to the activity of Munc18-1 (p67) as an activator of CDK5 (Shetty et al., 1995), which can phosphorylate Tau and coprecipitates with Munc18-1 and cytoskeletal components (Bhaskar et al., 2004). That pTau inclusions are found in the soma of neurons whereas overall levels of pTau are reduced in the Munc18-1−/− spinal cord agrees with results from CDK5-null mice, in which accumulations of phosphorylated neurofilaments can be seen in neuronal soma (Ohshima et al., 1996); such tangles also include neurofilaments other than Tau (Rudrabhatla et al., 2011). Although mice lacking CDK5 also undergo increased neurodegeneration, this occurs at a much lower rate than in Munc18-1−/− mutants (Verhage et al., 2000; Li et al., 2002). This fact, combined with the absence of cortical layering defects in Munc18-1 mutants (Ohshima et al., 1996; Verhage et al., 2000), suggests that a large component of the Munc18-1 phenotype is not mediated by reduced CDK5 activity.
The non-cell-autonomous suppression of degeneration of Munc18-1−/− neurons seems unexpected, but may be explained by the observation of amyloid plaque depositions in Munc18-1−/− spinal cords and the observation that loss of Munc18-1 may lead to aberrant APP processing (Sakurai et al., 2008). If processing of APP is abnormal in Munc18-1−/− neurons, then toxic species may be removed by control neurons. We hypothesize that the temporary nature of this protective effect is due to sufficiently high levels of αβ production in Munc18-1−/− neurons to preclude effective clearance by control neurons over the long term. Finally, Western blotting of brain samples from human Alzheimer's patients shows a specific reduction of the Munc18-1a splice form, but not others, in the brains of patients with advanced dementia (Ramos-Miguel et al., 2014), providing further evidence that loss of Munc18-1 correlates with Alzheimer's symptoms. Heterozygous mutations in STXBP1, the human Munc18-1 homolog, cause Ohtahara and Dravet's syndromes, which are characterized by early-onset epilepsy (Saitsu et al., 2008; Carvill et al., 2014); Munc18-1−/−-like neurodegeneration in these patients has not been yet reported. Although the degeneration occurring in Munc18-1−/− animals occurs prenatally and does not appear to have an ER stress component, given the evidence of amyloid plaques in the degenerating Munc18-1−/− spinal cord, the abnormal processing of APP in neurons lacking Munc18-1 (Sakurai et al., 2008), and the splice-specific reduction of Munc18-1a in Alzheimer's patients (Ramos-Miguel et al., 2014), we propose that the degeneration observed in Munc18-1−/− mice may provide novel insights into mechanisms underlying neurodegeneration in Alzheimer's disease.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research, the Canada Foundation for Innovation, Brain Canada, and the W. Garfield Weston Foundation (A.K.); the Natural Sciences and Engineering Research Council of Canada (M.L.); the National Institutes of Health (Grant R01 NS083998); and the Whitehall Foundation Research (J.A.K.). We thank Keith Murai for providing us with 3xTg-AD brain sections; Bennett Novitch, Tom Jessell, Keith Murai, Philip Barker, and Michel Cayouette for providing us with antibodies; Meirong Liang, Julie Cardin, Christine Jolicoeur, and Dominic Filion for technical assistance; and Isabelle Brisson, Joke Wortel, and Kim Kridsada for mouse husbandry and colony management.
The authors declare no competing financial interests.
References
- Atkin G, Paulson H. Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci. 2014;7:63. doi: 10.3389/fnmol.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Chivatakarn O, Bonanomi D, Lettieri K, Franco L, Xia C, Stein E, Ma L, Lewcock JW, Pfaff SL. Presenilin-dependent receptor processing is required for axon guidance. Cell. 2011;144:106–118. doi: 10.1016/j.cell.2010.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béchade C, Mallecourt C, Sedel F, Vyas S, Triller A. Motoneuron-derived neurotrophin-3 is a survival factor for PAX2-expressing spinal interneurons. J Neurosci. 2002;22:8779–8784. doi: 10.1523/JNEUROSCI.22-20-08779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendor JT, Logan TP, Edwards RH. The function of a-synuclein. Neuron. 2013;79:1044–1066. doi: 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. JSTOR: J of the Royal Statistical Society. 1995 Series B (Methodological), Vol. 57, No. 1 (1995), 289–300. J of the Royal Statistical Society: Series B (Statistical Methodology) [Google Scholar]
- Benjumeda I, Escalante A, Law C, Morales D, Chauvin G, Muça G, Coca Y, Márquez J, López-Bendito G, Kania A, Martínez L, Herrera E. Uncoupling of EphA/ephrinA signaling and spontaneous activity in neural circuit wiring. J Neurosci. 2013;33:18208–18218. doi: 10.1523/JNEUROSCI.1931-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar K, Shareef MM, Sharma VM, Shetty AP, Ramamohan Y, Pant HC, Raju TR, Shetty KT. Co-purification and localization of Munc18-1 (p67) and Cdk5 with neuronal cytoskeletal proteins. Neurochemistry International. 2004;44:35–44. doi: 10.1016/S0197-0186(03)00099-8. [DOI] [PubMed] [Google Scholar]
- Bittman KS, Panzer JA, Balice-Gordon RJ. Patterns of cell-cell coupling in embryonic spinal cord studied via ballistic delivery of gap-junction-permeable dyes. J Comp Neurol. 2004;477:273–285. doi: 10.1002/cne.20253. [DOI] [PubMed] [Google Scholar]
- Borodinsky LN, Root CM, Cronin JA, Sann SB, Gu X, Spitzer NC. Activity-dependent homeostatic specification of transmitter expression in embryonic neurons. Nature. 2004;429:523–530. doi: 10.1038/nature02518. [DOI] [PubMed] [Google Scholar]
- Bouchard JF, Horn KE, Stroh T, Kennedy TE. Depolarization recruits DCC to the plasma membrane of embryonic cortical neurons and enhances axon extension in response to netrin-1. J Neurochem. 2008;107:398–417. doi: 10.1111/j.1471-4159.2008.05609.x. [DOI] [PubMed] [Google Scholar]
- Bouwman J, Maia AS, Camoletto PG, Posthuma G, Roubos EW, Oorschot VM, Klumperman J, Verhage M. Quantification of synapse formation and maintenance in vivo in the absence of synaptic release. Neuroscience. 2004;126:115–126. doi: 10.1016/j.neuroscience.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Bouwman J, Spijker S, Schut D, Wächtler B, Ylstra B, Smit AB, Verhage M. Reduced expression of neuropeptide genes in a genome-wide screen of a secretion-deficient mouse. J Neurochem. 2006;99:84–96. doi: 10.1111/j.1471-4159.2006.04041.x. [DOI] [PubMed] [Google Scholar]
- Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/S0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Carvill GL, Weckhuysen S, McMahon JM, Hartmann C, Møller RS, Hjalgrim H, Cook J, Geraghty E, O'Roak BJ, Petrou S, Clarke A, Gill D, Sadleir LG, Muhle H, von Spiczak S, Nikanorova M, Hodgson BL, Gazina EV, Suls A, Shendure J, Dibbens LM, De Jonghe P, Helbig I, Berkovic SF, Scheffer IE, Mefford HC. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. 2014;82:1245–1253. doi: 10.1212/WNL.0000000000000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu-Wang IW, Oppenheim RW. Cell death of motoneurons in the chick embryo spinal cord. I. A light and electron microscopic study of naturally occurring and induced cell loss during development. J Comp Neurol. 1978;177:33–57. doi: 10.1002/cne.901770105. [DOI] [PubMed] [Google Scholar]
- Crisp SJ, Evers JF, Bate M. Endogenous patterns of activity are required for the maturation of a motor network. J Neurosci. 2011;31:10445–10450. doi: 10.1523/JNEUROSCI.0346-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnecki A, Le Corronc H, Rigato C, Le Bras B, Couraud F, Scain AL, Allain AE, Mouffle C, Bullier E, Mangin JM, Branchereau P, Legendre P. Acetylcholine controls GABA-, glutamate-, and glycine-dependent giant depolarizing potentials that govern spontaneous motoneuron activity at the onset of synaptogenesis in the mouse embryonic spinal cord. J Neurosci. 2014;34:6389–6404. doi: 10.1523/JNEUROSCI.2664-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasen JS, De Camilli A, Wang B, Tucker PW, Jessell TM. Hox repertoires for motor neuron diversity and connectivity gated by a single accessory factor, FoxP1. Cell. 2008;134:304–316. doi: 10.1016/j.cell.2008.06.019. [DOI] [PubMed] [Google Scholar]
- Demarque M, Spitzer NC. Activity-dependent expression of Lmx1b regulates specification of serotonergic neurons modulating swimming behavior. Neuron. 2010;67:321–334. doi: 10.1016/j.neuron.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppmann CD, Mihalas S, Sharma N, Lonze BE, Niebur E, Ginty DD. A model for neuronal competition during development. Science. 2008;320:369–373. doi: 10.1126/science.1152677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessaud E, Yang LL, Hill K, Cox B, Ulloa F, Ribeiro A, Mynett A, Novitch BG, Briscoe J. Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature. 2007;450:717–720. doi: 10.1038/nature06347. [DOI] [PubMed] [Google Scholar]
- Dieni S, Matsumoto T, Dekkers M, Rauskolb S, Ionescu MS, Deogracias R, Gundelfinger ED, Kojima M, Nestel S, Frotscher M, Barde YA. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J Cell Biol. 2012;196:775–788. doi: 10.1083/jcb.201201038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudok JJ, Groffen AJ, Toonen RF, Verhage M. Deletion of Munc18-1 in 5-HT neurons results in rapid degeneration of the 5-HT system and early postnatal lethality. PLoS One. 2011;6:e28137. doi: 10.1371/journal.pone.0028137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, La Spada AR. Intercellular (mis)communication in neurodegenerative disease. Neuron. 2012;73:886–901. doi: 10.1016/j.neuron.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TW, Oppenheim RW. Motor neuron trophic factors: therapeutic use in ALS? Brain Res Rev. 2011;67:1–39. doi: 10.1016/j.brainresrev.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TW, Burek MJ, Sosnowski JM, Prevette D, Oppenheim RW. The spatial-temporal gradient of naturally occurring motoneuron death reflects the time of prior exit from the cell cycle and position within the lateral motor column. Dev Biol. 1999;216:611–621. doi: 10.1006/dbio.1999.9490. [DOI] [PubMed] [Google Scholar]
- Haase G, Dessaud E, Garcès A, de Bovis B, Birling M, Filippi P, Schmalbruch H, Arber S, deLapeyrière O. GDNF acts through PEA3 to regulate cell body positioning and muscle innervation of specific motor neuron pools. Neuron. 2002;35:893–905. doi: 10.1016/S0896-6273(02)00864-4. [DOI] [PubMed] [Google Scholar]
- Hamburger V. Cell death in the development of the lateral motor column of the chick embryo. J Comp Neurol. 1975;160:535–546. doi: 10.1002/cne.901600408. [DOI] [PubMed] [Google Scholar]
- Haniu M, Talvenheimo J, Le J, Katta V, Welcher A, Rohde MF. Extracellular domain of neurotrophin receptor trkB: disulfide structure, N-glycosylation sites, and ligand binding. Arch Biochem Biophys. 1995;322:256–264. doi: 10.1006/abbi.1995.1460. [DOI] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Characterization of the circuits that generate spontaneous episodes of activity in the early embryonic mouse spinal cord. J Neurosci. 2003;23:587–600. doi: 10.1523/JNEUROSCI.23-02-00587.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Normal patterns of spontaneous activity are required for correct motor axon guidance and the expression of specific guidance molecules. Neuron. 2004;43:687–701. doi: 10.1016/j.neuron.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harmeier A, Wozny C, Rost BR, Munter LM, Hua H, Georgiev O, Beyermann M, Hildebrand PW, Weise C, Schaffner W, Schmitz D, Multhaup G. Role of amyloid-beta glycine 33 in oligomerization, toxicity, and neuronal plasticity. J Neurosci. 2009;29:7582–7590. doi: 10.1523/JNEUROSCI.1336-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SD, Broadie K, van de Goor J, Rubin GM. Mutations in the Drosophila Rop gene suggest a function in general secretion and synaptic transmission. Neuron. 1994;13:555–566. doi: 10.1016/0896-6273(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Hata Y, Südhof TC. A novel ubiquitous form of Munc-18 interacts with multiple syntaxins: use of the yeast two-hybrid system to study interactions between proteins involved in membrane traffic. J Biol Chem. 1995;270:13022–13028. doi: 10.1074/jbc.270.22.13022. [DOI] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeroma JH, Plomp JJ, Roubos EW, Verhage M. Development of the mouse neuromuscular junction in the absence of regulated secretion. Neuroscience. 2003;120:733–744. doi: 10.1016/S0306-4522(03)00258-6. [DOI] [PubMed] [Google Scholar]
- Heeroma JH, Roelandse M, Wierda K, van Aerde KI, Toonen RF, Hensbroek RA, Brussaard A, Matus A, Verhage M. Trophic support delays but does not prevent cell-intrinsic degeneration of neurons deficient for Munc18-1. Eur J Neurosci. 2004;20:623–634. doi: 10.1111/j.1460-9568.2004.03503.x. [DOI] [PubMed] [Google Scholar]
- Helmbacher F, Schneider-Maunoury S, Topilko P, Tiret L, Charnay P. Targeting of the EphA4 tyrosine kinase receptor affects dorsal/ventral pathfinding of limb motor axons. Development. 2000;127:3313–3324. doi: 10.1242/dev.127.15.3313. [DOI] [PubMed] [Google Scholar]
- Hinckley CA, Alaynick WA, Gallarda BW, Hayashi M, Hilde KL, Driscoll SP, Dekker JD, Tucker HO, Sharpee TO, Pfaff SL. Spinal locomotor circuits develop using hierarchical rules based on motorneuron position and identity. Neuron. 2015;87:1008–1021. doi: 10.1016/j.neuron.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollyday M, Hamburger V. An autoradiographic study of the formation of the lateral motor column in the chick embryo. Brain Res. 1977;132:197–208. doi: 10.1016/0006-8993(77)90416-4. [DOI] [PubMed] [Google Scholar]
- Hory-Lee F, Frank E. The nicotinic blocking agents d-tubocurare and alpha-bungarotoxin save motoneurons from naturally occurring death in the absence of neuromuscular blockade. J Neurosci. 1995;15:6453–6460. doi: 10.1523/JNEUROSCI.15-10-06453.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/S1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Kao TJ, Kania A. Ephrin-mediated cis-attenuation of Eph receptor signaling is essential for spinal motor axon guidance. Neuron. 2011;71:76–91. doi: 10.1016/j.neuron.2011.05.031. [DOI] [PubMed] [Google Scholar]
- Kao TJ, Law C, Kania A. Eph and ephrin signaling: lessons learned from spinal motor neurons. Semin Cell Dev Biol. 2012;23:83–91. doi: 10.1016/j.semcdb.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Kastanenka KV, Landmesser LT. In vivo activation of channelrhodopsin-2 reveals that normal patterns of spontaneous activity are required for motoneuron guidance and maintenance of guidance molecules. J Neurosci. 2010;30:10575–10585. doi: 10.1523/JNEUROSCI.2773-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. doi: 10.1016/S0092-8674(05)80088-1. [DOI] [PubMed] [Google Scholar]
- Lance-Jones C. Motoneuron cell death in the developing lumbar spinal cord of the mouse. Brain Res. 1982;256:473–479. doi: 10.1016/0165-3806(82)90192-4. [DOI] [PubMed] [Google Scholar]
- Law C, Paquet M, Kania A. Emergence of motor circuit activity. PLoS One. 2014;9:e93836. doi: 10.1371/journal.pone.0093836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Li BS, Zhang L, Takahashi S, Ma W, Jaffe H, Kulkarni AB, Pant HC. Cyclin-dependent kinase 5 prevents neuronal apoptosis by negative regulation of c-Jun N-terminal kinase 3. EMBO J. 2002;21:324–333. doi: 10.1093/emboj/21.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek KW, Kurtz LM, Spitzer NC. cJun integrates calcium activity and tlx3 expression to regulate neurotransmitter specification. Nat Neurosci. 2010;13:944–950. doi: 10.1038/nn.2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen JM, Kaplan JM. UNC-18 promotes both the anterograde trafficking and synaptic function of syntaxin. Mol Biol Cell. 2008;19:3836–3846. doi: 10.1091/mbc.E08-02-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner LD, Landmesser LT. Cholinergic and GABAergic inputs drive patterned spontaneous motoneuron activity before target contact. J Neurosci. 1999;19:3007–3022. doi: 10.1523/JNEUROSCI.19-08-03007.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming G, Henley J, Tessier-Lavigne M, Song H, Poo M. Electrical activity modulates growth cone guidance by diffusible factors. Neuron. 2001;29:441–452. doi: 10.1016/S0896-6273(01)00217-3. [DOI] [PubMed] [Google Scholar]
- Morabito MA, Sheng M, Tsai LH. Cyclin-dependent kinase 5 phosphorylates the N-terminal domain of the postsynaptic density protein PSD-95 in neurons. J Neurosci. 2004;24:865–876. doi: 10.1523/JNEUROSCI.4582-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CP, Lewcock JW, Hanson MG, Gosgnach S, Aimone JB, Gage FH, Lee KF, Landmesser LT, Pfaff SL. Cholinergic input is required during embryonic development to mediate proper assembly of spinal locomotor circuits. Neuron. 2005;46:37–49. doi: 10.1016/j.neuron.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Neefjes J, van der Kant R. Stuck in traffic: an emerging theme in diseases of the nervous system. Trends Neurosci. 2014;37:66–76. doi: 10.1016/j.tins.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Nikoletopoulou V, Lickert H, Frade JM, Rencurel C, Giallonardo P, Zhang L, Bibel M, Barde YA. Neurotrophin receptors TrkA and TrkC cause neuronal death whereas TrkB does not. Nature. 2010;467:59–63. doi: 10.1038/nature09336. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hoshino A, Tsai L, Henley JR, Goshima Y, Tessier-Lavigne M, Poo MM, Hong K. Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature. 2003;423:990–995. doi: 10.1038/nature01751. [DOI] [PubMed] [Google Scholar]
- Novitch BG, Chen AI, Jessell TM. Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. Neuron. 2001;31:773–789. doi: 10.1016/S0896-6273(01)00407-X. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci U S A. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. Neurotrophic survival molecules for motoneurons: an embarrassment of riches. Neuron. 1996;17:195–197. doi: 10.1016/S0896-6273(00)80151-8. [DOI] [PubMed] [Google Scholar]
- Palmesino E, Rousso DL, Kao TJ, Klar A, Laufer E, Uemura O, Okamoto H, Novitch BG, Kania A. Foxp1 and lhx1 coordinate motor neuron migration with axon trajectory choice by gating Reelin signalling. PLoS Biol. 2010;8:e1000446. doi: 10.1371/journal.pbio.1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevsner J, Hsu SC, Braun JE, Calakos N, Ting AE, Bennett MK, Scheller RH. Specificity and regulation of a synaptic vesicle docking complex. Neuron. 1994a;13:353–361. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- Pevsner J, Hsu SC, Scheller RH. n-Sec1: a neural-specific syntaxin-binding protein. Proc Natl Acad Sci U S A. 1994b;91:1445–1449. doi: 10.1073/pnas.91.4.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman R, Oppenheim RW. Cell death of motoneurons in the chick embryo spinal cord. IV. Evidence that a functional neuromuscular interaction is involved in the regulation of naturally occurring cell death and the stabilization of synapses. J Comp Neurol. 1979;187:425–446. doi: 10.1002/cne.901870210. [DOI] [PubMed] [Google Scholar]
- Pittman RH, Oppenheim RW. Neuromuscular blockade increases motoneurone survival during normal cell death in the chick embryo. Nature. 1978;271:364–366. doi: 10.1038/271364a0. [DOI] [PubMed] [Google Scholar]
- Prasad T, Wang X, Gray PA, Weiner JA. A differential developmental pattern of spinal interneuron apoptosis during synaptogenesis: insights from genetic analyses of the protocadherin- gene cluster. Development. 2008;135:4153–4164. doi: 10.1242/dev.026807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchtler H, Sweat F, Levine M. On the binding of Congo Red by amyloid. 1961. [Accessed April 24, 2015]. Available from: http://jhc.sagepub.com/content/10/3/355.full.pdf.
- Ramos-Miguel A HC, Beasley CL, Barr AM, Schneider JA, Bennett DA HW. 2014 Neuroscience Meeting Planner. Washington, DC: Society for Neuroscience; 2014. Differential regulation of cortical Munc18–1 splice variants in Alzheimer's disease: correlations with the severity of cognitive decline and neuropathology in a community-based aging study. Program No. 43.19/F2. [Google Scholar]
- Raposo G, van Santen HM, Leijendekker R, Geuze HJ, Ploegh HL. Misfolded major histocompatibility complex class I molecules accumulate in an expanded ER-Golgi intermediate compartment. J Cell Biol. 1995;131:1403–1419. doi: 10.1083/jcb.131.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reale MA, Hu G, Zafar AI, Getzenberg RH, Levine SM, Fearon ER. Expression and alternative splicing of the deleted in colorectal cancer (DCC) gene in normal and malignant tissues. Cancer Res. 1994;54:4493–4501. [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10:S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Rousso DL, Gaber ZB, Wellik D, Morrisey EE, Novitch BG. Coordinated actions of the forkhead protein Foxp1 and Hox proteins in the columnar organization of spinal motor neurons. Neuron. 2008;59:226–240. doi: 10.1016/j.neuron.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudrabhatla P. Regulation of neuronal cytoskeletal protein phosphorylation in neurodegenerative diseases. J Alzheimers Dis. 2014;41:671–684. doi: 10.3233/JAD-130794. [DOI] [PubMed] [Google Scholar]
- Rudrabhatla P, Jaffe H, Pant HC. Direct evidence of phosphorylated neuronal intermediate filament proteins in neurofibrillary tangles (NFTs): phosphoproteomics of Alzheimer's NFTs. FASEB J. 2011;25:3896–3905. doi: 10.1096/fj.11-181297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, Uruno K, Kumada S, Nishiyama K, Nishimura A, Okada I, Yoshimura Y, Hirai SI, Kumada T, Hayasaka K, Fukuda A, Ogata K, Matsumoto N. De novo mutations in the gene encoding STXBP1 (Munc18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40:782–788. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Kaneko K, Okuno M, Wada K, Kashiyama T, Shimizu H, Akagi T, Hashikawa T, Nukina N. Membrane microdomain switching: a regulatory mechanism of amyloid precursor protein processing. J Cell Biol. 2008;183:339–352. doi: 10.1083/jcb.200804075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Schulze KL, Littleton JT, Salzberg A, Halachmi N, Stern M, Lev Z, Bellen HJ. rop, a Drosophila homolog of yeast Sec1 and vertebrate n-Sec1/Munc-18 proteins, is a negative regulator of neurotransmitter release in vivo. Neuron. 1994;13:1099–1108. doi: 10.1016/0896-6273(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Shetty KT, Kaech S, Link WT, Jaffe H, Flores CM, Wray S, Pant HC, Beushausen S. Molecular characterization of a neuronal-specific protein that stimulates the activity of Cdk5. J Neurochem. 1995;64:1988–1995. doi: 10.1046/j.1471-4159.1995.64051988.x. [DOI] [PubMed] [Google Scholar]
- Spitzer NC. Electrical activity in early neuronal development. Nature. 2006;444:707–712. doi: 10.1038/nature05300. [DOI] [PubMed] [Google Scholar]
- Taylor AR, Gifondorwa DJ, Robinson MB, Strupe JL, Prevette D, Johnson JE, Hempstead B, Oppenheim RW, Milligan CE. Motoneuron programmed cell death in response to proBDNF. Dev Neurobiol. 2012;72:699–712. doi: 10.1002/dneu.20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellam JT, McIntosh S, James DE. Molecular identification of two novel Munc-18 isoforms expressed in non-neuronal tissues. J Biol Chem. 1995;270:5857–5863. doi: 10.1074/jbc.270.11.5857. [DOI] [PubMed] [Google Scholar]
- Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Toonen RF, Verhage M. Munc18-1 in secretion: lonely Munc joins SNARE team and takes control. Trends Neurosci. 2007;30:564–572. doi: 10.1016/j.tins.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Toonen RF, de Vries KJ, Zalm R, Südhof TC, Verhage M. Munc18-1 stabilizes syntaxin 1, but is not essential for syntaxin 1 targeting and SNARE complex formation. J Neurochem. 2005;93:1393–1400. doi: 10.1111/j.1471-4159.2005.03128.x. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Ensini M, Morton SB, Baldassare M, Edlund T, Jessell TM, Pfaff SL. Topographic organization of embryonic motor neurons defined by expression of LIM homeobox genes. Cell. 1994;79:957–970. doi: 10.1016/0092-8674(94)90027-2. [DOI] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115–e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallstedt A, Muhr J, Pattyn A, Pierani A, Mendelsohn M, Sander M, Jessell TM, Ericson J. Different levels of repressor activity assign redundant and specific roles to Nkx6 genes in motor neuron and interneuron specification. Neuron. 2001;31:743–755. doi: 10.1016/S0896-6273(01)00412-3. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci U S A. 2002;99:9037–9042. doi: 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Sons MS, Plomp JJ, Brose N. Aberrant morphology and residual transmitter release at the Munc13-deficient mouse neuromuscular synapse. Mol Cell Biol. 2005;25:5973–5984. doi: 10.1128/MCB.25.14.5973-5984.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Visel A, Thaller C, Eichele G. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 2004;32:D552–D556. doi: 10.1093/nar/gkh029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Südhof TC, Neher E, Verhage M. Munc18-1 promotes large dense-core vesicle docking. Neuron. 2001;31:581–591. doi: 10.1016/S0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- Wang S, Polo-Parada L, Landmesser LT. Characterization of rhythmic Ca2+ transients in early embryonic chick motoneurons: Ca2+ sources and effects of altered activation of transmitter receptors. J Neurosci. 2009;29:15232–15244. doi: 10.1523/JNEUROSCI.3809-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XH, Poo MM. Potentiation of developing synapses by postsynaptic release of neurotrophin-4. Neuron. 1997;19:825–835. doi: 10.1016/S0896-6273(00)80964-2. [DOI] [PubMed] [Google Scholar]
- Warp E, Agarwal G, Wyart C, Friedmann D, Oldfield CS, Conner A, Del Bene F, Arrenberg AB, Baier H, Isacoff EY. Emergence of patterned activity in the developing zebrafish spinal cord. Curr Biol. 2012;22:93–102. doi: 10.1016/j.cub.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Henderson CE. Patterns of programmed cell death in populations of developing spinal motoneurons in chicken, mouse, and rat. Dev Biol. 1999;214:60–71. doi: 10.1006/dbio.1999.9413. [DOI] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997;378:135–157. [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]